DOI:
10.1039/D2RA04015H
(Paper)
RSC Adv., 2022,
12, 26895-26907
In vitro and computational investigations of novel synthetic carboxamide-linked pyridopyrrolopyrimidines with potent activity as SARS-CoV-2-MPro inhibitors†
Received
29th June 2022
, Accepted 12th September 2022
First published on 22nd September 2022
Abstract
An essential target for COVID-19 is the main protease of SARS-CoV-2 (Mpro). With the objective of targeting this receptor, a novel set of pyrido[1,2-a]pyrrolo[2,3-d]pyrimidines with terminal carboxamide fragments was designed, synthesized, and considered as an initial motif for the creation of effective pan-coronavirus inhibitors. Accordingly, nine derivatives (21–29) have been introduced for in vitro assay to evaluate their antiviral activity and cytotoxicity effect against COVID-19 virus using Vero cells. The obtained data revealed that the majority of these derivatives showed potent cellular anti-COVID-19 activity and prevent viral growth by more than 90% at two different concentrations with weak or even no detectable cytotoxic effect on Vero cells. Extensive molecular docking simulations highlighted proper non-covalent interaction of new compounds within the binding pocket of Mpro as a potential target for their antiviral activity. In vitro assay for all the synthesized derivatives against the viral Mpro target indicated that compounds 25 and 29 have promising inhibitory activity with IC50 values at low micromolar concentrations. The molecular dynamic simulation results predicted the stability of compound 29 in the binding cavity of SARS-CoV-2 Mpro and hence supported the high inhibitory activity shown by the In vitro assay. These results suggested that compounds 25 and 29 merit further investigations as promising drug candidates for the management of SARS-CoV-2.
Introduction
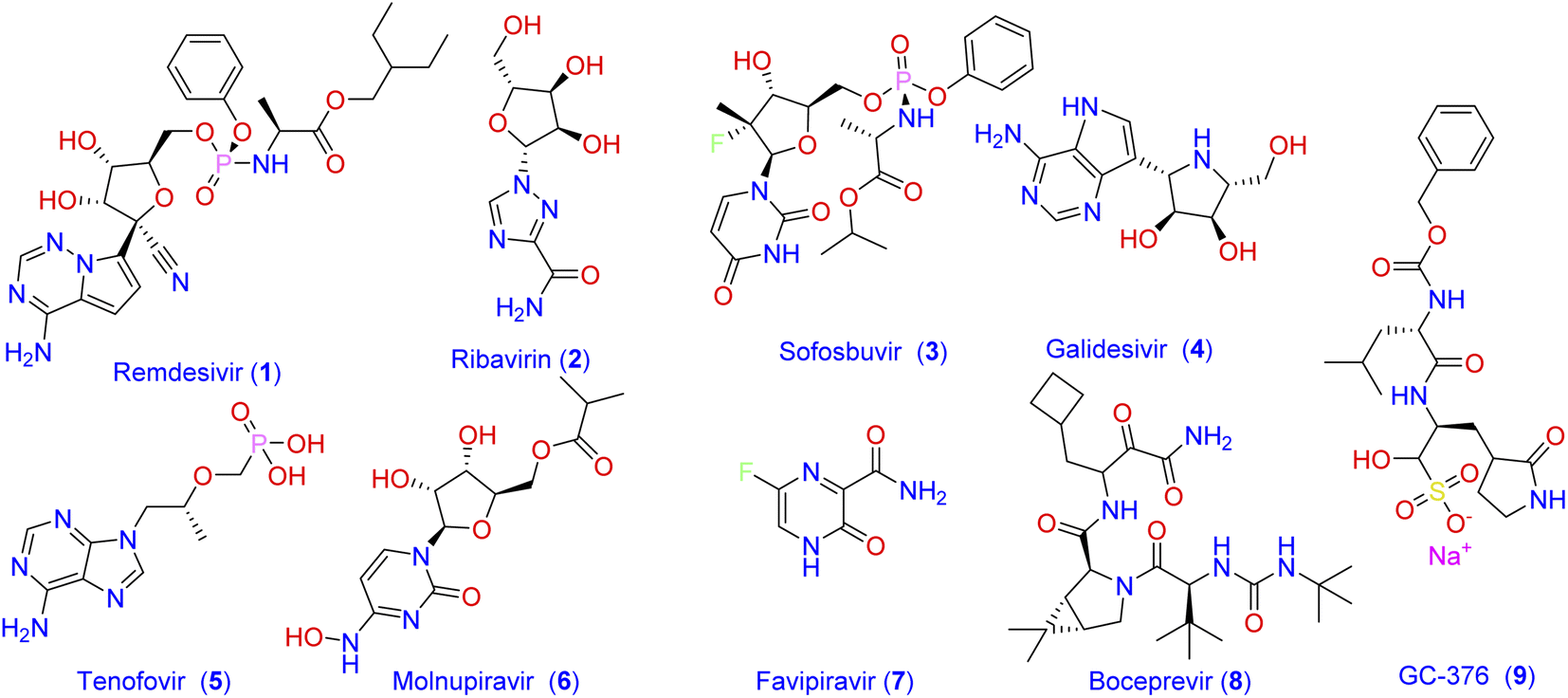
Coronavirus disease (COVID-19) is a life-threatening infectious disease caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), which was discovered first in Wuhan city in China and subsequently spread worldwide.1 Based on the epidemiological report of the WHO on 21 June 2022 there have been more than 537![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 confirmed cases of COVID-19, including 6319395 confirmed deaths globally.2 Until the end of 2021, there was no approved specific antiviral drug for treatment, and all options were based on symptomatic treatment and oxygen therapy to manage respiratory impairment. Recently, the Food and Drug Administration (FDA) has authorized the combination of nirmatrelvir and ritonavir for the emergency use only in mild-to-moderate cases of COVID-19. Consequently, there is still unmet needs for effective treatment options against the SARS‐CoV‐2 virus. One strategy that has been followed by scientists to combat the widespread of new viral strains is to use the already approved antiviral medications.3–5 Most of the currently used antiviral drugs are nucleoside analogs6 such as Remdesivir (1; Fig. 1), Ribavirin (2), Sofosbuvir (3), Galidesivir (4), and Tenofovir (5).
500000 confirmed cases of COVID-19, including 6319395 confirmed deaths globally.2 Until the end of 2021, there was no approved specific antiviral drug for treatment, and all options were based on symptomatic treatment and oxygen therapy to manage respiratory impairment. Recently, the Food and Drug Administration (FDA) has authorized the combination of nirmatrelvir and ritonavir for the emergency use only in mild-to-moderate cases of COVID-19. Consequently, there is still unmet needs for effective treatment options against the SARS‐CoV‐2 virus. One strategy that has been followed by scientists to combat the widespread of new viral strains is to use the already approved antiviral medications.3–5 Most of the currently used antiviral drugs are nucleoside analogs6 such as Remdesivir (1; Fig. 1), Ribavirin (2), Sofosbuvir (3), Galidesivir (4), and Tenofovir (5).
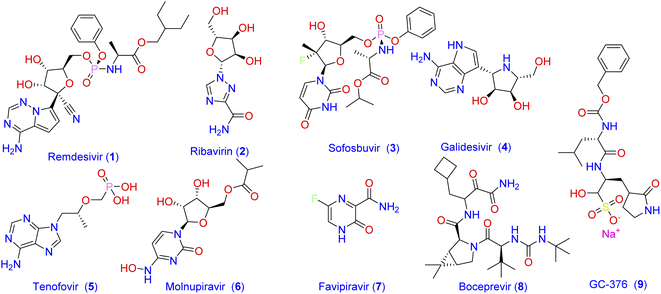 |
| | Fig. 1 Frequently prescribed antiviral drugs and recently identified molecules with prominent activity against SARS-CoV-2. | |
Despite the recommendation of such antiviral agents together with the antibacterial and other antiviral medications azithromycin, molnupiravir (6), and favipiravir (7) to manage the emergency cases, no effective drug has yet been introduced to treat specific structures of viral components of SARS-CoV-2.7 The main protease Mpro (also called 3CLpro) of coronavirus is a key mediator in viral replication and transcription. This functional importance, together with the absence of a closely related analogous target in humans, made the SARS-CoV-2 main protease Mpro an interesting target for the design of antiviral agents.5,8,9 Consequently, many attempts have been adopted to develop Mpro inhibitor-based antiviral medication. As a result, a number of antiviral agents with prominent activity against SARS-CoV-2 were discovered. These include boceprevir (8) and GC-376 (9) which were verified as inhibitors of Mpro through binding to its catalytic active site.10
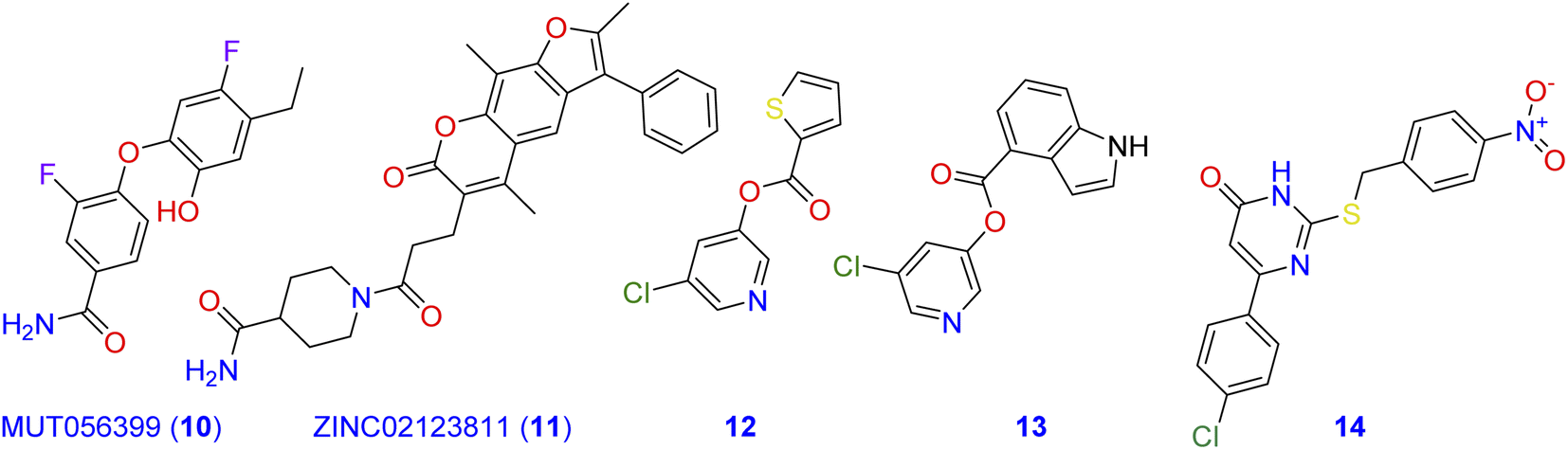
Computational methods play an important role in the design of novel Mpro antagonistic drugs.4,11–14 In this context, a structure-based virtual screening was conducted by Jin and co-workers to evaluate a library of more than 10000 well-known compounds as potential inhibitors of Mpro.15 Among compounds of the tested library, six inhibited Mpro with IC50 values range of 0.67–21.4 µM.16 Additionally, MUT056399 (10; Fig. 2) has been identified as a carboxamide-incorporating inhibitor of SARS-CoV-2-MPro with an EC50 value of 38.24 µM with the preferential quality of low cytotoxicity (CC50 > 100 µM). The carboxamide group of this latter forms two hydrogen bonds with the amino acid residues His163 and Phe140 in the S1 subsite of SARS-CoV-2-MPro.17,18 ZINC02123811 (11) is another carboxamide derivative derived from a natural source and has the affinity to bind with the active pocket of SARS-CoV-2 Mpro.19 The root-mean-square fluctuation (RMSF) analysis indicates that the complex of Mpro with this carboxamide derivative is pretty stable all over the simulation course.
