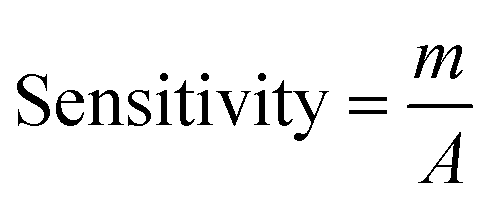DOI:
10.1039/D2RA03918D
(Paper)
RSC Adv., 2022,
12, 29491-29502
Sensitive electrochemical sensor based on nickel/PDDA/reduced graphene oxide modified screen-printed carbon electrode for nitrite detection†
Received
25th June 2022
, Accepted 30th September 2022
First published on 14th October 2022
Abstract
A simple, rapid method of the determination of nitrite in food samples is reported by using a highly sensitive electrochemical sensor based on nickel, poly(diallyldimethylammonium chloride) (PDDA), reduced graphene oxide (rGO) and a disposable screen-printed carbon electrode (SPCE). The method is based on a modification of the electrode to enhance the sensitivity and selectivity of the disposable and applicable SPCE, which is essential for the present analytical challenge. The nitrite determination was performed by using a cyclic voltammetry (CV) method under optimum conditions. Ni/PDDA/rGO/SPCE showed a linear working range of 6 to 100 μM of nitrite concentration. The limit of detection and limit of quantification were 1.99 μM (S/N = 3) and 6.6 μM (S/N = 10), respectively. The sensitivities were 0.453 μA μM−1 cm−2 for the lower concentration range and 0.171 μA μM−1 cm−2 for the higher concentration range. The Ni/PDDA/rGO sensor also showed excellent anti-interference ability and good long-term stability. The purposed disposable sensor was successfully applied to determine nitrite in sausages and pickled vegetable samples with satisfactory recovery.
Introduction
Nitrite is a well-known preservative to cure meat in the food industry. It inhibits the growth of several undesirable bacteria; however, nitrite may convert to carcinogenic nitrosamines when it reacts with secondary amine in acidic conditions. The long-term consumption of foods containing nitrite is associated with some cancer health risks.1,2 The Directive of the Scientific Committee on Food and the European Food Safety Authority has suggested that 50–100 mg nitrites per kilogram of meat may be safe to use for many meat products.3 The determination of nitrite has attracted a lot of interest owing to its harmful effects on human health and the environment. Therefore, accurate, economical and rapid quantitative determinations are required for monitoring the amount of nitrite to supervise the safety of food and water.
Several analytical techniques have been developed for quantifying the amount of nitrite. Chromatography,4,5 chemiluminescence,6 colorimetry,7 and spectroscopy8,9 techniques offer good specificity and sensitivity; however, these techniques require the use of expensive instruments with complicated procedures and long response times. Among the numerous nitrite-detection techniques, the electrochemical technique is widely used for nitrite determination due to its rapid response, simplicity, high sensitivity, high selectivity and low detection limit.10,11 Among electrochemical techniques, cyclic voltammetry is an electrochemical technique that is frequently used since it provides useful information about the redox system, including analyte concentrations, rate constants, number of transferred electrons and diffusion coefficients.2
Electrochemical sensors are a good candidate for the determination of various analytes due to their simplicity, sensitivity, and selectivity with affordable cost. The screen-printed carbon electrode (SPCE) is an electrochemical sensor that has received considerable attention in recent years because it can be modified and tailored to make it appropriate for the demands of the end user, offering high potential for commercial applications. However, an unmodified SPCE suffers from poor sensitivity and high overpotential, leading to inevitable surface fouling over time. It is still a challenge to improve the analytical responses of SPCEs. The modification of the electrode surface is an important issue in the electrochemical detection of nitrite. The main purpose of electrode modification is to facilitate electron transfer between the electrode surface and electroactive species. To achieve this purpose, carbon-based nanostructured materials, such as multi-walled carbon nanotubes,12 graphene13 and graphene nanocomposites,14,15 have been applied on the electrode surface. In addition, precious metals (Au,16 Pt,1 Pd,17), non-precious metals (Ni18 and Fe8) and metal oxides (Co3O4 (ref. 19) and Fe2O3 (ref. 20)) have been incorporated with carbon-based materials to fabricate electrodes for the electrochemical detection of nitrite.
Reduced graphene oxide (rGO), a derivative of graphene, has attracted tremendous attention due to its properties, such as its high surface area, and its excellent electrochemical properties. Chemical reduction is one of the preferred methods for preparing rGO from graphene oxide. However, the limited conductivity of rGO is still a challenging factor for an rGO-modified electrode. The decoration of metals or non-precious metals on rGO has been widely applied to improve the conductivity of rGO.21,22 Among non-precious metals, nickel has been widely used due to its low price and relatively high abundance. Therefore, nickel can be applied as an ideal substitute for precious metals to enhance the electrocatalytic activity of electrochemical sensors. However, nickel nanoparticles (Ni NPs) on the electrode surface are easily oxidized, which causes poor stability and a reduced active surface area of the electrochemical sensor. Some literature has reported that the strong interaction between Ni NPs and carbon supporting materials can prevent the oxidation of Ni NPs during operation.23
Poly(diallyldimethylammonium chloride), PDDA, is a linear cationic polyelectrolyte that has been reported to be attractive for the surface functionalization of a nanomaterial.24 It can also act as a stabilizer, disperser and linker in a nanocomposite.25 Its positive surface charge can provide additional active sites and enable electrostatic attraction for the adsorption of an analyte. These approaches can significantly improve the electrochemical performance of electrochemical sensors.
In this work, we aim to develop disposable electrochemical sensors by incorporation of nickel, rGO and PDDA for nitrite determination. Ni/PDDA/rGO was prepared by chemical reduction and its electrocatalytic activity on nitrite reduction was investigated by cyclic voltammetry. The prepared electrode showed high sensitivity, a wide linear range, a low detection limit, high reproducibility and good practicability for the detection of nitrite in real samples.
Experimental section
Chemicals and solutions
Graphene oxide dispersion in water 2 mg mL−1 (GO), poly(diallyldimethylammonium chloride) (PDDA), anhydrous sodium acetate (C2H3NaO2) and nickel(II) acetate tetrahydrate were purchased from Sigma-Aldrich (St. Louis, USA). Standard tested interfering substances, such as D-(+)-glucose, D-(−)-fructose (C6H12O6) with ≥99% purity, anhydrous zinc(II) chloride (ZnCl2) with ≥97% purity, copper(II) chloride (CuCl2), ammonium chloride (NH4Cl), iron(II) chloride tetrahydrate (FeCl2·4H2O) and iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), were obtained from Sigma-Aldrich (St. Louis, USA). Potassium chloride (KCl) was purchased from Carlo Erba Reagents (Barcelona, Spain). The electrolyte solution was a mixture of potassium dihydrogen phosphate (KH2PO4) and dipotassium hydrogen phosphate (K2HPO4) obtained from Sigma-Aldrich (St. Louis, USA). Carbon paste and Ag/AgCl paste were purchased from SunChemical (Torfaen, UK).
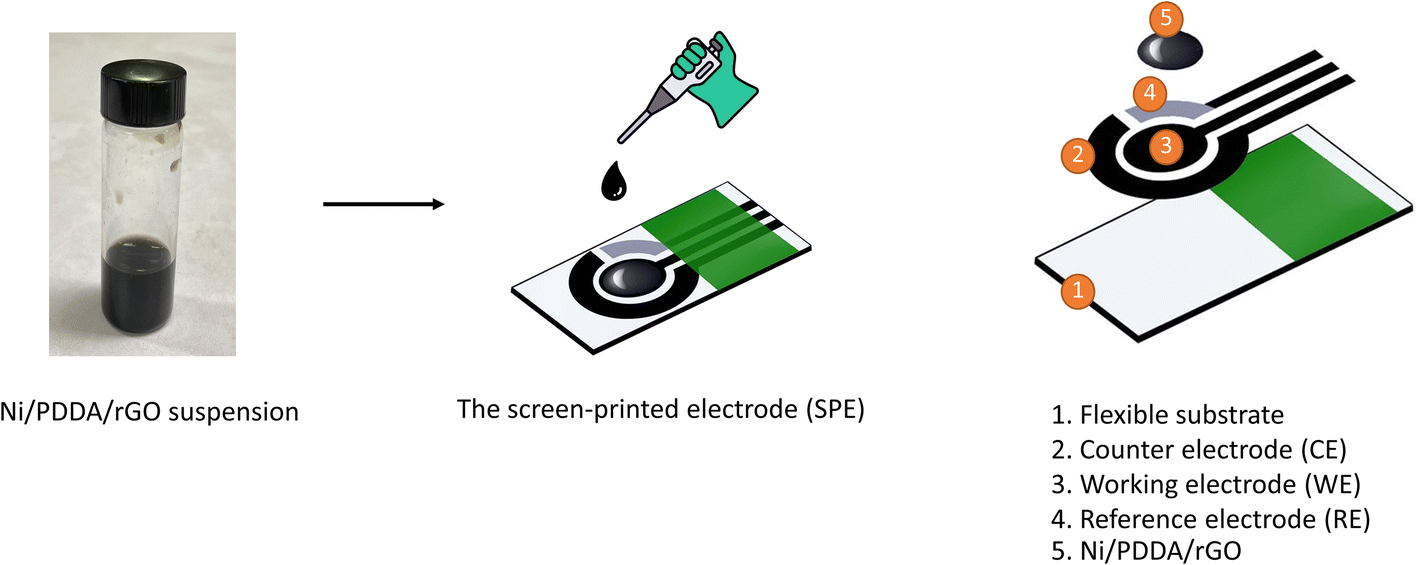
Preparation of nickel, reduced graphene oxide and PDDA screen-printed carbon electrode (Ni/PDDA/rGO/SPCE)
A screen-printing technique was applied to make the electrodes for electrochemical measurement. Carbon paste was screen-printed onto a 10 mm × 25 mm polyvinyl chloride (PVC) sheet and dried at 60 °C for 30 min to form a working electrode 3 mm in diameter and a counter electrode. After that, Ag/AgCl paste was applied onto the pre-screened PVC sheet to form a reference electrode and dried at 60 °C overnight. An insulating ink was finally screen-printed onto the three electrodes. The printed carbon electrode was dried at room temperature and kept in a desiccator.
