
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetoelectric core–shell CoFe2O4@BaTiO3 nanorods: their role in drug delivery and effect on multidrug resistance pump activity in vitro†
Sadaf Mushtaq ab,
Khuram Shahzadc,
Muhammad Rizwanc,
Anwar Ul-Hamidd,
Bilal Haider Abbasia,
Waqas Khalidc,
Muhammad Atifc,
Nafees Ahmadb,
Zulqurnain Alic and
Rashda Abbasi*b
ab,
Khuram Shahzadc,
Muhammad Rizwanc,
Anwar Ul-Hamidd,
Bilal Haider Abbasia,
Waqas Khalidc,
Muhammad Atifc,
Nafees Ahmadb,
Zulqurnain Alic and
Rashda Abbasi*b
aDepartment of Biotechnology, Quaid-i-Azam University Islamabad, Pakistan. E-mail: r.abbasi@daad-alumni.de; Fax: +92 51 9106283; Tel: +92 51 9106281
bInstitute of Biomedical and Genetic Engineering, G-9/1, Islamabad, Pakistan
cFunctional Materials Lab, Department of Physics, Air University Sector E-9, Islamabad, Pakistan
dCore Research Facilities, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
First published on 1st September 2022
Abstract
Nanoparticle mediated targeted drug delivery has become a widespread area of cancer research to address premature drug delivery problems. We report the synthesis of magneto-electric (ME) core–shell cobalt ferrite-barium titanate nanorods (CFO@BTO NRs) to achieve “on demand” drug release in vitro. Physical characterizations confirmed the formation of pure CFO@BTO NRs with appropriate magnetic and ferroelectric response, favorable for an externally controlled drug delivery system. Functionalization of NRs with doxorubicin (DOX) and methotrexate (MTX) achieved up to 98% drug release in 20 minutes, under a 4 mT magnetic field (MF). We observed strong MF and dose dependent cytotoxic response in HepG2 and HT144 cells and 3D spheroid models (p < 0.05). Cytotoxicity was characterized by enhanced oxidative stress, causing p53 mediated cell cycle arrest, DNA damage and cellular apoptosis via downregulation of Bcl-2 expression. In addition, MF and dose dependent inhibition of Multidrug Resistance (MDR) pump activity was also observed (p < 0.05) indicating effectivity in chemo-resistant cancers. Hence, CFO@BTO NRs represent an efficient carrier system for controlled drug delivery in cancer nanotherapeutics, where higher drug uptake is a prerequisite for effective treatment.
1. Introduction
With a tremendous impact on human mortality rate and expected increase in its burden, cancer remains an active area of research focusing on novel therapeutic interventions and nano drug delivery systems.1 The major limitation faced by conventional chemotherapy is a lack of selective cancer cell targeting, which equally harms healthy cells and causes undesired side effects such as fatigue, hair-loss, nausea, vomiting, infertility, organ damage and lower immunity.2 Furthermore, development of multidrug resistance (MDR) in cancer cells such as increased drug efflux (over expression of P-glycoprotein), variable drug metabolism and activation of DNA repair pathways also hamper chemotherapeutic efficacy.3Passive targeting of tumors has been implicated in several nanoparticle (NP) based drug delivery systems, relying on the Enhanced Permeability and Retention (EPR) effect. Functionalized NPs accumulate at tumor sites due to their leaky vasculature and poor lymphatic system. However, the percentage of accumulated NPs in tumors is <1% even when EPR is high.2,4 To overcome this problem, automation of NPs to target malignant cells via external or internal stimuli or target ligand provides a better approach. It has significant potential to improve the treatment efficacy and addressing problems of chemo-resistance, lower bioavailability, biodistribution and stability of drugs.1,5–7 Many researchers have acknowledged enhanced effect of chemotherapeutic drugs, delivered via NPs with increased bioavailability and decreased side effects.8–10
Most common nano drug carriers are designed to be temperature or intracellular pH responsive. But, problems occur in terms of pre-mature drug release where, a small change in physiological conditions (pH and temperature) of the body may result in off-target drug release.11–13 To eliminate this limiting factor and deliver drug only at target site while sparing normal cells, without depending on physiological conditions, magnetoelectric nanoparticles (MENPs) were introduced.14
MENPs are class of multiferroic materials where magnetic and electric fields are coupled with each other.15 The magnetic moments of magnetic material have direct relation with electric dipoles of ferroelectric material which induce intrinsic electric potential.16 This intrinsic electric field can be used to distinguish between normal and cancer cells and release bound drug at target sites using external magnetic field assistance.14,17 The magnitude of induced electric potential mainly depends on magneto electric coefficient i.e., interphase interaction between magnetic and piezoelectric phases18 for which core–shell structures are favorable.19 Cobalt ferrite–barium titanate core–shell nanostructures (CFO@BTO) are promising magnetoelectric nanocomposites with large magnetoelectric coefficient.20 Moreover, uniform layer of BTO shell on CFO core reduces cytotoxicity and makes them well suited for biological applications.10,21
Besides composition, shape of NPs also has huge impact on cellular uptake efficiency and targeted drug delivery. Several studies suggest that rod shaped NPs with high aspect ratio exhibit enhanced cellular uptake and drug delivery compared to spherical counterparts.22–24 Zhang et al. reported 1.5× increased internalization of gold nanorods than nanospheres in vitro.25 Similarly, Meng et al. also reported higher cellular internalization of silica nanorods with aspect ratio (2.1–2.5) than silica nanospheres in human cervical and human lungs cancer cell lines.26 Another study by Agarwal et al. based on polymer based (polyethylene glycol diacrylate) hydrogel nanorods showed higher cellular penetration than spherical nanospheres.27 Rod-shaped NPs have higher cellular retention compared to spherical NPs, due to their ability to escape endosomes before formation of lysosomes,28 enabling longer stay in cytoplasm. Besides, nanorods with diameter (<40 nm) can cross nuclear membrane and deliver their cargo inside the nucleus more efficiently than spherical NPs.29,30
In our previous research, we have studied drug delivering efficiency of spherical CFO@BTO NPs in vitro using MF assistance.10 Here, we aim at determining role of rod shape in anticancer drug delivery, cytotoxicity, and biocompatibility in MF assisted, drug functionalized cobalt ferrite–barium titanate nanorods (CFO@BTO NRs) in vitro. To the best of our knowledge, CFO@BTO NRs have not been reported yet for drug delivery applications in vitro.
2. Experimental
2.1. Materials
Titanium isopropoxide [C12H28O4Ti], ethanol, cetrimonium bromide ([(C16H33)N(CH3)3]Br (CTAB), sodium hydroxide (NaOH), trichloroacetic acid (TCA), fetal bovine serum (FBS), ethanol, orange G, dibutylphthalate polystyrene xylene (DPX) and trypan blue were purchased from Merck Germany. Dulbecco's Modified Eagle's Medium (DMEM), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), Triton X-100, sulforhodamine B (SRB), L-glutamine, sodium pyruvate, penicillin, streptomycin, ethidium bromide, 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA), trizma base, sodium dodecyl sulfate (SDS), neutral red, dimethyl sulfoxide (DMSO), acridine orange (AO), propidium iodide (PO), acetic acid, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), doxorubicin (DOX), methotrexate (MTX), cobalt nitrate [Co(NO3)2·6H2O], barium nitrate (BaNO3), iron nitrate [Fe(NO3)3·9H2O] and nitric acid (HNO3) were obtained from Sigma Aldrich (USA). Agarose (normal and low melting) and DNA ladder (100 bp) were purchased from Thermo Fischer Scientific. Ki-67 (clone MIB-1), p53 (clone DO-7), Bcl-2 (clone 124) mouse monoclonal antibodies, peroxidase blocker (DM821), diaminobenzidine (DAB), hematoxylin (K8018), chromogen (DM827) and secondary antibody (DM822) conjugated with horseradish peroxidase (HRP) were purchased from Agilent Technologies, Inc. (USA).2.2. Synthesis of CFO@BTO core shell nanorods
Core–shell CFO@BTO NRs were synthesized using solvothermal protocol.31 In brief, 0.2 M solutions of cobalt nitrate and iron nitrate with molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were dissolved in 100 ml deionized water. Afterwards, 0.1 M solution of CTAB (prepared in 20 ml ethylene glycol) was added with sonication for one hour at pH 11. The solution was then transferred to a three-neck round bottom flask for solvothermal process. The reaction was carried out at 180 °C for 10 hours with constant stirring. After complete reaction, formed precipitates were washed 3–4 times with deionized water and etched with 0.2 M HNO3 solution for 2 hours on sonication bath. In the next step, 0.2 M solutions of barium nitrate (in 50 ml water) and titanium isopropoxide (in 50 ml absolute ethanol) were mixed with constant stirring and transferred to three-neck round bottom flask for solvothermal process followed by addition of 2 M NaOH and etched CFO nanorods. The reaction was carried out at 150 °C for 3 hours with constant stirring. Precipitates formed after complete reaction were washed 3–4 times with deionized water to remove impurities. Sample was further dried at 100 °C in oven and annealed at 800 °C in furnace.
:2 were dissolved in 100 ml deionized water. Afterwards, 0.1 M solution of CTAB (prepared in 20 ml ethylene glycol) was added with sonication for one hour at pH 11. The solution was then transferred to a three-neck round bottom flask for solvothermal process. The reaction was carried out at 180 °C for 10 hours with constant stirring. After complete reaction, formed precipitates were washed 3–4 times with deionized water and etched with 0.2 M HNO3 solution for 2 hours on sonication bath. In the next step, 0.2 M solutions of barium nitrate (in 50 ml water) and titanium isopropoxide (in 50 ml absolute ethanol) were mixed with constant stirring and transferred to three-neck round bottom flask for solvothermal process followed by addition of 2 M NaOH and etched CFO nanorods. The reaction was carried out at 150 °C for 3 hours with constant stirring. Precipitates formed after complete reaction were washed 3–4 times with deionized water to remove impurities. Sample was further dried at 100 °C in oven and annealed at 800 °C in furnace.
2.3. Physical characterizations
Surface morphology, elemental composition, particle size distribution, and crystalline size of CFO@BTO NRs were assessed by scanning electron microscopy (SEM: TESCAN-VEGA3), fitted with energy dispersive spectroscopy (EDS), high resolution transmission electron microscopy (HR-TEM: JEM 2100F) and X-ray diffraction (XRD: D8-Advance Bruker AXS) diffractometer with CuKα radiation (k = 1.54178 Å) operating at 25 °C, respectively. Physical Property Measurement System (PPMS Quantum Design, USA) was used for determining magnetic behavior of MENPs. Ferroelectric properties were measured using Precision Premier II (Radiant Technologies, Inc.). Drug loading and drug release analysis was carried out by UV-Vis spectroscopy (Thermo Scientific™ Evolution) and colloidal stability was assessed by Zeta sizer Nano ZS 90 with a 633 nm wavelength He–Ne laser at 25 °C.2.4. Preparation of colloidal NRs suspension and polymer coating
To achieve hydrophilicity, phase change of CFO@BTO NRs was performed in two steps: formation of colloidal NRs suspension and surface functionalization with amphiphilic polymer.10 In the first step, freshly synthesized CFO@BTO NRs (0.1 M) were etched with 0.2 M HNO3 (15 ml) for 3 hours on sonication bath. The solution was covered with aluminum foil and heated at 80 °C for one hour. Sample was washed 3–4 times with methanol and re-dispersed in chloroform, forming stable colloidal suspension.Colloidal NRs were further functionalized with 0.8 M synthesized amphiphilic polymer; polyisobutylene-alt-maleic anhydride (PMA)32 as calculated by following equation:
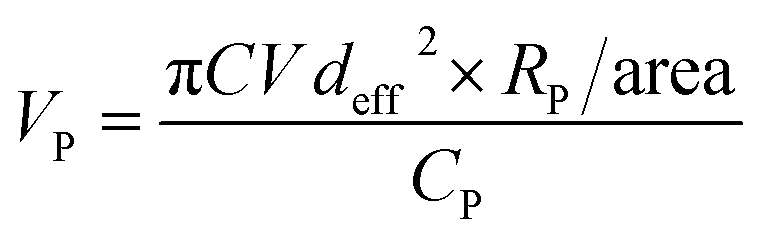 | (1) |
| A = πCVNAdeff2 |
| A = (3.1416) × (11 μM) × (500 μl) × (6.01 × 1023 mol−1) × (15 nm)2 |
| A = (2.31) × (1018) nm2 |
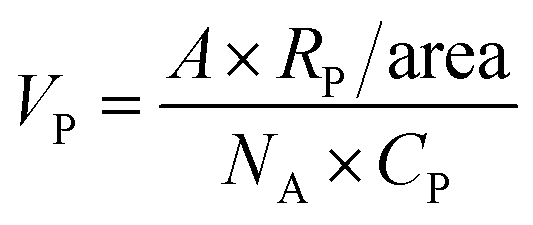
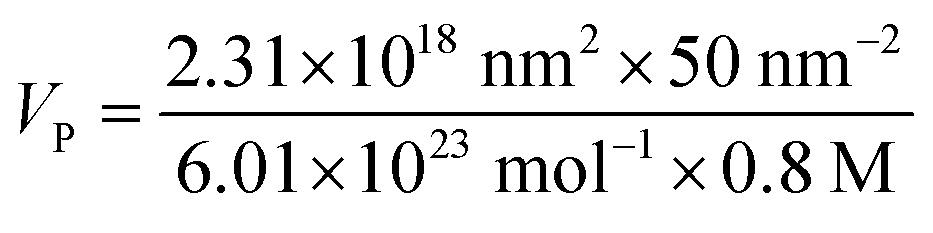
| VP = 240.4 μl |
Functionalization of CFO@BTO NRs (500 μl) was performed by adding 240 μl PMA with stirring at 60 °C using a rotary evaporator system (200 rpm) for 40 minutes. Sample was vacuum dried, washed with chloroform twice and re-dispersed in sodium borate buffer (SBB) at pH 9. PMA coated CFO@BTO NRs (NRs-PMA) were initially filtered using 0.2 μm syringe filters and later concentrated by using centrifugal filters (Amicon Ultra-4).
2.5. Drug loading and encapsulation efficiency
PMA coated CFO@BTO NRs were post functionalized with 0.5 mM anticancer drugs: doxorubicin (DOX) and methotrexate (MTX) using EDC chemistry.10 1 μM samples (200 μl) were incubated with optimized (0.096 M) EDC for 1 hour followed by addition of 0.5 mM (100 μl) DOX and MTX for 2 hours at room temperature. The amphiphilic polymer was synthesized by conjugating hydrophobic alkyl amines (dodecylamine) onto hydrophilic maleic anhydride rings via spontaneous amide linkage, which converts one maleic anhydride into one corresponding amide and one free carboxylic acid32 as shown in Fig. 1(a). The Hydrophobic domains (dodecylamine) on the polymer coat intercalate with the hydrophobic tails of the surfactant molecules (oleic acid) present on the surface of NPs and the hydrophilic backbone stabilizes the NPs by electrostatic repulsion.32 By using EDC with (PMA) functionalized NRs, carboxylic groups on surface of NRs become more reactive and form an amide linkage with primary amines present in anti-cancer drugs (DOX/MTX). The EDC reacts with carboxylic acid to form an amine-reactive O-acyl isourea intermediate33 as shown in Fig. 1(b). In the absence of amine group, these intermediates will regenerate carboxyl group, but in the presence of amine groups, an amide bond linkage34 is formed (Fig. 1(b)).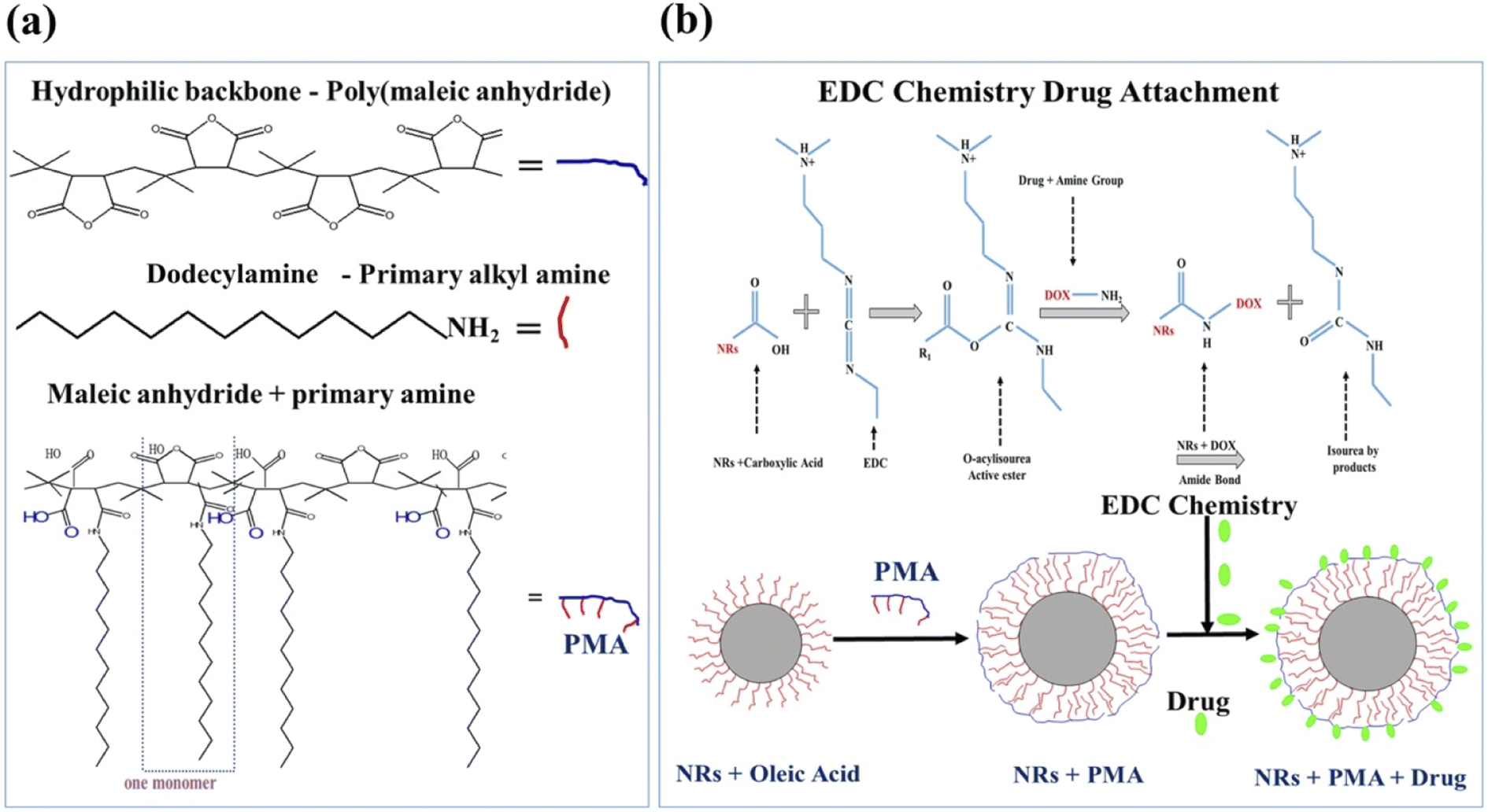 | ||
| Fig. 1 (a) Schematic diagram showing synthesis of amphiphilic polymer (PMA). (b) Coating of NRs with PMA and drug attachment via EDC chemistry. | ||
Drug loading was confirmed b UV-Vis spectroscopy and unbound drug was removed by centrifugal filters. DOX and MTX titration curves were used to determine concentration of unbound drugs in waste and drug loading and encapsulation efficiency (eqn (2) and (3)).