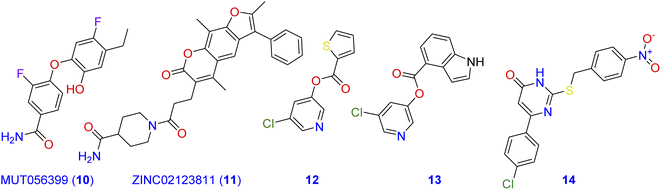 |
| | Fig. 2 Carboxamide, pyridine, and pyrimidine incorporating molecules with potent SARS-CoV-2-MPro antagonistic activity. | |
On the other hand, pyridine, pyrimidine, and pyrrole have long been considered privileged fragments in the construction of antiviral candidates and potential inhibitors of SARS-CoV-2-MPro in particular.20,21 Compound 12 is a pyridine derivative that has been evaluated for its activity against the Frankfurt-1 strain of SARS-CoV by MTT assay in Vero E6 cells. Results of the antiviral evaluation displayed a good potency and selectivity of 12 toward SARS-CoV-2-MPro with an IC50 value of 0.50 µM.22 In silico docking studies were also performed on compound 13 against the binding pocket of SARS-CoV-2-MPro. Results suggested that it is a promising anti-SARS-CoV agent (IC50 = 30 nM), and revealed a good fitting within the active site of the protein target through the formation of three hydrogen bonds with the amino acid residues Cys145, Ser144, and Gly143.23 Investigating the inhibitory potency of the pyrimidine-based molecule 14 against SARS-CoV-MPro demonstrated its good activity with an IC50 value of 6.10 µM.
Rationale and aim of the work
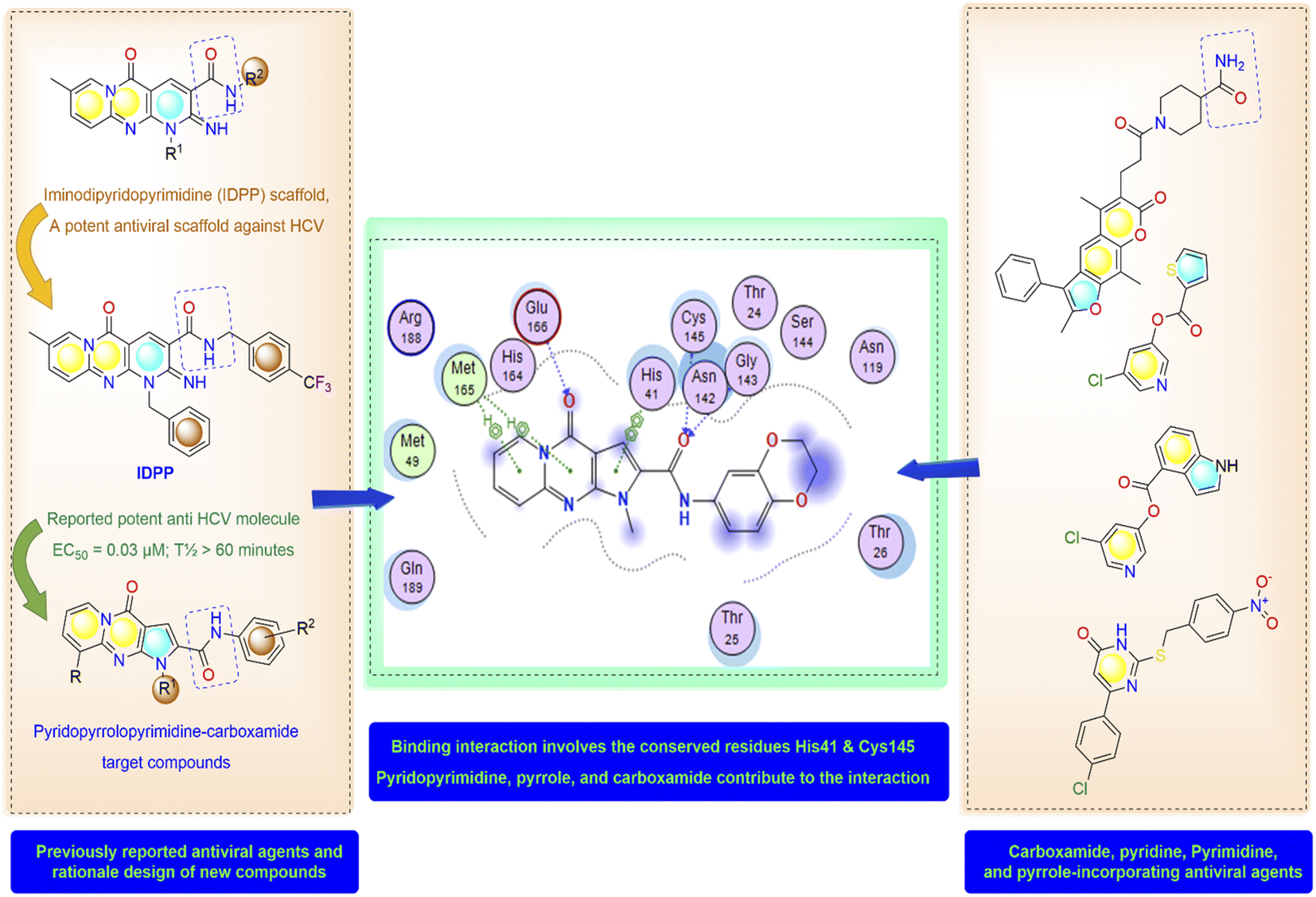
High-throughput screening of a library of small synthetic molecules against the infectious hepatitis C virus (HCV) identified the iminodipyridinopyrimidine (IDPP; Fig. 3) scaffold as a potent inhibitor in the early and the late steps of the HCV lifecycle.24,25 This scaffold showed also high levels of safety and metabolic stability. The 3D structure of HCV protease has a high level of similarity with the SARS-CoV-2-MPro (Z score = +8.40).26,27 Like Mpro, the HCV protease also has a double β-barrel fold, with relatively similar orientations to those of the SARS-CoV-2 Mpro, and a substrate-binding site located in a shallow cleft between its two six-to eight-stranded antiparallel β-barrels.28 The superimposition of these two proteases suggested the superimposition of their substrate binding pockets and their active-site catalytic residues.28,29 Accordingly, the IDPP scaffold is suggested to have a broad antiviral spectrum against both HCV and SARS-CoV-2-MPro. In continuation to our recent reports on the design of suggested bioactive molecules,30–33 the objective of the current research is to modify the structure of the reported HCV antiviral agents IDPP into pyridopyrrolopyrimidine new scaffolds and to assess the potential of new candidates to inhibit SARS-CoV-2-MPro. Hence, while keeping the carboxamide functionality linked to the new scaffold, two structural modifications of IDPP analog were conducted to enhance the ligand binding and consequently the potency: the six-membered pyridine-imine fragment was replaced with the pyrrole aromatic system; the benzyl substituent attached to the pyridine-nitrogen was replaced with a smaller hydrophobic moiety. Accordingly, the new scaffold possesses the advantages of incorporating the privileged fragments pyridine, pyrimidine, pyrrole, and carboxamide which have been reported as pharmacophoric groups in certain SARS-CoV-2-MPro inhibitors.20,21,34 A set of in vitro biological evaluation studies has been performed. The effect of new compounds to inhibit the viral replication of SARS-CoV-2 and antagonize its main protease was evaluated. As well, cell growth inhibition after treatment with new ligands has been assessed. A structure-based approach was utilized using molecular docking simulation of the reported active IDPP analog compared to the newly designed pyridopyrrolopyrimidines within the Mpro protease target of SARS-CoV; PDB 7L11. The pre-docking screening of designed ligands within the protease binding pocket (Fig. 3) showed three promising findings: (1) an edge-to-face aryl–aryl interaction with the catalytic residue His41; (2) terminal amide carbonyl is directed well at the NH of Cys145; (3) the carbonyl of the fused system appears with proper placement with the polar environment, including the side chains of Met165, His164, and Glu166. In contrast, the behavior of IDPP analog with pyridine-imine part fails to participate in the good orientation of fused system through these major and conserved interactions within the pocket due to loss of aromaticity and false ordinations of terminal amide and the carbonyl group of fused system. Moreover, there is an unfavorable benzyl substitution attached to the pyridine-N that is located at the polar binding pocket. Molecular dynamic simulations were used for confirming these findings and measuring the stability within the pocket.
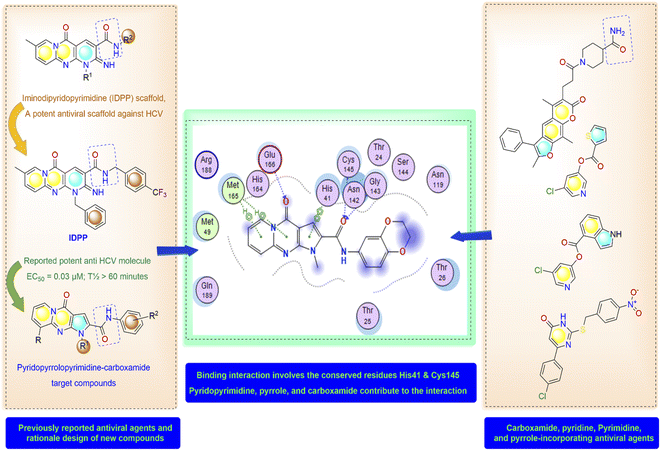 |
| | Fig. 3 Molecular design and rationale analysis for the binding interactions of novel pyridopyrrolopyrimidines within active pocket of SARS-CoV-2-MPro. | |
Results and discussion
Chemistry
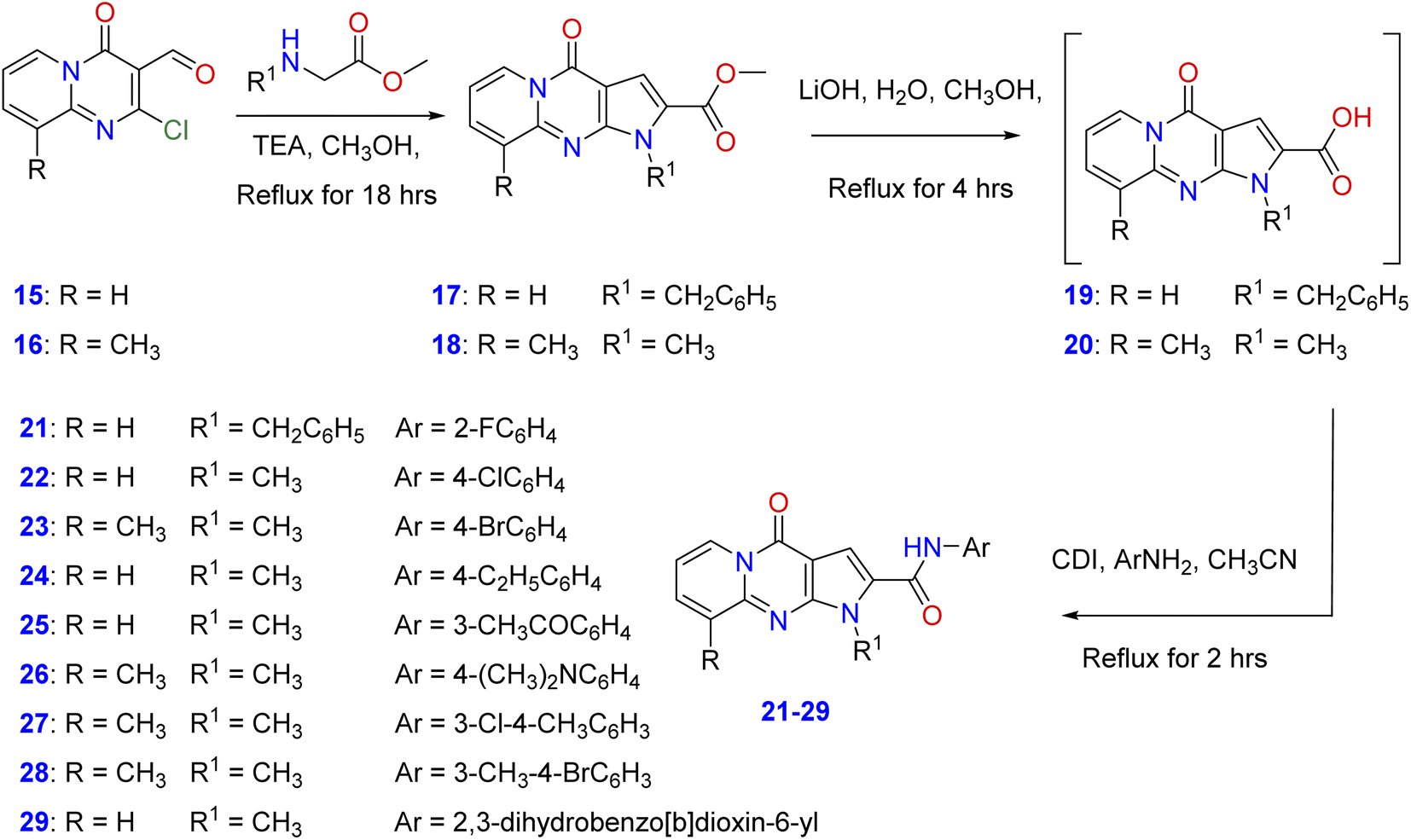
In order to elucidate the scope and features of the cyclization reaction, the starting methyl-1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-b]pyrrolo[2,3-d]pyrimidine-2-carboxylate and its 9-methyl derivatives (17, 18) were prepared in moderate to good yields by the reaction of 2-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbaldehyde35 and its 9-methyl derivatives (15, 16) with amino acid methyl esters namely, methyl-N-methyl(N-benzyl)glycinate or methyl methyl-N-benzyl glycinate in a mixture of methanol and triethylamine under reflux as shown in Scheme 1. Hydrolysis of the ester derivatives 17, 18 by aqueous lithium hydroxide affords the corresponding intermediate, 1-benzyl/methyl-4-oxo-1,4-dihydro-pyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxylic acid and its 9-methyl derivatives (19, 20). Condensation of these carboxylic acid intermediates with appropriate aniline derivatives in the presence of 1,1′-carbonyldiimidazole (CDI) and acetonitrile gives the target N-aryl-1-benzyl/methyl-4-oxo-1,4- dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamides (21–29) as shown in Scheme 1 with different substitutions on the original scaffold at N-1 and carboxamide functionality.
