
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContra-thermodynamic halolactonization of lactam-tethered 5-aryl-4(E)-pentenoic acids for the flexible and stereocontrolled synthesis of fused lactam-halolactones†
Timothy K. Beng *,
Claire Borg and
Morgan J. Rodriguez
*,
Claire Borg and
Morgan J. Rodriguez
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 10th October 2022
Abstract
Halolactonization of alkenoic acids enables the construction of oxygen-heterocycles via intramolecular halonium-induced nucleophilic addition. Although the literature is currently inundated with halolactonizations of 5-aryl-4(E)-pentenoic acids that predictably afford the 6-endo cyclization adducts, methods that reliably alter the innate regioselectivity bias to instead deliver the thermodynamically less favored 5-exo cyclization products are relatively rare. Here, we attempt to bridge this gap and have found mild conditions for contra-thermodynamic halolactonization of lactam-tethered 5-aryl-4(E)-pentenoic acids that lead to the formation of trans-fused lactam-γ-lactones. The natural proclivity for these 5-aryl-4(E)-pentenoic acids to undergo 6-endo cyclization is overridden and 5-exo-trig cyclization predominates. The success of the approach hinges on the use of N,N-dimethylformamide (DMF) as the solvent and N-methylmorpholine oxide as the catalyst. The lactam-lactone products are synthesized in high diastereoselectivity, modularity, and chemoselectivity. Notably, most of the bicycles contain one benzylic quaternary stereocenter as well as an α-alkoxy quaternary stereocenter.
Introduction
Functionalized lactones constitute the core of several natural products and pharmaceuticals.1 They are frequently used as versatile building blocks for accessing other oxygen-containing heterocycles and carboxylic acid derivatives.2 Meanwhile, lactams are ubiquitous in drug discovery and medicinal chemistry programs.3 In addition to their well-studied antibiotic activity,4 several functionalized lactams act as opioid receptor agonists,5 HIV-1 integrase inhibitors,6 anticancer,7 antidepressant,8 and anti-inflammatory agents.9 Importantly, sp3-rich fused lactam-lactones are resident in bioactive molecules such as neooxazolomycin and UCS 1025 A (Fig. 1). It has previously been articulated that the presence of carbon stereocenters correlate with success as compounds transition from discovery to drugs.10 Accordingly, medicinal chemists have become increasingly keen on exploring 3D-structural space, which makes sp3-rich fused lactam-lactones important targets for pharmaceutical companies. As depicted in Fig. 2A and B, some reliable tactics for the construction of fused lactam-lactones have been reported by Wee11 and Burton.12 | ||
| Fig. 1 Examples of C-fused bioactive γ-lactam-γ-lactones. | ||
 | ||
| Fig. 2 (A and B) Prior syntheses of fused lactam-γ-lactones, (C) our proposed plan for contra-thermodynamic 5-exo cyclization of 1, (D) our recently reported approach for thermodynamic 6-endo cyclization of 1. | ||
Among existing methods for the construction of common-ring lactones,13 halogen-initiated cyclization of unsaturated carboxylic acids is an attractive and direct approach for the stereoselective synthesis of halogenated lactones.14,15 The transformation is very versatile given that the generated halogen and lactone motifs are versatile linchpins for late-stage diversification. Moreover, over 4700 halogen-containing natural products exist,16 which has fueled numerous target-oriented synthesis efforts that employ halolactonization protocols. Although the literature is replete with halolactonizations of 5-aryl-4(E)-pentenoic acids that predictably afford the 6-endo cyclization adducts (mainly due to unsymmetrical build-up of positive charge next to the aryl group in the transition state17), the development of methods that reliably alter the innate regioselectivity bias to instead deliver the thermodynamically less favored 5-exo cyclization products is still at the incipient stages. For instance, Nicewicz and co-workers expertly developed a synergistic protocol that accomplished halolactonization of 5-aryl-4(E)-pentenoic acids with opposite regioselectivity.18 The authors articulated that even under their photocatalytic conditions, the use of N-halosuccinimides failed to override the innate 6-endo cyclization pathway.