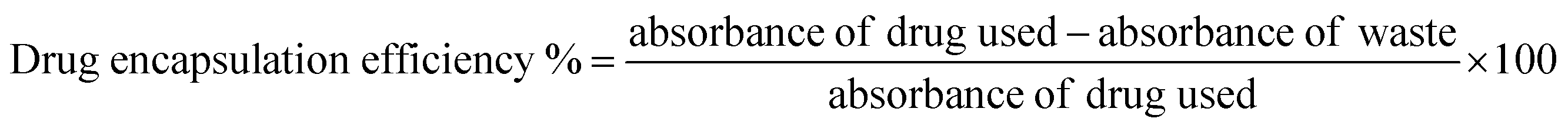 | (2) |
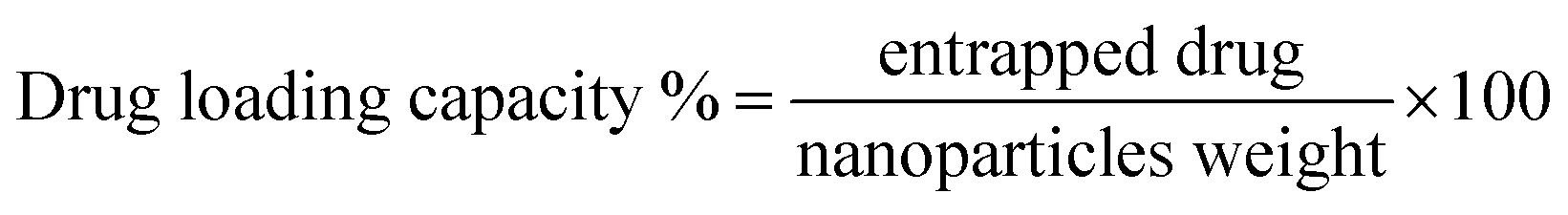 | (3) |
2.6. Time and MF dependent drug release kinetics of CFO@BTO NRs
Time and a.c. MF dependent drug release behavior of CFO@BTO NRs was determined as previously described10 by designing a Helmholtz coil having 600 turns (N), diameter of 8 cm and frequency 50 Hz. Drug loaded (DOX and MTX) CFO@BTO NRs were dispersed in phosphate buffer saline (PBS, pH 7.2) and exposed to varying a.c. MF (1–7 mT) for 0–60 minutes at room temperature. After every timepoint, samples were centrifuged at 5000 rpm for 5 minutes and supernatants were analyzed spectrophotometrically to determine amount of released drug with the help of DOX and MTX titration curves. % Drug release was calculated by the formula given in eqn (4):
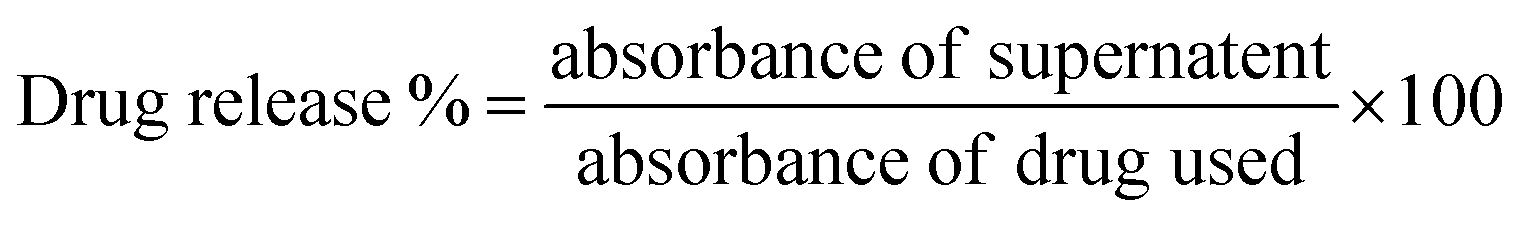 | (4) |
2.7. Cell culture
In vitro biological experiments were carried out using Human Hepatocellular Carcinoma (HepG2, ATCC®HB-8065™) and Human Malignant Melanoma (HT144, ATCC® HTB-63™) cell lines, grown in supplemented DMEM containing 10% FBS and 1% GPPS (L-glutamine 2 mM, sodium pyruvate 1 mM, penicillin 100 U ml−1 and streptomycin 100 μg ml−1). Cultures were maintained in humidified incubator at 37 °C with 10% CO2 and harvested by 0.5 mM trypsin/EDTA at room temperature for 1 minute.2.8. Cytotoxicity screening: sulforhodamine B (SRB) assay
NRs + DOX, NRs + MTX and NRs-PMA were evaluated for cytotoxicity in vitro using SRB assay.10 Pre-seeded HepG2 and HT144 cells (>90% viability, 1.5 × 105 cells per ml: Falcon® 96-well, flat bottom, clear microplate) were exposed to 1 μg ml−1 of drug functionalized NRs for 24 hours at standard culture conditions. Untreated cells (NTC) and free drug controls (DOX and MTX = 0.01 μM) equivalent to total drug attached with NRs at 1 μg ml−1 dose were also included. An external MF (4 mT) was applied for 20 minutes using Helmholtz coil. After treatment, cells were fixed with TCA (50%) for 1 hour at 4 °C, washed with deionized water thrice and air dried. Cells were stained with 0.05% SRB dye for 30 minutes at room temperature, followed by washings (5×) with 1% acetic acid to remove excess. Air dried plates were then used to take photographs at 200× using Olympus CK2 light microscope with attached camera (Optika C-B10 digital camera). Photographs were analyzed using Optika Pro View software (Version: x86, 3.7.13977.20190224). Experiment was performed twice with triplicates of all samples. For better comparison, treatment groups without MF assistance, were also included in the study.2.9. Quantitative assessment of apoptosis and necrosis in treated cells: acridine orange and propidium iodide (AOPI) staining
Quantitative assessment of necrotic and apoptotic cells in HepG2 and HT144 cells treated with 1 μg ml−1 of drug loaded CFO@BTO NRs with and without MF assistance (4 mT, 20 minutes) was done using AOPI staining.10 Cells (>90% viability, 1 × 105 cells per ml) were exposed to NRs + DOX and NRs + MTX for 4 hours. NRs-PMA (1 μg ml−1), NTC, free DOX and MTX (5 μM each) were included as controls. After treatment, cells were washed with PBS, stained with AOPI solution (100:32 μg ml−1) and observed under fluorescent microscope (Nikon, MicroPhot-SA). Viable cells exhibit green, necrotic cells exhibit red and apoptotic cells show yellow to orange fluorescence. Quantitative analysis of viable, apoptotic, and necrotic cells was performed using Optika Pro View (Version: x86, 3.7.13977.20190224) software and compared to NTC.
2.10. IC50 assessment of drug functionalized NRs: neutral red assay
Neutral red assay was performed to calculate half maximal inhibitory concentrations (IC50) of drug functionalized NRs against cancer cell lines (HepG2 and HT144) and freshly isolated lymphocytes.35 For lymphocytes, fresh blood (5 ml) was collected from healthy individuals (with informed consent) in EDTA vacutainers and diluted (1:3) with RBCs lysis buffer (155 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA; pH 7.2). Samples were incubated for 5 minutes and centrifuged at 4000 rpm for 10 minutes to separate out RBCs. The process was repeated 4–5 times until clear pellet of lymphocytes was obtained. Lymphocytes were then resuspended in fortified DMEM, and viability was assessed by trypan blue method.
To determine IC50, HepG2, HT144 and lymphocytes cells (>90% viability; 1.5 × 105 cells per ml) were treated with different concentrations of drug functionalized (0.0005–100 μg ml−1) for 24 hours at standard culture conditions. Experiment included MF assisted (4 mT, 20 minutes) and non-assisted treatment groups. Controls included NTC and free drugs (DOX and MTX = 5 × 10−6 to 1 μM) at doses equivalent to drug attached with NRs at tested doses. NRs-PMA and non-cellular controls (media only and NRs only) were also included. For free drugs, we calculated IC20, IC50 and IC80 values (μM) by treating HepG2 & HT144 cells with (5 × 10−6 to 30 μM) and lymphocytes with (0.001–100 μM) concentrations of DOX and MTX for 24 hours (ESI Fig. S2(b)†). After treatment, media was removed and replaced with neutral red medium (fortified DMEM containing 40 μg ml−1 neutral red) and incubated for 3 hours. Following incubation, neutral red medium was removed, cells were washed with PBS to remove excess dye and neutral red de-stain (49% deionized water, 50% of 96% ethanol and 1% glacial acetic acid) solution was added. Plates were shaken on microplate shaker for 10 minutes and absorbance readings were taken at 540 nm using FLUOstar Omega microplate reader BMG LABTECH. Experiment was performed twice with triplicates for all samples. Following formula was used to calculate percent viabilities:
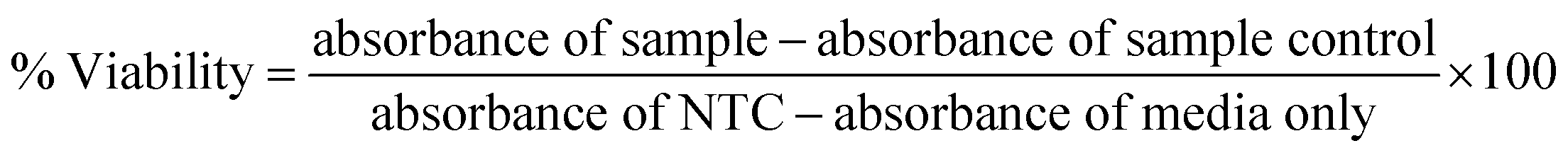 | (5) |
IC50 values were determined by using non-linear regression curve fitting (GraphPad Prism: 9.4.0).
2.11. Estimation of time dependent oxidative stress in treated cells
Oxidative stress in treated HepG2 and HT144 cells was calculated in time dependent manner (1–45 minutes) using H2-DCFDA (2′,7′-dichlorodihydrofluorescein diacetate) assay.36 In pre-seeded cells (>90% viability; 1.5 × 105 cells per ml), media was replaced by PBS containing 25 μM H2-DCFDA and 2% FBS with 45 minutes incubation. Cells were then treated with drug functionalized NRs at 1 μg ml−1 dose with and without MF assistance (4 mT, 20 minutes). Fluorescent intensities (emission and excitation = 355/590 nm; gain = 700) were recorded at different time points using microplate reader (FLUOstar Omega microplate reader BMG LABTECH). Controls included free DOX and MTX = 0.5 μM, NRs-PMA = 5 μg ml−1 and NTC. Non-cellular controls (DCF only and NRs only) was also included. Experiment was performed in triplicates.2.12. Effect of drug functionalized NRs on cell cycle
Pre-seeded HepG2 and HT144 cells (>90% viability; 1.5 × 105 cells per ml) were analyzed by flow cytometry37 after treatment with IC50 doses of drug functionalized NRs for 24 hours with and without MF assistance (4 mT, 20 minutes). Controls of the experiment included free DOX and MTX = 0.05 μM each and NTC. Treated cells were collected, washed (PBS) and fixed (70% ethanol, 20% deionized water and 10% PBS) at 4 °C. Prior to flow cytometry, fixed cells were washed, resuspended in PBS, and stained with DAPI for 5 minutes at room temperature in dark. At least 10000 cells per sample were analyzed using flow cytometer (CytoFLEX LX flow cytometer, Beckman Coulter Life Sciences) and CytExpert software (Version 2.4).
2.13. Assessment of genotoxicity in cells treated with drug functionalized NRs
Genotoxicity in cells was assessed by comet assay.37 Pre-seeded HepG2 and HT144 cells (>90% viability; 1.5 × 105 cells per ml) were treated with IC50 doses of drug loaded NRs in the presence and absence of MF (4 mT, 20 minutes) for 1 hour at optimum culture conditions. Untreated cells (NTC) and free drugs (DOX and MTX = 0.05 μM each) were included as experimental controls. After treatment, cells were harvested and counted. Cell suspension of 2.7 × 105 per ml was embedded in 0.7% low melting agarose and spotted on glass slides at 4 °C. Slides were dipped overnight in cell lysis buffer (100 mM Na2-EDTA, 10 mM Trizma base pH 10, 2.5 M NaCl, 1% Triton X-100 and 1% sodium sarcosinate) at 4 °C, followed by 20 minutes immersion in pre-chilled alkaline solution (0.3 M NaOH, 1 M Na2-EDTA; pH 13) to allow DNA unwinding. Slides were then electrophoresed at 25 V and 300 mA for 20 minutes and air dried. Cells were stained with propidium iodide (PI, 5 μg ml−1) for 5 minutes and visualized under fluorescent microscope (200×; Nikon, MicroPhot-SA). Analysis of 150–200 cells per sample was performed using ImageJ software and median olive tail moments (OTM) were calculated relative to NTC.2.14. Assessment of DNA fragmentation in cells treated with drug functionalized NRs: ladder assay
Apoptotic DNA fragmentation was assessed via DNA ladder assay.38 HepG2 and HT144 cells (>90% viability, 1 × 105 per ml) were treated with IC50 doses of drug functionalized NRs for 24 hours with and without MF assistance. Controls of the experiment included NTC, free DOX and MTX (30 μM each). After treatment, harvested cells were lysed with DMSO (100 μl), followed by addition of equal volume of TE buffer containing 2% SDS (pH 7.4). Samples were centrifuged at 12000 rpm for 10 minutes to separate out low molecular weight DNA fragments (180 bp or multiples) and quantified by Nanodrop 2000C. Equal volumes of DNA (with Orange G dye) were electrophoresed on 2% agarose gel containing 50 μg per ml ethidium bromide, at 50 V for 2 hours. DNA bands were visualized using UV transilluminator.
