
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of flexible Co nanowires from bulk precursors†
Victoria
Petrova
 a,
Adam A.
Corrao
b,
Shen
Wang
a,
Yuxuan
Xiao
d,
Karena W.
Chapman
b,
Eric E.
Fullerton
d,
Peter G.
Khalifah
bc and
Ping
Liu
*a
a,
Adam A.
Corrao
b,
Shen
Wang
a,
Yuxuan
Xiao
d,
Karena W.
Chapman
b,
Eric E.
Fullerton
d,
Peter G.
Khalifah
bc and
Ping
Liu
*a
aDepartment of Nanoengineering, University of California-San Diego, La Jolla, California 92093, USA. E-mail: piliu@ucsd.edu
bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA
cChemistry Division, Brookhaven National Laboratory, Upton, New York 11973, USA
dCenter for Memory and Recording Research, University of California San Diego, La Jolla, CA 92093-0401, USA
First published on 1st August 2022
Abstract
This work reports a method of producing flexible cobalt nanowires (NWs) directly from the chemical conversion of bulk precursors at room temperature. Chemical reduction of Li6CoCl8 produces a nanocomposite of Co and LiCl, of which the salt is subsequently removed. The dilute concentration of Co in the precursor combined with the anisotropic crystal structure of the hcp phase leads to 1D growth in the absence of any templates or additives. The Co NWs are shown to have high saturation magnetization (130.6 emu g−1). Our understanding of the NW formation mechanism points to new directions of scalable nanostructure generation.
Introduction
1D nanostructures offer a unique opportunity to study the electronic, plasmonic, magnetic, electrical, mechanical, and thermal properties of materials to enable a wide range of applications. Metal nanowires (NWs) have electrical properties and a flexible structure that can be utilized for flexible solar cells and touchscreens, while NWs with magnetic properties, as well as oxide-based magnetic materials, can be used for thin film magnets with high energy density, spintronic circuits and sensors.1–9 Metal NWs can also be used to complement polymer NWs to serve as photodetectors and waveguides.10–12 Additionally, research on metal-based NWs shows the importance of NW geometries on their magnetic properties.13,14The synthesis of 1D nanostructures generally employs either a top-down or a bottom-up approach. A top-down approach uses templates such as anodized alumina to confine the growth inside the channels.15 In bottom-up solution-based synthesis, ligands or surfactants are employed to encourage the growth of the solid phase in a specific direction while vapor-phase growth involves the use of catalysts to seed the growth.2,16 Examples of bottom-up synthesis include using sol–gel approaches and even rhizome plant extracts.17,18 Although the scalability of these methods has advanced significantly in recent years, it would be highly desirable to synthesize 1D nanostructures from bulk, solid precursors without the need of additional structural guiding agents. Such processes would be highly scalable and robust, with the potential to offer low-cost 1D nanomaterials in large quantities.
Examples of nanowire (NW) formation from decomposition of bulk precursors without assistance from templates, ligands, or catalysts are rare.19 Previously, aluminum metal–organic NWs were formed by exposing AlLi alloy particles to alcohol, dissolving out the Li alkoxide and forming Al(EtOH)3 NWs.20,21 The proposed formation mechanism is a minimization of strain energy at the boundary of the chemical reaction front. However, there are so far no simple methods of easily producing metal NWs from bulk precursors. Most fcc metals have a strong tendency to reduce their total surface energy by taking a highly symmetric shape during growth, and therefore do not grow in nanowire forms without templates or structural directing agents.
We report a simple, scalable method of producing flexible Co NWs directly from bulk precursors at room temperature. We have previously shown a process in which a metal halide undergoes a conversion reaction with n-butyllithium to form a nanocomposite of the metal and halide salt. The salt is then removed through dissolution in methanol, leaving behind an isotropic nanoporous metal.22 Here, we show that diluting the precursor metal halide by using the Suzuki phase Li6CoCl8 results in a new NW morphology. When the Suzuki phase reacts with n-butyllithium, a nanocomposite of Co in a LiCl matrix is formed. Upon removing the LiCl salt with methanol, the Co atoms migrate and are extruded from the matrix as NWs (eqn (1), Fig. 1).
| Li6CoCl8 + 2BuLi → 8LiCl + Co ⇒ Co (NWs) | (1) |
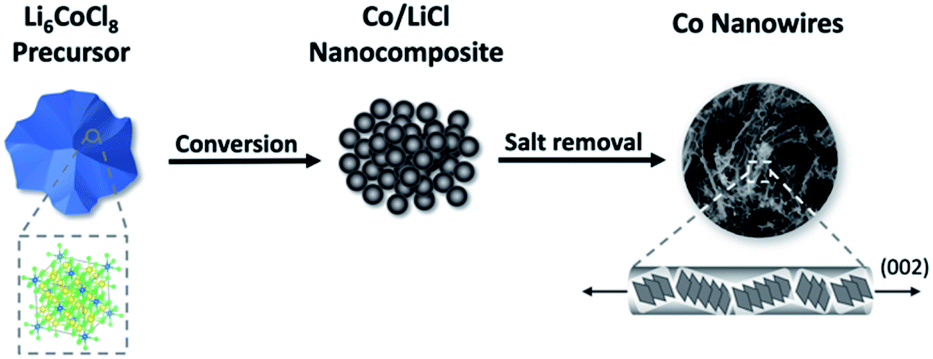 | ||
| Fig. 1 Schematic of synthesis procedure for producing Co NWs. Li6CoCl8 first undergoes a conversion reaction with n-BuLi to form a nanocomposite, from which the salt is removed to leave Co NWs. | ||
This work introduces a more efficient and simple synthesis procedure of producing Co NWs from bulk precursors. Understanding how to control morphology based on material properties can provide design guidelines to synthesize and tailor the morphology of nanomaterials.