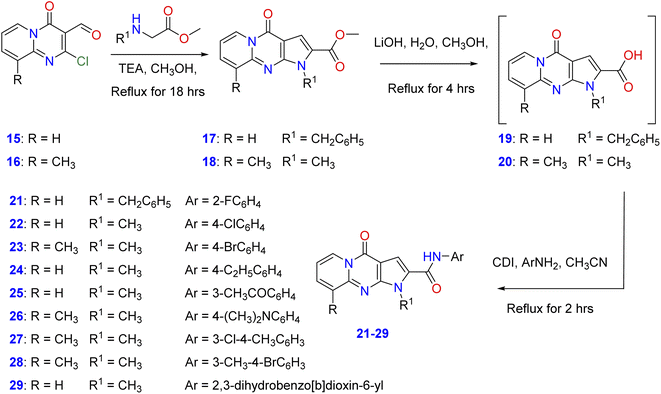 |
| | Scheme 1 Synthetic protocol of pyrido[1,2-a]pyrrolo[2,3-d]pyrimidine derivatives (21–29). | |
Spectroscopic data
The 1H-NMR spectra of 21–29 showed singlet signals attributable to NH proton of the carboxamide functionality at the regions of 10.85–11.12 ppm. Moreover, compounds 21–29 exhibited the signals of the methylene and N-methyl group protons at δ 5.88 and δ 4.01–4.09, respectively. In addition, the elemental analysis and mass spectra of compounds 21–29 confirmed their proposed structures, see ESI†.
Biological screening
Evaluation of antiviral activity against SARS-CoV-2. The antiviral activity of compounds (17, and 21–29) was screened in plated Vero E6 cells (CCL-8; ATCC origin provided by the Virology Sector, VACSERA, Giza, Egypt; https://www.vacsera.com/) and infected later with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.01. The % inhibition of viral replication was measured applying two analogs' concentrations: 1 and 10 µM. In addition, the concentrations required to inhibit virus replication by 50% effective inhibitory concentration (EC50) were calculated using five different concentrations by fitting a sigmoidal curve to the data following the logarithmic transformation of the drug concentration. In addition, cell growth inhibition or cytotoxicity was determined for all analogs (17, and 21–29) and remdesivir as a positive control. All the obtained data are presented in Table 1. It was found that remdesivir showed potent antiviral activity EC50 = 0.0099 µM with prominent cytotoxicity to Vero E6 cells with an IC50 value of 43.1 µM and SI index of 1481. As a preliminary evaluation step for the precursor compound 17 with ester group; it has no cytotoxic effect with weak antiviral activity compared to the target compounds by EC50 = 22.1 µM. Thus, the chemical modification of such compound into 21–29 substituted amides revealed a good effect on the antiviral activity profile with an acceptable MTT cytotoxicity effect. Moreover, all tested compounds showed cytotoxicity with IC50 values over that of remdesivir except for 24 which showed the highest cytotoxicity equal to 21.60 ± 1.64 µM with good Mpro inhibitory activity. The pyridopyrrolopyrimidine derivatives 22, 29, and 28 showed the lowest MTT IC50 cytotoxicity (193.0, 151.0, and 139.0) with EC50 values of 0.109, 0.051, and 0.108 µM towards the infected Vero cells. Interestingly, analog 29 with benzodioxan terminal part had a potent antiviral profile with percent inhibition values of 94.10 and 96.39% at 1 and 10 micromolar concentrations, respectively which were found to be very close to results of the positive control remdesivir (94.23 and 97.58%) at the same concentrations. The benzodioxan derivative was found to have the preferential of the lowest cytotoxic action (IC50 = 151.00 µM). In addition, 26, 21, and 23 analogs exhibited promising antiviral effects with percent inhibition values of 94.50, 95.92, and 92.92% at 10 µM, respectively. The problem with 21 and 23 is their elevated cytotoxic tendency to Vero cells.
Table 1 Antiviral activity and cytotoxicity assay of novel non-covalent SARS-CoV-2 Mpro inhibitors by propagation in Vero E6 cells
| Comp. ID |
Cytotoxicity IC50 (µM ± SEa) |
Antiviral assay (qPCR) |
SIb |
| Inhibition % |
EC50 and EC90 (µM ± SEa) |
| 1 µM |
10 µM |
EC50 |
EC90 |
| SE represents standard error from triplicate readings. Selectivity index (SI) was calculated by dividing of IC50 value for Vero cell cytotoxicity by the EC50 of the antiviral assay. |
| 17 |
>200 |
22.25 |
42.51 |
22.1 ± 1.012 |
36.13 ± 2.31 |
9 |
| 21 |
48.8 ± 3.72 |
83.49 |
95.92 |
0.0138 ± 0.00198 |
3.60 ± 0.42 |
3544 |
| 22 |
193.0 ± 14.7 |
88.16 |
94.10 |
0.1093 ± 0.01105 |
1.738 ± 0.54 |
1766 |
| 23 |
47.6 ± 3.62 |
78.96 |
92.92 |
0.0216 ± 0.00656 |
3.11 ± 1.61 |
2201 |
| 24 |
21.6 ± 1.64 |
94.01 |
96.22 |
0.0762 ± 0.00973 |
3.437 ± 0.55 |
283.3 |
| 25 |
121.0 ± 9.21 |
92.48 |
97.08 |
0.1066 ± 0.00115 |
2.50 ± 0.841 |
1135 |
| 26 |
87.3 ± 6.65 |
87.61 |
94.05 |
0.0376 ± 0.00423 |
3.801 ± 0.041 |
2323 |
| 27 |
64.4 ± 4.91 |
94.36 |
96.52 |
0.1035 ± 0.00344 |
2.601 ± 1.02 |
622.2 |
| 28 |
139.0 ± 10.6 |
92.71 |
95.60 |
0.108 ± 0.00304 |
1.41 ± 0.91 |
1278 |
| 29 |
151.0 ± 11.5 |
94.10 |
96.39 |
0.0519 ± 0.00512 |
8.953 ± 2.01 |
2909 |
| Remdesivir |
43.1 ± 3.28 |
94.23 |
97.58 |
0.0099 ± 0.00175 |
0.0835 ± 1.88 |
7195 |
The selectivity analysis of these compounds compared to their cytotoxic effect has also been investigated. Good antiviral candidate should be highly selective with a minimum SI value of 5 or higher.36–39 Almost all the new pyridopyrrolopyrimidines showed good SI values and proved the prominent antiviral activity. Comparing the performance of novel compounds to the reference remdesivir, SI was calculated and showed 4000 to 283 ranges with promising antiviral selectivity. Some of the designed derivatives did not function positively in the antiviral activity in vitro, and their relative abilities to inhibit viral replication did not always correlate directly with in vitro inhibition parameters toward Mpro.40
SARS-CoV-2 Mpro inhibition assay. Both the crystal structures of human coronavirus Mpro and for an inhibitor complex of porcine coronavirus Mpro were determined and led to a construction of a homology model for SARS-CoV-2-MPro.16 The main protease, Mpro in SARS-CoV-2 is considered a viable drug target because of its important role in the cleavage of the virus polypeptide.41–43 Several Mpro inhibitors have been reported as covalent or non-covalent inhibitors. The covalent inhibitors work by modifying the catalytic Cys145 with potent enzymatic inhibition and cellular antiviral activity, including GC376, PX-12, and carmofur, each containing a reactive nitrogen heterocycle as pyrrolidone, imidazole, or pyrimidine that mimics the glutamine P1 position.44–46 Interestingly, many non-covalent inhibitors of SARS-CoV-2 Mpro were developed and validated such as ML188.47Herein, N-aryl-1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide derivatives were tested by in vitro assay for inhibition of SARS-CoV-2 Mpro activity and compared with the known references Lopinavir (non-specific) and GC376 (specific) Mpro inhibitors.48 As shown in Table 2, intermediate 17 seems to be functionally inactive against the Mpro enzyme as seen from its IC50 value. However, both 25 and 29exhibited high SARS-CoV-2 Mpro inhibition activity with IC50 values of 5.42 and 3.22 µM, respectively, which are more potent than the reference drugs Lopinavir and GC376 (82.17, 12.85 µM). On the other hand, introducing a halogen substituent to the terminal aryl ring, as in cases of 27, 28, 23 and 22, leads to a dramatical fall in the activity (IC50 values = 165.20 µM, 45.96 µM, 65.92 µM and 236.50 µM, respectively). As well, compound 26 with N,N-dimethylamino group at the para position of the terminal aryl showed a dramatic reduction in the inhibition activity with (IC50 values 407.50 µM). In combination with results presented in Table 1, this indicates that the 3,4-dihydrodioxane or 3-acetyl group attached to the phenyl ring is necessary for activity against Mpro of these compounds. The results all partially conveyed the proposed inhibitory effect of these derivatives on the Mpro, but more mechanistic investigations are still needed to specify the major mechanism of action whether it is competitive on non-competitive.