2.15. Evaluation of cancer biomarker expression (Ki-67, p53 and Bcl-2) in treated cells: immunocytochemistry (ICC)
Expression of Ki-67, p53 and Bcl-2 proteins was evaluated by ICC (Dako EnVision™ FLEX detection system) in treated cells.39 HepG2 and HT144 cells (>90% viability; 1.5 × 105 cells per ml) were cultured on sterile coverslips, treated with IC50 doses of drug functionalized NRs for 24 hours with or without MF assistance (4 mT, 20 minutes), and fixed with absolute methanol. Untreated cells (NTC) and free drugs (DOX and MTX = 0.05 μM each) were included as experimental controls. Antigen retrieval was performed at 95 °C for 45 minutes and endogenous peroxidases were blocked by addition of peroxidase blocker for 10 minutes. Cells were incubated with mouse monoclonal antibodies; Ki-67 (clone MIB-1; working dilution = 1:150), p53 (clone DO-7; working dilution = 1:50) and Bcl-2 (clone 124; working dilution = 1:100) overnight at 4 °C. Following day, HRP conjugated secondary antibody (rabbit, polyclonal) was added for 30 minutes and stained with DAB chromogen (dark brown stain) with several washings in between the process. Counter staining was performed with hematoxylin for 5–6 minutes. Cells were dehydrated, mounted, and visualized under light microscope (Nikon, MicroPhot-SA with digital camera Optika C-B10; 200×). Images were analyzed with Optika Pro View software (Version: x86, 3.7.13977.20190224) and percentage of antibody positive cells was calculated in each sample replicate using following formula:
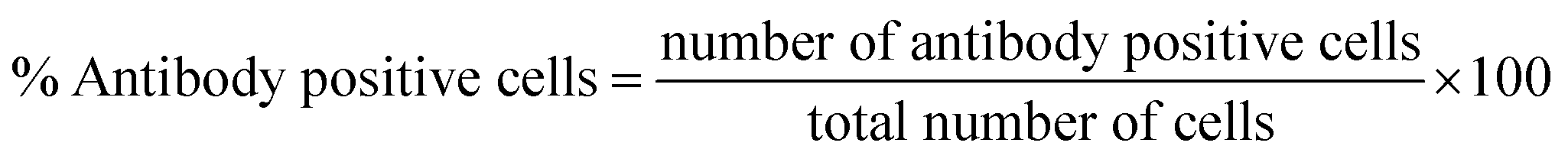 | (6) |
2.16. Determination of multidrug resistance (MDR) pump activity in cancer cells after treatment with drug functionalized NRs
Both HepG2 and HT144 cell lines have active ABC transporters40,41 responsible for MDR. MDR1 and MRP1 activity in cells was determined using a fluorometric MDR assay kit (ab 112142, Abcam, Cambridge, MA, USA) using manufacturer's protocol.42 Pre-seeded HepG2 and HT144 cells (15000/well) were exposed to NRs + DOX and NRs + MTX at 1 and 5 μg ml−1 concentrations with and without MF assistance (4 mT, 20 minutes) for 24 hours. NTC, free DOX and MTX (0.1 μM each) were included as controls whereas cyclosporine A (Cyc A: 10 μM) was used as positive control (inhibitor of MDR pump). Next, dye loading solution (100 μl per well) was added, and plates were incubated in dark for 3 hours at room temperature. Fluorescent intensity of cells was determined by plate reader (FLUOstar Omega microplate reader BMG LABTECH) at excitation and emission wavelengths of 485 and 530 nm, respectively. Fluorescent intensity inside the cells was determined relative to NTC after subtracting drug only background. Higher fluorescence inside cells indicated inhibition of MDR pump activity. Experiment was performed in triplicates.2.17. Cytotoxicity screening of drug functionalized NRs in 3D spheroids
3D spheroid cultures represent a more suitable approach towards screening of therapeutic agents mainly due to their close resemblance to in vivo tumors. In the present study, 3D spheroids of HepG2 and HT144 cell lines were grown for 72 hours using 1.5% agarose coated (50 μl per well) sterile 96 well plates (Falcon® 96-well, flat bottom, clear Microplate) with plating density of 5000 cells per well (>90% viable). Plated cells were accumulated in agarose meniscus by centrifugation for 10 minutes at 2500 rpm and incubated at 37 °C.2 Spheroids were treated with drug functionalized NRs for 14 days at 5 μg ml−1 concentration with and without MF assistance (4 mT, 20 minutes). Controls included NTC and free drugs (DOX and MTX = 0.05 μM each). Media was replenished after every 48 hours. At each time point, photographs were taken using Olympus CK2 light microscope at 100× magnification with attached camera (Optika C-B10 digital camera) and Optica Pro View software (Version: x86, 3.7.13977.20190224). ImageJ software was used to calculate average diameter of spheroids covering lowest and highest diameter ranges of treated and untreated spheroids.At 14th day, viability of spheroids was determined in triplicates using trypan blue method.43 Spheroids were washed with PBS and trypsinized for 5 minutes to allow formation of single cell suspension.
3. Results and discussion
3.1. Physical characterizations
The XRD data was analyzed using Rietveld refinement techniques by Fullprof Suit program. The data for CFO@BTO NRs composite was refined according to space groups (Fd3m, R3m) and Crystallographic Information File (CIF) for cobalt ferrite and barium titanate with chi-squared (χ2) value 2.89. The Rietveld refined XRD pattern of (CFO@BTO NRs) is marked as triangle indicating experimental data with red solid line marked as calculated intensities. The difference between two intensities was marked with blue line at the bottom of graph and Bragg's peak positions are marked as vertical lines according to their space groups. All the observed peaks are allowed the Bragg's 2θ positions. The background was refined using pseudo-voigt function by taking atomic fractional positions as fixed parameters during refinement, however some factors such as lattice constant, isothermal parameters, scale, and shape factors are considered as free parameters.X-ray diffraction analysis of CFO@BTO NRs has confirmed the formation of crystalline material as shown in Fig. 2(a). Peaks positions are well matched when compared with JCPDS no. (22-1086 & 05-0626) for CFO and BTO, respectively. The average crystalline size was calculated by Debye–Scherrer formula and found to be 38 nm. Moreover, tetragonal phase of BTO was confirmed by peak's split at 2θ = 45.9°. The inset image in Fig. 2(a) indicates the tetragonal phase of barium titanate. Barium titanate has different phases depending upon Curie temperature and distortion in Centro symmetry of titanium atom. Tetragonal phase and piezoelectric behavior of BTO are prime objectives for biological studies. Phase can be recognized through XRD pattern by identifying the peak's split at specific angles.44
 | ||
| Fig. 2 Physical characterizations of CFO@BTO NRs. (a) Rietveld refined XRD pattern of CFO@BTO NRs, inset image shows the tetragonal phase of BTO. (b) SEM images at different magnifications confirming formation of CFO@BTO NRs and EDS spectrum indicating major elemental composition. (c) Bright field transmission electron microscopy (TEM) image of CFO@BTO NRs with High Resolution TEM (HR-TEM) and corresponding selected area electron diffraction (SAED) patterns of CFO@BTO NRs. (d) MH loops at 300 K of CFO and CFO@BTO NRs under 2 T applied field. (e) ZFC-FC magnetization curve measured in the temperature range of 0–360 K under applied cooling field (HCF) of 0.5 T. (f) TB vs. H (T) of ZFC-FC magnetization curve measured in the temperature range of 10–300 K under applied cooling field (HCF) of 0.5 T to 7 T. | ||
Surface morphology and presence of major elemental composition of core–shell nanorods was analyzed using scanning electron microscopy and energy dispersive spectroscopy. SEM images have shown the formation of core–shell nano rods (Fig. 2(b)). High resolution SEM image (500 nm) indicates that ferromagnetic CFO core is uniformly surrounded by piezoelectric BTO shell. The formation of ferromagnetic CFO nanorod core depends upon CTAB concentration. It is a surfactant having bromine that produces stable elongated micelles in synthesis solution. The bromine of CTAB forms complex bonds with CFO that controls its growth in single axial direction, resulting in lengthened CFO crystals. Average length and width of core–shell nanorods was found to be (length = 78 nm, width = 30 nm, aspect ratio = 2.6) using image J software. The pH and CTAB concentration can be adjusted to control the length and thickness of nanorods. EDS spectrum has also confirmed the major elemental presence with negligible impurity peaks in the sample.
The high resolution TEM image in Fig. 2(c) explain the crystalline structure with edges of double grain of nanoparticles. Calculated inter planner distance for both phases (0.252 nm, and 0.398 nm) with marked lattice fringes with their respective planes (311, and 100) have confirmed the presence of CFO and BTO respectively. These planes are well matched with the inter planner distance of diffraction patterens standards obtained from their standard JCPDS database. Crystalline nature of composite was studied by selected area electron diffraction (SAED) patterns. The SAED images explain the position of crystalline system upon diffraction with concentric rings for both CFO and BTO with their respective hkl planes.
The magnetization vs. applied field (MH) loops of samples were obtained at room temperature using cycling fields ± 2 T (Fig. 2(d)). Ferromagnetic behavior was observed at room temperature with saturation magnetization Ms of (66 emu g−1) and (47.4 emu g−1) respectively for CFO and CFO@BTO NRs. CFO has higher value of Ms than CFO@BTO due to having large magnetic contents as compared to CFO@BTO NRs composite. Further, the coercivity Hc values for CFO@BTO NRs (1396 Oe) is greater than CFO (488 Oe) due to formation of core–shell morphology that enhanced the interphase exchange coupling in composite.45 Moreover for core–shell nanorods, the rise in coercivity in the samples can be attributed to the presence of BTO layer at surface which gives rise to strong uniaxial anisotropy originating from the interaction of core and surface atoms.46
The temperature dependent magnetization (0 K to 360 K) was studied in the presence of field cooling (FC) and zero field cooling (ZFC) at different static magnetic fields (0.5–7 T). From Fig. 2(e) and (f), the blocking temperature TB showed inverse relation with applied field. Both FC and ZFC magnetization shows qualitatively similar behavior, however at all HCF there is transition between ferromagnetic and superparamagnetic regimes. Below TB the curves show ferromagnetic behavior and above TB superparamagnetic behavior was observed. The competition between magneto crystalline anisotropy energy and thermal energy generates the splitting between FC–ZFC curves.47 However, at lower applied field of 0.5 T, blocking temperature (TB) became very high (345 K) due to core–shell morphology of samples.
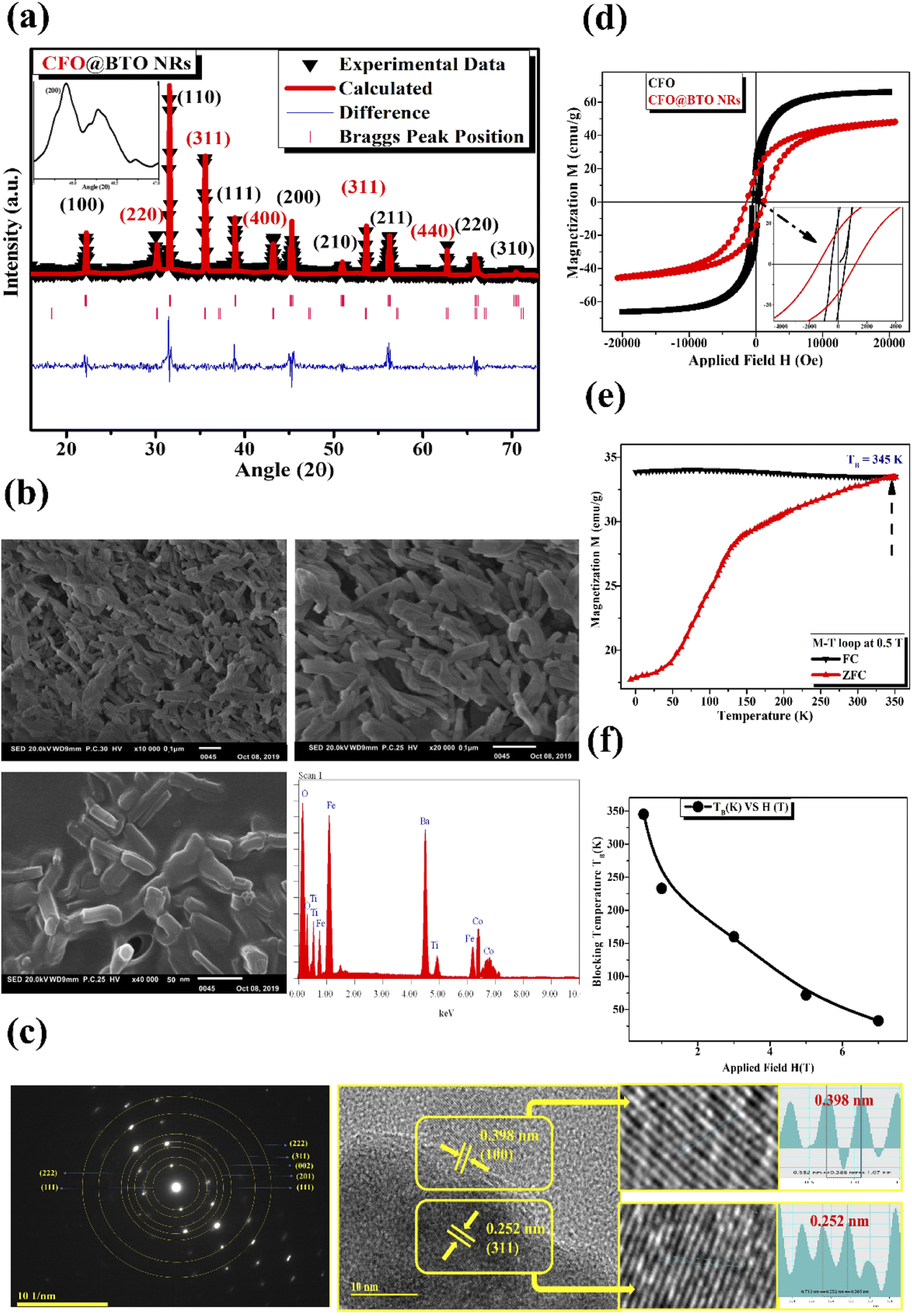
Piezoelectric behavior was studied using ferroelectric hysteresis loop (P–E) at 200 V electric field (Fig. 3(a)) that shows the formation of a ferroelectric loop. The coercivity and remnant polarization was found in order of 1.29 kV cm−1, and 3.26 μC cm−2. Core–shell nanorods have large value of remnant polarization and coercivity than core–shell spherical NPs as reposted in literature,10 due to large surface and interacting area between two phases. Moreover, large value of ferroelectric measurements has confirmed the formation of pure tetragonal (BTO) phase.
 | ||
| Fig. 3 (a) Polarization (μC cm−2) vs. electric field (kV cm−1) hysteresis loop at 200 V measured at room temperature. (b and c) Zeta potential and hydrodynamic size (nm) of CFO@BTO NRs in different suspension media (deionized water, PBS, SBB and DMEM). (d and e) UV-Vis absorbance plot of DOX and MTX with NRs-PMA, sample waste, NRs + DOX and NRs + MTX respectively. (f and g) Drug release kinetics of DOX and MTX functionalized CFO@BTO NRs using different magnetic field intensities (0–7 mT) at different exposure times (1, 5, 10, 20, 40 and 60 minutes) respectively. *p < 0.05, ■p < 0.01, ✦p < 0.005, ★p < 0.001 and ▲p < 0.0005 (paired two-tailed t-test when drug release in the presence of MF was compared to 0 mT at all time points). | ||
The colloidal stability (hydrodynamic size, surface charge, and polydispersity index PDI) of polymer coated and drug functionalized NRs was determined by dynamic light scattering (DLS). The samples were dispersed in different suspension media (deionized water, SBB pH 9.0, PBS pH 7.4 and DMEM). Potential value in SBB and PBS indicated indirect surface charge on NPs surface. Whereas, deionized water suspension was analyzed to determine electrolyte influence on NPs stability, and DMEM was included as representative of biological assays.48 The results as shown in Fig. 3(b) and (c) and Table 1 revealed better colloidal stability indicated by higher value of zeta potential (−37 mV to −23 mV) in all suspension media. The slightly lower value of zeta potential in DMEM was due to interaction of serum proteins with NPs.10 The hydrodynamic size measurements emphasized on successful drug attachment at NPs surface. Polymer coated NRs had lower hydrodynamic size value (76–117 nm) compared to drug attached samples (109–171 nm). The hydrodynamic size of all samples increased in DMEM due to serum proteins–NPs interactions (up to 171 ± 8 nm) which is considered suitable for biological applications.10,49 The uniform distribution of all samples in all suspension media was confirmed from lower values (0.13–0.33) of polydispersity index (PDI) as shown in Table 1.