Experimental methods
Synthesis of Li6CoCl8, Li2CoCl4, and Li6NiCl8
Li6CoCl8, Li2CoCl4, and Li6NiCl8 precursors are synthesized through a solid-state process as described previously.23–25 Stoichiometric ratios of anhydrous CoCl2 or NiCl2 (98% Sigma-Aldrich) and LiCl (99% Sigma-Aldrich) are ground using a mortar and pestle for 10 min in an argon glovebox and then vacuum sealed in a glass ampoule. The combined CoCl2 and LiCl powders are heated at 400 °C and 315 °C for seven days to form Li6CoCl8 and Li2CoCl4 respectively. The combined NiCl2 and LiCl powder is heated at 540 °C for seven days to form Li6NiCl8. The Li6CoCl8, Li2CoCl4 and Li6NiCl8 precursors are then cooled to room temperature and stored in an argon glovebox (<10 ppm O2). It is imperative that the precursors are pure and dry since both chloride salts are very hygroscopic and can easily introduce impurities if exposed to atmospheric conditions.The precursor is converted into a nanocomposite following a published procedure.22 The precursor is first ground with mortar and pestle and sieved so that particles are homogenous and smaller than 150 µm. 1 g of precursor is then reacted with ×1.5 excess 0.05 M n-butyllithium (from 1.6 M in hexane, Sigma-Aldrich and further diluted with hexane, HPLC, Fisher Scientific) for seven days. The resulting nanocomposite powder is isolated by rinsing with hexane with a filter funnel under vacuum and allowed to dry overnight at room temperature. The nanocomposite is further rinsed with methanol (HPLC, sparged with argon for 1 h and desiccated with molecular sieves, Type 3A, Sigma-Aldrich) five times to remove the LiCl salt. Since methanol has a high volatility, drying the final product overnight at room temperature in the glovebox is sufficient to obtain a fully dry final product. The conversion and salt removal processes take place in a glovebox. The writing of these methods is structured so that natural language processing can extract data and easily create a synthesis flow chart for future applications in building databases for materials synthesis machine learning.26
Laboratory and synchrotron X-ray diffraction
Lab XRD scans are done on a benchtop powder diffractometer (Bruker D2-Phaser) using Cu Kα radiation (λ = 1.5406 Å) in a 2θ range from 10° to 80° with scanning rate 0.02° s−1 and step size 0.05°. Samples are prepared in an argon glovebox using Kapton tape to cover the powders to mitigate oxidation during scanning. Diffraction peaks are indexed using X'Pert Highscore Plus based on reference structures from the Inorganic Crystal Structure Database (ICSD).Samples are prepared for synchrotron XRD data collection in an argon glovebox. Powders are ground, packed in Kapton capillaries (Cole Palmer, polyimide), and sealed with epoxy to mitigate oxidation during transport and data collection. Synchrotron X-ray diffraction data are collected in transmission geometry on beamline 17-BM-B of the Advanced Photon Source (APS) at Argonne National Laboratory using an incident beam energy of 27.439 keV (λ = 0.45185 Å).
Diffraction images were recorded on an amorphous Si 2D area detector (Varex 4343CT, 2880 × 2880 pixel array, 150 × 150 µm pixel size) at a sample-to-detector distance of 400 mm. Data were collected using 25 separate 0.8 second frames that were summed for a total collection time of 20 seconds. The resulting 2D images were integrated to 1D patterns using pyFAI.27 The instrument geometry (sample-to-detector distance, detector tilts) was calibrated using data from a LaB6 standard (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, space group #221, a = 4.15682 Å). Phase identification was done using Jade,28 version 9, against reference patterns in the International Centre for Diffraction Data (ICDD) database.29 Pawley and Rietveld refinements were done in TOPAS (Version 6, Bruker-AXS).30–32 The instrumental contribution to peak profiles was modelled using a 6-term pseudo-Voigt function with parameters determined through Pawley fitting of the LaB6 standard and then fixed for subsequent sample refinements.
m, space group #221, a = 4.15682 Å). Phase identification was done using Jade,28 version 9, against reference patterns in the International Centre for Diffraction Data (ICDD) database.29 Pawley and Rietveld refinements were done in TOPAS (Version 6, Bruker-AXS).30–32 The instrumental contribution to peak profiles was modelled using a 6-term pseudo-Voigt function with parameters determined through Pawley fitting of the LaB6 standard and then fixed for subsequent sample refinements.
SEM and TEM characterization
SEM images are acquired with a Zeiss Sigma 500 scanning electron microscope operating at 3 keV EHT voltage. Samples are loaded into a sample holder, heat sealed in an aluminum-lined blue bag, and brought to the SEM for rapid transfer with minimal air exposure. Images are analyzed using ImageJ software to determine NW length.TEM images are obtained at a 200 keV acceleration voltage on a Thermo Fisher Talos 200X system. Small amounts of sample powder are dropped on a copper-coated TEM grid which is loaded into the sample holder in an argon glovebox, heat sealed in an aluminum-lined blue bag, brought to the TEM and inserted under argon gas flow. Images, lattice parameters and electron diffraction patterns are analyzed using the Gatan Micrograph software and fast Fourier transform (FFT) tools.