The chemical reduction method was used to synthesize the Ni/PDDA/rGO nanoparticles by mixing 0.5 mL of GO, 0.3 mL of ultrapure water and 400 μL of 0.01 M nickel(II) acetate tetrahydrate and sonicating for 30 min. 1 mL of 0.01 M sodium borohydride was then added to reduce the mixture. The mixture was further sonicated for 30 min and left without sonication for 2 h. After that, the PDDA solution was added into the mixture and stirred for 30 min. The mixture was centrifuged twice at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The Ni/PDDA/rGO nanoparticles were redispersed in a solution of ethanol:ultrapure water in 1:1 ratio to form catalyst ink. 4 μL of Ni/PDDA/rGO catalyst ink was drop-casted onto the working electrode of the bare SPCE and dried at room temperature. The preparation process of Ni/PDDA/rGO/SPCE is shown in Scheme 1.
000 rpm for 30 min. The Ni/PDDA/rGO nanoparticles were redispersed in a solution of ethanol:ultrapure water in 1:1 ratio to form catalyst ink. 4 μL of Ni/PDDA/rGO catalyst ink was drop-casted onto the working electrode of the bare SPCE and dried at room temperature. The preparation process of Ni/PDDA/rGO/SPCE is shown in Scheme 1.
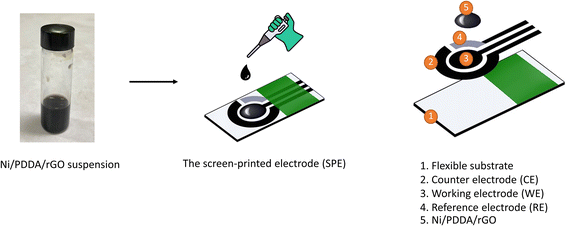 |
| | Scheme 1 Schematic illustration of Ni/PDDA/rGO/SPCE. | |
Electrochemical measurement
A 100 μM nitrite solution was prepared and used as a stock solution. The stock solution was then diluted with 0.1 M PBS solution (pH 6.0) to obtain nitrite standard solutions in a concentration range of 6 to 100 μM. For the cyclic voltammetric measurement in the study of nitrite determination, 100 μL of 0.1 M PBS electrolyte at pH 6.0 was dropped onto Ni/PDDA/rGO/SPCE. The performance of the electrode was tested by applying a potential ranging from 0.2 to 1.0 V for 30 scans at a scan rate of 50 mV s−1. The PBS electrolyte was removed. 100 μL of the nitrite standard solution in 0.1 M PBS pH 6.0 was then dropped onto the electrode surface for the determination of nitrite at a scan rate of 50 mV s−1. The potential of the electrode was controlled by an Autolab PGstat20 potentiostat (Metrohm-Autolab, Netherlands).
Preparation of real samples
As-prepared Ni/PDDA/rGO/SPCE was used to test the electrochemical determination of nitrite in food samples. Sausage and pickled vegetable samples obtained from a local market in Bangkok were used in this study. The extraction of nitrite from the samples was undertaken by mixing 5 g of a crushed sample with 100 mL of ultrapure water. The mixture was heated at 70 °C for 2 h. The filtrate was filtered and cooled down before mixing with PBS.
A standard addition method was adopted to measure nitrite in food samples. 100 μM of nitrite standard solution was spiked into the stock solution of food samples to give concentrations of 5, 10, 15 and 20 μM in 0.1 M PBS solution (pH 6.0). The CV measurements of spiked and non-spiked samples were carried out under optimum conditions at a scan rate of 50 mV s−1. The obtained results were used to calculate the percentage recovery (%R) as follows:
| | |
% R = (CS − CU) × 100/CA
| (1) |
where
CS is the concentration of nitrite in the spiked sample,
CU is the concentration of nitrite in the non-spiked sample and
CA is the concentration of nitrite in the non-spiked sample. Calibration curves in the range of 6 to 100 μM were used to calculate nitrite concentration in the food samples. The experiment was repeated three times for each concentration level and the relative standard deviation (RSD) was calculated.
Results and discussion
Characterizations
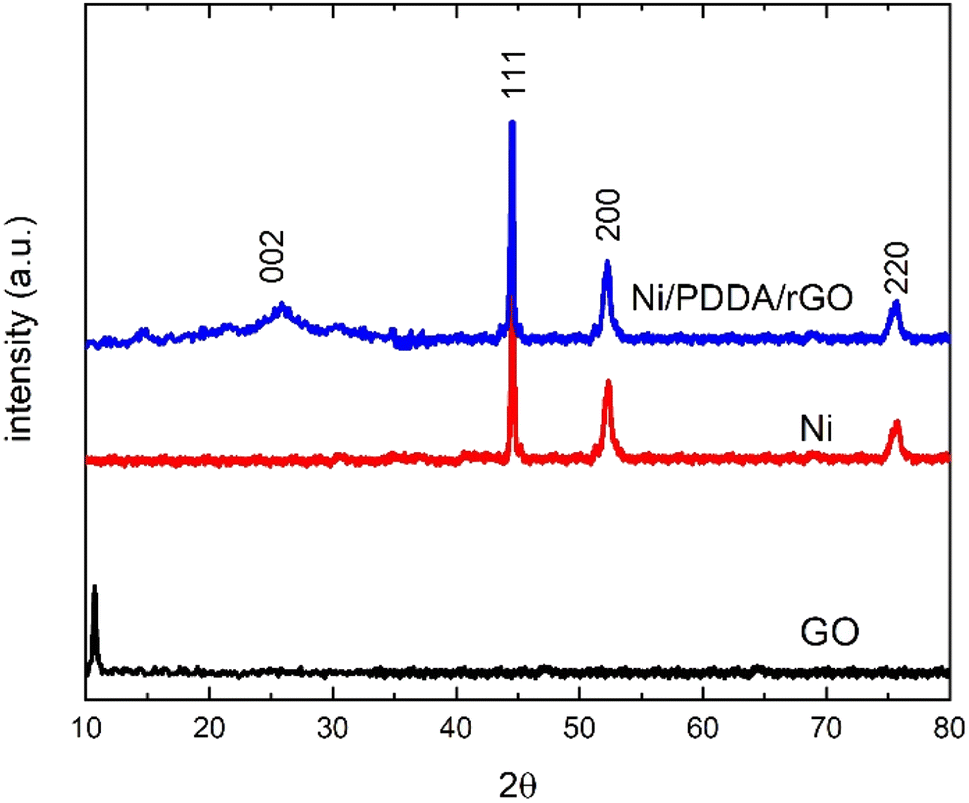
The phase structure of the Ni/PDDA/rGO catalyst ink was firstly determined by XRD. Fig. 1 shows XRD patterns of GO, Ni and Ni/PDDA/rGO. Within the 2θ range investigated, GO showed the characteristic peak at 2θ of around 10.9°, which corresponded to the (0 0 1) reflection of graphene oxide and existence of oxygen-rich groups on the GO sheet.26 For Ni, the peaks at 44.3°, 51.9°, and 76.3° arose from the Ni (111), (200), and (220) crystal planes, respectively, of face-centered cubic (fcc) Ni, in good agreement with the standard for Ni (JCPDS 04-0850).27 As for Ni/PDDA/rGO, after chemical reduction of GO a broad peak can be seen at 23.2°, which was attributed to the (002) diffraction of rGO. The presence of the broad peak for (002) indicated that the crystal plane of rGO was rearranged randomly and oxygen-rich groups on graphene oxide in Ni/PDDA/rGO were reduced. The peaks arising from Ni were also present in the Ni/PDDA/rGO spectrum, in good agreement with those of the Ni spectrum.
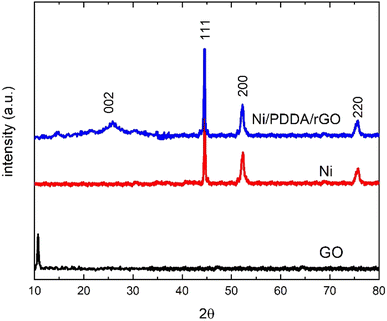 |
| | Fig. 1 XRD patterns of GO, Ni and Ni/PDDA/rGO. | |
The surface morphologies of rGO/SPCE and Ni/PDDA/rGO/SPCE were characterized using SEM and EDS analysis. Fig. 2(a) and (b) show SEM images of rGO/SPCE and Ni/PDDA/rGO/SPCE, respectively. rGO/SPCE showed a cloudy and wrinkled texture of rGO sheets that fully covered the SPCE surface. The wrinkled texture was due to the removal of oxygen functional groups of the GO sheets which prevented the stacking of the GO sheets. Ni/PDDA/rGO/SPCE showed spherical-shaped particles of Ni distributed on the surface of the rGO sheets. It was interesting that the cloudy and wrinkled texture of the rGO sheets became stretched and smooth after the introduction of Ni nanoparticles, since the nanoparticles acted as a nanospacers between the rGO layers. Fig. 2(c) and (d) show EDS analysis of rGO/SPCE and Ni/PDDA/rGO/SPCE, respectively. The spectra of carbon and oxygen at 0.27 and 0.52 keV were present in both rGO/SPCE and Ni/PDDA/rGO/SPCE, confirming the elemental composition of rGO. The spectrum of N at 0.39 keV and that of Ni at 0.85 keV were due to the presence of PDDA and Ni particles, respectively, on Ni/PDDA/rGO/SPCE. The EDX analysis of Ni/PDDA/rGO/SPCE showed well-defined elemental peaks for carbon, nitrogen, oxygen and nickel, which further confirmed the elemental purity of the Ni/PDDA/rGO catalyst ink. Fig. S1† shows an average elemental composition of Ni/PDDA/rGO/SPCE, which predicted the presence of elemental C (55.11 wt%), N (5.05 wt%), O (17.83 wt%) and Ni (22.01 wt%). The uniform distribution of elements in Ni/PDDA/rGO/SPCE was confirmed by EDX mapping analysis, as shown in Fig. 2(e)–(h). It was evident that carbon, nitrogen, oxygen and nickel were evenly distributed throughout the electrode surface.