Table 2 Inhibitory data of the target compounds against SARS-CoV-2 Mpro
| Compound's ID |
IC50 (µM ± SEa) |
Compound's ID |
IC50 (µM ± SE*) |
| SE represents standard deviation from triplicate readings. |
| 17 |
329.5 ± 15.1 |
26 |
407.50 ± 3.44 |
| 21 |
25.36 ± 2.40 |
27 |
165.20 ± 16.70 |
| 22 |
236.50 ± 4.29 |
28 |
45.96 ± 8.62 |
| 23 |
65.92 ± 12.30 |
29 |
3.22 ± 2.83 |
| 24 |
82.17 ± 15.0 |
Lopinavir |
82.17 ± 7.66 |
| 25 |
5.42 ± 21.30 |
GC-376 |
12.85 ± 0.74 |
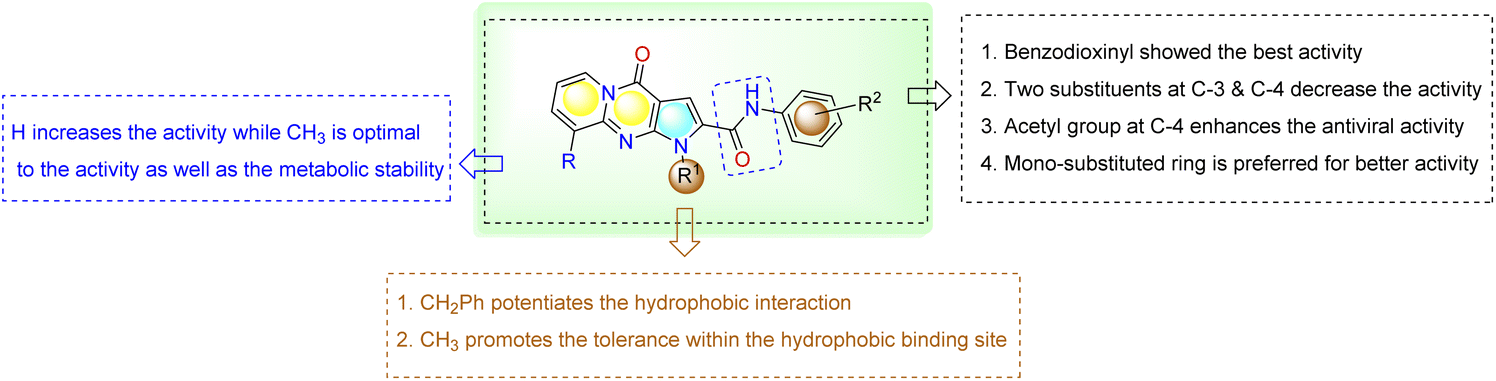
SAR analysis. The antiviral assay results indicated that the target compounds showed excellent antiviral activity against SARS-CoV-2-MPro. Results in Table 2 showed that when the aryl substituent is 3,4-dihydrobenzo[b]dioxin-6-y (29), 3-acetylC6H4 (25) groups, on the corresponding target compounds, it exhibited good protection activity. Moreover, the results showed that the increase in the hydrogen bond accepter and hydrophobicity increases the interaction within pocket and consequently improves the activity. Steric hindrance effect on aromatic rings in the pocket well decrease activity for antiviral activity at the same position confirmed by the following activity order; 26 (Ar = 4-N,N-dimethyl-Ph) > 22 (Ar = 4-Cl-Ph) > 27 (Ar = 3-Cl-4-CH3-Ph) > 24 (Ar = 4-C2H6-Ph) > 23 (Ar = 4-Br-Ph) > 28 (Ar = 3-CH3, 4-Br-Ph), > 21 (Ar = 2-F-Ph). The activity of these analogs could be attributed in general to the 5-membered aromatic pyrrole ring which correctly and precisely orients the amide bond to the molecule within the binding site. A summary of the SAR analysis is presented in Fig. 4.
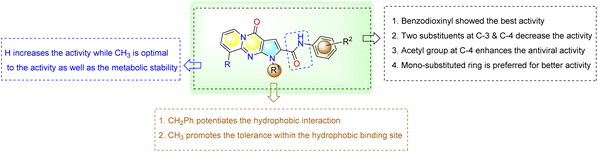 |
| | Fig. 4 SAR analysis of target compounds correlated with Mpro protease activity. | |
Computational investigations
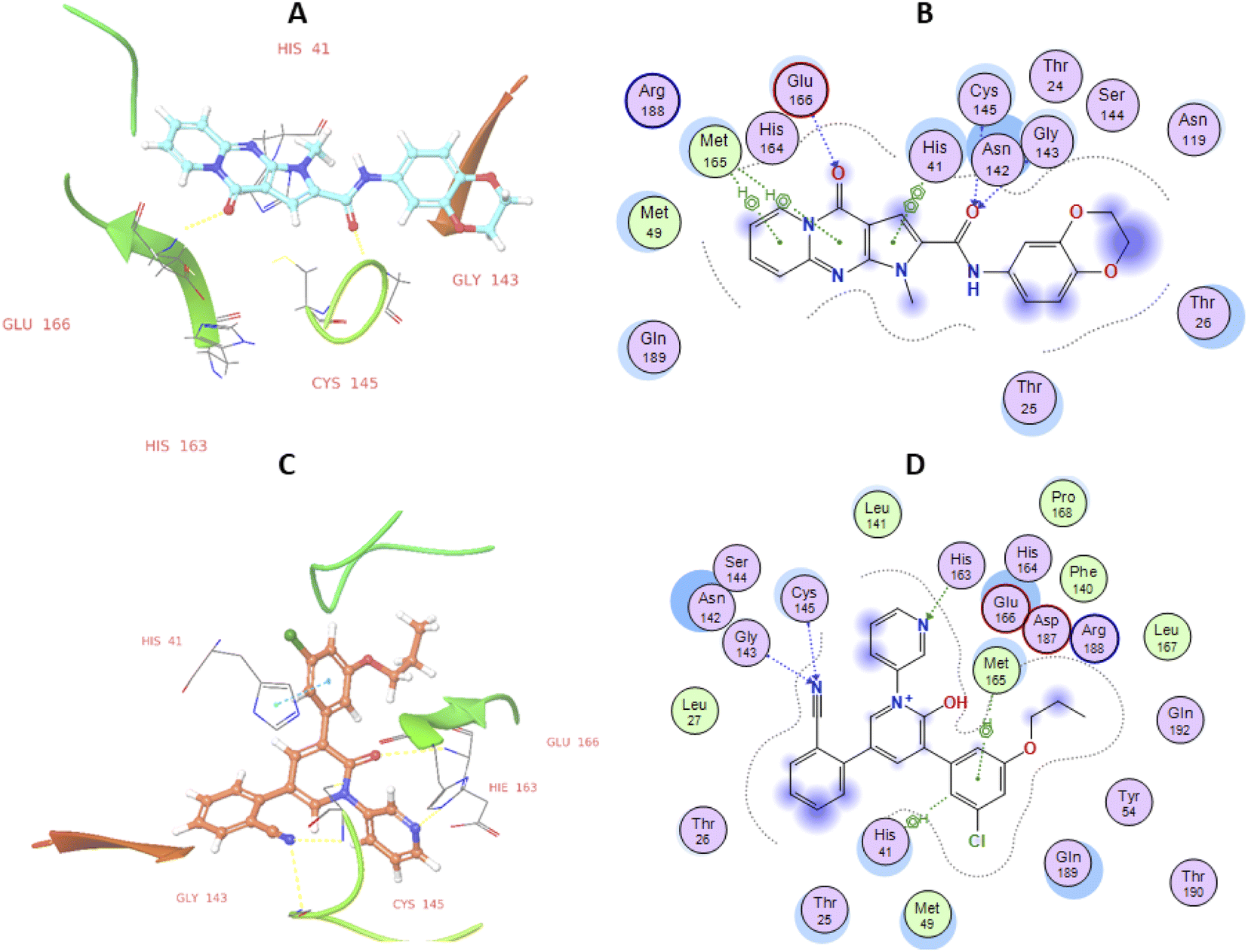
Molecular docking study. To gain better understanding on the potency of the studied compound, to explicate the plausible mechanism by which it can induce antiviral activity and guide further SAR studies, we proceeded to examine the interaction of the most active pyrido[1,2-a]pyrrolo[2,3-d]pyrimidine derivative (29) with SARS-CoV-2 Mpro protein. Here, the structure of SARS-CoV-2 Mpro in complex with a potent bipyridine-benzonitrile inhibitor (XF1) crystal structure (PDB: 7L11) was selected as the binding model.49 This molecular docking study was performed using the default protocol associated with the glide module of the Schrodinger modelling suite.50–52 Results showed that compound 29 was appropriately oriented in the active site of SARS-CoV-2 Mpro protein with a significant docking score (−7.45 kcal Mol−1) and displayed three hydrogen binding interactions with amino acids Gly143, Cys145, and Glu166 as depicted in Fig. 5A and B. The co-crystallized ligand exhibited two hydrogen binding interactions with the first couple of amino acids while the third one interacted hydrophobically with XF1. The binding affinity exhibited by compound 29 is attributed to significant bonded and non-bonded interactions with residues lining the active site of the SARS-CoV-2 Mpro which is supported by the following finding: an arene–arene interaction with catalytic residue His41; hydrogen bond interaction between amide carbonyl and the NH of Cys145; as well as the hydrogen bond interaction between the carbonyl of the fused system with Glu166. An additional bi-directional hydrophobic interaction arose between both six-membered rings of the new pyridopyrrolopyrimidine-2-carboxamide with the crucial amino acid residue Met165. The binding pocket per residue interaction analysis for the active compound 29 showed that the molecule is embedded into the active site through a series of significant van der Waals interactions (Table 3). Docking score of the compound 29 was close to those of Co-Crystalized ligand (−10.07 kcal mol−1) showing that this compound may bind to SARS-CoV-2 Mpro catalytic site with good affinity. To validate results of our conducted docking experiment, we a used docking control in which the internal co-crystallized ligand XF1 was re-docked into the binding pocket of the chosen crystal structure. Results of this validation process revealed an acceptable superimposition of the re-docked XF1 conformer over that of the crystal structure and showed a RMSD value of 1.49 (Fig. S1; see ESI†). As well, a set of almost matching hydrogen bonding and hydrophobic interactions with that observed in the case of compound 29 was obtained (Fig. 5C and D).