Magnetic measurements
The magnetization of Co nanowire powder was characterized at 300 K by using Quantum Design vibrating sample magnetometry (VSM). The powder was packed in sample holder in an argon glovebox and quickly transferred in an argon-gas sealed bag to the VSM. Before measuring the magnetic hysteresis loop, the powder was saturated at 25 kOe. Then the moment vs. field loop was swept from 25 kOe to −25 kOe and back to 25 kOe while keeping the sample chamber under vacuum to minimize oxidation. Later, the measured moment was normalized to magnetization by dividing the weight of powder. Note that linear diamagnetic background was subtracted.Results and discussion
Crystal structure and phase identification
The synthesized Suzuki phase Li6CoCl8 has a rock salt crystal structure (Fmm) confirmed by X-ray diffraction (Fig. 2a) and is a superstructure of LiCl with lattice parameters twice those of LiCl and an ordered arrangement of cations and vacancies over octahedral sites.
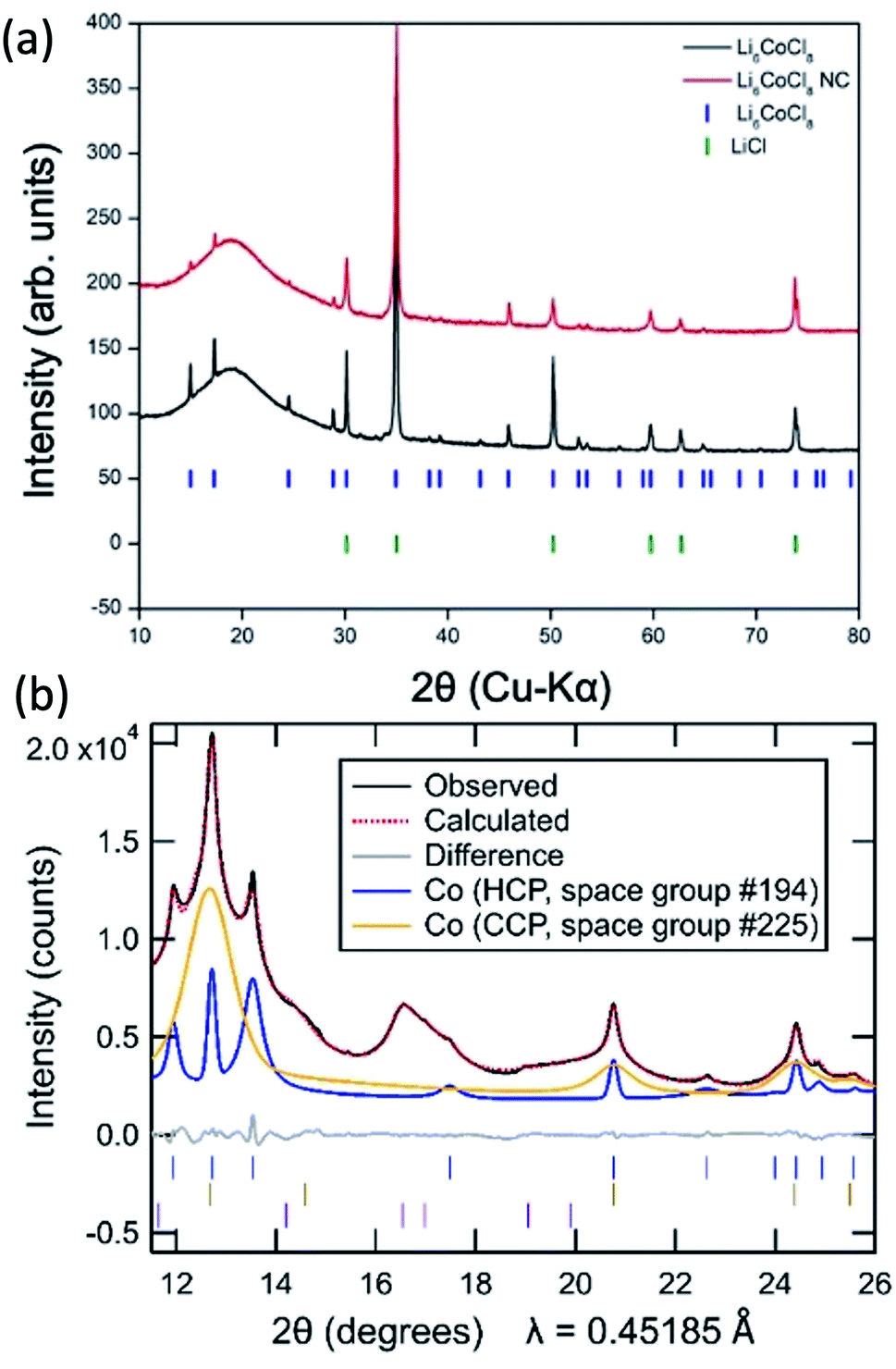 | ||
| Fig. 2 (a) Laboratory XRD patterns of synthesized Li6CoCl8 (black) and nanocomposite (red) with markers of literature peak positions of Li6CoCl8 (blue) and LiCl (green). (b) Synchrotron diffraction pattern of Co NWs (black) with the simulated pattern (red) of an hcp (26%, blue) and ccp (74%, orange) Co mixture. Markers are shown for six minor unindexed peaks (purple), with further details in Fig. S4.† | ||
The nanocomposite formed after reaction with n-butyllithium for seven days shows the disappearance of certain Suzuki peaks (2θ = 38.2, 39.2, 43.1), the significant decrease in intensity of other Suzuki peaks as well as the appearance of peaks of LiCl (Fig. 2a). Because the Suzuki phase is a superstructure of LiCl, the Li6CoCl8 sub-cell peaks correspond to the same interplanar distances as the LiCl peaks and are nearly exactly overlapped, thus reaction progress is primarily determined by changes in the Suzuki peaks. While the minor residual Suzuki peaks indicate the reaction may not have proceeded to completion, any precursor left in the nanocomposite is soluble and removed in the methanol dissolution step and therefore does not affect the formation of NWs. Further experiments on longer reaction times (30 days) show a fully converted precursor with the same resulting NW morphology (ESI S1†). Full conversion is exceedingly slow due to the diluted Co precursor as Li6CoCl8; this slow rate is likely a result of the small volume change that affects the reaction propagation. Estimation of volume change from precursor to nanocomposite based on eqn (1) is 3.5%. In comparison, CoCl2 experiences a 23% volume expansion and full conversion occurs on the order of hours.22 The small volume change will discourage a reaction mechanism involving particle fracture and infusion of butyllithium. During the lithiation process, the reaction front is hypothesized to occur through extrusion, similar to the lithiation of FeF3 in which Li insertion and Fe extrusion occur concurrently but the fluoride lattice remains coherent.33 Here the virtually identical structure of LiCl and Li6CoCl8 makes a similar mechanism also likely.
While laboratory XRD is sufficient to determine the Suzuki phase and analyze changes during conversion, synchrotron XRD is required to fully resolve the cobalt phases in the NWs, as is shown previously on ZnO/Co nanoporous composites.34 Lab XRD of the Co NWs only shows a broad Co peak due to the extremely small nanoscale Co domains (ESI S2†).