 |
| | Fig. 2 SEM images of (a) rGO/SPCE, (b) Ni/PDDA/rGO/SPCE, EDX spectrum of (c) rGO/SPCE, (d) Ni/PDDA/rGO/SPCE and (e)–(h) EDX mappings of Ni/PDDA/rGO/SPCE. | |
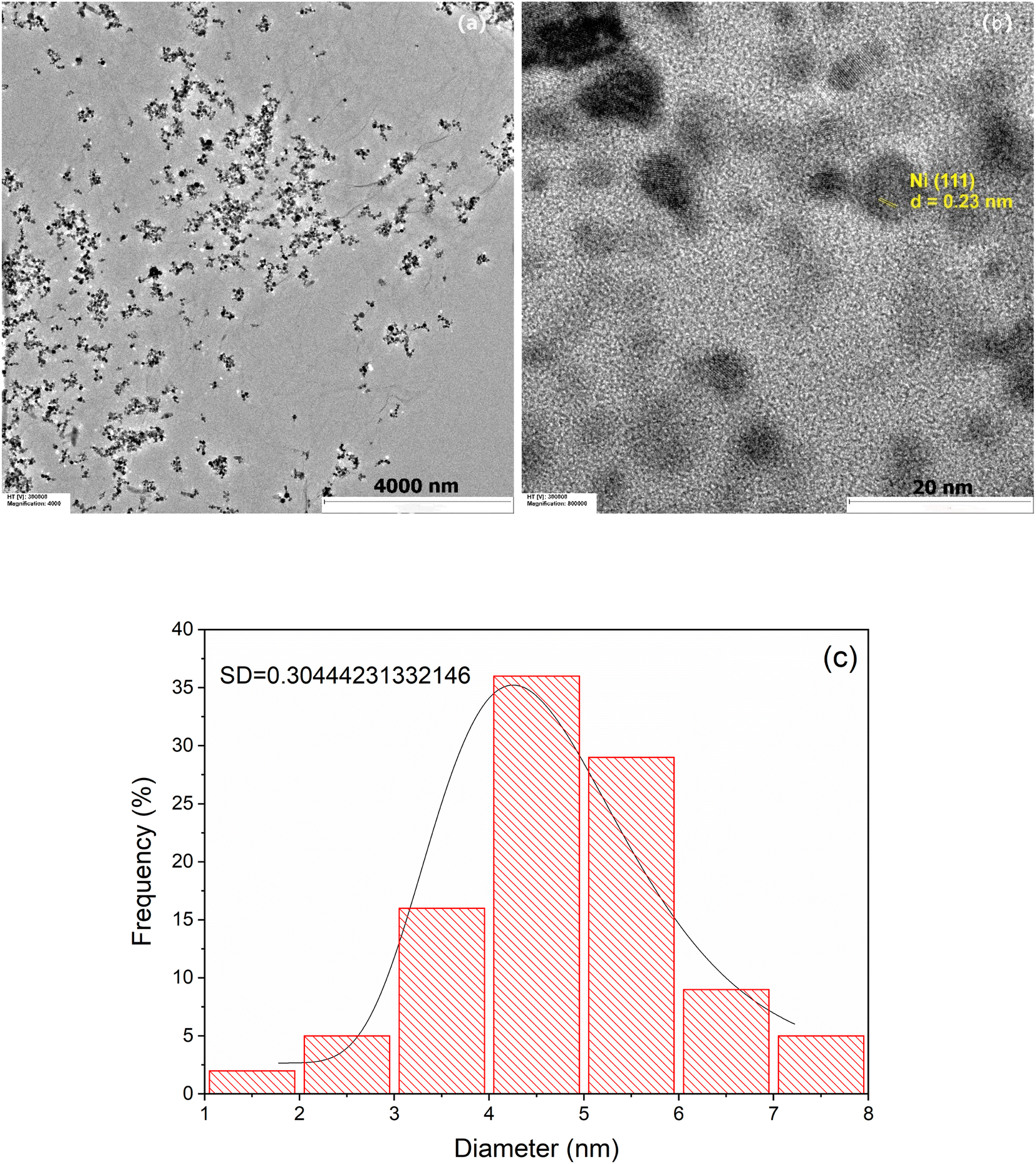
Fig. 3 shows representative TEM images at lower (a) and higher (b) magnification of the prepared Ni/PDDA/rGO catalyst ink. The deposited nanoparticles were well distributed on the rGO sheets. Fig. 3(b) shows a lattice fringe of selected nanoparticles. The D-spacing of the crystal was 0.23 nm, corresponding to the (111) crystal plane of metallic Ni.28 The Ni nanoparticles were uniformly dispersed over the rGO sheet due to the crystal growth of nickel nanoparticles during chemical reduction. The rGO sheets acted as protective layers for the growth of Ni nanoparticles. The size of the nanoparticles was carefully determined by selecting 100 nanoparticles to obtain the most predominant diameter of the particles. The histogram of the size of the Ni nanoparticles on the rGO sheet shows a narrow size distribution with an average diameter of 4.33 ± 2.16 nm, as shown in Fig. 3(c).
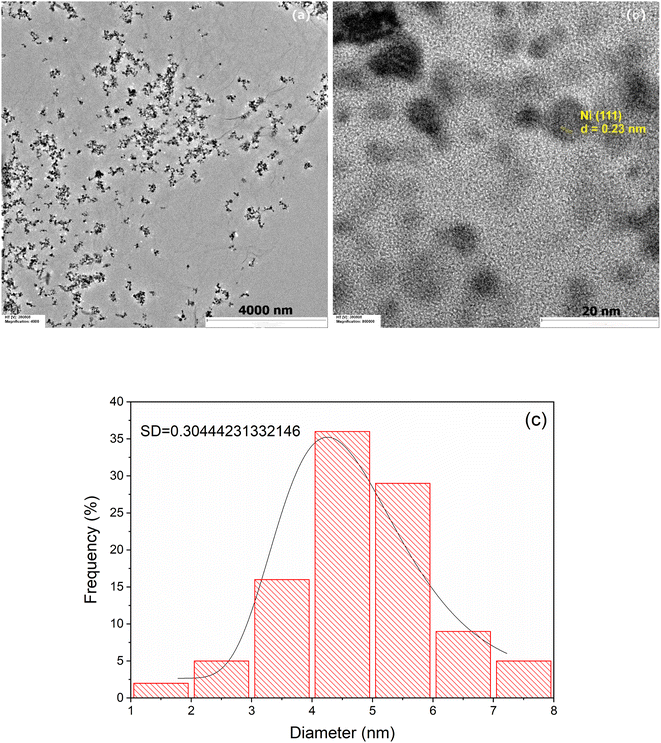 |
| | Fig. 3 TEM images of Ni/PDDA/rGO at (a) low magnification, (b) high magnification and (c) the histogram of the size of Ni nanoparticles on the surface of the rGO sheet. | |
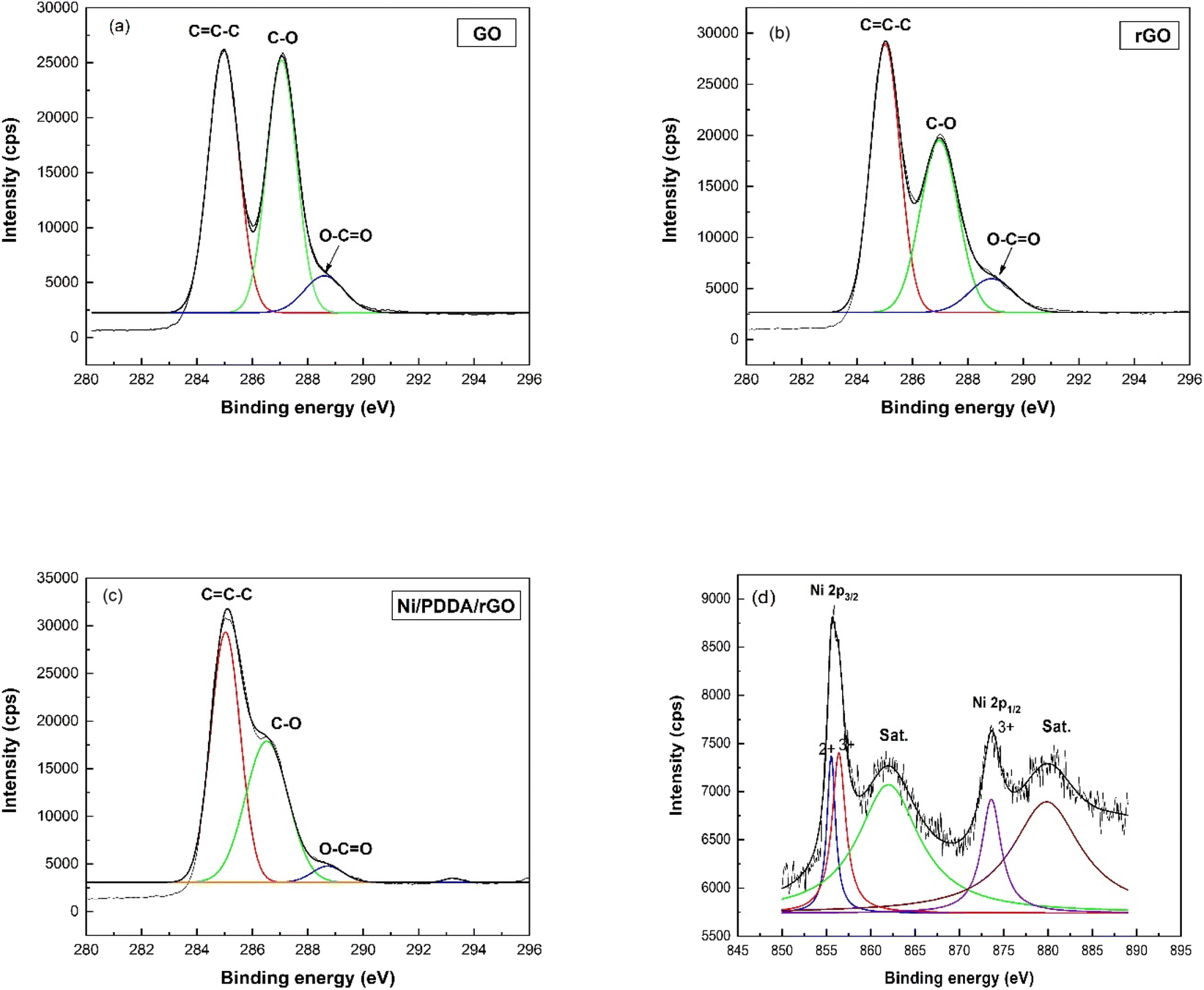
The XPS technique was used to confirm the surface composition and electronic valence states of the prepared catalyst ink. The C1s XPS spectra of GO, rGO and Ni/PDDA/rGO are shown in Fig. 4(a)–(c), respectively, to demonstrate the successful reduction of GO to rGO by chemical reduction. The C1s spectrum of GO was de-convoluted into three peaks located at 284.8 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C), 286.8 eV(C–O) and 289.0 eV (O–CO). The C1s XPS spectra of rGO and Ni/PDDA/rGO showed similar components to GO. However, the peaks attributed to oxygen-functionalized C were much weaker than those of GO, indicating that most of the oxygen-containing functional groups were deoxygenated during chemical reduction. Fig. 4(d) shows the Ni 2p spectrum of Ni/PDDA/rGO. The Ni 2p spectrum showed two spin–orbital doublets at 855.6 eV and 873.6 eV, attributed to Ni 2p3/2 and Ni 2p1/2, respectively.29,30 In addition, shakeup satellites at around 862.0 eV and 879.8 eV, characteristic of Ni in the form of oxide and hydroxide phases, were observed. The peaks shifted to higher binding energy compared to those of standard binding energy, which were at 856.0 and 873.0 eV. The shift of the peaks was due to interaction between Ni particles and the rGO sheet. The formation of nickel oxide also had an effect on the shift of binding energy, while the peaks at higher energy (≥855.0 eV) were associated with Ni2O3.31,32
C–C), 286.8 eV(C–O) and 289.0 eV (O–CO). The C1s XPS spectra of rGO and Ni/PDDA/rGO showed similar components to GO. However, the peaks attributed to oxygen-functionalized C were much weaker than those of GO, indicating that most of the oxygen-containing functional groups were deoxygenated during chemical reduction. Fig. 4(d) shows the Ni 2p spectrum of Ni/PDDA/rGO. The Ni 2p spectrum showed two spin–orbital doublets at 855.6 eV and 873.6 eV, attributed to Ni 2p3/2 and Ni 2p1/2, respectively.29,30 In addition, shakeup satellites at around 862.0 eV and 879.8 eV, characteristic of Ni in the form of oxide and hydroxide phases, were observed. The peaks shifted to higher binding energy compared to those of standard binding energy, which were at 856.0 and 873.0 eV. The shift of the peaks was due to interaction between Ni particles and the rGO sheet. The formation of nickel oxide also had an effect on the shift of binding energy, while the peaks at higher energy (≥855.0 eV) were associated with Ni2O3.31,32