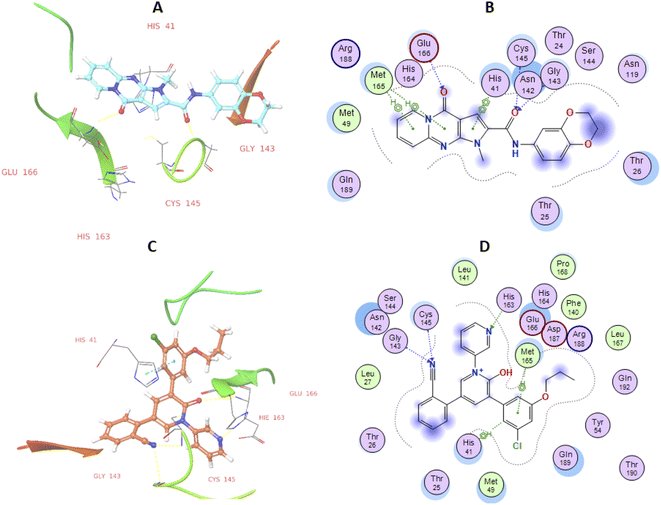 |
| | Fig. 5 2D and 3D binding interaction data of active 29 analog (A and B) compared to Co-Crystalized ligand (C and D) in active site of SARS-CoV 2 Mpro enzyme with PDB entry code 7L11. Color-coding scheme for interaction includes polar hydrogen bonding interaction as blue or green arrows and non-polar aromatic type as green lines mediated with small aromatic ring. | |
Table 3 Comparative analysis of interaction data of 29 and reference bound compounds
| IDs |
Score |
Binding pocket residue interaction |
| His41 |
Cys145 |
Gly143 |
His163 |
Glu166 |
| VWDa |
D |
VWD |
D |
VWD |
D |
VWD |
D |
VWD |
D |
| VWD = van der Waals interactions (kcal mol−1) D = Distance (Å). |
| 29 |
−7.45 |
−3.86 |
2.23 |
−2.81 |
3.44 |
−2.48 |
2.11 |
−0.44 |
4.88 |
−2.57 |
2.16 |
| Ref. |
−10.1 |
−4.71 |
3.01 |
−3.47 |
2.32 |
−1.66 |
2.61 |
−1.11 |
2.02 |
−4.85 |
2.17 |
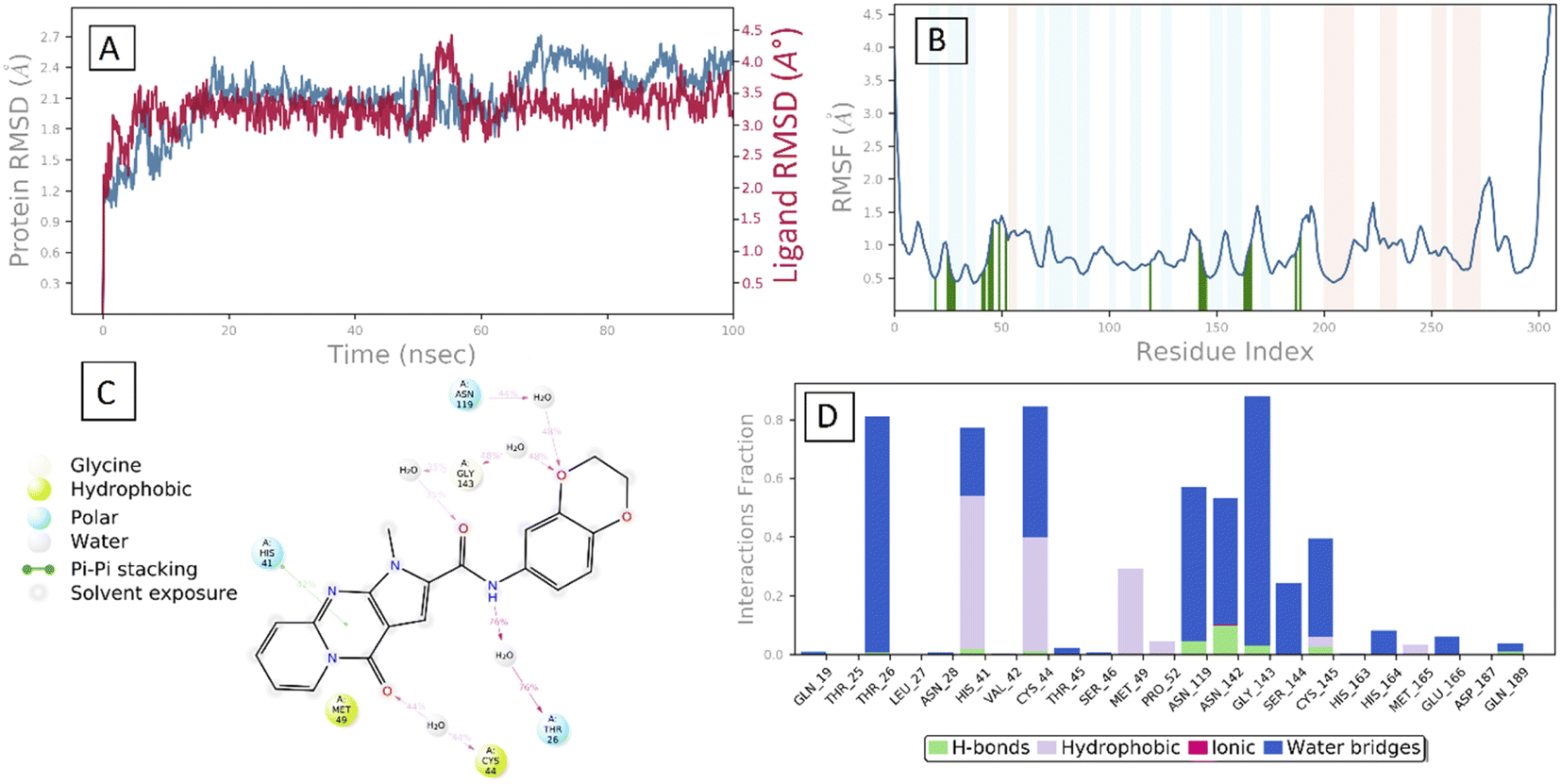
Molecular dynamic (MD) simulation. MD simulations can be used to determine the dynamic and thermodynamic parameters of living systems under specific conditions of physiological environments,53,54 which is very computationally demanding process, hence we used MD simulations to explore the binding stability and conformational behavior of the 29 SARS-CoV-2 Mpro complex in physiological environments through time. The MD simulation of 100 ns time in an explicit solvent system was performed using the Desmond software. The simulated complexes' trajectories were examined for various conventional simulation parameters, such as backbone root-mean standard deviations (RMSD) for α-carbon atoms. Furthermore, the root-mean square fluctuations (RMSF) of particular amino acid residues, as well as ligand protein contact mapping, were assessed. A lower RMSD value throughout the MD simulation indicates the protein–ligand complex is more stable, whereas a higher RMSD value reveals that the protein–ligand complex is less stable. Fig. 6B shows the RMSD of the Cα backbone (blue) of SARS-CoV-2 Mpro protein throughout the simulation based on the reference frame backbone. The maximum RMSD value of Cα backbone of SARS-CoV-2 Mpro protein is 2.7 Å, which indicates that the 29–SARS-CoV-2 Mpro complex was retained consistently during the simulation time.
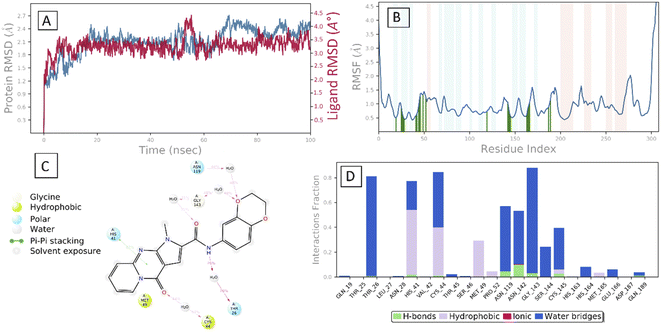 |
| | Fig. 6 MD simulation analysis of 29–SARS-CoV-2 Mpro (A) RMSD (Protein RMSD is presented in grey while RMSD of compound 29 are presented in red color) (B) protein amino acids RMSF (C) 2D ligand interaction diagram and (D) protein–ligand contact analysis of MD trajectory. | |
After the early fluctuation due to the equilibration, the RMSD of the compound 29 remained constant with 3.5 Å till 55 ns, after which minor RMSD fluctuation was noticed and remained stable for the rest of the simulation. The mobility of amino acid residues was assessed throughout time simulation using the protein RMSF in comparison to a reference structure. In the protein RMSF plot, α-helical and β-strand regions are represented in red and blue colors, respectively, while in the white color, the loop region is represented. Protein α-helical and β-strands regions are stiffer than the unstructured region of protein and as a result, fluctuate less than loop regions. If the active site and main chain atoms fluctuated insignificantly, it indicated that the conformational change was minimal, implying that the reported lead compound was tightly bound within the cavity of the SARS-CoV-2 Mpro protein binding pocket. The RMSF plot of 29–SARS-CoV-2 Mpro is displayed in Fig. 3B. According to the RMSF plot, it was observed that compound 29 contacted 23 amino acids of SARS-CoV-2 Mpro binding pocket, namely, Gln19, Thr25, Thr26, Leu27, Asn28, His41, Val42, Cys44, Thr45, Ser46, Met49, Pro52, Asn119, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Asp187, and Gln189. All this interacted residue is marked in green-colored vertical bars, having a RMSF value of less than 1.5 Å. Compound 29 was able to tightly bind to the active site through multiple interactions, including 42% simulation time of π–π stacking with catalytic dyed residue His 41 (Fig. 6C). Fig. 3D indicates that Compound 29 forms hydrophobic and water mediated hydrogen bonding with Tyr25, Asn28, Cys44, Met49, Asn119, Asn142, Gly143, Ser144 and Cys145. It visible that compound 29 made all three types (hydrogen, hydrophobic and water mediated hydrogen bonding) of interaction with crucial residues His 41 and Cys145. All the amino acid interactions identified during docking studies of the target molecule were also displayed during the dynamic study.
Conclusion
In conclusion, we have designed a new set of novel pyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamides and evaluated their antiviral activity against COVID-19. Most of the designed compounds exhibited potent antiviral activity. SAR analysis of the novel scaffold was estimated based on biological and theoretical results obtained for these compounds. The benzodioxan derivative 29 showed very potent anti-SARS-CoV-2 activity at the concentrations of 1 and 10 µM by 94.10 and 96.39% inhibition of the viral growth with EC50 = 0.0519 µM. These novel derivatives were subjected to Mpro protease inhibitory assay and molecular docking study to better understand their underlying mechanism. Compound 29 strongly inhibited the activity of Mpro with an IC50 value of 3.22 µM compared to 12.85 µM for the reference control GC-376. Results of molecular docking study exhibited that compound 29 works through the non-covalent interaction with the main protease of SARS-CoV. Also, a molecular dynamic simulation study has been performed. Docking and dynamic simulations studies proved the prominent interaction behaviour and stability of 29 within the binding pocket through the conserved His41, Glu166, and Cys145 residues. More mechanistic investigations are still needed to decide whether this compound is a competitive or non-competitive inhibitor of the SARS-CoV Mpro.
Materials and method
General
All chemicals were purchased from Sigma-Aldrich Chemical Co. (Sigma-Aldrich Corp., St. Louis, MO, USA). All the melting points were measured with a Stuart Scientific Co. Ltd apparatus, which are uncorrected. The 1H (500 MHz) NMR spectra were measured on a BRUKER AV 500 MHz spectrometer in DMSO-d6, a solvent, using the tetramethylsilane (TMS) as an internal standard. The mass spectra were determined on Agilent 1100 LC MSD Model G1315B Mass Spectrometer (Agilent Technologies, United States). The elemental analysis was carried out at the Regional Centre for Mycology and Biotechnology (RCMP), Al-Azhar University, Cairo, Egypt, and the results were within ± 0.25%. The reaction courses and product mixtures were routinely monitored by the thin layer chromatography (TLC) on silica gel pre-coated F254 Merck plates.
General procedure for synthesis of methyl 1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxylate and 9-methyl derivative (17, 18)
A reaction mixture of 2-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbaldehyde or its 9-methyl derivative (15, 16) (0.01 mol)55 with methyl-N-methyl(N-benzyl)glycinate or methyl methyl-N-benzyl glycinate (0.01 mol) in methanol (20 ml) and triethylamine (0.5 ml) was heated under reflux conditions for 4 h. After the reaction reached completion, the reaction mixture was cooled to room temperature, and the precipitated solid was filtered off, washed with cold methanol, and recrystallized from ethanol/benzene to give compounds 17 and 18 are as previously described.12
General procedure for synthesis of 1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxylic acid (19, 20) and N-aryl-1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (21–29)
A reaction mixture of methyl 1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]-pyrimidine-2-carboxylate or its 9-methyl derivatives (17, 18) (0.01 mol) in aq. lithium hydroxide (0.01 mol) in methanol (20 ml) was refluxed for 2 h. After the reaction reached completion, the reaction mixture was cooled to room temperature, andthe methanol was removed under reduced pressure till dryness, the obtained oil intermediate, 1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxylic acid or its 9-methyl derivatives (19, 20) was then washed with 10% HCl then H2O and the remaining oil was extracted by acetonitrile. The latter was reacted withvarious aromatic amines (0.01 mol) in the presence of 1,1′-carbonyldiimidazole (CDI) (0.01 mol) and acetonitrile (20 ml) under reflux for 4 h. The resultant precipitates were filtered off and the filtrate was evaporated to dryness. The residue was subjected to a column chromatography on silica gel with hexane/ethyl acetate (EtOAc) (3:1) as an eluent to afford the target N-aryl-1-benzyl/methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (21–29). The physical and spectral data of compounds (21–29) are as follows:
1-Benzyl-N-(2-fluorophenyl)-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (21). Colorless needles from ethanol/benzene; yield 87%; mp. 215–217 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.14 (bs, 1H, NH), 8.97, 8.94 (d, 1H, J = 11.6 Hz, pyridine, H-6), 7.78 (s, 2H, Ar, H-3,5), 7.62, 7.60 (d, 1H, J = 9.2 Hz, pyridine, H-8), 7.52 (t, 1H, J = 7.6 Hz, pyridine, H-7), 7.31–7.15 (m, 9H, pyridine H-9, pyrrole H-3 and Ar-H), 5.88 (s, 2H, CH2); 13C NMR (DMSO-d6) δ: 168.72 (CO), 165.72 (CO), 161.72 (C-10a), 159.79 (C-2, Ph), 158.00 (C-9a), 151.94 (C-1, PhCH2), 147.21 (C-8), 143.94 (C-1, Ph), 141.94 (C-4, Ph), 131.58 (C-3,5, Ph), 130.00 (C-2,6, Ph), 129.04 (C-6), 128.97 (C-2), 125.99 (C-4, PhCH2), 120.42 (C-9, C-6, Ph), 116.84 (C-3), 115.43 (C-3, Ph), 115.25 (C-7), 114.04 (C-1, Ph), 103.16 (C-3a), 55.11 (CH2); LCMS (electrospray) m/z (M+ + 1) 413; Anal. Calcd for C24H17FN4O2 (412.42): C, 69.89; H, 4.15; N, 13.59. Found: C, 69.95; H, 4.19; N, 13.63%.