Synchrotron X-ray powder diffraction studies show that the final nanowire product contains a mixture of two different cobalt polymorphs as well as a minor contribution from at least one as-yet unindexed phase with all three components exhibiting broad peaks (Fig. 2b). While bulk Co is typically found in its cubic close packed (ccp) form, a hexagonal close packed (hcp) polymorph has previously been reported to be more stable at low temperatures, and nanoparticles of cobalt commonly exhibit a mixture of hcp and ccp polymorphs,35 one or both of which may contain stacking faults.36,37 Powder diffraction methods are frequently used to obtain quantitative insights into phase fractions (based on the pattern of peak intensities) and into sample size and strain (based on the peak profiles), information which is of interest for the present nanowire samples.
Initial efforts to model the Co nanowires diffraction pattern through Rietveld refinements where the peak shapes of hcp Co and ccp Co follow conventional models of isotropic size and/or strain broadening resulted in poor fits (ESI S3a†), as evidenced by the large refinement Rwp of 11.9% for the optimal parameter choices. The problems in the fitting persisted even when utilizing Pawley methods which allowed the peak intensities of these phases to be freely refined (Rwp = 2.5%, ESI S3b†), further supporting the conclusion that the primary challenge in the refinement is properly describing the sample peak shape. Attempts to improve the fitting of diffraction peaks in Rietveld refinements using anisotropic strain broadening38 and anisotropic size broadening39,40 models in which the peak shapes are hkl-dependent were similarly unsuccessful.
We therefore developed a novel approach for Rietveld refinement in which the pseudo-Voigt shape of each hkl peak was independently refined using custom TOPAS macros written for this purpose. This allowed the intensities of all the peaks for the hcp and ccp polymorphs to be fully constrained by their conventional structural model (with the isotropic displacement parameter of Co additionally constrained to be the same in both phases). Even within these strong constraints, it was possible to obtain a far better fit (Rwp = 1.9%, ESI S3c†) than the other conventional approaches. The refined lattice parameters and crystallographic parameters of the two Co phases are provided in the ESI, together with information in Table S2† about the unindexed peaks that were utilized in the same manner for all the fits shown in ESI S3.†
The need to use independent peak shape parameters for each peak suggests that the nanoscale domains of the hcp and ccp have shapes which are strongly anisotropic or irregular. The much broader peaks of the ccp phase indicate that its domain size is significantly smaller than that of the hcp phase. While the shape anisotropy suggests that the size cannot be captured using a single dimension, the refined size parameters of 5 nm (hcp) and 1.6 nm (ccp) approximated from Pawley fits using an isotropic size broadening model (ESI Table S3†) provide a sense of the relative scale of the domains of these two Co polymorphs. The large breadths of the unindexed peaks (ESI S4†) are comparable to that of the ccp Co phase, suggesting that these unindexed peaks may be associated with a thin surface layer on Co associated with surface oxidation or another decomposition reaction. A thin surface layer may be constrained by the underlying metal, perhaps preventing this phase from adopting a known structure type and thus hindering efforts to index it.