| Sample | NRs-PMA | NRs + DOX | NRs + MTX | |
|---|---|---|---|---|
| Zeta potential (mV) ± SD | Water | −37 ± 1.1 | −33 ± 1.2 | −31 ± 1.5 |
| SBB | −32 ± 1.3 | −30 ± 1.4 | −30 ± 1.4 | |
| PBS | −29 ± 1.9 | −26 ± 1.1 | −28 ± 1.9 | |
| DMEM | −24 ± 2.1 | −23 ± 1.8 | −23 ± 1.9 | |
| Hydrodynamic size (nm) ± SD | Water | 76 ± 3 | 109 ± 3 | 109 ± 3 |
| SBB | 81 ± 4 | 135 ± 5 | 127 ± 5 | |
| PBS | 82 ± 4 | 130 ± 2 | 126 ± 3 | |
| DMEM | 118 ± 5 | 171 ± 8 | 169 ± 6 | |
| PDI | Water | 0.20 | 0.19 | 0.20 |
| SBB | 0.26 | 0.22 | 0.21 | |
| PBS | 0.21 | 0.29 | 0.28 | |
| DMEM | 0.41 | 0.47 | 0.40 |
3.2. Drug loading
Doxorubicin (DOX) and methotrexate (MTX) drugs were loaded on PMA functionalized CFO@BTO NRs and confirmed via UV-Vis spectroscopy, with distinct peaks at 480 and 370 nm respectively (Fig. 3(d) and (e)). Centrifugal filters were used to remove unbound drug from samples and quantified using drug titration curves as shown in Fig. 3(d) and (e) insets. NRs drug encapsulation was 87 and 91%, whereas drug loading concentration was found to be 61 and 67% for DOX and MTX, respectively.3.3. Drug release kinetics (time and MF dependent)
MF and time dependent (1–7 mT, 1–60 minutes) drug release experiment was performed to determine ideal MF and time point where maximum drug release is achieved. The obtained results clearly indicated direct relationship between % drug release and applied MF with time (Fig. 3(f) and (g)). Optimized MF and timepoint which achieved 98% drug (DOX and MTX) release was 4 mT and 20 minutes, respectively which was implicated in biological assays in vitro. The drug release mechanism of CFO@BTO NRs can be explained as controlled release by a passive mechanism of action using external MF. Upon exposure of drug functionalized NRs to external MF, electric dipoles are formed due to magnetoelectric effect50,51 which will generate dipole moment that breaks the original symmetry of the charge in the CFO@BTO shell. On further increase in MF up to 4 mT, the dipole charge density becomes comparable to the ionic charge density in the shell and weakens nanoparticles drug bond. The produced charge density will breakdown nanoparticles drug bond on one side and strengthen the bond in the reverse direction.52 Further, the bond symmetry in the reverse direction can be broken by applying a magnetic field in the reverse direction.14,17 This phenomenon ideally sweeps all bond orientation with a.c. MF and enhance drug release efficiency.103.4. Drug loaded NRs cause enhanced cytotoxicity with MF assistance
Cell membrane is an electrically polarizable medium due to presence of several ion channels and transporters which allow permeability to ions such as K+, Na+, Cl−, and Ca2+. Therefore, a membrane potential (Vm) arises due to unequal distribution of these ions in intracellular and extracellular environment.53 Literature suggests that Vm is associated with cell cycle progression, wound healing, cellular differentiation, development and regeneration.53,54Since ion channels are actively involved in cancer progression, Vm therefore has an active role in cancer pathology. According to Cone's theory, malignant transformation of cells is characterized by significant membrane depolarization.55 Cancer cells maintain depolarized Vm characterized by high intracellular Na+ concentration which favors mitosis and alters membrane porosity.56 Studies suggest that normal cells are hyperpolarized (Vm = −60 mV to −100 mV). For instance, Vm of normal human lymphocytes is −70 mV.56,57 Whereas cancer cells are depolarized with Vm varying from −5 mV to −50 mV. For example, Vm of HepG2 cells is −9.8 ± 0.5 compared to normal hepatocytes (−35 mV).58
Since Vm in cancer cells is consistently weaker than healthy counterparts, the electric field across membrane of a cancer cell will also be reduced.56 Magneto-electric nanoparticles alter membrane porosity by electroporation (in response to electric field). MENRs have negative surface charge like that of plasma membrane. However, their cellular translocation is made possible via electrostatic interactions and ionic charge imbalance.59–61 Under the influence of external MF, NPs generate sufficient electric field (magnetoelectric effect) in the vicinity of plasma membrane, generating pores for NPs entry and drug delivery.17 MF required for electroporation of cancer cells is much lower (around 3 mT) due to depolarized Vm, compared to normal cells (20 mT or greater) allowing target specificity.10,17,62,63
Apart from MF assisted targeting, shape of NPs has also a key role in cellular toxicity and internalization. Several studies suggest better cellular uptake of rod-shaped NPs compared to spherical counterparts.28–30
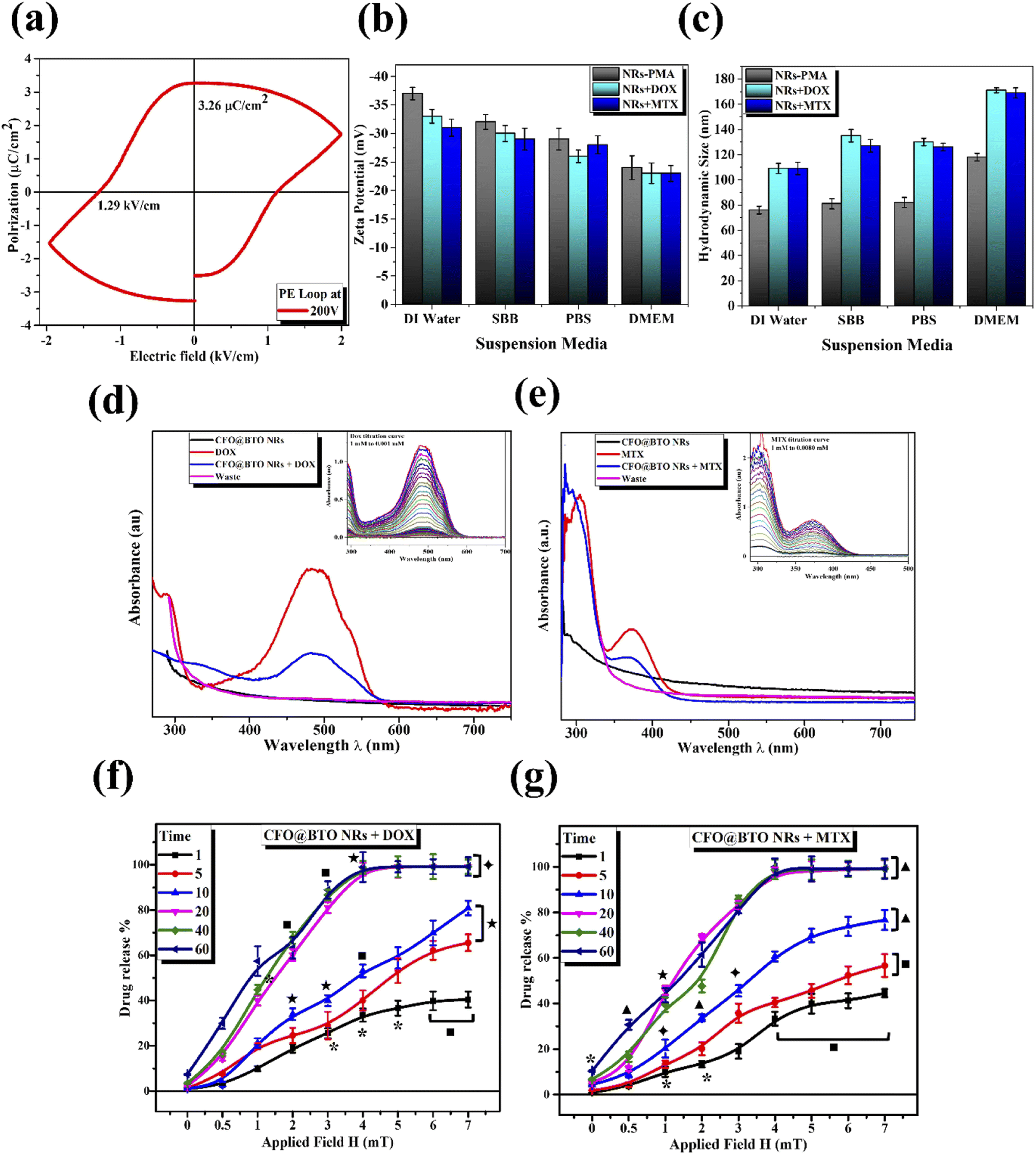
Cytotoxic ability of drug loaded NRs was tested in vitro using HepG2 and HT144 cell lines. Cells were treated with 1 μg ml−1 of NRs + DOX, NRs + MTX and NRs-PMA for 24 hours with and without MF assistance in the presence of free drug controls (DOX and MTX = 0.01 μM each) and NTC. Obtained results (Fig. 4(a)) indicated prominent difference in cytotoxicity when compared to NTC. Cellular viability in the absence of MF was around 60% which decreased prominently in the presence of MF, resulting in <50% viability in both cell lines with morphological alterations.64 Cytotoxicity was also higher than equivalent free DOX and MTX controls where 70–80% cells were viable. NRs-PMA showed minimal cytotoxicity (approx. 5–10%) in the presence and absence of MF indicating suitability for functionalization and safety in biological systems. We also observed no significant difference in NTC viability in the presence of MF, compared to NTC alone (ESI Fig. S1(a)†).
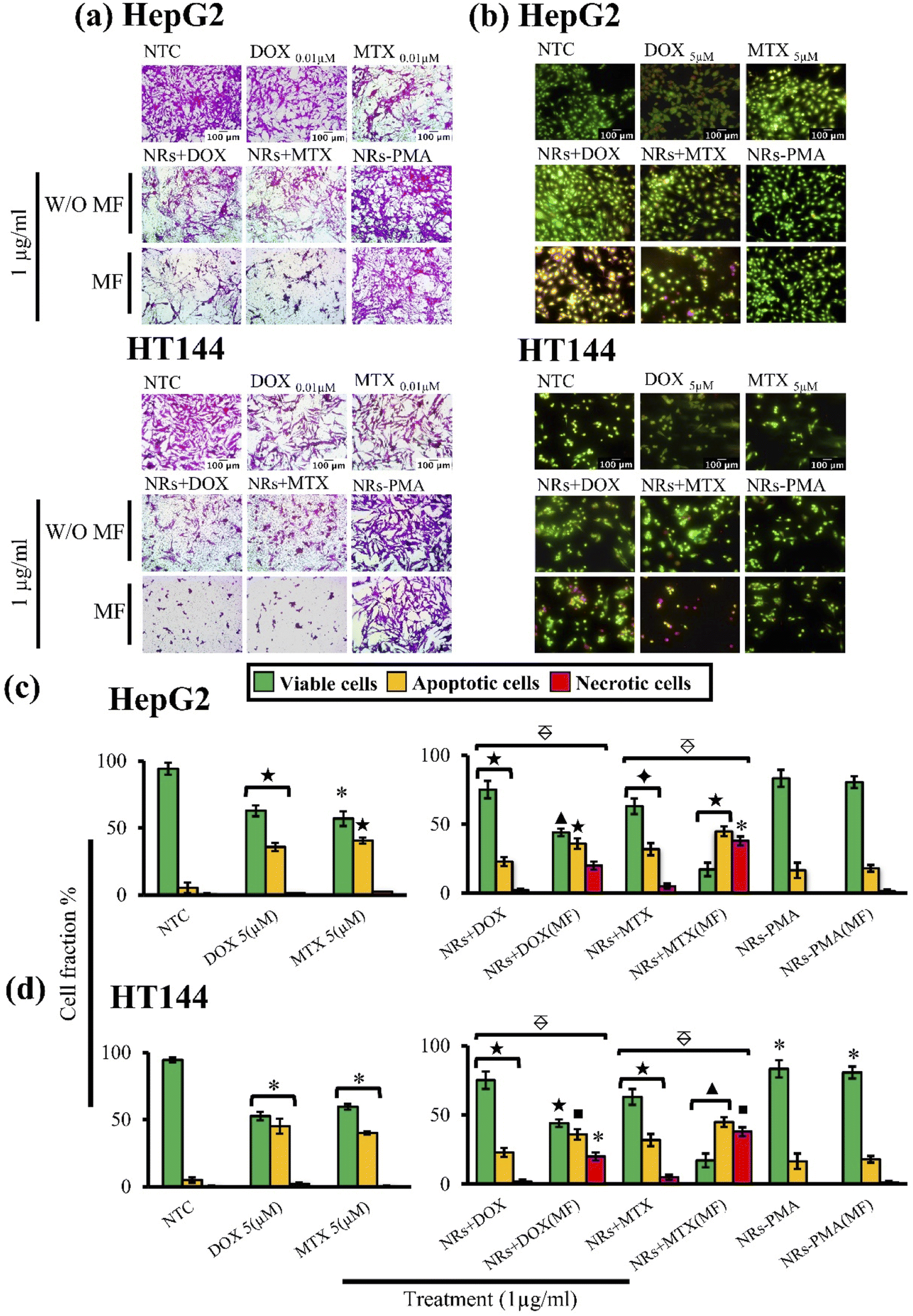 | ||
Fig. 4 (a) Cytotoxicity screening (SRB) and (b) fluorescent microscopic images (AOPI) of HepG2 and HT144 cells (magnification = 200×, scale bar = 100 μm) treated with drug functionalized (DOX and MTX) CFOBTO NRs (1 μg ml−1) for 24 hours in the presence and absence of external magnetic field (4 mT; 20 min). Controls of the study included untreated HepG2 and HT144 cells (NTC), free DOX and MTX (0.01 μM each in SRB and 5 μM each in AOPI) and NRs-PMA (1 μg ml−1). (c and d) Bar charts indicating percent fractions of live, necrotic, and apoptotic cells after treatment in HepG2 and HT144 cells, respectively. *p < 0.05, ■p < 0.01, ✦p < 0.005, ★p < 0.001 and ▲p < 0.0005 (paired two-tailed t test when compared to NTC).  p < 0.005 (paired two-tailed t test) when MF assisted, and un-assisted samples were compared. Experiment was repeated twice with triplicates for all samples. p < 0.005 (paired two-tailed t test) when MF assisted, and un-assisted samples were compared. Experiment was repeated twice with triplicates for all samples. | ||
3.5. Drug loaded NRs induce higher apoptosis with MF assistance
AOPI staining was performed to quantitatively determine fractions of live, apoptotic, and necrotic cells relative to NTC in treated HepG2 and HT144 cells (Fig. 4(c) and (d)). Upon staining with AOPI, live, apoptotic, and necrotic cells appeared green, orangish-yellow, and red respectively (Fig. 4(b)). Results indicated significant increase in apoptotic and necrotic cellular fractions when external MF was applied.In HepG2 cells, NRs + DOX produced an apoptotic fraction of 26.66 ± 1.9% at 1 μg ml−1 which increased up to 2× (63.7 ± 4.7%) when MF was applied. Similarly, NRs + MTX produced 19.6 ± 3.7% apoptotic and 3.0 ± 1.1% necrotic cells which increased up to 3× when MF was applied (60.1 ± 3.6% apoptotic and 10.1 ± 1.51% necrotic cells). Results were significant (p < 0.005) when both MF assisted, and unassisted samples were compared. NRs-PMA however, did not produce significant apoptosis (percent viable cells around 87% in MF assisted and unassisted samples). Free DOX and MTX controls (0.01 μM each) produced 35.8 ± 3.2% and 40.5 ± 2.3% apoptotic cells respectively (p < 0.001 compared to NTC). Whereas necrotic fraction was non-significant (1.4 ± 0.1% and 2.5 ± 0% respectively).
In HT144 cells, NRs + DOX produced 22.9 ± 3.3% apoptotic and 2.0 ± 1.2% necrotic cells. Application of MF caused 1.5× increase in apoptotic fraction (36.0 ± 3.8%) and 10× increase in necrotic fraction (20.0 ± 2.94%). Treatment with NRs + MTX produced an apoptotic and necrotic fraction of 31.9 ± 4.4% and 5.04 ± 1.9% respectively. With the assistance of MF, up to 1.4- and 7.5-folds increase was observed in apoptotic (44.8 ± 3.5%) and necrotic (37.9 ± 3.23%) fractions respectively. In both samples, MF assisted samples showed significantly higher (p < 0.005) effect compared to unassisted samples. MF assisted and unassisted NRs-PMA produced minimal cytotoxicity with percent viable cells around 83.3%. Free DOX and MTX controls (0.01 μM each) produced 45.1 ± 5.5% and 40 ± 1.2% (p < 0.05 compared to NTC) apoptotic fraction with non-significant necrosis (2.2 ± 0.9% and 0.5 ± 0.3%, respectively).
In both cell lines, we observed no significant difference in NTC and NTC (MF) (ESI Fig. S1(b)†), indicating that a low MF (4 mT, 20 min) had no effect on cellular viability of untreated cells.