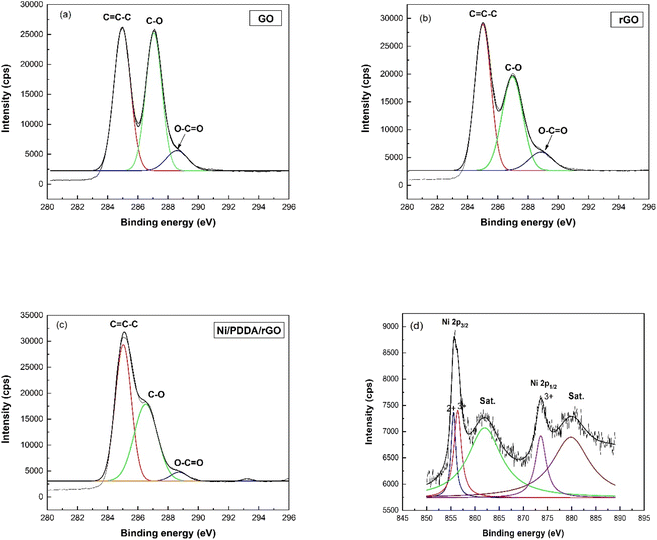 |
| | Fig. 4 XPS C1s spectra of (a) GO, (b) rGO, and (c) Ni/PDDA/rGO. (d) Ni2p spectra of Ni/PDDA/rGO. | |
Electrochemical measurement
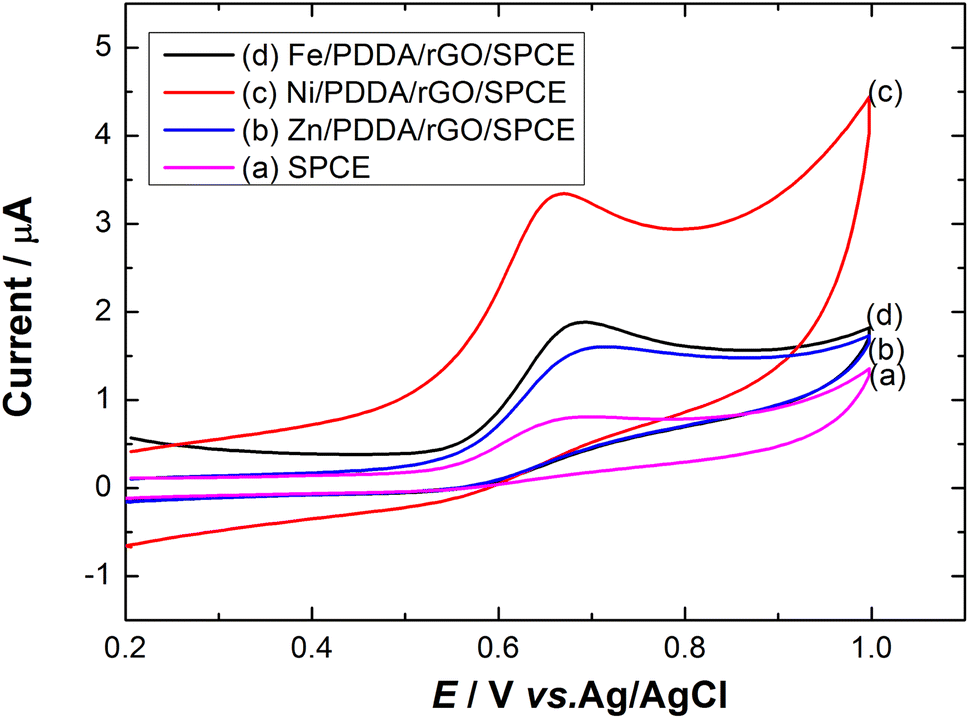
Effect of metals. To investigate the effect of metal on nitrite detection, different metals were used to modify the surface of the SPCE. Fig. 5 shows cyclic voltammograms of Zn, Ni and Fe/PDDA/rGO/SPCE in N2-saturated 20.0 μM NaNO2 and 0.1 M PBS (pH 6.00). The anodic peak current of Ni/PDDA/rGO/SPCE was higher than that of Fe/PDDA/rGO/SPCE, Zn/PDDA/rGO/SPCE or SPCE, indicating that Ni can facilitate faster electron transfer between the electrode surface and nitrite ions. Hence, Ni was chosen for nitrite detection in this work.
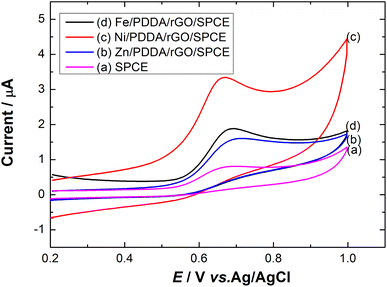 |
| | Fig. 5 Cyclic voltammograms of (a) SPCE, (b) Zn/PDDA/rGO/SPCE, (c) Ni/PDDA/rGO/SPCE and (d) Fe/PDDA/rGO/SPCE in 20.0 μM NaNO2 + 0.1 M PBS (pH 6.00) at a scan rate of 50 mV s−1. | |
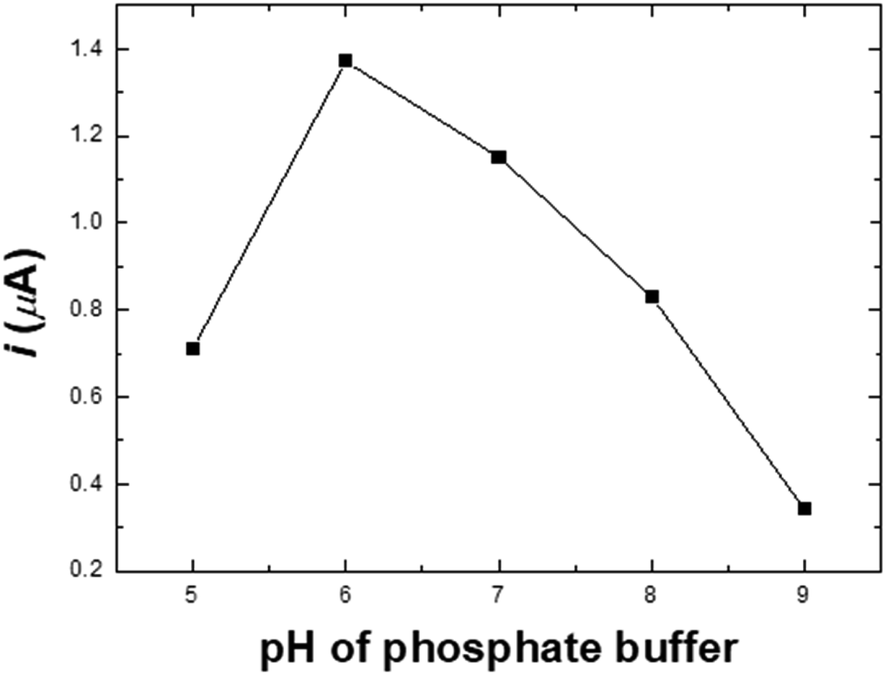
Effect of pH. A study of how pH affects the determination of nitrite in the pH range of 5.0–9.0 in PBS solution containing 20.0 μM NaNO2 on the Ni/PDDA/rGO/SPCE is presented in Fig. S2.† The anodic peak currents of nitrite increased with rising pH and reached a maximum at pH 6.0, dropping afterwards, as shown in Fig. 6. The instability of the nitrite ion was activated when the pH was lower than 7.0 due to its decomposition, as shown in eqn (2). The NO2− ion can be decomposed into NO and NO3−, resulting in lower peak current.13 However, nitrite oxidation was more difficult at a pH higher than 7 since the pKa of HNO2 was 3.3. Most nitrite ions were protonated in the acidic solutions. It was possible that the shortage of protons decreased the catalytic reaction of nitrite; therefore, the anodic peak current increased with the decrease in solution pH.33–35| | |
2H+ + 3NO2− → 2NO + NO3− + H2O
| (2) |
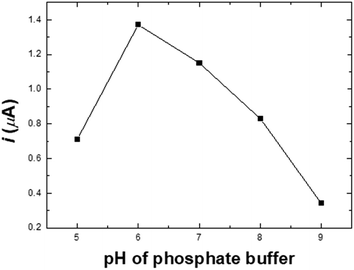 |
| | Fig. 6 The oxidation peak currents of nitrite vs. pH in 20.0 μM NaNO2 + 0.1 M PBS at a scan rate of 50 mV s−1. | |
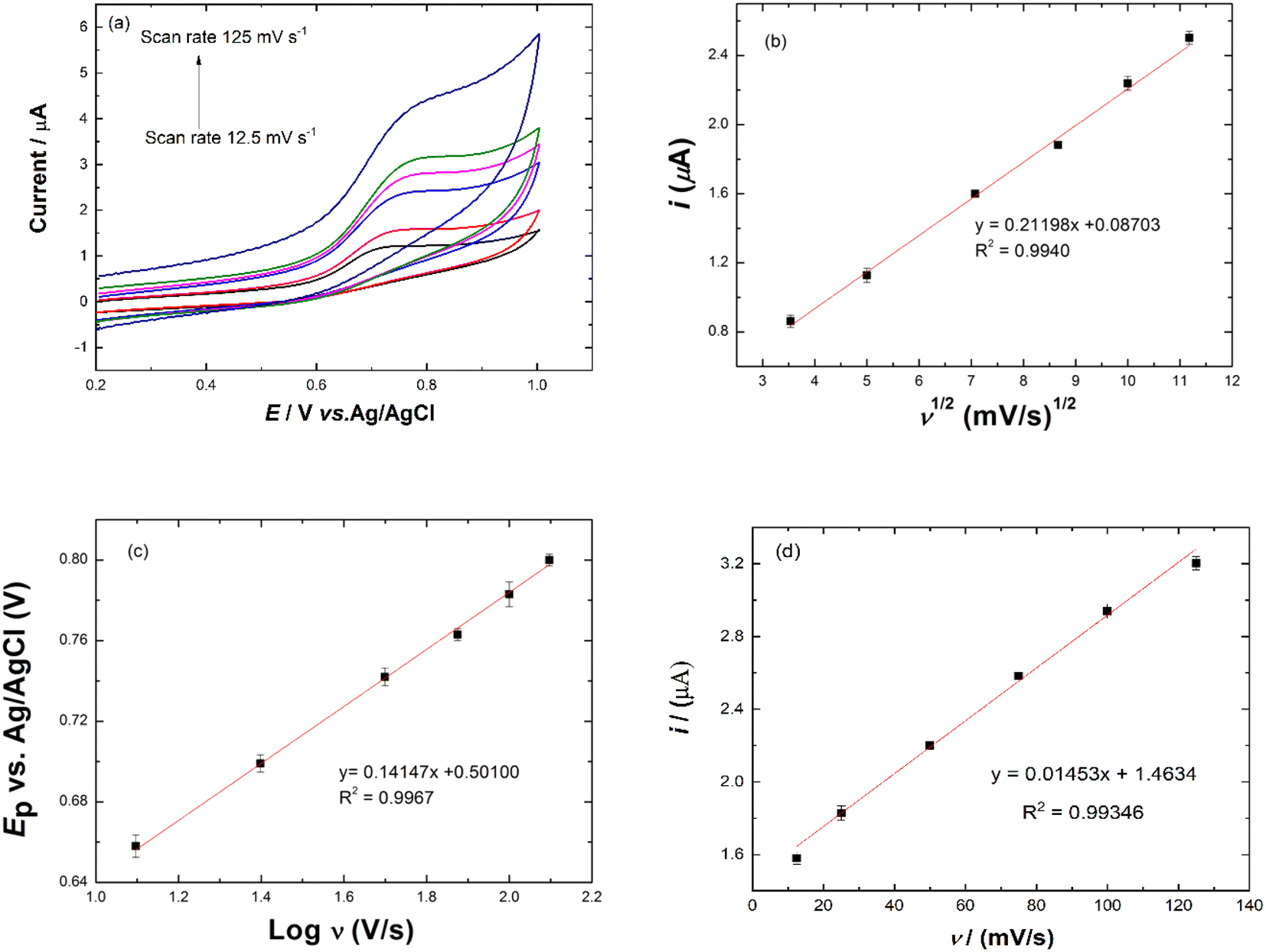
Effect of scan rate. The effect of scan rate on peak potential of nitrite oxidation was investigated. Fig. 7(a) shows the CVs of scan rates ranging from 12.5 to 125.0 mV s−1 in 20.0 μM NaNO2 and PBS solution pH 6.0. The peak potential of nitrite oxidation shifted towards positive potential as the scan rate increased. The linear regression equation for square root of scan rate vs. currents was obtained with the following equation: ipa (μA) = 0.21198ν1/2 (mV s−1)1/2 + 0.08703 with a correlation coefficient of 0.9940, as shown in Fig. 7(b), suggesting that nitrite oxidation was a diffusion-controlled process. The logarithms of scan rates and peak potentials are shown in Fig. 7(c). The regression equation was Epa (V) = 0.14147logν (V s−1) + 0.50100 (R2 = 0.9967). The value of αn was calculated from the slope of the Ep vs. logν curve and eqn (3). The slope was 0.14147, so the value of αn was 0.40560. The value of α was calculated from eqn (4). The value of α was 0.4057. The number of electrons (n) transferred in the electro-oxidation of nitrite was 0.99 ≈ 1, which means the electron transfer process was involved in the rate-determining step. The catalytic mechanism of the Ni/PDDA/rGO for oxidation of nitrite in eqn (5) and (6) showed that the reaction involved one electron.36| | |