The explicit use of structural models allows the relative fractions of the hcp phase (26%) and ccp phase (74%) to be extracted from the refinements in a manner that would be impossible if Pawley fits were used, though some uncertainty remains in these phase fractions due to the overlapping intensity of unindexed peaks with the dominant 111 peak of the ccp Co phase at 2θ = 12.67°.
Morphology and property characterization
SEM micrographs (Fig. 3a) show large micron sized particles of NWs. Higher magnification (Fig. 3b) shows the NWs are long (with an average length of 273 nm), flexible and homogenous; they appear to grow from the surface of the particle outwards and are very well intermixed. To obtain further information on the phases present and the direction of growth, the NWs are also characterized using TEM (Fig. 4). Co NWs reported previously tend to be made of single crystal hcp Co growing continuously in the 002 direction,5,41,42 however this new morphology shows an unassisted growth mechanism that produces curved, flexible NWs. | ||
| Fig. 3 SEM micrographs showing micron sized particles of Co NWs (a and b). Higher magnification (b) shows flexible, homogenous wires with an average length of 273 nm. Scale bars are 200 nm. | ||
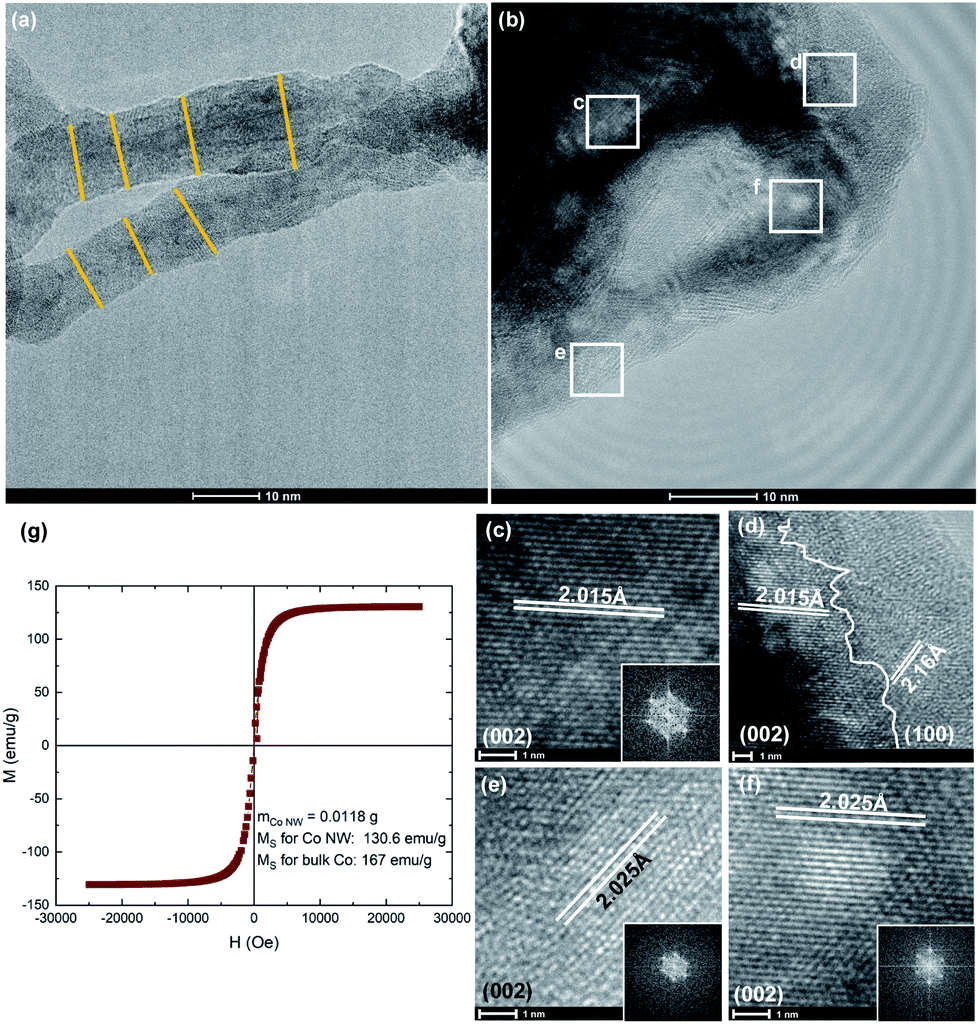 | ||
| Fig. 4 Clockwise from top left: lower resolution TEM micrograph of Co NWs shows an average NW diameter of ∼11 nm (a); higher resolution TEM micrographs of Co NWs (b) with SAED insets of regions showing predominantly hcp phase (c–f) as well as the intersection of grains (d). Magnetic measurements show the Ms of NWs is close to the value of bulk cobalt value (g). | ||
Low resolution TEM images show the NWs have an average diameter of ∼11 nm (Fig. 4a). Further details on NW geometry can be found in ESI S5.† Multi-crystalline regions of intersecting phases are revealed by high resolution images. Selected area electron diffraction (SAED) micrographs show lattice spacings of 2.02 Å corresponding to that of (002) direction of hcp phase cobalt (Fig. 4c–f) as well as an instance of 2.16 Å corresponding to the (100) direction (Fig. 4d). This corresponds well with previous reports of Co NWs synthesized by a polyol process where cobalt nanorods are obtained by reduction of carboxylate CoII salts in which the dominant direction is (002).5,41 Growth in this direction is likely due to a thermodynamic driving force to minimize the total surface free energy by exposing lower surface free energy facets and therefore growing along the higher energy (002) facet.43 Other regions also show the ccp phase is present with lattice spacings of 2.05 Å corresponding to the (111) facet (ESI S6†). These observations are consistent with the results from the XRD analysis. An intersection of these phases shows that the wires do not grow continuously in one direction but are rather a multigrain product which contributes to their flexible morphology. This most closely resembles a coalescence attachment growth in which nanoparticles serve as building blocks to form winding polycrystalline chains.44,45 We note that the phase fractions derived from synchrotron XRD fittings differ slightly from TEM observations because larger hcp domains are easier to resolve than small ccp domains and TEM is a local probe not meant for quantifying average behavior.