3.6. Drug loaded NRs have lower IC50 with MF assistance in cancer cells and are biocompatible to normal lymphocytes
Half maximal inhibitory concentration (IC50) of functionalized NRs was determined by neutral red assay. Cells (HepG2, HT144 and lymphocytes) were exposed to several concentrations (0.0005–100 μg ml−1) of NRs + DOX, NRs + MTX and NRs-PMA for 24 hours with and without MF assistance. Controls of the experiment included free DOX and MTX in concentrations (0.000005–1 μM) equivalent to drug attached with tested doses of NRs and untreated cells (NTC). At all tested doses, percentage viabilities were plotted (Fig. 5(a)) and IC50 concentrations were calculated (Table 2). IC20, IC50 and IC80 concentrations of free drugs were also calculated (ESI Fig. S2(b) and Table S1†).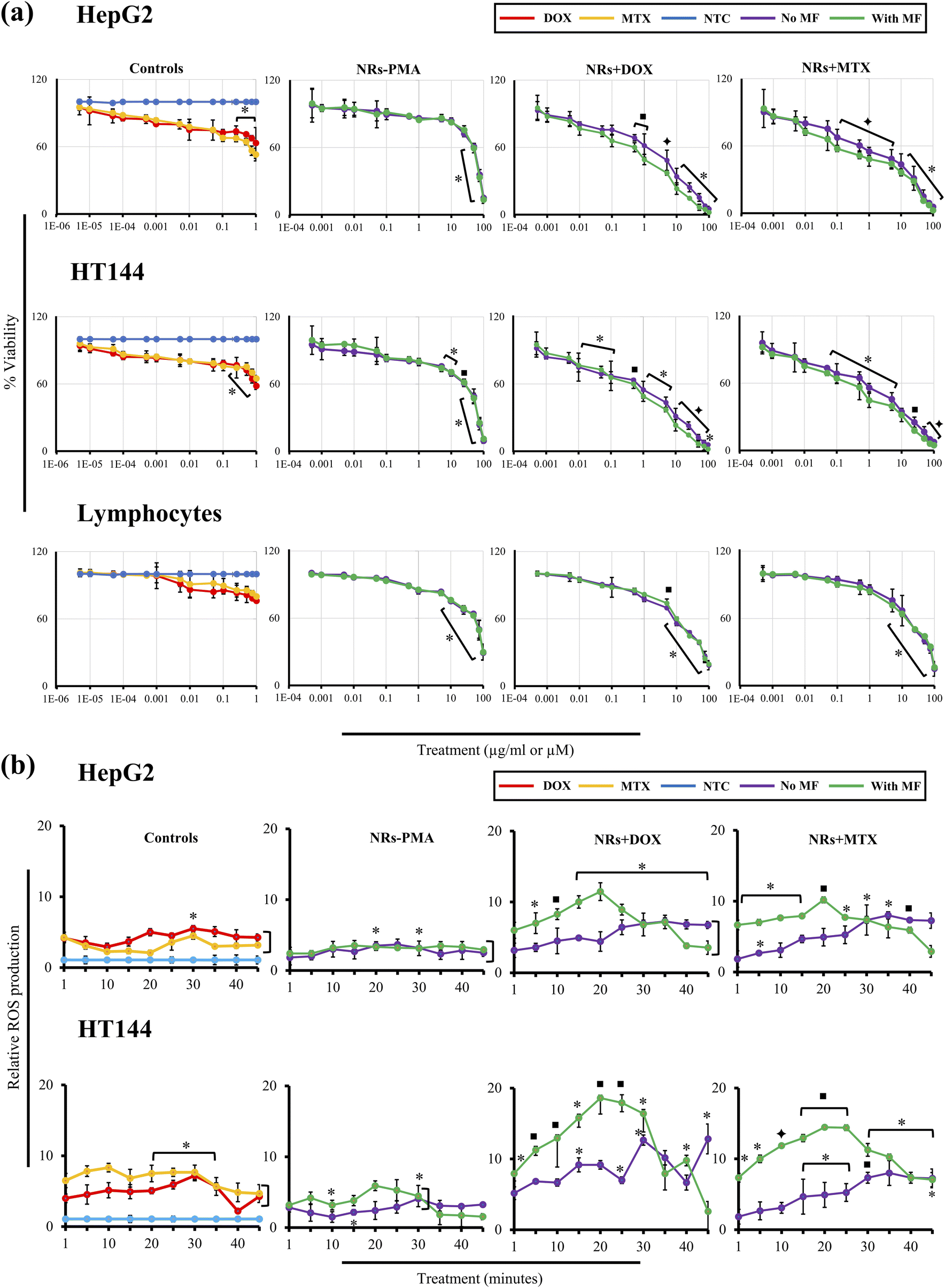 | ||
| Fig. 5 (a) Dose dependent cytotoxicity of drug functionalized CFO@BTO NRs in HepG2, HT144 and lymphocytes cells. (Left to right): Cells were treated with free DOX and MTX (0.000005–1 μM), NRs-PMA, NRs + DOX and NRs + MTX (0.0005–100 μg ml−1) for 24 hours at in the absence and presence of MF assistance (4 mT, 20 min). Untreated cells (NTC) were also included as controls. Plotted data represents mean ± SD of three independent triplicates. Doses on x-axis are plotted as log values. *p < 0.05, ■p < 0.01, and ✦p < 0.005 (paired two tailed t-test when compared to NTC). Non-linear regression curve analysis (GraphPad Prism 9.4.0) was used to determine IC50 concentrations (μg ml−1). (b) Measurement of time dependent (1–45 minutes) intracellular oxidative stress in HepG2 and HT144 cells. (Left to right): Cells were treated with free drug controls (DOX and MTX = 0.5 μM each), NRs-PMA = 5 μg ml−1, NRs + DOX and NRs + MTX at 1 μg ml−1 dose with and without MF assistance (4 mT, 20 min). ROS production at each time point was calculated relative to untreated cells (NTC). Data is represented as mean ± SD of experimental triplicates. *p < 0.05, ■p < 0.01 and ✦p < 0.005 (two tailed t-test when samples were compared to NTC). | ||
| Sample | IC50 (μg ml−1) | |||||
|---|---|---|---|---|---|---|
| HepG2 | HT144 | Lymphocytes | ||||
| w/o MF | MF | w/o MF | MF | w/o MF | MF | |
| NRs + DOX | 3.83 | 1.06 | 2.45 | 0.76 | 14.84 | 17.55 |
| NRs + MTX | 3.85 | 0.76 | 2.46 | 0.86 | 23.78 | 21.29 |
| NRs-PMA | 56.37 | 56.2 | 39.27 | 38.7 | 60.34 | 60.7 |
In treated HepG2 cells, MF assisted, and non-assisted samples started showing significant difference in cytotoxicity even at a low dose of 0.1 μg ml−1. NRs + DOX and NRs + MTX showed viability of 79.0 ± 2.9% and 71.26 ± 5.1% (p < 0.01) respectively in the absence of MF. However, external MF assistance caused viability values to significantly (p < 0.05) drop up to 1.2 folds (65.76 ± 5.2% and 57.6 ± 2.6% respectively). This difference in cytotoxicity increased up to 2.3 folds at higher doses indicating better internalization of drug functionalized NRs with MF assistance.
Similarly, in HT144 cells, treatment with 0.1 μg ml−1 of NRs + DOX and NRs + MTX reduced cellular viability to 73.95 ± 9.6% and 80.0 ± 2.95% (p < 0.05) respectively. However, MF assistance significantly enhanced the cytotoxic potential (p < 0.05) up to 1.2× (cellular viabilities = 60.4 ± 2.49% and 64.21 ± 4.03%, respectively) which increased up to 1.7× at higher doses.
In freshly isolated human lymphocytes, no significant difference was observed in MF assisted and un-assisted samples indicating selective targeting of cancer cells. NRs + DOX showed significant cytotoxicity at 5 μg ml−1 and higher doses (% viability range = 69.81 ± 0.67% to 18.60 ± 4.1%). They were biocompatible at lower doses (% viability range = 100 ± 1.06% to 81.84 ± 0.92%). Whereas NRs + MTX caused significant (p < 0.05) cytotoxic response at 10 μg ml−1 and higher doses (% viability range = 66.94 ± 7.53% to 15.05 ± 6.57%) with non-significant effect at lower doses (% viability range = 100 ± 5.70% to 76.03 ± 6.14%). Obtained IC50 values in lymphocytes (Table 2) were up to 28× and 9.7× higher compared to that of MF assisted and unassisted NRs in cancer cell lines respectively.
In HepG2, HT144 and lymphocytes cells, NRs-PMA showed minimal cytotoxicity at 0.1 μg ml−1 dose. Percent viabilities were 91.3 ± 3.2%, 85.36 ± 2.65% and 94.85 ± 1.2% respectively. Cytotoxicity was negligible at doses lower than 1 μg ml−1. However, higher doses (5–100 μg ml−1) produced significant (p < 0.05) cytotoxicity in HT144 cells with viability values ranging from 75.9 ± 1.76% to 9.43 ± 2.3%. Whereas viability in HepG2 cells ranged from 86.2 ± 3.2% to 13.53 ± 3.36%. NRs-PMA showed maximum biocompatibility in lymphocytes with significant decrease in viability (p < 0.05) at 10 μg ml−1 and higher doses (% viability range = 75.24 ± 0.49% to 29.2 ± 6.36%). Free drug controls (DOX and MTX) produced significant cytotoxicity in both cell lines at higher doses 0.25–1 μM (p < 0.05: % viability range = 78.5 ± 2.7% to 52.91 ± 5.63%) which indicates that cytotoxicity is enhanced when drugs are loaded on NRs. Non-significant results were obtained in lymphocytes, showing up to 80% viability at 1 μM dose of free drugs.
In all untreated cells (HepG2, HT144 & lymphocytes), we observed >99% cellular viability with or without MF influence (4 mT, 20 min) validating harmless role of MF in cellular viability (ESI Fig. S2(a)†).
3.7. Drug functionalized NRs produce time dependent oxidative stress in the presence of MF
Oxidative stress is one of the predominant mechanisms of nanotoxicity causing impairment of cellular redox regulation, lipid peroxidation, generation of protein radicals, DNA strand breaks, initiation of inflammatory responses and apoptosis. NPs mediated ROS production is dependent on several physical and chemical parameters such as shape, size, surface area, oxidation status, solubility, surface ligands, particle agglomeration and aggregation.65,66Generally, shape of NPs is not considered a primary determinant of oxidative stress. However, it has significant role in cellular uptake. For example, rod-shape NPs have higher cellular uptake compared to spherical, cylindrical, and cubical shapes. Moreover, higher the aspect ratio of NRs, higher the cellular uptake due to increased surface area causing enhanced ROS production.67,68
Drug functionalized NRs were tested for production of oxidative stress in HepG2 and HT144 cells in a time dependent manner (0–45 minutes) at 1 μg ml−1 dose with and without MF assistance. NRs-PMA (5 μg ml−1), free drugs (DOX and MTX = 0.5 μM each) and NTC were included as controls (Fig. 5(b)). NRs-PMA caused up to 3.5× to 6× (p < 0.05) increased oxidative stress in HepG2 and HT144, respectively at 5 μg ml−1. Sen et al. also reported similar findings where polymeric coatings reduced cytotoxicity but had no effect on cellular ROS production. However, increased expression of antioxidant enzymes (SOD, CAT and GPx1) was reported in cell lines upon insult with polymer coated NPs.69
Results indicated clear enhancement of ROS production with MF assistance. In both cell lines, NRs + DOX and NRs + MTX produced significant ROS (p < 0.05) from 1 minute onward and reached maximum at 20 minutes time interval under MF. ROS declined after 20 minutes indicating that MF released maximum drugs within 20 minutes. In non-assisted samples, no significant increase in relative ROS production was observed for first 15 minutes of exposure and slight to moderate increase was observed after 15 minutes and reached maximum level at 30 minutes. Maximum ROS produced under MF was up to 2 folds higher in HepG2 and 3 folds higher in HT144 compared to non-assisted samples which emphasizes upon efficient drug delivery and release under MF assistance. Also, HT144 appeared to be more sensitive towards drug functionalized NRs as ROS produced under MF was 1.6 folds higher than that of HepG2. Lower sensitivity of HepG2 can be explained by presence of intrinsic antioxidant and xenobiotic detoxification mechanisms.70,71
Furthermore, upon comparison with free drug controls (DOX and MTX), NRs + DOX and NRs + MTX produced higher ROS (1.6–2.3× in HepG2 and 1.9–2.6× in HT144) indicating better performance of anticancer drugs when delivered via MF assisted CFO@BTO NRs. Our results also indicated non-significant oxidative stress in NTC, exposed to MF (4 mT, 20 minutes) compared to NTC alone (ESI Fig. S3†).
3.8. Drug functionalized NRs induce cell cycle arrest at a much lower dose with MF assistance
Effect on cell cycle progression was determined by flow cytometry after treating HepG2 and HT144 cells with IC50 doses (Table 2) of NRs + DOX and NRs + MTX for 24 hours with and without MF assistance. Obtained results were compared to NTC(MF) and NTC respectively (Fig. 6(a)). No significant difference was observed between NTC and NTC(MF) in both cell lines (ESI Fig. S4(a)†).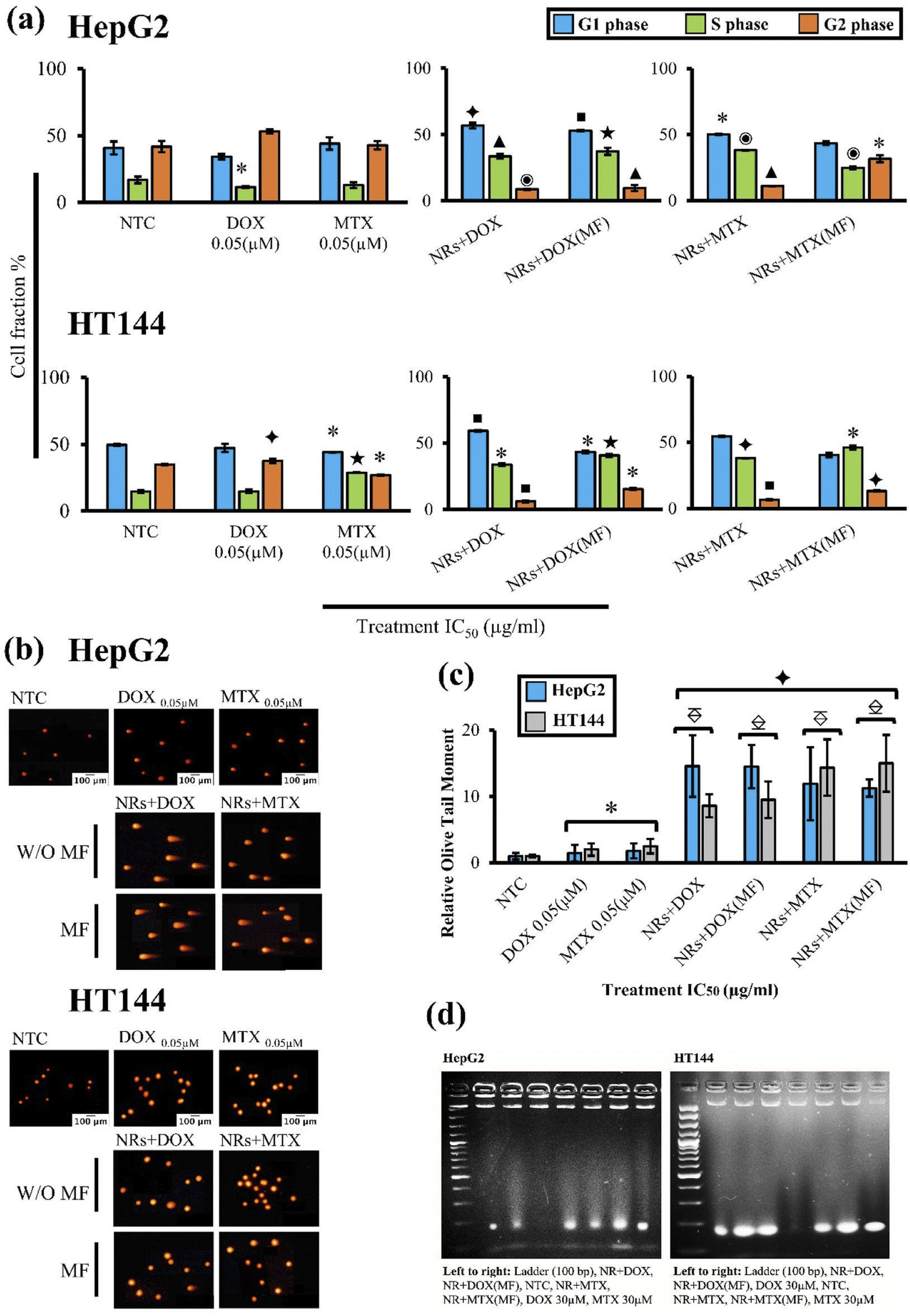 | ||
Fig. 6 (a) Flow cytometric analysis of cell cycle progression and (b) fluorescent microscopic images (magnification = 200×, scale bar = 100 μm) after treatment of HepG2 and HT144 cells with drug functionalized (DOX and MTX) CFO@BTO NRs. HepG2 and HT144 cells were treated with IC50 doses (IC50 values with MF = ∼1 μg ml−1, IC50 values W/O MF = 2.45–3.85 μg ml−1 for both cell lines) for 24 and 1 hour respectively with and without MF assistance (4 mT, 20 minutes). Controls of the experiment included untreated cells (NTC), free DOX and MTX (0.05 μM each). (c) Genotoxicity was determined by measuring olive tail moments relative to NTC. Data represents mean ± SD of three experimental replicates. *p < 0.05, ■p < 0.01, ✦p < 0.005, ★p < 0.001, ▲p < 0.0005 and ◉p < 0.0001(two tailed t-test when samples were compared to NTC).  p < 0.005 and p < 0.005 and  p < 0.001 (two-tailed t test when samples were compared to free drug controls). (d) DNA laddering (180 bp) in HepG2 and HT144 cells after 24 hours treatment with MF assisted and un-assisted NRs + DOX and NRs + MTX at IC50 doses (IC50 values with MF = ∼1 μg ml−1, IC50 values W/O MF = 2.45–3.85 μg ml−1 for both cell lines). Free DOX and MTX = 30 μM each and NTC were included as controls. DNA samples were electrophoresed with DNA ladder (100 bp) at 50 V for 2 hours using 2% agarose gel. p < 0.001 (two-tailed t test when samples were compared to free drug controls). (d) DNA laddering (180 bp) in HepG2 and HT144 cells after 24 hours treatment with MF assisted and un-assisted NRs + DOX and NRs + MTX at IC50 doses (IC50 values with MF = ∼1 μg ml−1, IC50 values W/O MF = 2.45–3.85 μg ml−1 for both cell lines). Free DOX and MTX = 30 μM each and NTC were included as controls. DNA samples were electrophoresed with DNA ladder (100 bp) at 50 V for 2 hours using 2% agarose gel. | ||
In HepG2 cells, free DOX caused G2 arrest with significantly lower (p < 0.05) cell number in S phase. Free MTX slightly effected G1 and G2 phases however the effects were non-significant. Both MF assisted and non-assisted NRs + DOX and NRs + MTX caused significant G1 (p < 0.05) and S phase (p < 0.001) arrest with lower cell population in G2 phase (p < 0.05). When compared to free DOX control (equivalent to drug attached with IC50 concentration of NRs without MF), both MF assisted and non-assisted NRs + DOX caused enhanced G1 (1.6× higher) and S phase (3.2× higher) arrest. However, when compared to free MTX control (equivalent to drug attached with IC50 concentration of NRs without MF), G1 arrest caused by NRs + MTX and NRs + MTX (MF) was 1.1× and S phase was 3× higher. These results indicate efficient drug delivery via NRs and early cell cycle arrest compared to free drugs.