N-(4-Chlorophenyl)-1-methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (22). Colorless needles from ethanol/benzene; yield 89%; mp. 245–247 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.18 (bs, 1H, NH), 8.93, 8.91 (d, 1H, J = 8 Hz, pyridine, H-6), 8.83, 8.81 (d, 2H, J = 8 Hz, Ar-H-2,6), 7.73 (s, 1H, pyrrole, H-3), 7.70, 7.86 (d, 1H, J = 8 Hz, pyridine, H-8), 7.54, 7.52 (d, 1H, J = 8 Hz, pyridine, H-9), 7.30, 7.28 (d, 2H, J = 8 Hz, Ar-H-3,5), 7.06 (t, 1H, J = 8 Hz pyridine, H-7), 4.09 (s, 3H, CH3); 13C NMR (DMSO-d6) δ: 169.74 (CO), 168.17 (CO), 162.74 (C-10a), 160.17 (C-9a), 147.24 (C-1, Ph), 144.70 (C-8), 131.56 (C-4, Ph), 131.11 (C-3,5, Ph), 130.03 (C-6), 129.32 (C-2), 128.60, 128.29 (C-2,6, Ph), 125.97 (C-9), 120.37 (C-3), 114.40 (C-7), 103.11 (C-3a), 37.32 (CH3); LCMS (electrospray) m/z (M+ + 1) 353, m/z (M+ + 3) 355; Anal. Calcd for C24H17ClN4O2 (352.77): C, 61.28; H, 3.71; N, 15.88. Found: C, 61.21; H, 3.65; N, 15.83%.
N-(4-Bromophenyl)-1-methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (23). Colorless needles from ethanol/benzene; yield 86%; mp. 278–280 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.32 (bs, 1H, NH), 8.83, 8.81 (d, 1H, J = 7.6 Hz, pyridine, H-6), 7.77, 7.75 (d, 2H, J = 6 Hz, Ar-H-2,6), 7.71 (s, 1H, pyrrole, H-3), 7.66, 7.65 (d, 2H, J = 8.4 Hz, Ar-H-3,5), 7.54, 7.51 (d, 1H, J = 8.8 Hz, pyridine, H-8), 7.02 (t, 1H, J = 12 Hz pyridine, H-7), 4.02 (s, 3H, CH3), 2.25 (s, 3H, CH3); 13C NMR (DMSO-d6) δ: 168.68 (CO), 165.04 (CO), 160.24 (C-10a), 147.24 (C-9a), 145.12 (C-1, Ph), 131.55 (C-8), 131.52 (C-3,5, Ph), 130.05 (C-6), 129.39 (C-2), 128.28 (C-9), 125.97 (C-2,6, Ph), 120.37 (C-4, Ph), 119.61 (C-3), 114.05 (C-7), 103.11 (C-3a), 37.39 (CH3); LCMS (electrospray) m/z (M+ + 1) 413, m/z (M+ + 3) 415; Anal. Calcd for C24H17BrN4O2 (411.25): C, 55.49; H, 3.68; N, 13.62. Found: C, 55.43; H, 3.61; N, 13.57%.
N-(4-Ethylphenyl)-1-methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (24). Colorless needles from ethanol/benzene; yield 86%; mp. 260–262 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.18 (bs, 1H, NH), 8.91, 8.89 (d, 1H, J = 8.0 Hz, pyridine, H-6), 7.78, 7.76 (d, 2H, J = 8.0 Hz, Ar-H-2,6), 7.76 (s, 1H, pyrrole, H-3), 7.68, 7.66 (d, 2H, J = 8.0 Hz, Ar-H-3,5), 7.59, 7.57 (d, 1H, J = 8.0 Hz, pyridine, H-8), 7.19, 7.17 (d, 1H, J = 8.0 Hz, pyridine, H-9), 7.12 (t, 1H, J = 8.0 Hz pyridine, H-7), 4.01 (s, 3H, CH3), 2.58 (q, 2H, J = 8.0, CH2), 1.71 (s, 3H, J = 8.0, CH3); LCMS (electrospray) m/z (M+ + 1) 347; Anal. Calcd for C20H18N4O2 (346.38): C, 69.35; H, 5.24; N, 16.17. Found: C, 69.29; H, 5.19; N, 16.11%.
N-(3-Acetylphenyl)-1-methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]pyrimidine-2-carboxamide (25). Yellow needles from ethanol/benzene; yield 81%; mp. 238–240 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.41 (bs, 1H, NH), 8.94, 8.92 (d, 1H, J = 8.0 Hz, pyridine, H-6), 8.38 (s, 1H, Ar-H-2), 8.08, 8.06 (d, 1H, J = 8.4 Hz, pyridine, H-8), 7.77 (s, 1H, pyrrole, H-3), 7.72, 7.70 (d, 1H, J = 8.0 Hz, Ar-H-6), 7.61, 7.59 (d, 1H, J = 8.0 Hz, Ar-H-4, pyridine, H-9), 7.51 (t, 1H, J = 8.0 Hz, Ar-H-5), 7.13 (t, 1H, J = 7.2 Hz pyridine, H-7), 4.03 (s, 3H, CH3), 2.59 (s, 3H, COCH3); LCMS (electrospray) m/z (M+) 360; Anal. Calcd for C20H16N4O3 (360.37): C, 66.66; H, 4.48; N, 15.55. Found: C, 66.61; H, 4.44; N, 15.50%.
N-(4-(Dimethylamino)phenyl)-1,9-dimethyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]-pyrimidine-2-carboxamide (26). Yellow needles from ethanol/benzene; yield 81%; mp. 267–269 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 9.99 (bs, 1H, NH), 8.82, 8.80 (d, 1H, J = 8.0 Hz, pyridine, H-6), 7.63, 7.61 (d, 2H, J = 8.0 Hz, Ar-H-2,6), 7.60 (s, 1H, pyrrole, H-3), 7.60,7.57 (d, 1H, J = 12.0 Hz, pyridine, H-8), 7.00 (t, 1H, J = 12 Hz pyridine, H-7), 6.75, 6.73 (d, 2H, J = 8.0 Hz, Ar-H-3,5), 4.01 (s, 3H, CH3), 2.85 [s, 6H, N(CH3)2], 2.50 (s, 3H, CH3); 13C NMR (DMSO-d6) δ: 168.68 (CO), 165.04 (CO), 160.24 (C-10a), 147.24 (C-9a), 145.12 (C-1, Ph), 131.55 (C-8), 131.52 (C-3,5, Ph), 130.05 (C-6), 129.39 (C-2), 128.28 (C-9), 125.97 (C-2,6, Ph), 120.37 (C-4, Ph), 119.61 (C-3), 114.05 (C-7), 103.11 (C-3a), 37.39 (CH3); LCMS (electrospray) m/z (M+ + 1) 376, (M+ + 2) 377; Anal. Calcd for C21H21N5O2 (375.42): C, 67.18; H, 5.64; N, 18.65. Found: C, 67.24; H, 5.69; N, 18.70%.
N-(3-Chloro-4-methylphenyl)-1,9-dimethyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]-pyrimidine-2-carboxamide (27). Pale yellow needles from ethanol/benzene; yield 81%; mp. 280–282 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.22 (bs, 1H, NH), 8.83, 8.81 (d, 1H, J = 7.2 Hz, pyridine, H-6), 7.79 (s, 1H, Ar, H-2), 7.70 (s, 1H, pyrrole, H-3), 7.66, 7.64 (s, 1H, J = 8.0 Hz, Ar, H-6), 7.57, 7.55 (s, 1H, J = 8.4 Hz, Ar, H-5), 7.52,7.50 (d, 1H, J = 8.4 Hz, pyridine, H-8), 7.03 (t, 1H, J = 7.6 Hz pyridine, H-7), 4.02 (s, 3H, CH3), 2.50 (s, 3H, CH3), 2.29 (s, 3H, CH3); LCMS (electrospray) m/z (M+) 381, (M+ + 2) 383, (M+ + 3) 384; Anal. Calcd for C20H17ClN4O2 (380.83): C, 63.08; 4.50; N; 14.71. Found: C, 63.01; 4.44; N; 14.67%.
N-(4-Bromo-3-methylphenyl)-1,9-dimethyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo[2,3-d]-pyrimidine-2-carboxamide (28). Yellow needles from ethanol/benzene; yield 82%; mp. 230–232 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.22 (bs, 1H, NH), 8.83, 8.81 (d, 1H, J = 7.2 Hz, pyridine, H-6), 7.79 (s, 1H, Ar, H-2), 7.03 (s, 1H, pyrrole, H-3), 7.66, 7.64 (s, 1H, J = 8.0 Hz, Ar, H-6), 7.57, 7.55 (s, 1H, J = 8.4 Hz, Ar, H-5), 7.52,7.51 (d, 1H, J = 6.4 Hz, pyridine, H-8), 7.03 (t, 1H, J = 7.6 Hz pyridine, H-7), 4.02 (s, 3H, CH3), 2.52 (s, 3H, CH3), 2.34 (s, 3H, CH3); LCMS (electrospray) m/z (M) 424, (M++1) 425, (M++4) + 428; Anal. Calcd for C20H17BrN4O2 (425.28): C, 56.48; H, 4.03; N; 13.17. Found: C, 56.41; H, 4.00; N; 13.12%.
N-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1-methyl-4-oxo-1,4-dihydropyrido[1,2-a]pyrrolo-[2,3-d]pyrimidine-2-carboxamide (29). Pale yellow needles from ethanol/benzene; yield 81%; mp. 215–217 °C; 1H NMR (DMSO-d6, 400 MHz) δ: 10.08 (bs, 1H, NH), 8.94, 8.92 (d, 1H, J = 8.0 Hz, pyridine, H-6), 8.80 (s, 1H, Ar-H-2), 7.76, 7.74 (d, 1H, J = 8.0 Hz, pyridine, H-8), 7.65 (s, 1H, pyrrole, H-3), 7.61, 7.59 (d, 1H, J = 8.0 Hz, Ar-H-9), 7.21, 7.19 (d, 1H, J = 8.8 Hz, Ar-H-6), 7.12 (t, 1H, J = 8.0 Hz pyridine, H-7), 6.84, 6.81 (t, 1H, J = 8.8 Hz, Ar-H-5), 4.33 (s, 4H, 2CH2), 4.03 (s, 3H, CH3); LCMS (electrospray) m/z (M+ + 1)377; Anal. Calcd for C20H16N4O3 (376.37): C, 63.82; H, 4.28; N, 14.89. Found: C, 63.88; H, 4.34; N, 14.94%.
Biological evaluation studies
Antiviral assay. To determine the effect of target compounds on SARS-CoV-2 viral load (SARS-CoV-2 isolate EGY/WAT-2 VACCERA), qualitative detection of SARS-CoV-2 viral RNA was carried out using a Real-Time PCR assay.56 Total RNA was extracted using genesig® Coronavirus SARS-CoV-2 Real-Time PCR Assay kit (Primer design TM Ltd, Southampton, United Kingdom) according to the manufacturer's protocol.57 Briefly, a monolayer of Vero cells was prepared in a 96-well plate. The cells were then treated with a single non-toxic dose of each compound in a triplicate and infected with SARS-CoV-2 at a MOI of 0.01 with incubation period of 1 h at 37 °C followed by removal of inoculum and overlaying with cell culture media containing the test compound. As a vehicle control, cells in three wells were treated with 0.1% DMSO diluted with cell culture media. The plate was kept for 48 h at 37 °C in the presence of 5% CO2, followed by performing a virus yield inhibition assay using qRT-PCR on harvested supernatants from the wells. This kit contains a master mix, primers, and probe designed for reverse transcription of the extracted RNA and Real-Time PCR for the detection of SARS-CoV-2. The assay was performed using the Rotor-Gene Q instrument (Qiagen, Germany) under the following amplification conditions: 10 min Reverse Transcription at 55 °C, 2 min initial activation at 95 °C, followed by 45 cycles of 10 s denaturation at 95 °C and 60 s annealing and extension at 60 °C (see ESI†). The target compounds were further assessed using a dose response assay by adding to cells at five different concentrations of 10, 1, 0.1, 0.01, and 0.001 µM. EC50 and EC90 values that produce virus-induced cell death were measured. Selectivity index (SI) was calculated as a ratio of drug's IC50 and EC50 values (SI = IC50/EC50).