The magnetic properties of the Co NWs are measured using vibrating sample magnetometry (Fig. 4g). The saturation magnetization (Ms) is found to be 130.6 emu g−1, achieving 78% the value of bulk cobalt. Such a high magnetization exceeds recorded values previously reported for cobalt nanoparticles and nanorods (102 and 113 emu g−1 respectively).5,46 Co is known to be a ferromagnetic material and is often used in hard magnets to raise the Curie temperature and improve magnetic properties due to an enhanced interdiffusion across interfaces.46,47 The NWs' strong magnetic properties combined with their curved morphology make them a promising candidate for applications such as spintronic circuits and magnetic sensors.6,7 Additionally, intergrowths and stacking faults in the cobalt nanoparticle catalysts have shown high catalytic activities in the Fischer–Tropsch synthesis.4 It is worth noting that grain size as well as doping may also affect magnetic properties,48–51 and the addition of a second phase can enhance electronic properties.52,53
Formation mechanisms
Based on these observations, we hypothesize that the anisotropic hcp phase in a dilute precursor is responsible for NW formation and next examine its validity by using other related precursors. Li2CoCl4, which has an orthorhombic crystal structure and is a less dilute system than Li6CoCl8, can be used to probe whether the dilution or crystal structure of the precursor plays a more dominant role in the formation mechanism of NWs. Li2CoCl4 was synthesized according to report by Kanno et al.24 and XRD results show the formation of the Li2CoCl4 phase as well as the conversion to the nanocomposite of LiCl and Co (ESI S7†). SEM images show the same flexible, thin and long NWs as those produced from the Suzuki phase precursor (Fig. 5b). These contrast with a nanoporous metal morphology when CoCl2 was used as a precursor (Fig. 5c).22 While the exact mechanism is unclear, the comparison of various dilutions of the precursor metal halide from CoCl2 to Li2CoCl4 to Li6CoCl8 shows that dilution is necessary to produce NWs. This may be because of a combination of small volume change and an extrusion-like process during lithiation. The formation of NWs from a less dilute precursor as Li2CoCl4 also aids in a more efficient synthesis procedure since less LiCl is required and shows that the crystal structure of the precursor is not as important as the dilution of Co in the LiCl matrix. | ||
| Fig. 5 SEM images comparing the effect of different precursors: Li6CoCl8 and Li2CoCl4 produce NWs (a and b), however less dilution ((c), CoCl2) and a different ccp metal ((d), Li6NiCl8) lead to nanoporous morphology. | ||
Additionally, Li6NiCl8 was synthesized to isolate the effect of a dilute salt matrix by keeping the same metal to LiCl ratio as the Li6CoCl8 precursor but using a different transition metal that does not have an anisotropic phase. The XRD pattern shows the Suzuki peaks in the Ni system, as well as conversion to the nanocomposite and the final product (ESI S8†). SEM images of the final product, however, reveal a nanoporous morphology similar to that derived from previously reported NiCl2 precursors (Fig. 5d).22 This result suggests that a critical parameter for forming NWs is the anisotropic hcp phase that leads to NWs due to the lower energy barrier of growing in a preferential direction. Since Ni is only present in the fcc phase, there is no driving force to produce anisotropic morphology.
Several key parameters that help achieve 1D growth in metal NWs are highlighted: (1) a precursor with a low metal concentration experiences a small volume change during lithiation and encourages a slow, extrusion type of phase conversion process; the resultant small size leads to a mixture of hcp vs. ccp phases, (2) the precursor metal must be able to exist in an anisotropic phase which allows growth in a preferred orientation. Additionally, the mixture of both Co phases with the larger hcp phase being smaller than the NW dimensions and possible stacking faults due to a coalescence attachment growth mechanism may contribute to the curved morphology.44,54
Conclusions
This work presents a novel method of synthesizing cobalt nanowires from bulk precursors without the addition of catalysts or external stimuli. By using a dilute Suzuki phase precursor, Li6CoCl8, reacting with n-butyllithium to create a nanocomposite, and then dissolving the resulting salt, a cobalt nanowire morphology is achieved. This is enabled by both the dilute salt matrix which affects the cobalt crystallite size as well as the significant presence of the hcp phase. Ultimately, the anisotropic hcp phase allows for preferred growth in the (002) direction. The same NW morphology can be achieved with a less dilute precursor such as Li2CoCl4, however using a ccp metal such as Ni in Li6NiCl8 results in a nanoporous morphology. A high saturation magnetization value of 130.6 emu g−1 shows the Co NW's magnetic properties exceed those of previously reported cobalt nanoparticles and nanorods; a property important for applications such as flexible electronics, magnetic sensors and catalysts.5–7,46 This work provides a scalable synthesis procedure at ambient conditions that can help pave the way for future designs of nanowires.Author contributions
V. P. fabricated samples, performed and analyzed lab X-ray diffraction data and analysed TEM data with guidance from P. L. A. C. obtained and analysed mail-in X-ray diffraction data collected at beamline 17-BM of the Advanced Photon Source with guidance from P. K. S. W. assisted in collection of TEM data. Y. X. assisted in collecting magnetic measurements with guidance from E. F. A. C., P. K. and P. L. assisted in understanding the data and discussing formation mechanisms behind Co NWs. K. C. contributed the idea of testing different diluted precursors such as the Suzuki phase. V. P. wrote the manuscript with input from all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of GENESIS: A Next Generation Synthesis Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award number DE-SC0019212. Partial support for AAC was provided by an NRT QuADS Fellowship (Quantitative Analysis of Dynamic Structures) under NSF DGE-1922639. This research used the mail-in program at Beamline 17-BM of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542148). We gratefully acknowledge discussions with Alan Coelho about extending the capabilities of the TOPAS software.References
- L. Chen, Y. Liu and Y. Zhao, et al. , J. Phys. D: Appl. Phys., 2018, 51, 113002 CrossRef.