In HT144 cells, free DOX caused significant (p < 0.005) G2 arrest and free MTX caused accumulation of cells in S phase (p < 0.001) with significantly lower (p < 0.05) cell population in G1 phase. Upon treatment with NRs + DOX, significant G1 (p < 0.01) and S (p < 0.05) phase arrest was observed. MF assisted NRs + DOX produced highly significant S phase arrest (p < 0.001) with decreased cellular population in G1 and G2 phases (p < 0.05). In NRs + MTX, both MF assisted, and non-assisted samples produced significant S phase arrest (p < 0.05) with decreased cellular population in G2 (p < 0.01). Upon comparison with free DOX control, NRs + DOX enhanced G1 arrest by 1.2 folds and MF assisted NRs + DOX enhanced S arrest by 2.7 folds. Similarly, S phase arrest was up to 1.6× higher in NRs + MTX (MF assisted and non-assisted) compared to free MTX control which indicates efficient drug delivery via NRs indicating halting of DNA synthesis.
In both cell lines, we obtained similar results with MF assisted and unassisted NRs at IC50 doses (Table 2). However, this finding indicates that same magnitude of cytotoxic response can be achieved at dose ∼1 μg ml−1 with the help of MF assistance, compared to unassisted samples at doses 2.45–3.85 μg ml−1. Thereby, validating an active cancer targeting strategy where dose is minimized to obtain optimum results.
3.9. Drug functionalized NRs induce DNA strand breaks
Comet assay was performed to determine drug induced genotoxicity in HepG2 and HT144 cells (Fig. 6(b)). Cells were treated with drug functionalized NRs for 1 hour at IC50 doses with and without MF assistance. Free drugs (DOX and MTX = 0.05 μM each) and NTC were included as controls. An Olive tail moment (OTM) as determinant of genotoxicity was measured for each sample relative to NTC (Fig. 6(c)).In HepG2 cells, MF assisted and non-assisted NRs + DOX produced maximum significant (p < 0.005) genotoxicity with relative OTM of 14.5 ± 3.95 approximately. When compared to free DOX control (relative OTM = 1.43 ± 1.27), NRs + DOX had significantly (p < 0.005) enhanced (10 folds) genotoxic activity. Similarly, MF assisted and non-assisted NRs + MTX also had significantly (p < 0.005) enhanced (up to 7×) genotoxicity (relative OTM = 11.26 ± 1.3 and 11.91 ± 5.53 respectively) compared to free MTX control, where relative OTM of 1.78 ± 1.12 (p < 0.05) was observed.
Similar trend was observed in HT144 cells. MF assisted and non-assisted NRs + DOX produced significant (p < 0.01) DNA damage with relative OTM of 9.5 ± 2.75 and 8.6 ± 1.71, respectively. Obtained results were significantly (p < 0.005) improved (up to 5×) when compared with free DOX control having relative OTM of 2.0 ± 0.94 (p < 0.05) only. In case of NRs + MTX, both MF assisted, and non-assisted samples showed significant (p < 0.005) genotoxicity, having relative OTM of 15 ± 4.28 and 14.35 ± 4.26, respectively. Results were significantly (p < 0.005) enhanced (up to 6×) when compared to free MTX control having relative OTM of 2.5 ± 0.94 (p < 0.05) only.
Obtained results indicated no significant differences between MF assisted and non-assisted samples at IC50 doses (Table 2: ∼1 μg ml−1 and 2.45–3.85 μg ml−1 for MF assisted and unassisted samples respectively), indicating efficient NRs mediated drug delivery controlled by external MF, resulting in optimal DNA damage at doses ∼1 μg ml−1 in both cell lines. Furthermore, applied MF had no significant impact on genotoxicity in NTC (MF) and NTC controls of both cell lines (ESI Fig. S4(b) and (c)†).
3.10. Drug functionalized NRs cause apoptotic DNA fragmentation
DNA laddering is a hallmark of apoptotic cell death. Upon treatment of HepG2 and HT144 cells with drug functionalized CFO@BTO NRs (IC50 doses, 24 hour treatment) with and without MF assistance, fragments of DNA (around 180 bp) were observed on 2% agarose gel (Fig. 6(d)). These results suggest apoptotic cellular death upon treatment with NRs + DOX and NRs + MTX.3.11. Drug functionalized NRs cause alterations in Ki-67, p53 and Bcl-2 expressions in treated cells
Ki-67 is a proliferative cancer biomarker localized in nucleus and involved in rRNA synthesis. It has prognostic characteristic for determining survival rates in cancer patients. Higher the expression of Ki-67, lower the survival rate. It has low expression rate in G1 and S phases and higher expression in G2 phase of cell cycle.72,73Whereas p53, a tumor suppressor protein has diverse gene regulating functions ranging from DNA repair, apoptosis, and cell cycle. Elevated expression of p53 is mainly associated with stress-based provocation such as oxidative stress, hypoxia, ionizing radiations, and carcinogens.74–76
Bcl-2, the proto-oncogene is involved in inhibition of cell death and is overly expressed in cancer cells. However, it is downregulated indirectly via p53 by activation of Bax protein, causing cells to undergo apoptosis in response to certain stress stimuli.77 However, Bcl-2 promoter also contains a p53 negative response element which may indicate its direct trans-repression via p53.78
Present study included immunocytochemical assessment of Ki-67, p53 and Bcl-2 cancer biomarkers in HepG2 and HT144 cells after treatment with drug functionalized CFO@BTO NRs at IC50 doses for 24 hours with and without MF assistance (Fig. 7(a) and (b)).
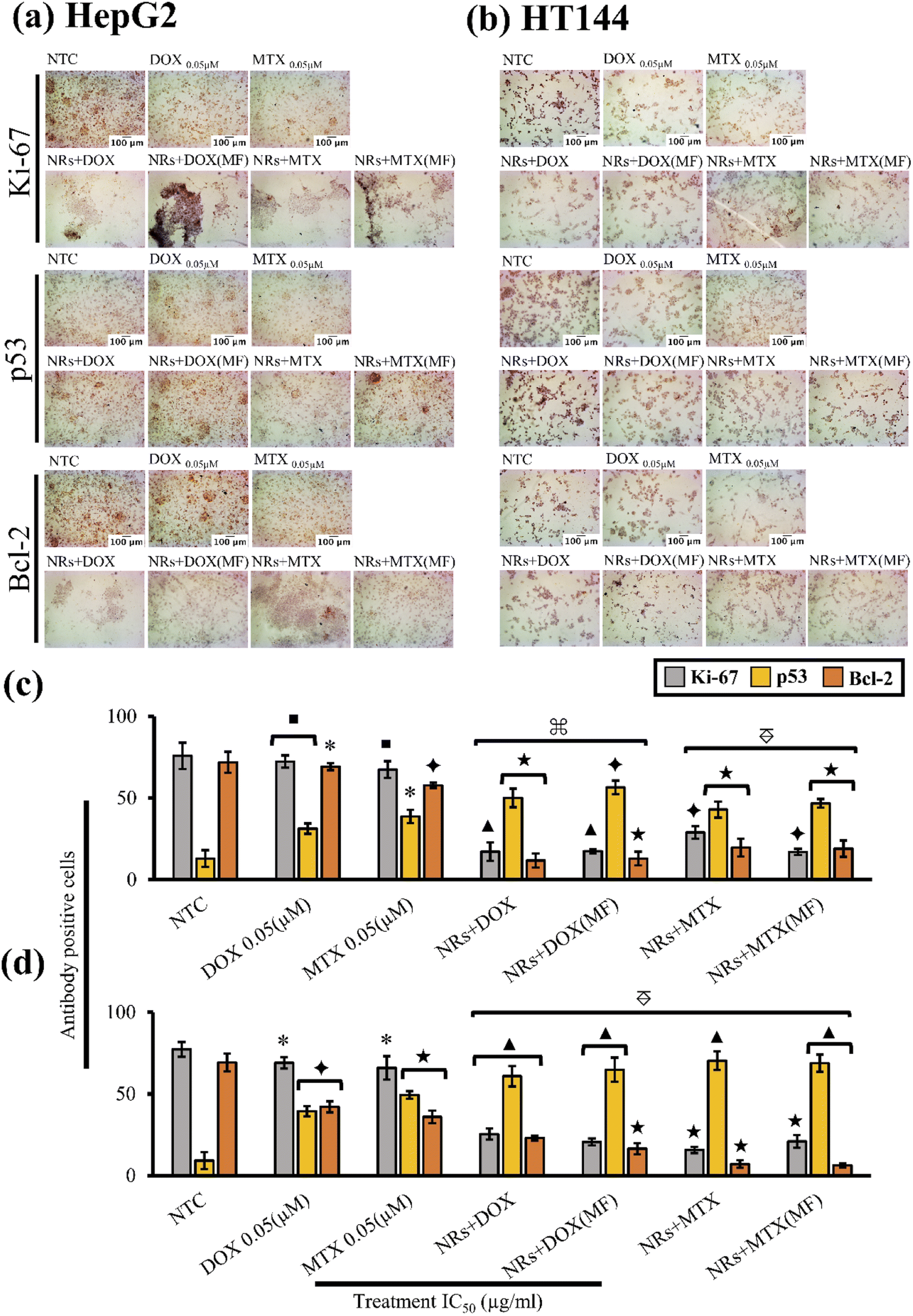 | ||
Fig. 7 (a and b) Microscopic images (magnification = 200×, scale bar = 100 μm) of immunocytochemical (ICC) assessment of Ki-67, p53 and Bcl-2 cancer biomarkers in HepG2 and HT144 cells respectively, treated with IC50 doses (IC50 values with MF = ∼1 μg ml−1, IC50 values W/O MF = 2.45–3.85 μg ml−1 for both cell lines) of NRs + DOX and NRs + MTX for 24 hours with and without MF assistance (4 mT, 20 minutes). Controls included free DOX and MTX (0.05 μM each) and NTC. (c and d) Quantitative analysis of ICC (HepG2 and HT144 respectively) with antibody positive cells counted and plotted as percentages (mean ± SD). *p < 0.05, ■p < 0.01, ✦p < 0.005, ★p < 0.001 and ▲p < 0.0005 (paired two-tailed t-test when compared to NTC).  p < 0.005, p < 0.005,  p < 0.001 and p < 0.001 and  p < 0.0005 (paired two-tailed t-test when samples were compared to free drug controls). p < 0.0005 (paired two-tailed t-test when samples were compared to free drug controls). | ||
In HepG2 cells (Fig. 7(c)), NTC had positive Ki-67 expression of 75.8 ± 8.1% whereas, p53 and Bcl-2 expressing cells were 12.9 ± 5.1% and 71.9 ± 6.4% respectively. Upon treatment with both MF assisted and un-assisted NRs + DOX and NRs + MTX, a 4× significant reduction (p < 0.005 compared to NTC) in Ki-67 expression was observed with % antibody positive cells ranging from 17.11 ± 5.67% to 28.88 ± 3.83%. Similarly, significant (p < 0.005 compared to NTC) increase in p53 expression (4×) was observed with % p53 positive cells ranging from 42.9 ± 4.9% to 56.5 ± 4.1%. Increased p53 expression also led to decreased Bcl-2 expression (6×), where antibody positive cells were recorded at 11.71 ± 4.27% to 19.71 ± 5.46% (p < 0.001). Obtained results were also significantly enhanced (p < 0.005) when compared with free drug controls where expression levels of Ki-67, p53 and Bcl-2 were 72.3 ± 3.8%, 31.25 ± 3.2% and 69.2 ± 2.1% (p < 0.05) in free DOX and 67.4 ± 5.1%, 38.75 ± 4.1% and 57.8 ± 1.6% (p < 0.05) in free MTX, respectively. This indicates enhanced cellular cytotoxic response when anticancer drugs were delivered via magnetoelectric CFO@BTO NRs.
Similar findings were obtained in HT144 cells (Fig. 7(d)). Untreated cells positively expressing Ki-67, p53 and Bcl-2 were 77.3 ± 4.5%, 9.16 ± 5.2% and 69.2 ± 5.5% respectively. Treatment with MF assisted and un-assisted NRs + DOX and NRs + MTX significantly reduced (p < 0.001 compared to NTC) Ki-67 expression up to 4×, ranging from 15.7 ± 1.9% to 25.4 ± 3.4%. Likewise, elevation in p53 (7×) and inhibition of Bcl-2 expression (3×) was observed with antibody expression ranging from 60.8 ± 6.3% to 70.3 ± 5.7% (p < 0.0005) in p53 and 6.25 ± 1.3% to 23 ± 1.5% (p < 0.001) in Bcl-2. Upon comparison with free drugs, all samples showed significantly enhanced (p < 0.005) effects. Expression levels of Ki-67, p53 and Bcl-2 were 69.03 ± 3.5%, 39.4 ± 3.1% and 42.1 ± 3.4% (p < 0.05) in free DOX and 65.9 ± 7.2%, 49.4 ± 2.3% and 35.9 ± 3.9% (p < 0.05) in free MTX, respectively.
In both cell lines, drug functionalized NRs reduced expression of Ki-67 protein, indicating possible role in inhibition of cellular proliferation. However, flow cytometric results also indicated G1 and S phase arrests in both cell lines. Since expression rate of Ki-67 is already low at G1 and S phases of cell cycle,72,73 it is unclear whether NRs had a role in inhibition of Ki-67 protein. Therefore, further investigations are required to validate these findings.
The results indicated no significant difference between MF assisted and un-assisted samples at IC50 doses (Table 2) in both cell lines. This finding suggests that application of external MF can produce similar results at a much lower dose (∼1 μg ml−1) compared to un-assisted samples (2.45–3.85 μg ml−1: Table 2) due to enhanced drug release and internalization of NRs. We also report no significant difference in expression levels of Ki-67, Bcl-2 and p53 proteins in NTC (MF) compared to NTC alone in both cell lines (ESI Fig. S5(a) and (b)†).