Ep = A + (2.303RT)/αnFlogν
| (3) |
| | |
Ep/2 − Ep = 1.857(RT/αF)
| (4) |
| | |
Ni3+ + NO2 + H2O → Ni2+ + NO3− + 2H +
| (6) |
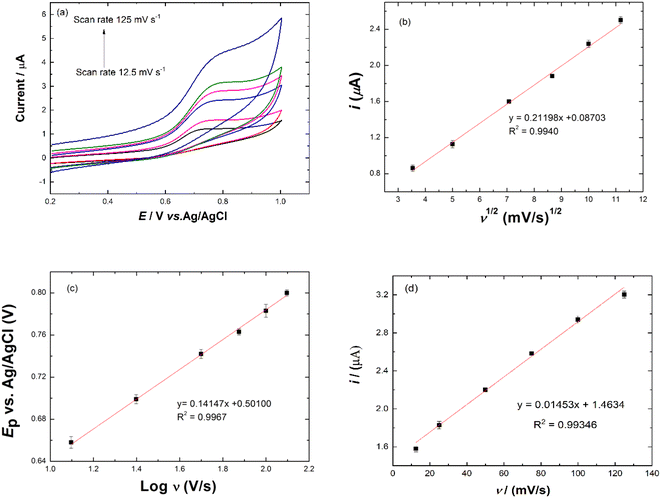 |
| | Fig. 7 (a) Cyclic voltammograms of Ni/PDDA/rGO/SPCE in 20.0 μM NaNO2 and PBS solution pH 6.0 at different scan rates from 12.5 to 125 mV s−1. (b) Plot of square root scan rate and Ip. (c) Plot of logarithm of the scan rate vs. Ep and (d) plot of scan rate vs. Ip. | |
The average surface concentration (Γ) of the electroactive sites of the Ni/PDDA/rGO/SPCE surface can be estimated from the slope of Ip vs. scan rate, as shown in Fig. 7(d), using the Brown–Anson model37,38 based on eqn (7):
where
n is the number of electrons transferred,
F is the Faraday constant (96
485 C mol
−1),
A is the surface area of the electrode (0.07065 cm
2),
R is the gas constant (8.314 J mol
−1 K
−1),
T is the absolute temperature (298 K) and
Ip/
ν is the slope of the calibration plot (scan rate value). The average surface concentration (
Γ) of the electroactive sites of the Ni/PDDA/rGO/SPCE surface was 4.33 × 10
−9 mol cm
−2.
The electroactive surface area of Ni/PDDA/rGO/SPCE was analyzed using cyclic voltammetry at various scan rates in a solution of 5 mM Fe(CN)63−/4− in 0.1 M KCl, as shown in Fig. S3(a).† The anodic peak current (Ipa) linearly increased with the square root of scan rate (ν1/2), as shown in Fig. S3(b),† suggesting that the redox reaction on the electrode surface was a diffusion-controlled process. The electrochemically active surface area (ECSA) of Ni/PDDA/rGO/SPCE was calculated using the Randles–Sevcik equation for a reversible process:39
| | |
Ip = (2.69 × 105)n3/2AD1/2υ1/2C0
| (8) |
where
Ip is the anodic peak current,
n is the number of electrons transferred in the redox event,
A is the electrochemically active surface area (cm
2),
D is the diffusion coefficient (7.6 × 10
−6 cm
2 s
−1 for Fe(CN)
63−/4−),
C0 is the bulk concentration of the analyte (mol cm
−3), and
υ is the scan rate (V s
−1). Based on the slope of the plot of
Ip vs. υ1/2, the calculated ECSA was 9.07 × 10
−2 cm
2 for Ni/PDDA/rGO/SPCE.
Electrochemical impedance spectroscopy
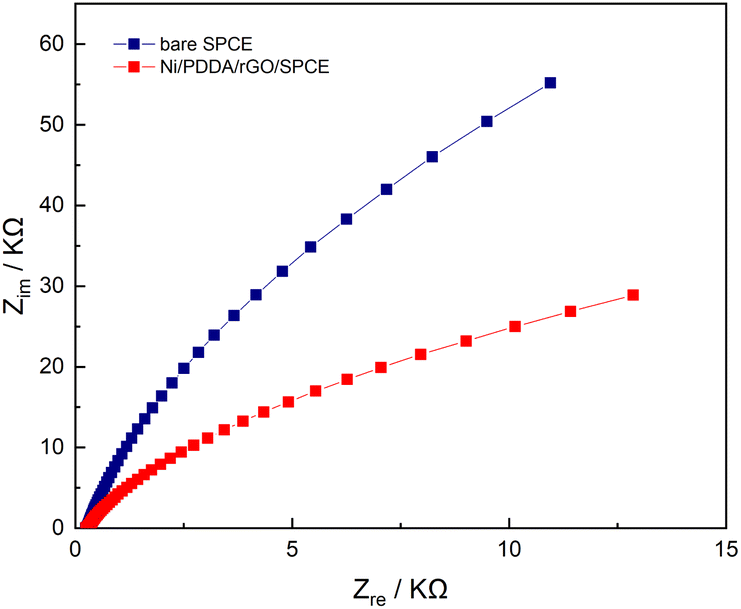
Electrochemical impedance spectroscopy (EIS) is a powerful method for studying the electrochemical properties between interfaces on a working electrode. EIS measurements were performed on both a bare SPCE and an Ni/PDDA/rGO-modified SPCE in a solution of 0.5 mM [Fe(CN)6]3−/4− in 0.1 M KCl. The frequencies used in the EIS measurement were 5 Hz to 100 kHz with 10 mA perturbation and no applied direct potential. As can be seen in Fig. 8, the well-defined semicircle at a higher frequency of the Nyquist plot decreased after introducing nickel, PDDA and rGO onto the bare SPCE. A simple Randles circuit was used to fit the EIS data. We found that the charge transfer resistance (Rct) of the SPCE decreased from 220 kΩ to 59 kΩ after modification with nickel, PDDA and rGO. This could be ascribed to the increase in electrical conductivity of the modified electrode. In addition, the double layer capacitance (Cdl) of the SPCE increased from 489.4 nF to 727.1 nF, indicating a boost in active electrochemical sites on the SPCE after modification. The EIS results confirm the enhancement of electron transfer efficiency between the active species in the solution and the SPCE by modification with nickel, PDDA and rGO.