- D. Huo, M. J. Kim, Z. Lyu, Y. Shi, B. J. Wiley and Y. Xia, Chem. Rev., 2019, 119, 8972–9073 CrossRef CAS PubMed.
- K. Gandha, K. Elkins, N. Poudyal, X. Liu and J. P. Liu, Sci. Rep., 2014, 4, 5345 CrossRef CAS PubMed.
- S. W. T. Price, D. J. Martin, A. D. Parsons, W. A. Sławiński, A. Vamvakeros, S. J. Keylock, A. M. Beale and J. F. W. Mosselmans, Sci. Adv., 2017, 3, 3 Search PubMed.
- Y. Soumare, C. Garcia, T. Maurer, G. Chaboussant, F. Ott, F. Fiévet, J. Piquemal and G. Viau, Adv. Funct. Mater., 2009, 19, 1971–1977 CrossRef CAS.
- G. Muscas, P. E. Jönsson, I. G. Serrano, Ö. Vallin and M. V. Kamalakar, Nanoscale, 2021, 13, 6043–6052 RSC.
- S. Ota, A. Ando and D. Chiba, Nat. Electron., 2018, 1, 124–129 CrossRef.
- A. v. Trukhanov, V. O. Turchenko, I. A. Bobrikov, S. v. Trukhanov, I. S. Kazakevich and A. M. Balagurov, J. Magn. Magn. Mater., 2015, 393, 253–259 CrossRef CAS.
- A. v. Trukhanov, S. v. Trukhanov, L. v. Panina, V. G. Kostishyn, D. N. Chitanov, I. S. Kazakevich, A. v. Trukhanov, V. A. Turchenko and M. M. Salem, Ceram. Int., 2017, 43, 5635–5641 CrossRef CAS.
- X. Yang, D. Bao and B. Li, RSC Adv., 2015, 5, 60770–60774 RSC.
- X. Yang, Y. Liu, H. Lei and B. Li, Nanoscale, 2016, 8, 15529–15537 RSC.
- X. Yang, L. Wen, J. Yan, Y. Bao, Q. Chen, A. Camposeo, D. Pisignano and B. Li, J. Phys. Chem. Lett., 2021, 12, 7034–7040 CrossRef CAS PubMed.
- A. v. Trukhanov, S. S. Grabchikov, S. A. Sharko, S. v. Trukhanov, K. L. Trukhanova, O. S. Volkova and A. Shakin, Mater. Res. Express, 2016, 6, 65010 CrossRef.
- S. A. Sharko, A. I. Serokurova, T. I. Zubar, S. v. Trukhanov, D. I. Tishkevich, A. A. Samokhvalov, A. L. Kozlovskiy, M. v. Zdorovets, L. v. Panina, V. M. Fedosyuk and A. v. Trukhanov, J. Alloys Compd., 2020, 846, 156474 CrossRef CAS.
- L. Wen, R. Xu, Y. Mi and Y. Lei, Nat. Nanotechnol., 2017, 12, 244–250 CrossRef CAS PubMed.
- N. Wang, Y. Cai and R. Q. Zhang, Mater. Sci. Eng., R, 2008, 60(1–6), 1–51 Search PubMed.
- A. Thakur, N. Sharma, M. Bhatti, M. Sharma, A. v. Trukhanov, S. v. Trukhanov, L. v. Panina, K. A. Astapovich and P. Thakur, Nano-Struct. Nano-Objects, 2020, 24, 100599 CrossRef CAS.
- N. A. Algarou, Y. Slimani, M. A. Almessiere, F. S. Alahmari, M. G. Vakhitov, D. S. Klygach, S. v. Trukhanov, A. v. Trukhanov and A. Baykal, J. Mater. Res. Technol., 2020, 9, 5858–5870 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- D. Lei, J. Benson, A. Magasinski, G. Berdichevsky and G. Yushin, Science, 2017, 355, 267–271 CrossRef CAS PubMed.