3.12. Drug functionalized NRs cause dose and MF dependent inhibition of MDR pump activity in treated cells
Multidrug resistance (MDR) to chemotherapeutic drugs imparts a major hurdle in cancer therapy leading to poor patient outcome. Over the course of chemotherapy, cancer cells acquire genetic alterations which cause changes in drug action mechanisms (changes in apoptotic signaling, over expression of drug efflux pumps and enhanced repair mechanisms), making them desensitized to structurally and functionally similar or unrelated drugs.79–81In addition, poor pharmacokinetics, limited half-life, limited biodistribution and non-specific uptake of drugs at tumor sites also contribute to MDR development. Since, chemotherapeutic agents cause adverse effects in normal cells as well, the dosage of these drugs is restricted due to which incomplete treatment occurs.82
Strategies to combat MDR include development of novel pharmaceuticals or targeted drug delivery at tumor sites. From drug delivery standpoint, MDR can be suppressed by loading anticancer drugs on NPs such as carbon-based NPs, liposomes, polymeric or non-polymeric NPs, dendrimers, and quantum dots.83 The EPR effect along with active targeting (ligand or stimulus based) favors NPs internalization via endocytosis, escaping P-gp pumps and MDR transporter proteins, resulting in improved half-life and prevention of pre-mature drug release.84 It is also reported that functionalization of NPs surface with polymer forms a hydration layer which causes steric inhibition of electrostatic and hydrophobic interactions with plasma proteins, causing escape from reticuloendothelial system (RES). As a result, bioavailability and biodistribution profiles of the drug are improved significantly.80
Overexpression of P-glycoprotein (P-gp), a protein efflux pump (MDR1 gene product), belonging to ABCB1 family of ATP-binding cassette (ABC) proteins, is majorly involved in pumping out several chemotherapeutic agents such as doxorubicin, vincristine, etoposide, paclitaxel and rhodamine-123.85
However, Multidrug Resistance-associated Protein (MRP1) belonging to ABCC1 family is also linked with development of MDR in cancer. MRP1 is majorly involved in pumping out hydrophobic molecules and several xenobiotics such as antimetabolites (folate-based), antiandrogens, anthracyclines, heavy metals, and vinca-alkaloids. Overexpression of MRP1 leads to elimination of therapeutic agents from cancer cells, hindering therapeutic efficacy.86 Increased expression of MRP1 protein is also associated with metastasis.87
In the present study, HepG2 and HT144 cells were treated with NRs + DOX and NRs + MTX at 1 and 5 μg ml−1 doses with and without MF assistance for 24 hours and inhibition of MDR pump activity was estimated via fluorometric determination of dye retention inside the cells relative to NTC (Fig. 8). In HepG2 cells, samples showed dose and MF dependent inhibition of MDR pump. NRs + DOX at 1 μg ml−1 caused significant inhibition of MDR pump by 2.4 folds (p < 0.05) relative to NTC which increased significantly (p < 0.05) up to 8.2 folds (p < 0.01) when MF was used. NRs + MTX at 1 μg ml−1 caused up to 6 folds (p < 0.05) increase in MDR pump inhibition under MF assistance compared to 1.1× in non-assisted sample. Inhibition of MDR pump increased at higher dose and fold difference between MF assisted and un-assisted samples reached up to 5 folds (p < 0.005). Free DOX and MTX had low dye retention (2.2 and 1.42×) relative to NTC indicating active P-gp pump activity in HepG2 cells. Whereas Cyc A (positive control) inhibited MDR pump up to 4 folds (p < 0.05).
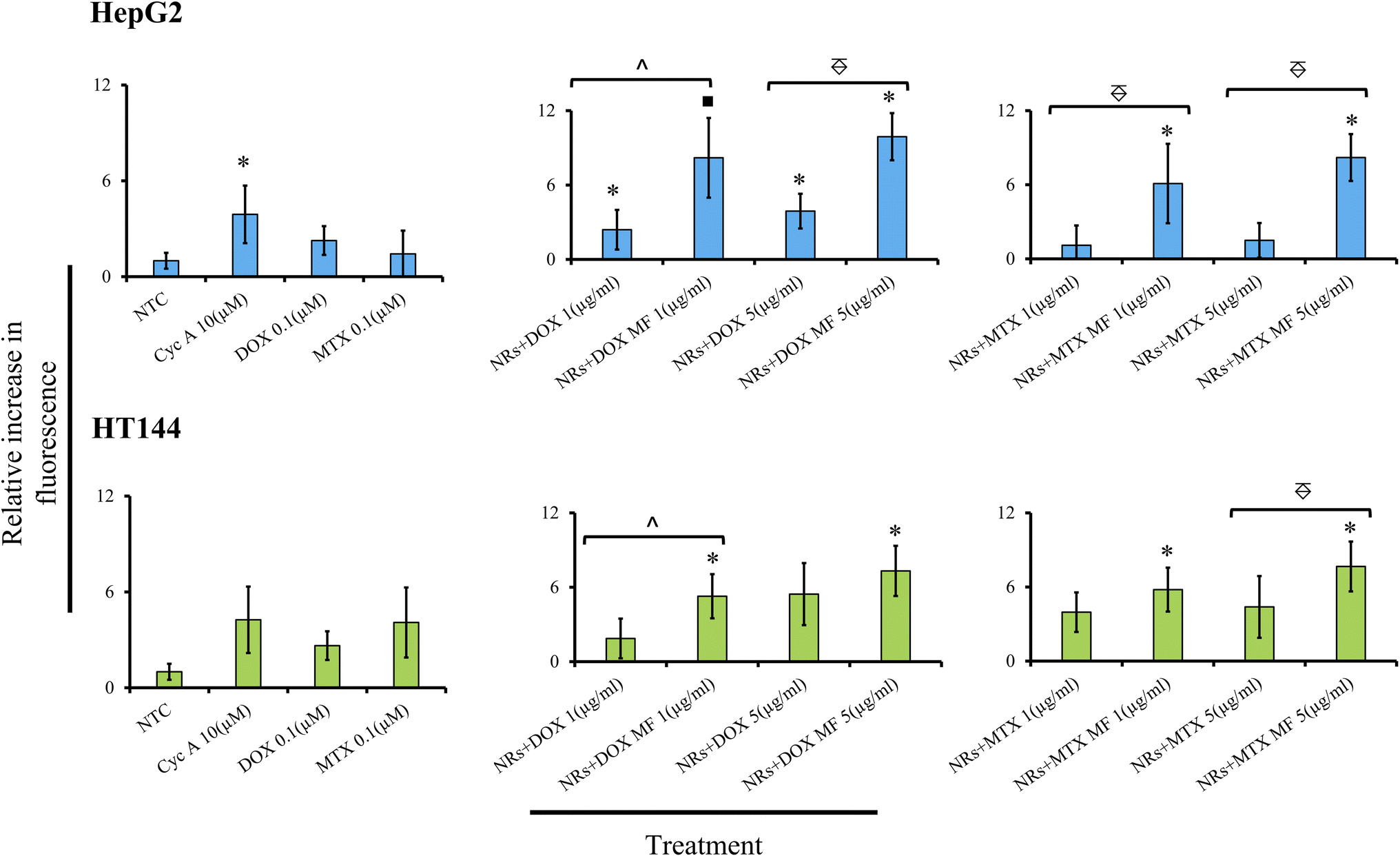 | ||
Fig. 8 Inhibition of MDR pump activity in HepG2 and HT144 cells after treatment with NRs + DOX and NRs + MTX at 1 and 5 μg ml−1 concentrations for 24 hours with and without MF assistance (4 mT, 20 minutes). Cyclosporine A (Cyc A, 10 μM) was used as positive whereas, DOX, MTX (0.1 μM each) and NTC were included as negative controls. Increase in fluorescence indicating increased MDR pump inhibition, was determined relative to NTC (mean ± SD). Experiment was performed in triplicates. *p < 0.05 and ■p < 0.01 (paired two-tailed t-test when compared to NTC). ^p < 0.05 and  p < 0.005 (paired two-tailed t-test when MF assisted, and un-assisted samples were compared). p < 0.005 (paired two-tailed t-test when MF assisted, and un-assisted samples were compared). | ||
In HT144 cells, similar dose and MF dependent behavior was observed. NRs + DOX at 1 μg ml−1 caused pump inhibition by 1.8 folds relative to NTC. The effect significantly increased up to 5.2 folds (p < 0.05) with MF assistance. Similarly, drug efflux inhibition in NRs + MTX (1 μg ml−1) was up to 4 folds relative to NTC, which increased up to 6 folds (p < 0.05) in the presence of MF. At 5 μg ml−1 dose, both samples showed increased activity against MDR pump (up to 5.4×) which amplified up to 7.6× with MF assistance. Cyc A caused inhibition of MDR pump by 4 folds. Free DOX and MTX had lower retention in cells (2.63 and 4.08× respectively). DOX and MTX have also been reported as substrates of P-gp pump, having inducing effect on MDR.88,89 In both cell lines, MF exposure had no significant effect on MDR pump activity in NTC compared to unexposed NTC (ESI Fig. S6†).
3.13. Drug functionalized NRs cause MF dependent cytotoxicity in 3D spheroids
Drug functionalized NRs were screened for cytotoxicity on 3D spheroid models of HepG2 and HT144 cells with and without MF assistance. Spheroids share close resemblance towards in vivo phenotype (drug resistance, hypoxia and denser extracellular matrix) and allow better understanding of pathobiological aspects of human cancers.90Untreated HepG2 and HT144 spheroids had diameters of 393 ± 39.8 μm and 494 ± 136.44 μm after 72 hours which reached to maximum diameters of 426.52 ± 33.21 μm and 591.94 ± 60.7 μm (Fig. 9(a) and (b)) at 14th day with % viability of 81.2 ± 3.3% and 78.5 ± 5.5% respectively (Fig. 9(c)). Free DOX and MTX caused up to 1.2× (p < 0.001) and 1.6× (p < 0.005) reduction in spheroid diameter in HepG2 and HT144 respectively at 14th day of treatment with cellular disaggregation observed in HT144. Up to 78.4 ± 1.5% cells were viable in HepG2 spheroids treated with free drugs whereas, HT144 spheroids showed maximum viability of 76.2 ± 2.9%.
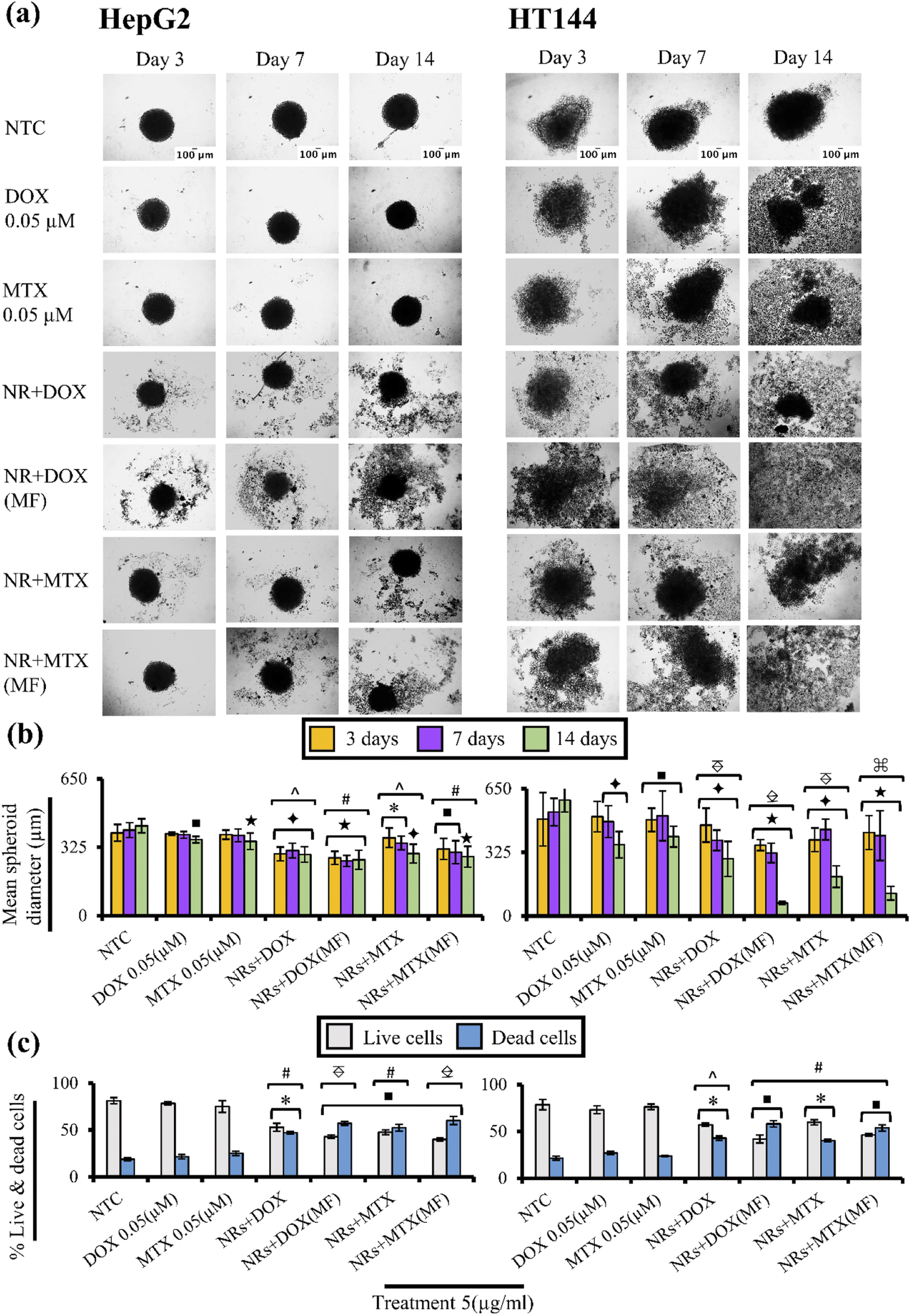 | ||
Fig. 9 (a) Microscopic images representing cytotoxic effects of NRs + DOX and NRs + MTX on HepG2 and HT144 3D spheroids. Spheroids were treated with drug functionalized NRs for 14 days with and without MF assistance (4 mT, 20 minutes) at 5 μg ml−1 dose. Controls included untreated spheroids (NTC) and free drugs (DOX and MTX = 0.05 μM each). Magnification = 100×, scale bar = 100 μm. (b) Bar charts indicating average changes in (left to right) HepG2 and HT144 spheroids diameter after treatment with drug functionalized NRs for 3, 7 and 14 days. Mean ± SD spheroid diameters were calculated using ImageJ software at each time point. Multiple regions were covered to include all maximum and minimum diameter ranges of spheroids. (c) Determination of cellular viability via trypan blue assay at 14th day in (left to right) HepG2 and HT144 spheroids after treatment with drug functionalized NRs. Data presents mean ± SD of three replicates. *p < 0.05, ■p < 0.01, ✦p < 0.005 and ★p < 0.001 (paired two-tailed t-test when compared to NTC). ^p < 0.05, #p < 0.01,  p < 0.005, p < 0.005,  p < 0.001 and p < 0.001 and  p < 0.0005 (paired two-tailed t-test when compared to free drug controls). p < 0.0005 (paired two-tailed t-test when compared to free drug controls). | ||
In HepG2 spheroids (14th day), treatment with NRs + DOX and NRs + MTX reduced spheroid diameter up to 1.47 folds (p < 0.005) with viability up to 53 ± 4.2% (p < 0.05) compared to NTC (% viability = 81.2 ± 3.3%). Whereas MF assistance increased cytotoxicity by reducing spheroid diameter up to 1.6× (p < 0.001) with enhanced spheroid disaggregation, having strong reduction in % viability (43 ± 1.9%; p < 0.01). Significantly strong results (p < 0.005) were obtained compared to free drug controls, indicating better performance of MF assisted NRs as drug carriers.
In HT144 spheroids, NRs + DOX and NRs + MTX reduced spheroid diameter up to 2.9 folds (p < 0.005) at 14th day of treatment with viability up to 57.14 ± 1.7% (p < 0.05) compared to NTC (% viability = 78.5 ± 5.5%). Use of MF assistance significantly upregulated cytotoxicity with up to 9× (p < 0.001) decrease in spheroid diameter and almost complete disaggregation of spheroids. Viability values up to 41.9 ± 4.2% (p < 0.01) were obtained. MF assisted NRs + drugs were significantly (p < 0.01) efficient in reducing cellular viability and spheroid diameter, compared to free drug controls indicating better penetration of NRs inside spheroids.
In both cell lines, MF exposure had no prominent effect on spheroid diameter and viability in NTC compared to NTC alone, indicating non cytotoxic effects of low intensity MF (ESI Fig. S7(a)–(c)†).