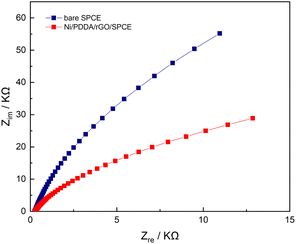 |
| | Fig. 8 Nyquist plots of bare SPCE and Ni/PDDA/rGO/SPCE in a solution of 0.5 mM of [Fe(CN)6]3−/4− in 0.1 M KCl with the frequency ranging from 5 Hz to 100 kHz. | |
Electrochemical determination of nitrite (n = 3)
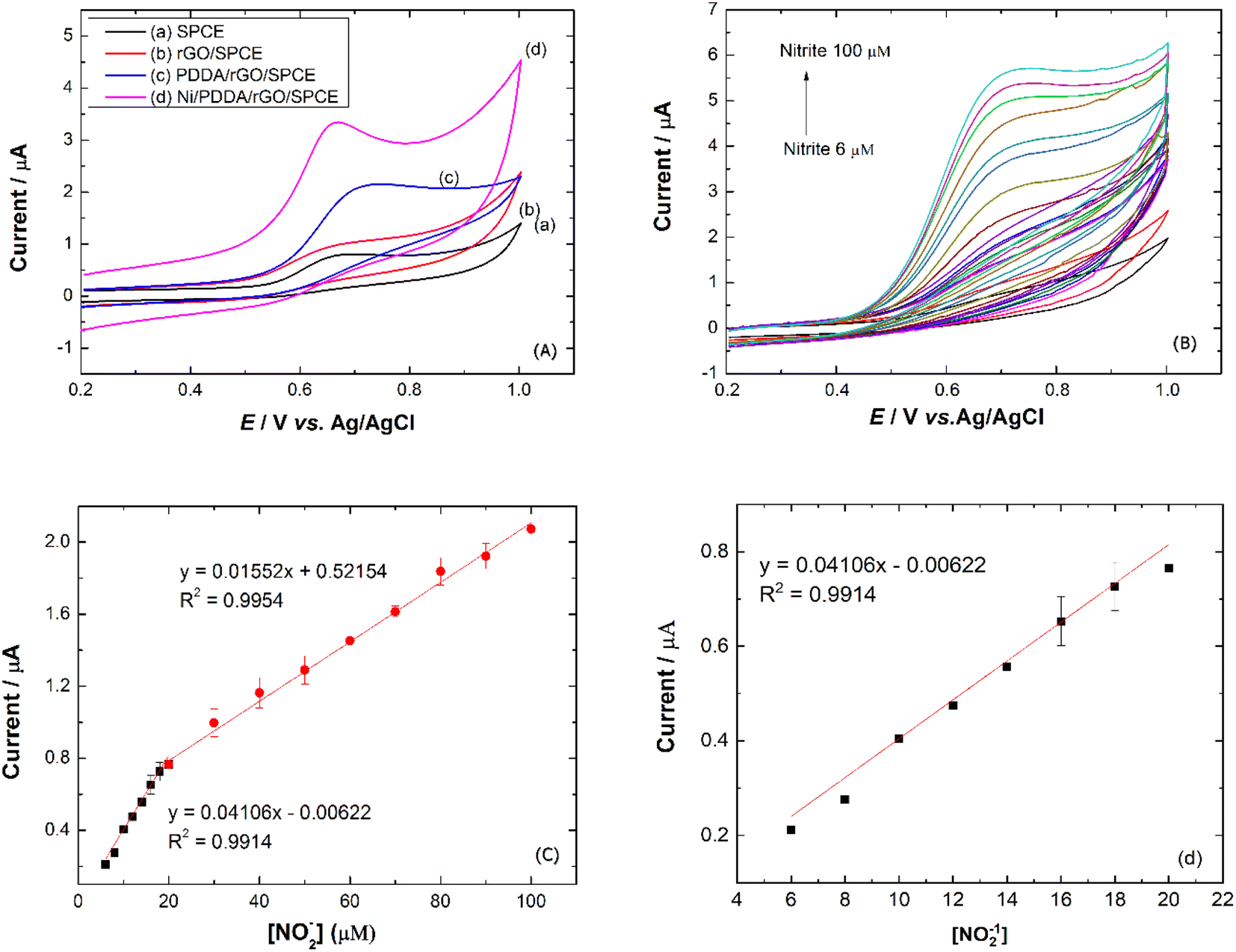
To study the electrochemical performance of the prepared SPCE, CV was used to determine nitrite in PBS buffer solution. The cyclic voltammograms of (a) SPCE, (b) rGO/SPCE, (c) PDDA/rGO/SPCE and (d) Ni/PDDA/rGO/SPCE in PBS solution containing 20.0 μM NaNO2 at a scan rate of 50 mV s−1 are shown in Fig. 9(a). The CV signal of the electrodes showed peaks at Epa = 0.70 V, which were attributed to oxidation of nitrite. Compared with SPCE, rGO/SPCE and PDDA/rGO/SPCE, Ni/PDDA/rGO/SPCE showed the highest anodic peak current and a lower overpotential. The high electrocatalytic activity of Ni/PDDA/rGO/SPCE towards nitrite oxidation could possibly be attributed to the uniform size and good dispersion of Ni over the rGO sheet, which resulted in the active sites on the prepared SPCE.
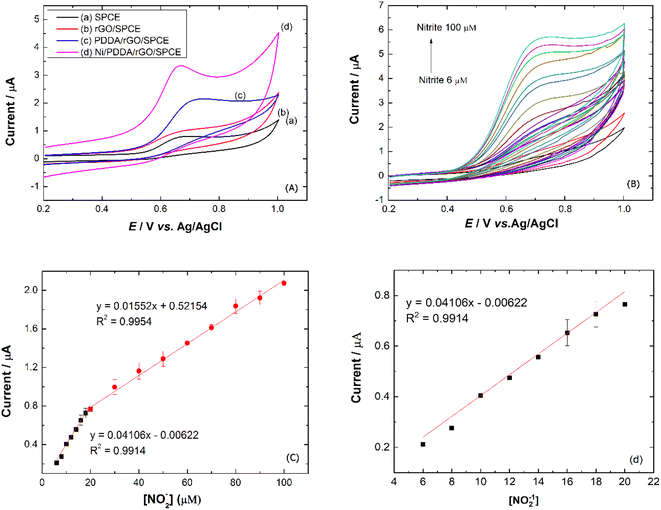 |
| | Fig. 9 (a) CVs of prepared electrodes in 20.0 μM NaNO2 and PBS pH 6.0. (b) CVs of Ni/PDDA/rGO in 0.1 M PBS pH 6.0 with concentrations from 6 to 100 μM of NaNO2 at a scan rate of 50 mV s−1. (c) Plots of Ipa vs. [NO2−] and (d) plots of Ipa vs. [NO2−] at lower concentration. | |
A relationship between the anodic peak current and concentration of nitrite by Ni/PDDA/rGO/SPCE was investigated by using cyclic voltammetry under the optimal conditions. Fig. 9(b) shows CVs obtained from different concentrations of standard nitrite solution ranging from 6 to 100 μM. A well-defined peak at +0.70 V corresponded to the peak of nitrite oxidation. The peak currents of nitrite oxidation increased as the nitrite concentration increased. Fig. 9(c) shows a linear calibration plot of nitrite concentration and anodic peak currents measured using Ni/PDDA/rGO/SPCE. The linear correlation between anodic current and the concentration of nitrite ranges from 6 μM to 20 μM and from 20 μM to 100 μM. The linear regression of the lower range was Ipa (μA) = 0.04106C (μM) − 0.00622 with R2 = 0.9914, as shown in Fig. 9(d), and that of the higher range was Ipa (μA) = 0.01552C (μM) + 0.52154 with R2 = 0.9954. The sensitivity was calculated to be 0.453 μA μM−1 cm−2 for the lower concentration range and 0.171 μA μM−1 cm−2 for the higher concentration range based on eqn (9):39
| |
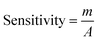 | (9) |
where
m is the slope of the calibration plot (μA μM
−1),
A is the active surface area of the working electrode (cm
2). The two linear ranges of nitrite detection on Ni/PDDA/rGO/SPCE could be explained by the rapid conversion of nitrite into nitrate on the electrode surface in the low concentration range. Nitrite adsorption on the surface of the electrode was inevitable, resulting in hindered nitrite diffusion in the high concentration range. Therefore, the sensitivity of the electrode in the low concentration range of nitrite was higher than that in the high concentration range of nitrite.
40 The limit of detection (LOD) and the limit of quantification (LOQ) were 1.99 μM (S/N = 3) and 6.6 μM (S/N = 10), respectively. The purposed electrode is comparable with previously reported results given in
Table 1. The LOD and linear range of the purposed Ni/PDDA/rGO/SPCE sensor showed promising quantitative performance in nitrite determination, especially in comparison with other precious metal modified SPCEs.