- F. Wang, K. Turcheniuk, B. Wang, A.-Y. Song, X. Ren, A. Vallamattam, A. Park, K. Hanley, T. Zhu and G. Yushin, J. Am. Chem. Soc., 2018, 140, 12493–12500 CrossRef CAS PubMed.
- C. Coaty, H. Zhou, H. Liu and P. Liu, ACS Nano, 2018, 12, 432–440 CrossRef CAS.
- R. Kanno, Y. Takeda, A. Takahashi, O. Yamamoto, R. Suyama and M. Koizumi, J. Solid State Chem., 1987, 71, 189–195 CrossRef CAS.
- R. Kanno, Y. Takeda, A. Takahashi, O. Yamamoto, R. Suyama and S. Kume, J. Solid State Chem., 1987, 71, 196–204 CrossRef CAS.
- H. D. Lutz, P. Kuske and K. Wussow, Z. Anorg. Allg. Chem., 1987, 553, 172–178 CrossRef CAS.
- O. Kononova, H. Huo, T. He, Z. Rong, T. Botari, W. Sun, V. Tshitoyan and G. Ceder, Sci. Data, 2019, 6, 1–11 CrossRef PubMed.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed.
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS.
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- X. Hua, A. S. Eggeman, E. Castillo-martínez, R. Robert, H. S. Geddes, Z. Lu, C. J. Pickard, W. Meng, K. M. Wiaderek, N. Pereira, G. G. Amatucci, P. A. Midgley, K. W. Chapman, U. Steiner, A. L. Goodwin and C. P. Grey, Nat. Mater., 2021, 20, 841–850 CrossRef CAS PubMed.
- C. M. Coaty, A. A. Corrao, V. Petrova, T. Kim, D. P. Fenning, P. G. Khalifah and P. Liu, Nanoscale, 2021, 13, 8242–8253 RSC.
- R. Dehghan, T. W. Hansen, J. B. Wagner, A. Holmen, E. Rytter, Ø. Borg and J. C. Walmsley, Catal. Lett., 2011, 141, 754–761 CrossRef CAS.
- W. A. Sławiński, E. Zacharaki, H. Fjellvåg and A. O. Sjåstad, Cryst. Growth Des., 2018, 18, 2316–2325 CrossRef.
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13849–13854 CrossRef CAS.
- P. W. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- D. Ectors, F. Goetz-Neunhoeffer and J. Neubauer, J. Appl. Crystallogr., 2015, 48, 1998–2001 CrossRef CAS.
- D. Ectors, F. Goetz-Neunhoeffer and J. Neubauer, J. Appl. Crystallogr., 2015, 48, 189–194 CrossRef CAS.
- D. Ciuculescu, F. Dumestre, M. Comesaña-Hermo, B. Chaudret, M. Spasova, M. Farle and C. Amiens, Chem. Mater., 2009, 21, 3987–3995 CrossRef CAS.
- K. Maaz, S. Karim, M. Usman, A. Mumtaz, J. Liu, J. L. Duan and M. Maqbool, Nanoscale, 2010, 5, 1111–1117 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- H. G. Liao, L. Cui, S. Whitelam and H. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- V. Chaudhary, Y. Zhong, H. Parmar, V. Sharma, X. Tan and R. v. Ramanujan, ChemistryOpen, 2018, 7, 590–598 CrossRef CAS PubMed.
- C. D. Cullity and B. D. Graham, Introduction to Magnetic Materials, 2009, vol. 12 Search PubMed.
- S. V. Trukhanov, I. O. Troyanchuk, V. V. Fedotova, V. A. Ryzhov, A. Maignan, D. Flahaut, H. Szymczak and R. Szymczak, Phys. Status Solidi B, 2005, 242, 1123–1131 CrossRef CAS.
- V. D. Doroshev, V. A. Borodin, V. I. Kamenev, A. S. Mazur, T. N. Tarasenko, A. I. Tovstolytkin and S. v. Trukhanov, J. Appl. Phys., 2008, 104, 93909 CrossRef.
- S. V. Trukhanov, L. S. Lobanovski, M. V. Bushinsky, I. O. Troyanchuk and H. Szymczak, J. Phys.: Condens. Matter, 2003, 15, 1783–1795 CrossRef CAS.
- S. v. Trukhanov, D. P. Kozlenko and A. v. Trukhanov, J. Magn. Magn. Mater., 2008, 320, 91–94 CrossRef.
- M. A. Almessiere, A. v. Trukhanov, Y. Slimani, K. Y. You, S. v. Trukhanov, E. L. Trukhanova, F. Esa, A. Sadaqati, K. Chaudhary, M. Zdorovets and A. Baykal, Nanomaterials, 2019, 9, 202 CrossRef CAS PubMed.
- A. V. Trukhanov, N. A. Algarou, Y. Slimani, M. A. Almessiere, A. Baykal, D. I. Tishkevich, D. A. Vinnik, M. G. Vakhitov, D. S. Klygach, M. V. Silibin, T. I. Zubar and S. V. Trukhanov, RSC Adv., 2020, 10, 32638–32651 RSC.
- O. Ducreux, B. Rebours, J. Lynch, M. Roy-Auberger and D. Bazin, Oil Gas Sci. Technol., 2009, 64, 49–62 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, further XRD measurements, SEM and TEM images and XRD refinement analysis. See https://doi.org/10.1039/d2ra03790d |
| This journal is © The Royal Society of Chemistry 2022 |