Application of a.c. MF causes production of pulsed electric field by MENRs which interacts with polar phosphate groups of plasma membrane bilayer. This interaction causes repulsion between negatively charged phosphate groups and pulse electric field of MENRs. As a result, repulsive forces cause dislocation of phospholipids, creating a nanopore on membrane surface which allows efficient permeation of MENRs through it.91
4. Conclusion
Magneto-electric core–shell nanorods (CFO@BTO NRs) were designed to amplify drug delivery and to achieve controlled drug release under external MF stimulation. CFO@BTO NRs were successfully synthesized by solvothermal method. Physical studies have confirmed the formation of pure core–shell ME nanostructures having average crystalline size ∼38 nm. Moreover, magnetic behavior of sample having saturation magnetization (47.4 emu g−1), coercivity (1396 Oe) and high blocking temperature (TB) 345 K at 0.5 T from MT curves also supports the formation of core–shell structures. Ferroelectric analysis has shown BTO tetragonal phase with large value of Remnant polarization 3.26 μC cm−2 and coercivity 1.29 kV cm−1 at 200 V. Synthesized CFO@BTO NRs were functionalized with PMA and anticancer drugs (DOX and MTX) using EDC chemistry with 87% and 91% drug loading efficiency, respectively. Maximum drug release (98%) was achieved at 4 mT MF applied for 20 minutes. In vitro cytotoxicity assay in HepG2 and HT144 cancer cell lines showed MF and dose dependent cytotoxicity with IC50 values ranging from 0.76–1 μg ml−1 in MF assisted and 2.45–3.85 μg ml−1 in non-assisted samples. Furthermore, augmentation in intracellular ROS production was observed up to 1.6 folds with MF. Cancer cells undergo apoptosis because of oxidative stress (p < 0.05) leading to genotoxicity (p < 0.005), p53 (elevated expression: p < 0.005) mediated cell cycle arrest (G1 and S phase: p < 0.05), and down-regulation of anti-apoptotic Bcl-2 protein (p < 0.005). In addition, drug functionalized NRs exhibited significant (p < 0.05) MDR pump inhibition activity, highlighting their potential role in combating chemo-resistance in cancer. Similarly, MF assisted NRs + Drugs caused significantly enhanced cell death and disaggregation (p < 0.01) in 3D spheroids of HepG2 and HT144 compared to free drug controls, indicating enhanced cellular permeation. With definitive results obtained with MF assistance at doses ∼1 μg ml−1, CFO@BTO NRs prove to be effective drug carriers, offering selective targeting of cancer cells without harming normal cells in vitro. However, further exploitation of cytotoxicity mechanisms and in vivo pharmacokinetics studies are required to elaborate their potential in cancer therapy.Author contributions
RA and ZA: conceptualization, funding acquisition, resources supervision, methodology, visualization, validation and reviewing & editing of manuscript. SM, KS, MR, MA and AUH: investigation and data curation. SM: funding acquisition. SM, KS, ZA, WK and RA: formal analysis. SM and KS: writing original draft. RA, ZA, NA, MA, WK and BHA: reviewing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Higher Education Commission (HEC) Pakistan (grant no. 9944/Federal/NRPU/R&D/HEC/2017 and indigenous scholarship for PhD (Phase 2, Batch 3), P. No. 315-4657-2BS3-052) is highly acknowledged.Notes and references
- Z. Edis, J. Wang, M. K. Waqas, M. Ijaz and M. J. Ijaz, Int. J. Nanomed., 2021, 16, 1313 CrossRef PubMed.
- L. Palanikumar, S. Al-Hosani, M. Kalmouni, V. P. Nguyen, L. Ali, R. Pasricha, F. N. Barrera and M. Magzoub, Commun. Biol., 2020, 3, 1–17 CrossRef PubMed.
- M. García-Aranda and M. Redondo, Cancers, 2019, 11, 1822 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- Z. Liu, S. Tabakman, K. Welsher and H. Dai, Nano Res., 2009, 2, 85–120 CrossRef CAS PubMed.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. J. Heller, Nat. Rev. Mater., 2021, 1–20 Search PubMed.
- K. J. Park, ACS Nano, 2013, 7, 7442–7447 CrossRef CAS PubMed.
- X. Zhang, Y. Li, M. Wei, C. Liu, T. Yu and J. Yang, Drug Delivery, 2019, 26, 129–136 CrossRef CAS PubMed.
- M. Li, X. Sun, N. Zhang, W. Wang, Y. Yang, H. Jia and W. Liu, Adv. Sci., 2018, 5, 1800155 CrossRef PubMed.
- K. Shahzad, S. Mushtaq, M. Rizwan, W. Khalid, M. Atif, F. U. Din, N. Ahmad, R. Abbasi and Z. Ali, Mater. Sci. Eng. C, 2021, 119, 111444 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. J. Amiji, J. Contr. Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- Y. H. Bae and K. J. Park, J. Contr. Release, 2011, 153, 198 CrossRef CAS PubMed.
- I. I. Lungu, M. Radulescu, G. D. Mogosanu and A. M. Grumezescu, J. Morphol. Embryol., 2016, 57, 23–32 Search PubMed.
- R. Guduru and S. Khizroev, Part. Part. Syst. Char., 2014, 31, 605–611 CrossRef CAS.
- K. Bhoi, H. Mohanty, M. F. Abdullah, D. K. Pradhan, S. N. Babu, A. Singh, P. Vishwakarma, A. Kumar, R. Thomas and D. K. Pradhan, Sci. Rep., 2021, 11, 1–17 CrossRef PubMed.
- P. M. Visakh and B. Raneesh, in Nanostructured Multiferroics, ed. B. Raneesh and P. M. Visakh, Wiley online library, 2021, ch. 1, pp. 1–2.2 Search PubMed.
- R. Guduru, P. Liang, C. Runowicz, M. Nair, V. Atluri and S. Khizroev, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- A. Sundaresan and N. J. Ter-Oganessian, J. Appl. Phys., 2021, 129, 060901 CrossRef CAS.
- P. Wang, E. Zhang, D. Toledo, I. T. Smith, B. Navarrete, N. Furman, A. F. Hernandez, M. Telusma, D. McDaniel and P. Liang, Nano Lett., 2020, 20, 5765–5772 CrossRef CAS PubMed.
- A. Nagesetti, A. Rodzinski, E. Stimphil, T. Stewart, C. Khanal, P. Wang, R. Guduru, P. Liang, I. Agoulnik and J. Horstmyer, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- S. Betal, A. K. Saha, E. Ortega, M. Dutta, A. K. Ramasubramanian, A. S. Bhalla and R. Guo, Sci. Rep., 2018, 8, 1–9 CAS.
- V. T. Cong, K. Gaus, R. D. Tilley and J. J. Gooding, Expet Opin. Drug Deliv., 2018, 15, 881–892 CrossRef CAS PubMed.
- N. P. Truong, M. R. Whittaker, C. W. Mak and T. P. Davis, Expet Opin. Drug Deliv., 2015, 12, 129–142 CrossRef CAS PubMed.
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- H. Lv, Y. Guo, Z. Yang, Y. Cheng, L. P. Wang, B. Zhang, Y. Zhao, Z. J. Xu and G. Ji, J. Mater. Chem. C, 2017, 5, 491–512 RSC.
- N. Hao, L. Li, Q. Zhang, X. Huang, X. Meng, Y. Zhang, D. Chen, F. Tang and L. Li, Microporous Mesoporous Mater., 2012, 162, 14–23 CrossRef CAS.
- R. Agarwal, V. Singh, P. Jurney, L. Shi, S. Sreenivasan and K. Roy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17247–17252 CrossRef CAS PubMed.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C.-Y. Fang, I. Rehor, P. Cigler, H.-C. Chang, G. Lin and R. Liu, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- W. Wang, K. Gaus, R. D. Tilley and J. Gooding, Mater. Horiz., 2019, 6, 1538–1547 RSC.
- R. Agarwal, P. Jurney, M. Raythatha, V. Singh, S. V. Sreenivasan, L. Shi and K. Roy, Adv. Healthc. Mater., 2015, 4, 2269–2280 CrossRef CAS PubMed.
- A. Yan, X. Liu, G. Qiu, H. Wu, R. Yi, N. Zhang and J. Xu, J. Alloys Compd., 2008, 458, 487–491 CrossRef CAS.
- C. A. J. Lin, R. A. Sperling, J. K. Li, T. Y. Yang, P. Y. Li, M. Zanella, W. H. Chang and W. J. Parak, Small, 2008, 4, 334–341 CrossRef CAS PubMed.
- M. C. Desai and L. M. S. Stramiello, Tetrahedron Lett., 1993, 34, 7685–7688 CrossRef CAS.
- Y. J. Pu, R. K. Vaid, S. K. Boini, R. W. Towsley, C. W. Doecke and D. Mitchell, Org. Process Res. Dev., 2009, 13, 310–314 CrossRef CAS.
- G. Repetto, A. Del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125 CrossRef CAS PubMed.
- X. Wang and M. G. Roper, Anal. Methods, 2014, 6, 3019–3024 RSC.
- R. Abbasi, T. Efferth, C. Kuhmann, T. Opatz, X. Hao, O. Popanda and P. Schmezer, Toxicol. Appl. Pharmacol., 2012, 259, 302–310 CrossRef CAS PubMed.
- S. Suman, A. Pandey and S. Chandna, Cytotechnology, 2012, 64, 9–14 CrossRef PubMed.
- R. Wan, Y. Mo, Z. Zhang, M. Jiang, S. Tang and Q. Zhang, Part. Fibre Toxicol., 2017, 14, 1–15 CrossRef PubMed.
- H. Roelofsen, T. A. Vos, I. J. Schippers, F. Kuipers, H. Koning, H. Moshage, P. Jansen and M. Muller, Gastroenterology, 1997, 112, 511–521 CrossRef CAS PubMed.
- W. Berger, E. Hauptmann, L. Elbling, M. Vetterlein, E. M. Kokoschka and M. Micksche, Int. J. Cancer, 1997, 71, 108–115 CrossRef CAS PubMed.
- X. Wang, Y. Li, Y. Qian, Y. Cao, P. Shriwas, H. Zhang and X. Chen, Oncotarget, 2017, 8, 87860 CrossRef PubMed.
- W. Strober, Curr. Protoc. Im., 1997, 21, A–3B Search PubMed.
- H. W. Lee, S. Moon, C. H. Choi and D. K. Kim, J. Am. Ceram. Soc., 2012, 95, 2429–2434 CrossRef CAS.
- J. P. Liu, E. Fullerton, O. Gutfleisch and D. J. Sellmyer, Nanoscale magnetic materials and applications, Springer, 2009 Search PubMed.
- E. Winkler, R. Zysler, M. V. Mansilla, D. Fiorani, D. Rinaldi, M. Vasilakaki and K. Trohidou, Nanotechnology, 2008, 19, 185702 CrossRef CAS PubMed.
- S. Chikazumi and C. D. Graham, Physics of Ferromagnetism 2e, Oxford University Press on Demand, 2009 Search PubMed.
- J.-K. Kim and D. F. Lawler, Bull. Korean Chem. Soc., 2005, 26, 1083–1089 CrossRef CAS.
- A. Mayer, M. Vadon, B. Rinner, A. Novak, R. Wintersteiger and E. Fröhlich, Toxicology, 2009, 258, 139–147 CrossRef CAS PubMed.
- M. Etier, V. V. Shvartsman, S. Salamon, Y. Gao, H. Wende and D. C. Lupascu, J. Am. Ceram. Soc., 2016, 99, 3623–3631 CrossRef CAS.
- V. Corral-Flores, D. Bueno-Baques and R. F. d. Ziolo, Acta Mater., 2010, 58, 764–769 CrossRef CAS.
- M. Nair, R. Guduru, P. Liang, J. Hong, V. Sagar and S. Khizroev, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- M. Yang and W. J. Brackenbury, Front. Physiol., 2013, 4, 185 CAS.
- V. R. Rao, M. Perez-Neut, S. Kaja and S. Gentile, Cancers, 2015, 7, 849–875 CrossRef CAS PubMed.
- C. D. Cone Jr, J. Theor. Biol., 1971, 30, 151–181 CrossRef CAS.
- Royalrife, http://www.royalrife.com/haltiwanger1.pdf, accessed June 2022.
- H. A. Wilson and T. M. Chused, J. Cell. Physiol., 1985, 125, 72–81 CrossRef CAS PubMed.
- D. Sun, Y. Gong, H. Kojima, G. Wang, E. Ravinsky, M. Zhang and G. Minuk, Am. J. Physiol. Gastrointest. Liver Physiol., 2003, 285, G12–G19 CrossRef CAS PubMed.
- S. H. Wright, Adv. Physiol. Educ., 2004, 28, 139–142 CrossRef PubMed.
- S. Ho and G. S. Mittal, Crit. Rev. Biotechnol., 1996, 16, 349–362 CrossRef CAS PubMed.
- H. Nakamura, K. Sezawa, M. Hata, S. Ohsaki and S. Watano, Phys. Chem. Chem. Phys., 2019, 21, 18830–18838 RSC.
- A. Rodzinski, R. Guduru, P. Liang, A. Hadjikhani, T. Stewart, E. Stimphil, C. Runowicz, R. Cote, N. Altman and R. Datar, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- A. Kaushik, R. Nikkhah-Moshaie, R. Sinha, V. Bhardwaj, V. Atluri, R. D. Jayant, A. Yndart, B. Kateb, N. Pala and M. Nair, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- O. T. Marisca, K. Kantner, C. Pfeiffer, Q. Zhang, B. Pelaz, N. Leopold, W. J. Parak and J. Rejman, Nanomaterials, 2015, 5, 1418–1430 CrossRef CAS PubMed.
- P. P. Fu, Q. Xia, H.-M. Hwang, P. C. Ray and H. Yu, J. Food Drug Anal., 2014, 22, 64–75 CrossRef CAS PubMed.
- G. Poli, G. Leonarduzzi, F. Biasi and E. Chiarpotto, Curr. Med. Chem., 2004, 11, 1163–1182 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- S. E. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- G. T. Sen, G. Ozkemahli, R. Shahbazi, P. Erkekoglu, K. Ulubayram and B. Kocer-Gumusel, Int. J. Toxicol., 2020, 39, 328–340 CrossRef CAS PubMed.
- Y. Yao, Y. Zang, J. Qu, M. Tang and T. Zhang, Int. J. Nanomed., 2019, 14, 8787 CrossRef CAS PubMed.
- J. Chiang, in Pathobiology of Human Disease, ed. L. M. McManus and R. N. Mitchell, Elsevier, San Diego, 2014, pp. 1770–1782, DOI:10.1016/B978-0-12-386456-7.04202-7.
- H. M. Ragab, N. Samy, M. Afify, N. Abd El Maksoud and H. M. Shaaban, Int. J. Genet. Eng. Biotechnol., 2018, 16, 479–484 CrossRef PubMed.
- S. Etemad-Moghadam, S. Fouladdel, E. Azizi and M. Alaeddini, J. Buon, 2013, 18, 1062–1068 CAS.
- K. Hientz, A. Mohr, D. Bhakta-Guha and T. Efferth, Oncotarget, 2017, 8, 8921 CrossRef PubMed.
- A. Efeyan and M. Serrano, Cell Cycle, 2007, 6, 1006–1010 CrossRef CAS PubMed.
- J. Pflaum, S. Schlosser and M. Müller, Front. Oncol., 2014, 4, 285 Search PubMed.
- M. Hemann and S. Lowe, Cell Death Differ., 2006, 13, 1256 CrossRef CAS PubMed.
- T. Miyashita, M. Harigai, M. Hanada and J. C. Reed, Cancer Res., 1994, 54, 3131–3135 CAS.
- G. Bonadonna, R. Zucali, S. Monfardini, M. de Lena and C. Uslenghi, Cancer, 1975, 36, 252–259 CrossRef CAS PubMed.
- C.-M. J. Hu and L. Zhang, Curr. Drug Metabol., 2009, 10, 836–841 CrossRef CAS PubMed.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- R. K. Jain, Sci. Am., 1994, 271, 58–65 CrossRef CAS PubMed.
- J. Ahmad, S. Akhter, M. Ahmed Khan, M. Wahajuddin, N. H. Greig, M. Amjad Kamal, P. Midoux and C. Pichon, Curr. Pharmaceut. Des., 2016, 22, 4360–4373 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nanosci. Technol., 2010, 239–250 Search PubMed.
- P. de León Valeria and B.-R. Raúl, Cancer Cell Int., 2005, 5, 1–13 CrossRef PubMed.
- M. Munoz, M. He`nderson, M. Haber and M. Norris, IUBMB Life, 2007, 59, 752–757 CrossRef CAS PubMed.
- S. Zöchbauer-Müller, M. Filipits, M. Rudas, R. Brunner, G. Krajnik, R. Suchomel, K. Schmid and R. Pirker, Anticancer Res., 2001, 21, 119–124 Search PubMed.
- K. Qin, K. Chen, W. Zhao, X. Zhao, J. Luo, Q. Wang, C. Gao, X. Li and C. Wang, J. Immunol. Res., 2018, 2018 Search PubMed.
- R. Silva, H. Carmo, R. Dinis-Oliveira, A. Cordeiro-da-Silva, S. C. Lima, F. Carvalho, M. de Lourdes Bastos and F. Remião, Arch. Toxicol., 2011, 85, 315–326 CrossRef CAS PubMed.
- S. Daunys, A. Janonienė, I. Januškevičienė, M. Paškevičiūtė and V. Petrikaitė, in Bio-Nanomedicine for Cancer Therapy, Springer, 2021, pp. 243–270 Search PubMed.
- S. Betal, M. Dutta, B. Shrestha, L. Cotica, L. Tang, A. Bhalla and R. Guo, Integrated Ferroelectrics, 2016, 174, 186–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03429h |
| This journal is © The Royal Society of Chemistry 2022 |