Table 1 Comparison of the efficiency of Ni/PDDA/rGO/SPCE with literature on modified electrodes for nitrite determination
| Modified electrodes |
Determination method |
LOD (μM) |
Linear range (μM) |
Sensitivity μA μM−1 cm−2 |
Ref. |
| Fe3O4/r-GO/GCE |
Amperometric |
0.3 |
1–210 |
— |
41 |
| Cu-NDs/rGO/GCE |
Amperometric |
0.4 |
1250–13000 |
0.214 |
21 |
| CTAB-GO/MWCNTs/GCE |
DPV |
1.5 |
5–800 |
— |
42 |
| Co3O4-rGO/CNTs |
Amperometric |
0.016 |
100–8000 and 8000–56000 |
0.408 |
19 |
| 0.153 |
| Pt/Ni(OH)2/MWCNTs/GC |
Amperometric |
0.13 |
0.4–5670 |
0.145 |
43 |
| Ni7S6/MWCNTs |
Amperometric |
0.30 |
1–4200 |
0.18504 |
44 |
| ErGO/Au NPs/SPCE |
CV |
0.13 |
1–6000 |
0.3048 |
45 |
| Graphite/CD/SPCE |
Amperometric |
0.26 |
0.7–2150 |
0.4762 |
46 |
| LIG/f-MWCNT-AuNPs/SPCE |
SWV |
0.9 |
10–140 |
— |
47 |
| AuNPs–Fe2O3 d |
CV |
0.07 |
1–1000 |
0.149 |
48 |
| Fe3O4/GO/COOH/GC |
DPV |
0.37 |
90–600 |
2.17 |
49 |
| Au/CaCO3–GCE |
CV |
0.0115 |
1–300 |
— |
50 |
| Ni/PDDA/rGO/SPCE |
CV |
1.99 |
6–20 and 20–100 |
0.453 |
This work |
Interference studies and real sample analysis
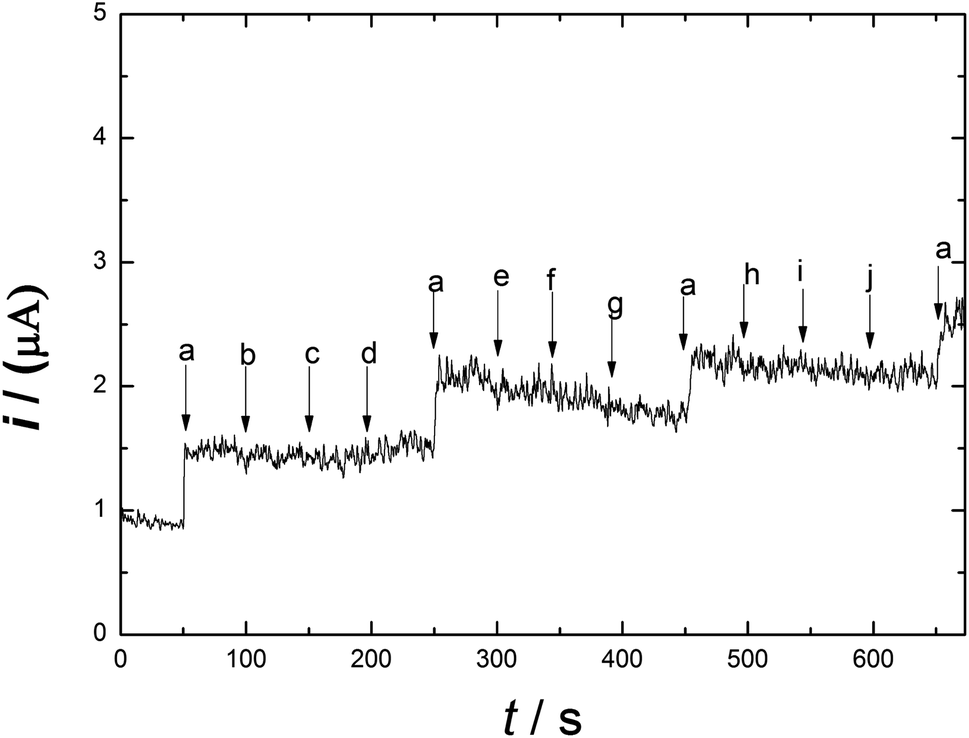
Meat products usually contain additives and chemical substances, such as Fe3+, Cu2+, Zn2+, K+, CH3COO−, Fe2+, NH4+, glucose and fructose. The selectivity and sensitivity of Ni/PDDA/rGO/SPCE for the determination of nitrite in the presence of possible sources of interference was examined by the amperometric method. Fig. 10 shows the amperometric i–t response of Ni/PDDA/rGO/SPCE with successive additions of nitrite, Fe3+, Cu2+, Zn2+, K+, CH3COO−, Fe2+, NH4+, glucose and fructose. An amperometric response of the modified electrode was observed at once with the addition of nitrite; however, the additions of the interference species did not obviously affect the current response. The modified electrode showed the same current response as in the first addition, indicating that the electrode has high selectivity towards nitrite and has anti-poisoning ability.
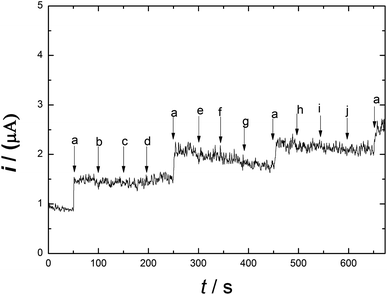 |
| | Fig. 10 Amperometric responses on Ni/PDDA/rGO for the successive additions of 1 μM NaNO2 (a) and each 100 μM addition of other interference species: Fe3+ (b), Cu2+ (c), Zn2+ (d), K+ (e), CH3COO− (f), Fe2+ (g), NH4+ (h), glucose (i) and fructose (j) in phosphate buffer solution (pH 6.00) at 0.90 V. | |
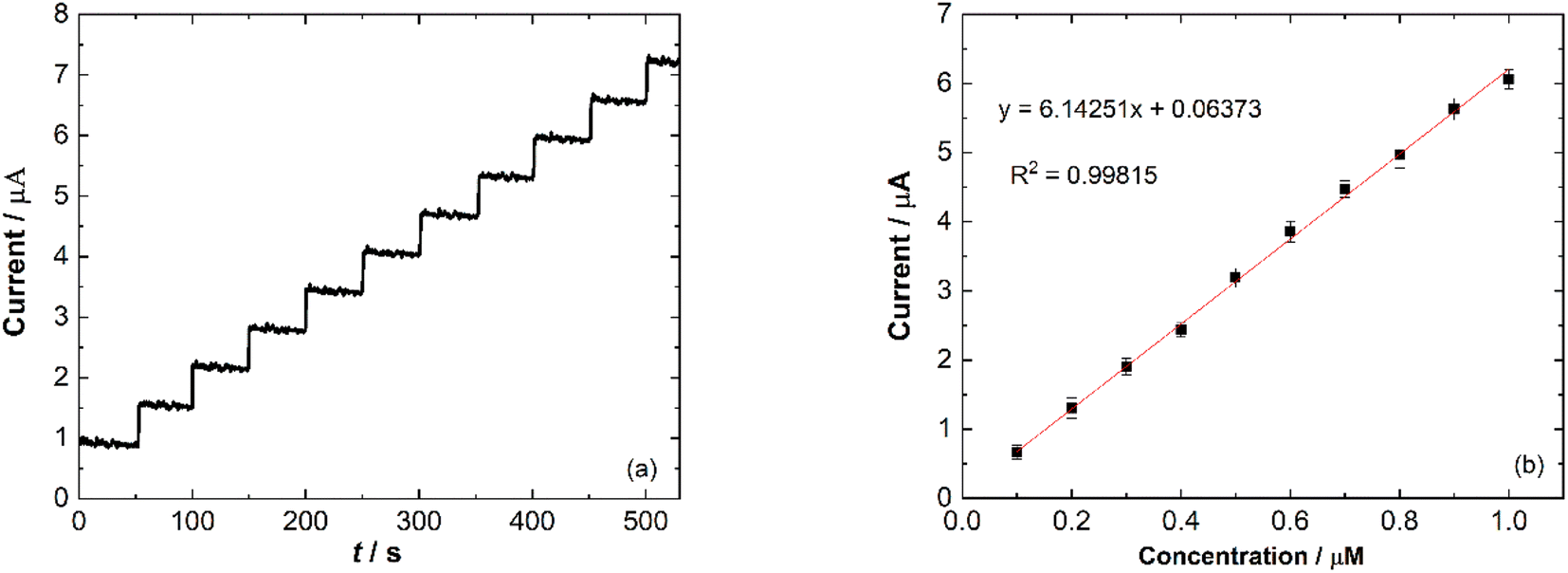
Amperometry detection of nitrite was carried out to measure the response of the modified electrode towards the addition of nitrite. The current response of catalytic current–time for Ni/PDDA/rGO/SPCE with consecutive additions of 1 μM NaNO2 (50 s) in phosphate buffer solution (pH 6.00) was measured at 0.90 V under conditions of constant stirring. Fig. 11(a) shows the typical amperometric curve where the anodic current increased along with the addition of nitrite. The corresponding linear relationship between the current response and nitrite concentration is shown in Fig. 11(b) with a correlation coefficient of 0.90815.
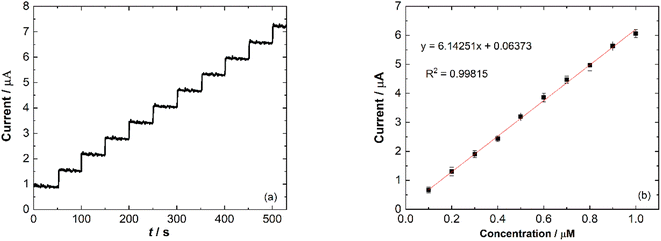 |
| | Fig. 11 (a) Amperometric response of Ni/PDDA/rGO/SPCE at 0.90 V with consecutive additions of 1 μM NaNO2 in phosphate buffer solution (pH 6.00) and (b) a calibration plot of Ip vs. concentration of nitrite. | |
Ni/PDDA/rGO/SPCE was used to evaluate its practicability for nitrite detection in real samples, such as sausages and pickled vegetables, by using a standard addition method. Measurement results of spiked and non-spiked samples are shown in Table 2. High recovery ranging from 98.94% to 100.59% was obtained. Triplicate analysis of the samples was achieved with reasonably good % RSDs of between 0.48% and 1.88%. The results confirmed a potential application of Ni/PDDA/rGO/SPCE for the determination of nitrite in food samples.
Table 2 The nitrite concentration detected by an Ni/PDDA/rGO-modified SPCE in real samples
| Sample |
Original value/μM |
Added (μM) |
Found (μM) |
RSD (%) |
Recovery (%) |
| Sausage |
22.32 |
10.00 |
32.50 ± 0.18 |
0.55 |
100.54 |
| 20.00 |
42.82 ± 0.21 |
0.48 |
101.17 |
| 30.00 |
51.77 ± 0.35 |
0.68 |
98.94 |
| Pickled vegetable |
5.35 |
5.00 |
10.41 ± 0.16 |
1.57 |
100.52 |
| 10.00 |
15.43 ± 0.29 |
1.88 |
100.48 |
| 15.00 |
20.47 ± 0.26 |
1.26 |
100.59 |
Reproducibility and stability of Ni/PDDA/rGO/SPCE
The reproducibility and stability of Ni/PDDA/rGO/SPCE were evaluated by the intra-assay and inter-assay precision (RSD), as shown in Fig. S4(a) and (b),† respectively. The intra-day measurement was taken for three independent determinations of 60 μM nitrite in a PBS buffer solution pH 6.0, resulting in an excellent RSD of 0.48%. Furthermore, the inter-day precision obtained by measuring the anodic peak current for three consecutive days in 60 μM nitrite solution was satisfactorily achieved at 1.12% RSD. In addition, the stability of the electrode was evaluated by measuring the anodic peak current of 60 μM nitrite over the period of a week, as shown in Fig. S4(c).† It was observed that the Ni/PDDA/rGO did not come off during measurement, providing assurance that the catalyst ink of Ni/PDDA/rGO was well adhered to the surface of the SPCE. The electrode retained 91% of the original response towards 60 μM nitrite in a PBS buffer solution pH 6.0 after storage in a desiccator at room temperature for a week. These results can be attributed to the excellent repeatability and stability of the proposed electrode.