Cytotoxicity evaluation using a viability assay. The cytotoxic activity was assessed using the MTT colorimetric assay as reported previously.58–62 In brief, tumour cell lines were suspended in medium at concentration 5 × 104 cell/well in Corning® 96-well tissue culture plates and then incubated for 24 h. The tested compounds with concentrations ranging from 0 to 50 µg ml−1 were then added into 96-well plates (in a triplicate) to achieve different concentrations for each compound. Vehicle controls with media or 0.5% DMSO were run for each 96 well plate as a negative control. After incubating for 24 h, the numbers of viable cells were determined.
SARS-CoV-2-MPro inhibition assay. Kits of SARS-CoV-2 Mpro with an internally quenched substrate, and assay buffer were purchased from BPS Bioscience (San Diego, CA, USA). The SARS-CoV-2-MPro enzyme assay was developed in 384-well black, medium binding microplates with a total volume of 20 µL and then reduced to 1536-well format as previously reported.63 In a 384-well plate format, 10 µL of SARS-CoV-2 Mpro in reaction buffer was added into each well, followed by the addition of 10 µL of the inhibitor compound.63,64 Fluorescent intensity was measured at different time intervals. Briefly, 10 µl of drugs (250 µM) were incubated for 1 hour with 4 ng Mpro-MBP tagged enzyme in 30 µl of assay buffer. By the end of the incubation time, 10 µl fluorescent substrate (250 µM) were added to initiate the enzymatic reaction. The final reaction volume was 50 µl and the final concentration of drugs and substrate in the reaction mixture was 50 µM. After a second incubation period of 16–18 h, fluorescence was measured at 360/40 nm excitation and 460/40 nm emission using Synergy HT fluorescent plate reader. Drugs were screened from 0 to 1000 µM dose range to calculate the IC50. To prepare positive control wells, 1% DMSO with 4 ng of enzyme and 50 µM of substrate were added without enzyme inhibitors. The standard lopinavir inhibitor was used at 50 µM to serve as a positive control. Wells containing 1% DMSO with 50 µM of substrate without enzyme served as blanks. All the values were subtracted from blank values (see ESI† for manufacturer's kit (BPS Biosciences CA)).64,65 The experiment was conducted at both room temperature (RT) and 37 °C.
Computational analysis methods
Molecular docking. To investigate the binding interaction of a target compound with the active site of SARS-CoV-2 Mpro enzyme, a molecular docking study was conducted using the Glide module of Schrodinger, associated with the graphical user interface program Maestro 8.50,66 SARS-CoV2 Mpro protein crystal structure in complex with bipyridine benzonitrile was acquired from the RCSB Protein Data Bank (PDB) as entry code 7L11.49 The protein preparation was completed in two steps: preparation and refinement, using Maestro 8.0's protein preparation wizard. After ensuring chemical correctness, water molecules in crystal structures were removed, and hydrogens were added wherever they were missing.67–69 Using the OPLS 2005 forcefield, the energy of the crystal structure was minimized to avert any electronic clash in the structure. The binding site was specified around the co-crystallized ligand (Bipyridine benzonitrile), and the receptor grid was created using Grid Generation tool. Molecular docking was accomplished in standard precision mode using the glide ligand docking module. A single best pose is produced as the output for a specific ligand.70
Molecular dynamics (MD) simulation study. The thermodynamic stability of a promising molecule in complex with SARS-CoV-2 Mpro was evaluated using MD simulations on the OPLS3e forcefield with the Desmond package (Desmond 2020-3, Schrodinger LLC).71 The ligand–protein complex was fixed in a solvent-soaked orthorhombic periodic box with a minimum distance of 10 Å between protein atoms and the box edges. The solvent was implemented using a single point charge (SPC) water model. The charge of the system was neutralized by adding Na+ and Cl−counter ions and 0.15 M NaCl Salt concentration was set corresponding to the physiological system using the Desmond ‘System Builder’ panel.72,73 Then, the built solvated system was minimized and relaxed utilizing OPLS3e force field parameters as the default protocol associated with Desmond. The isothermal isobaric ensemble (normal pressure and temperature/NPT) was set during the simulation, with a temperature of 300 K and an atmospheric pressure of 1.0315 bar using the Nose–Hoover chain thermostat method and the Martyna–Tobias–Klein barostat method, respectively.74,75 The MD simulation was carried out for a period of 100 ns, and trajectory snapshots were recorded at an interval of 20 ps. MD trajectories were investigated using Desmond's Simulation Interaction Diagram (SID) to predict the binding orientation of the ligand.
Author contributions
A. Aljuhani, H. E. A. Ahmed, and S. Ihmaid were responsible for the conception and rational design of the work. A. El-Agrody, A. M. Omar, M. F. Zayed, S. A. Salama, H. S. Abulkhair, and A. Aljuhani, were responsible for collection of data and synthesis of new compounds. S. Althagfan, Y. M. Alahmadi, S. Ahmed, and H. Ahmed, performed the molecular docking study. Molecular dynamic simulations have been conducted by H. E. A. Ahmed, S. Ihmaid, and A. A. Al-Karmalawy. A. El-Agrody, I. Ahma, M. A. Almikhlafi, S. H. Abulkhair, A. M. El-Agrody, S. A. Turkistani, M. Almaghrabi and, and H. S. Abulkhair were responsible for analyzing the spectral data and preparation of the ESI.† All authors read and approved the final form of the manuscript.
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgements
We greatly thank Taif University for providing fund for this work through Taif University Researcher Supporting Project number (TURSP-2020/52), Taif University, Taif, Saudi Arabia. We would also like to thank Professor Ali H. El-Far, Department of Biochemistry, Faculty of Veterinary Medicine, Damanhour University, Egypt, for his considerable help throughout revision stages of the manuscript.
References
- P. Gautret, J.-C. Lagier, P. Parola, V. T. Hoang, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira, H. Tissot Dupont, S. Honoré, P. Colson, E. Chabrière, B. La Scola, J.-M. Rolain, P. Brouqui and D. Raoult, Int. J. Antimicrob. Agents, 2020, 56, 105949 CrossRef CAS PubMed.
- World Health Organization, WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int/, (accessed 7 July 2022) Search PubMed.
- A. R. Maher, M. Maglione, S. Bagley, M. Suttorp, J.-H. Hu, B. Ewing, Z. Wang, M. Timmer, D. Sultzer and P. G. Shekelle, JAMA, 2011, 306, 1359 CrossRef CAS PubMed.
- A. Mahmoud, A. Mostafa, A. A. Al-Karmalawy, A. Zidan, H. S. Abulkhair, S. H. Mahmoud, M. Shehata, M. M. Elhefnawi and M. A. Ali, Heliyon, 2021, 7, e07962 CrossRef PubMed.
- M. I. A. Hamed, K. M. Darwish, R. Soltane, A. Chrouda, A. Mostafa, N. M. Abo Shama, S. S. Elhady, H. S. Abulkhair, A. E. Khodir, A. A. Elmaaty and A. A. Al-karmalawy, RSC Adv., 2021, 11, 35536–35558 RSC.
- A. J. Pruijssers and M. R. Denison, Curr. Opin. Virol., 2019, 35, 57–62 CrossRef CAS PubMed.
- G. G. Plata, The BMJ, 2022, o1282 CrossRef PubMed.
- A. Mahmoud, A. Mostafa, A. A. Al-Karmalawy, A. Zidan, H. S. Abulkhair, S. H. Mahmoud, M. Shehata, M. M. Elhefnawi and M. A. Ali, Heliyon, 2021, 7, e07962 CrossRef PubMed.
- O. Kutkat, Y. Moatasim, A. A. Al-Karmalawy, H. S. Abulkhair, M. R. Gomaa, A. N. El-Taweel, N. M. Abo Shama, M. GabAllah, D. B. Mahmoud, G. Kayali, M. A. Ali, A. Kandeil and A. Mostafa, Sci. Rep., 2022, 12, 12920 CrossRef CAS PubMed.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang, P. Niu, H. Song, Y. Shi, X. Li, W. Tan, J. Qi and G. F. Gao, Nat. Commun., 2020, 11, 4417 CrossRef CAS.
- M. M. Hammoud, M. Khattab, M. Abdel-Motaal, J. Van der Eycken, R. Alnajjar, H. S. Abulkhair and A. A. Al-Karmalawy, J. Biomol. Struct. Dyn., 2022 DOI:10.1080/07391102.2022.2082533 , https://www.tandfonline.com/doi/abs/10.1080/07391102.2022.2082533?journalCode=tbsd20.
- A. A. Gaber, A. M. El-Morsy, F. F. Sherbiny, A. H. Bayoumi, K. M. El-Gamal, K. El-Adl, A. A. Al-Karmalawy, R. R. Ezz Eldin, M. A. Saleh and H. S. Abulkhair, Arch. Pharm., 2021 DOI:10.1002/ardp.202100258 , https://onlinelibrary.wiley.com/doi/10.1002/ardp.202100258.
- C. Choudhury, J. Biomol. Struct. Dyn., 2021, 39, 3733–3746 CrossRef CAS PubMed.
- I. Antonopoulou, E. Sapountzaki, U. Rova and P. Christakopoulos, Comput. Struct. Biotechnol. J., 2022, 20, 1306–1344 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- S. Günther, P. Y. A. Reinke, Y. Fernández-García, J. Lieske, T. J. Lane, H. M. Ginn, F. H. M. Koua, C. Ehrt, W. Ewert, D. Oberthuer, O. Yefanov, S. Meier, K. Lorenzen, B. Krichel, J.-D. Kopicki, L. Gelisio, W. Brehm, I. Dunkel, B. Seychell, H. Gieseler, B. Norton-Baker, B. Escudero-Pérez, M. Domaracky, S. Saouane, A. Tolstikova, T. A. White, A. Hänle, M. Groessler, H. Fleckenstein, F. Trost, M. Galchenkova, Y. Gevorkov, C. Li, S. Awel, A. Peck, M. Barthelmess, F. Schlünzen, P. Lourdu Xavier, N. Werner, H. Andaleeb, N. Ullah, S. Falke, V. Srinivasan, B. A. França, M. Schwinzer, H. Brognaro, C. Rogers, D. Melo, J. J. Zaitseva-Doyle, J. Knoska, G. E. Peña-Murillo, A. R. Mashhour, V. Hennicke, P. Fischer, J. Hakanpää, J. Meyer, P. Gribbon, B. Ellinger, M. Kuzikov, M. Wolf, A. R. Beccari, G. Bourenkov, D. von Stetten, G. Pompidor, I. Bento, S. Panneerselvam, I. Karpics, T. R. Schneider, M. M. Garcia-Alai, S. Niebling, C. Günther, C. Schmidt, R. Schubert, H. Han, J. Boger, D. C. F. Monteiro, L. Zhang, X. Sun, J. Pletzer-Zelgert, J. Wollenhaupt, C. G. Feiler, M. S. Weiss, E.-C. Schulz, P. Mehrabi, K. Karničar, A. Usenik, J. Loboda, H. Tidow, A. Chari, R. Hilgenfeld, C. Uetrecht, R. Cox, A. Zaliani, T. Beck, M. Rarey, S. Günther, D. Turk, W. Hinrichs, H. N. Chapman, A. R. Pearson, C. Betzel and A. Meents, Science, 2021, 372, 642–646 CrossRef PubMed.
- I. Antonopoulou, E. Sapountzaki, U. Rova and P. Christakopoulos, Comput. Struct. Biotechnol. J., 2022, 20, 1306–1344 CrossRef CAS PubMed.
- D. S. Jairajpuri, A. Hussain, K. Nasreen, T. Mohammad, F. Anjum, M. Tabish Rehman, G. Mustafa Hasan, M. F. Alajmi and M. Imtaiyaz Hassan, Saudi J. Biol. Sci., 2021, 28, 2423–2431 CrossRef CAS PubMed.