Our interest in the synthesis and post-diversification of lactam-tethered alkenoic acids19 prompted us to explore a practical and flexible route toward sp3-rich fused lactam-lactones bearing quaternary and contiguous stereocenters. Toward this end, we sought to interrogate readily available lactam-bearing 5-aryl-4(E)-pentenoic acids of type 1,20a in a contra-thermodynamic and catalytic halolactonization protocol (Fig. 2C). Herein, we demonstrate that the natural proclivity of these 5-aryl-4(E)-pentenoic acids to undergo 6-endo cyclization can be overridden using solvent- and catalyst-controlled reactivity. When DMF is employed as the solvent and N-methylmorpholine oxide (NMO) as the catalyst, the 5-exo cyclization products are obtained in diastereoselective, chemoselective, modular, and scalable fashions. The method nicely complements our recently developed protocol for accessing the thermodynamic δ-lactone-fused lactams (Fig. 2D).20b
Results and discussion
During our previous attempts at thermodynamic 6-endo cyclization of lactam-bearing 5-aryl-4(E)-pentenoic acids of type 1, we noted that bromolactonization using N-bromosuccinimide (NBS, 1.1 equiv.) and Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S (5 mol%) failed to furnish the desired product when the solvent was switched from dichloromethane to N,N-dimethylformamide (DMF).20b Instead, the contra-thermodynamic 5-exo cyclization product was formed in modest yield. Having previously found an isolated example of catalyst-free regiodivergent bromolactonization on a valerolactam-tethered alkenoic acid,19f we set out to explore efficient and general conditions for contra-thermodynamic halolactonization of 1.
S (5 mol%) failed to furnish the desired product when the solvent was switched from dichloromethane to N,N-dimethylformamide (DMF).20b Instead, the contra-thermodynamic 5-exo cyclization product was formed in modest yield. Having previously found an isolated example of catalyst-free regiodivergent bromolactonization on a valerolactam-tethered alkenoic acid,19f we set out to explore efficient and general conditions for contra-thermodynamic halolactonization of 1.
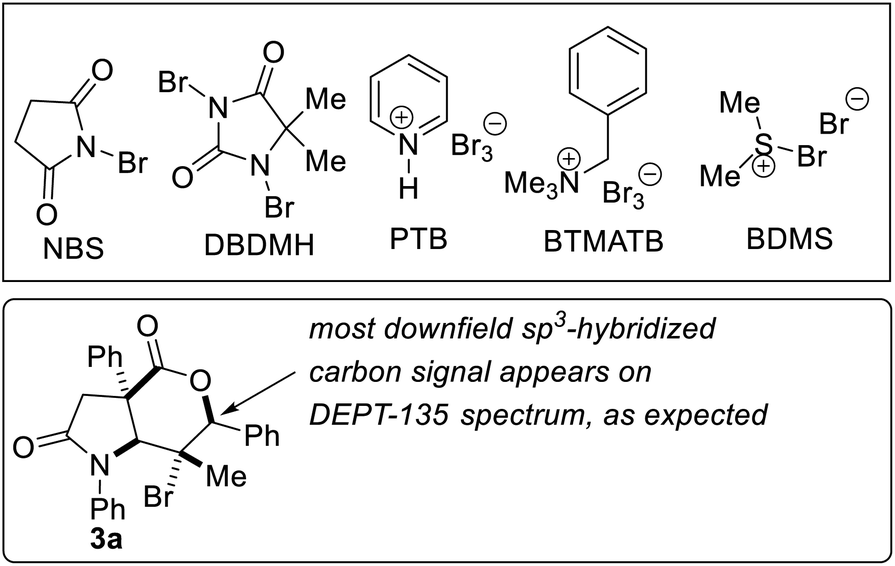
We benchmarked our optimization efforts for bromolactonization of alkenoic acid 1a with the reaction conditions described in Table 1. Trisubstituted alkenoic acid 1a was chosen as the model substrate in order to test the power of the methodology since halolactonization reactions of trisubstituted alkenes are known to be problematic from the standpoints of regioselectivity and stereospecificity.17 During the optimization studies, DMF indeed emerged as the solvent of choice (entries 1–4). Other electrophilic brominating agents did not perform as well as NBS (entries 5–9). N-methylmorpholine oxide (NMO) emerged as the most effective catalyst (entries 11–15). The 5-exo cyclization pathway also predominates in the absence of a catalyst, but longer reaction times (∼72 h) are required to attain full conversion and the exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :endo ratio is compromised (3:1). Under the optimized conditions, lactam-lactone 2a was obtained in good yield, high anti-stereoselectivity, and in impeccable 5-exo selectivity.