Conclusion
In the present work, we introduced an electrochemical sensor based on nickel and PDDA on a reduced graphene oxide sheet modified screen-printed carbon electrode for the determination of nitrite in food samples. The proposed sensors showed desirable analytical properties towards nitrite, including a low overpotential for electro-oxidation, a wide linear range, high sensitivity, high selectivity, a low detection limit, and great feasibility for various food samples with acceptable accuracy and recovery. The modification of the SPCE using Ni, PDDA and rGO significantly enhanced the response of nitrite oxidation, compared with the non-modified SPCE, due to an increase in the active surface area for oxidative reaction. Ni/PDDA/rGO/SPCE showed a synergetic catalytic effect for nitrite oxidation. The linear range was obtained between 6 and 100 μM nitrite concentration, and the detection limit of the compound was 1.99 μM (S/N = 3 by CV). The sensitivities were 0.453 μA μM−1 cm−2 for the lower concentration range and 0.171 μA μM−1 cm−2 for the higher concentration range. The percentage recovery of Ni/PDDA/rGO/SPCE for nitrite detection in real samples was in the range of 98.94–101.17%. Compared to other modified electrodes, the remarkable analytical sensitivity of Ni/PDDA/rGO/SPCE, together with its simple preparation of a lower-cost device, gives such a disposable electrode great potential application as a nitrite sensor for testing the safety of commercial food products. A further advantage of Ni/PDDA/rGO/SPCE was its single use, which eliminated the problem of fouling of the electrode surface. In the step of extracting nitrite from meat samples, some potential interfering species would be co-extracted. This problem was also obviated due to the selectivity of the electrode. These features clearly show that the proposed electrode successfully performed as an effective nitrite electrochemical sensor for the food industry at very low cost.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript prior to submission.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Kasetsart University Research and Development Institute (KURDI) contract number FF(KU) 32.61, and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education. The Department of Chemistry, Faculty of Science, Kasetsart University is also acknowledged for instruments and facilities support.
References
- M. S. Shailaja, R. Rajamanickam and S. Wahidulla, Environ. Pollut., 2006, 143(1), 174–177 CrossRef CAS PubMed.
- G. Yildiz, N. Oztekin, A. Orbay and F. Senkal, Food Chem., 2014, 152, 245–250 CrossRef CAS.
- S. Opinion, EFSA Supporting Publ., 2010, 8(5), 1–12 Search PubMed.
- H. Kodamatani, S. Yamazaki, K. Saito, T. Tomiyasu and Y. Komatsu, J. Chromatogr. A, 2009, 1216(15), 3163–3167 CrossRef CAS PubMed.
- E. Pagliano and Z. Mester, Food Chem., 2019, 286, 710–714 CrossRef CAS PubMed.
- A. F. Lagalante and P. W. Greenbacker, Anal. Chim. Acta, 2007, 590(2), 151–158 CrossRef CAS PubMed.
- E. Trofimchuk, Y. Hu, A. Nilghaz, M. Z. Hua, S. Sun and X. Lu, Food Chem., 2011, 316, 126396 CrossRef PubMed.
- J. Chen, S. Pang, L. He and S. R. Nugen, Biosens. Bioelectron., 2016, 85, 726–733 CrossRef CAS PubMed.
- Y. Deng, J. Qian, Y. Zhou and Y. Niu, RSC Adv., 2021, 11, 10922–10928 RSC.
- X. H. Pham, C. A. Li, K. N. Han, B. C. Huynh-Nguyen, T. H. Le, E. Ko, J. H. Kim and G. H. Seong, Sens. Actuators, B, 2014, 193, 815–822 CrossRef CAS.
- H. Chen, T. Yang, F. Liu and W. Li, Sens. Actuators, B, 2019, 286, 401–407 CrossRef CAS.
- T. Rębiś, M. Falkowski, M. Kryjewski, L. Popenda, L. Sobotta, S. Jurga, M. P. Marszall, J. Mielcarek, G. Milczarek and T. Goslinski, Dyes Pigm., 2019, 171, 107660 CrossRef.
- B. Yang, D. Bin, H. Wang, M. Zhu, P. Yang and Y. Du, Colloids Surf., A, 2015, 481, 43–50 CrossRef CAS.
- M. Shabani-Nooshabadi, M. Roostaee and F. Tahernejad-Javazmi, J. Mol. Liq., 2016, 219, 142–148 CrossRef CAS.
- V. Mani, A. P. Periasamy and S. M. Chen, Electrochem. Commun., 2012, 17(1), 75–78 CrossRef CAS.
- Q. Ding, L. Cao, M. Liu, H. Lin and D. P. Yang, RSC Adv., 2021, 11, 4112 RSC.
- D. Liu, Q. Guo, X. Zhang, H. Hou and T. You, J. Colloid Interface Sci., 2015, 450, 168–173 CrossRef CAS.
- A. M. Hengne, A. K. Samal, L. R. Enakonda, M. Harb, L. E. Gevers, D. H. Anjum, M. N. Hedhili, Y. Saih, K. W. Huang and J. M. Basset, ACS Omega, 2018, 3(4), 3688–3701 CrossRef CAS PubMed.
- Z. Zhao, J. Zhang, W. Wang, Y. Sun, P. Li, J. Hu, L. Chen and W. Gong, Appl. Surf. Sci., 2019, 485, 274–282 CrossRef CAS.
- M. Wang, M. Cui, M. Zhao and H. Cao, J. Electroanal. Chem., 2018, 809, 117–124 CrossRef CAS.
- D. Zhang, Y. Fang, Z. Miao, M. Ma, X. Du, S. Takahashi, J. I. Anzai and Q. Chen, Electrochim. Acta, 2013, 107, 656–663 CrossRef CAS.
- M. M. Shahid, P. Rameshkumar, A. Numan, S. Shahabuddin, M. Alizadeh, P. S. Khiew and W. S. Chiu, Mater. Sci. Eng., C, 2019, 100, 388–395 CrossRef CAS PubMed.
- W. Gao, Q. Li, M. Dou, Z. Zhang and F. Wang, J. Mater. Chem. B, 2018, 6, 6781–6787 RSC.
- J. Borowiec, K. Yan, C. C. Tin and J. Zhangb, J. Electrochem. Soc., 2015, 162(3), 164–169 CrossRef.
- J. J. Shi, G. H. Yanga and J. J. Zhu, J. Mater. Chem., 2011, 21, 7343–7349 RSC.
- Z. Ji, Y. Wang, X. Shen, H. Ma, J. Yang, A. Yuan and H. Zhou, J. Colloid Interface Sci., 2017, 487, 223–230 CrossRef CAS PubMed.
- Z. Wang, Y. Hu, W. Yang, M. Zhou and X. Hu, J. Sens., 2012, 12, 4860–4869 CrossRef CAS PubMed.
- J. Wang, Q. Zhao, H. Hou, Y. Wu, W. Yu, X. Ji and L. Shao, RSC Adv., 2017, 7, 14152 RSC.
- S. Dutta, A. Indra, Y. Feng, T. Song and U. Paik, ACS Appl. Mater. Interfaces, 2017, 9(39), 33766–33774 CrossRef CAS PubMed.
- A. M. Hengne, A. K. Samal, L. R. Enakonda, M. Harb, L. E. Gevers, D. H. Anjum, M. N. Hedhili, Y. Saih, K. W. Huang and J. M. Basset, ACS Omega, 2018, 3(4), 3688–3701 CrossRef CAS PubMed.
- K. S. Kim and N. Winograd, Surf. Sci., 1974, 43(2), 625–643 CrossRef CAS.
- D. Jesús, J. Carrazza, P. Pereira and F. Zaera, Surf. Sci., 1998, 397(1–3), 34–47 CrossRef.
- X. Huang, Y. Li, Y. Chen and L. Wang, Sens. Actuators, B, 2008, 134(2), 780–786 CrossRef CAS.
- Y. Wang, E. Laborda and R. G. Compton, J. Electroanal. Chem., 2012, 670, 56–61 CrossRef CAS.
- K. Zhao, H. Song, S. Zhuang, L. Dai and P. He, Electrochem. Commun., 2007, 9, 65–70 CrossRef CAS.
- M. B. González García and A. Costa García, Bioelectrochemistry, 1995, 38(2), 389–395 CrossRef.
- S. Khan, Akrema, S. Qazi, R. Ahmad, K. Raza and Rahisuddin, ACS Omega, 2021, 6(24), 16076–16085 CrossRef CAS PubMed.
- H. Bora, D. Mandal and A. Chandra, ACS Appl. Bio Mater., 2022, 5(4), 1721–1730 CrossRef CAS PubMed.
- P. Traiwatcharanon, W. Siriwatcharapiboon, O. Jongprateep and C. Wongchoosuk, RSC Adv., 2022, 12, 16079–16092 RSC.
- S. Zhang, B. Q. Li and J. B. Zheng, Anal. Methods, 2015, 7(19), 8366–8372 RSC.
- H. Teymourian, A. Salimi and S. Khezrian, Biosens. Bioelectron., 2013, 49, 1–8 CrossRef CAS PubMed.
- Y. J. Yang and W. Li, Biosens. Bioelectron., 2014, 56, 300–306 CrossRef CAS PubMed.
- Q. Sheng, D. Liu and J. Zheng, J. Electroanal. Chem., 2017, 796, 9–16 CrossRef CAS.
- W. Wu, Y. Li, J. Jin, H. Wu, S. Wang, Y. Ding and J. Ou, Microchim. Acta, 2016, 183(12), 3159–3166 CrossRef CAS.
- J. M. Jian, L. Fu, J. Ji, L. Li, X. Guo and T. L. Ren, Sens. Actuators, B, 2018, 262, 125–136 CrossRef CAS.
- S. Palanisamy, B. Thirumalraj and S. M. Chen, J. Electroanal. Chem., 2016, 760, 97–104 CrossRef CAS.
- S. Nasraoui, A. Al-Hamry, P. R. Teixeira, S. Ameur, L. G. Paterno, M. B. Ali and O. Kanoun, J. Electroanal. Chem., 2020, 880, 114893 CrossRef.
- S. Kang, H. Zhang, G. Wang, Y. Zhang, H. Zhao, H. Zhou and W. Cai, Inorg. Chem. Front., 2019, 6, 1432–1441 RSC.
- M. Rostami, G. Abdi, S. Habib and A. Alizadeh, J. Electroanal. Chem., 2019, 847, 113239 CrossRef CAS.
- Q. Ding, L. Cao, M. Liu, H. Lin and D. P. Yang, RSC Adv., 2021, 11, 4112–4117 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Wilai Siriwatcharapiboon
ab and
Wilai Siriwatcharapiboon