- A. K. Ghosh, G. Gong, V. Grum-Tokars, D. C. Mulhearn, S. C. Baker, M. Coughlin, B. S. Prabhakar, K. Sleeman, M. E. Johnson and A. D. Mesecar, Bioorg. Med. Chem. Lett., 2008, 18, 5684–5688 CrossRef CAS PubMed.
- Z. Jin, Y. Zhao, Y. Sun, B. Zhang, H. Wang, Y. Wu, Y. Zhu, C. Zhu, T. Hu, X. Du, Y. Duan, J. Yu, X. Yang, X. Yang, K. Yang, X. Liu, L. W. Guddat, G. Xiao, L. Zhang, H. Yang and Z. Rao, Nat. Struct. Mol. Biol., 2020, 27, 529–532 CrossRef CAS.
- M. Negi, P. A. Chawla, A. Faruk and V. Chawla, Bioorg. Chem., 2020, 104, 104315 CrossRef CAS PubMed.
- A. K. Ghosh, G. Gong, V. Grum-Tokars, D. C. Mulhearn, S. C. Baker, M. Coughlin, B. S. Prabhakar, K. Sleeman, M. E. Johnson and A. D. Mesecar, Bioorg. Med. Chem. Lett., 2008, 18, 5684–5688 CrossRef CAS PubMed.
- D.-S. Park, E. Jo, J. Choi, M. Lee, S. Kim, H.-Y. Kim, J. Nam, S. Ahn, J. Y. Hwang and M. P. Windisch, Eur. J. Med. Chem., 2017, 140, 65–73 CrossRef CAS PubMed.
- M. Lee, J. Yang, E. Jo, J.-Y. Lee, H.-Y. Kim, R. Bartenschlager, E.-C. Shin, Y.-S. Bae and M. P. Windisch, Sci. Rep., 2017, 7, 44676 CrossRef.
- K. Bafna, R. M. Krug and G. T. Montelione, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12153615.
- K. Bafna, K. White, B. Harish, R. Rosales, T. A. Ramelot, T. B. Acton, E. Moreno, T. Kehrer, L. Miorin, C. A. Royer, A. García-Sastre, R. M. Krug and G. T. Montelione, Cell Rep., 2021, 35, 109133 CrossRef CAS PubMed.
- K. Bafna, K. White, B. Harish, R. Rosales, T. A. Ramelot, T. B. Acton, E. Moreno, T. Kehrer, L. Miorin, C. A. Royer, A. García-Sastre, R. M. Krug and G. T. Montelione, Cell Rep., 2021, 35, 109133 CrossRef CAS.
- R. Oerlemans, A. J. Ruiz-Moreno, Y. Cong, N. Dinesh Kumar, M. A. Velasco-Velazquez, C. G. Neochoritis, J. Smith, F. Reggiori, M. R. Groves and A. Dömling, RSC Med. Chem., 2021, 12, 370–379 RSC.
- H. S. Abulkhair, A. Turky, A. Ghiaty, H. E. A. Ahmed and A. H. Bayoumi, Bioorg. Chem., 2020, 100, 103899 CrossRef CAS.
- M. H. El-Shershaby, K. M. El-Gamal, A. H. Bayoumi, K. El-Adl, H. E. A. Ahmed and H. S. Abulkhair, Arch. Pharm., 2021, 354, e2000277 CrossRef PubMed.
- A. M. Omar, S. Ihmaid, E. S. E. Habib, S. S. Althagfan, S. Ahmed, H. S. Abulkhair and H. E. A. Ahmed, Bioorg. Chem., 2020, 99, 103781 CrossRef CAS PubMed.
- E. M. Othman, E. A. Fayed, E. M. Husseiny and H. S. Abulkhair, New J. Chem., 2022, 46, 12206–12216 RSC.
- D. S. Jairajpuri, A. Hussain, K. Nasreen, T. Mohammad, F. Anjum, M. Tabish Rehman, G. Mustafa Hasan, M. F. Alajmi and M. Imtaiyaz Hassan, Saudi J. Biol. Sci., 2021, 28, 2423–2431 CrossRef CAS.
- M. Abass, M. Ismail, W. Abdel-Monem and A. Mayas, Chem. Pap., 2010, 64 DOI:10.2478/s11696-009-0100-0 , https://link.springer.com/article/10.2478/s11696-009-0100-0.
- M. F. Maioral, C. do N. Bodack, N. M. Stefanes, Á. Bigolin, A. Mascarello, L. D. Chiaradia-Delatorre, R. A. Yunes, R. J. Nunes and M. C. Santos-Silva, Biochimie, 2017, 140, 48–57 CrossRef CAS.
- M. H. El-Shershaby, A. Ghiaty, A. H. Bayoumi, A. A. Al-Karmalawy, E. M. Husseiny, M. S. El-Zoghbi and H. S. Abulkhair, Bioorg. Med. Chem., 2021, 42, 116266 CrossRef CAS PubMed.
- E. M. Othman, E. A. Fayed, E. M. Husseiny and H. S. Abulkhair, Bioorg. Chem., 2022, 123, 105762 CrossRef CAS PubMed.
- E. M. Othman, E. A. Fayed, E. M. Husseiny and H. S. Abulkhair, Bioorg. Chem., 2022, 127, 105968 CrossRef CAS PubMed.
- J. Osipiuk, S.-A. Azizi, S. Dvorkin, M. Endres, R. Jedrzejczak, K. A. Jones, S. Kang, R. S. Kathayat, Y. Kim, V. G. Lisnyak, S. L. Maki, V. Nicolaescu, C. A. Taylor, C. Tesar, Y.-A. Zhang, Z. Zhou, G. Randall, K. Michalska, S. A. Snyder, B. C. Dickinson and A. Joachimiak, Nat. Commun., 2021, 12, 743 CrossRef CAS PubMed.
- A. Abo Elmaaty, M. I. A. Hamed, M. I. Ismail, E. B. Elkaeed, H. S. Abulkhair, M. Khattab and A. A. Al-Karmalawy, Molecules, 2021, 26, 3772 CrossRef.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- C. Ma, Y. Hu, J. A. Townsend, P. I. Lagarias, M. T. Marty, A. Kolocouris and J. Wang, ACS Pharmacol. Transl. Sci., 2020, 3, 1265–1277 CrossRef CAS PubMed.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. A. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, Nat. Commun., 2020, 11, 4282 CrossRef CAS PubMed.
- H. Abul-Khair, S. Elmeligie, A. Bayoumi, A. Ghiaty, A. El-Morsy and M. H. Hassan, J. Heterocycl. Chem., 2013, 50, 1202–1208 CAS.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS PubMed.
- K. Sharun, R. Tiwari and K. Dhama, Ann. Med. Surg., 2021, 61, 122–125 CrossRef.
- C.-H. Zhang, E. A. Stone, M. Deshmukh, J. A. Ippolito, M. M. Ghahremanpour, J. Tirado-Rives, K. A. Spasov, S. Zhang, Y. Takeo, S. N. Kudalkar, Z. Liang, F. Isaacs, B. Lindenbach, S. J. Miller, K. S. Anderson and W. L. Jorgensen, ACS Cent. Sci., 2021, 7, 467–475 CrossRef CAS.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- A. A. Zaki, M. M. Y. Kaddah, H. S. Abulkhair and A. Ashour, RSC Adv., 2022, 12, 2980–2991 RSC.
- H. S. Abulkhair, S. Elmeligie, A. Ghiaty, A. El-Morsy, A. H. Bayoumi, H. E. A. Ahmed, K. El-Adl, M. F. Zayed, M. H. Hassan, E. N. Akl and M. S. El-Zoghbi, Arch. Pharm., 2021, 354, 2000449 CrossRef CAS PubMed.
- S. K. Ihmaid, A. Aljuhani, M. Alsehli, N. Rezki, A. Alawi, A. J. Aldhafiri, S. A. Salama, H. E. A. Ahmed and M. R. Aouad, J. Mol. Struct., 2022, 1249, 131568 CrossRef CAS.
- M. H. El-Shershaby, A. Ghiaty, A. H. Bayoumi, H. E. A. Ahmed, M. S. El-Zoghbi, K. El-Adl and H. S. Abulkhair, New J. Chem., 2021, 45, 11136–11152 RSC.
- A. De, S. Sarkar and A. Majee, Chem. Heterocycl. Compd., 2021, 57, 410–416 CrossRef CAS PubMed.
- Y.-S. Chung, N.-J. Lee, S. H. Woo, J.-M. Kim, H. M. Kim, H. J. Jo, Y. E. Park and M.-G. Han, Sci. Rep., 2021, 11, 14817 CrossRef CAS.
- Genesig, Coronavirus COVID-19 genesig® Real-Time PCR assay, https://www.genesig.com/assets/files/Path_COVID_19_CE_STED_IFU_Issue_500.pdf, (accessed 12 March 2022) Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. M. Mehany, H. S. Abulkhair and I. H. Eissa, Arch. Pharm., 2022, 355(7), 2200048 CrossRef CAS PubMed , https://onlinelibrary.wiley.com/doi/10.1002/ardp.202200048.
- K. El-Adl, M. K. Ibrahim, F. Khedr, H. S. Abulkhair and I. H. Eissa, Arch. Pharm., 2022, 355, 2100278 CrossRef CAS PubMed.
- F. Khedr, M. K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Arch. Pharm., 2021, 354, e2100201 CrossRef PubMed.
- K. El-Adl, H. Sakr, S. S. A. El-Hddad, A. G. A. El-Helby, M. Nasser and H. S. Abulkhair, Arch. Pharm., 2021, 354(7) DOI:10.1002/ardp.202000491 , https://onlinelibrary.wiley.com/doi/10.1002/ardp.202000491.
- W. Zhu, M. Xu, C. Z. Chen, H. Guo, M. Shen, X. Hu, P. Shinn, C. Klumpp-Thomas, S. G. Michael and W. Zheng, Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-throughput Screening, 2020 Search PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, ChemBioChem, 2020, 21, 730–738 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- K. El-Adl, A. G. A. El-Helby, H. Sakr, R. R. Ayyad, H. A. Mahdy, M. Nasser, H. S. Abulkhair and S. S. A. El-Hddad, Arch. Pharm., 2021, 354, e202000279 Search PubMed.
- H. G. Ezzat, A. H. Bayoumi, F. F. Sherbiny, A. M. El-Morsy, A. Ghiaty, M. Alswah and H. S. Abulkhair, Mol. Diversity, 2021, 25, 291–306 CrossRef CAS PubMed.
- R. G. Yousef, H. M. Sakr, I. H. Eissa, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and K. El-Adl, New J. Chem., 2021, 45, 16949–16964 RSC.
- I. Ahmad, M. Shaikh, S. Surana, A. Ghosh and H. Patel, J. Biomol. Struct. Dyn., 2022, 40, 3046–3059 CrossRef CAS PubMed.
- D. E. Shaw Research, 2021, https://www.schrodinger.com/products/desmond.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- I. Ahmad, H. Jadhav, Y. Shinde, V. Jagtap, R. Girase and H. Patel, In Silico Pharmacol., 2021, 9, 23 CrossRef PubMed.
- G. Kalibaeva, M. Ferrario and G. Ciccotti, Mol. Phys., 2003, 101, 765–778 CrossRef CAS.
- G. J. Martyna, Phys. Rev. C: Nucl. Phys., 1994, 50, 3234–3236 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04015h |
| ‡ These authors contributed equally to this work and share first authorship. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence h,
Iqrar Ahmadi,
Harun Pateli,
Sahar Ahmedbj,
Mohannad A. Almikhlafik,
Ahmed M. El-Agrodyl,
Mohamed F. Zayedgm,
Safaa Abdulrahman Turkistanim,
Shorouk H. Abulkhair
h,
Iqrar Ahmadi,
Harun Pateli,
Sahar Ahmedbj,
Mohannad A. Almikhlafik,
Ahmed M. El-Agrodyl,
Mohamed F. Zayedgm,
Safaa Abdulrahman Turkistanim,
Shorouk H. Abulkhair