:endo ratio is compromised (3:1). Under the optimized conditions, lactam-lactone 2a was obtained in good yield, high anti-stereoselectivity, and in impeccable 5-exo selectivity.
|
|---|
 |

The scope of the contra-thermodynamic bromolactonization has been explored by altering the three variables highlighted in alkenoic acid 1 (Scheme 1). Knowing that the nature of the nitrogen substituent present on a nitrogen heterocycle can have a dramatic effect on its biological activity and reactivity,21 the effect of the N-substituent on the sulfenolactonization was first explored. In the event, we find that N-aryl-, alkyl-, allyl-, and benzyl-substituted-γ-lactam-tethered trisubstituted alkenoic acids undergo successful contra-thermodynamic 5-exo bromolactonization (see 2a–m). The N-substituent has an effect on the diastereoselectivity in some cases (see 2j). The strategic construction of lactam-bromolactones harboring the N-phenethyl group (see 2k–n and 2p–t) is noteworthy given that the latter is often employed as a precursor to the indolizidine/quinolizidine/tetrahydroisoquinoline scaffolds through Pictet–Spengler-style cyclizations.22 The transformation displays excellent chemoselectivity given that a lactam-tethered alkenoic acid bearing an N-allyl substituent reacts with NBS to afford bicycle 2e, without complications arising from bromolactonization of the kinetically more accessible allyl group (see 2o as well). We attribute this chemoselective bromolactonization to conformational constraints and to the more activated nature of the styrenyl double bond.
 | ||
| Scheme 1 5-exo-bromolactonization of γ-lactam-tethered alkenoic acids. | ||
We have also explored the scope of the transformation with respect to the alkene portion resident in 1. Starting with the internal substituent on the alkene (i.e., R′), we find that this 5-exo-trig cyclization is quite sensitive to the steric environment given that the replacement of the internal methyl substituent with an unbranched hexyl group leads to a compromise in the efficiency of the annulation (2e vs. 2o or 2m vs. 2n). The situation is unsurprisingly exacerbated when the methyl group is replaced by more sterically imposing aryl groups (2k vs. 2q–t or 2m vs. 2u), thus, necessitating an increase in the temperature to 60 °C. Of note, products 2q–u are indeed the thermodynamic products since they result from the unsymmetrical build-up of positive charge on the tertiary benzylic position. Efforts to explore the effect of external aryl substituent on the alkene (i.e., the Ar group in 1) have revealed that lactam-tethered 5-aryl-4(E)-pentenoic acids bearing electron-rich aryl groups are as competent as their electron-deficient congeners (2v/x vs. 2w). The successful deployment of these lactam-tethered trisubstituted alkenoic acids has facilitated the stereocontrolled construction of lactam-γ-lactones bearing two tetrasubstituted stereocenters, which is noteworthy since, as noted previously, halolactonization reactions of trisubstituted alkenes are not often stereospecific.17 Finally, sterically less imposing 1,2-disubstituted styrenoic acids are competent substrates for the bromolactonization (see 2y/z).
Iodolactonization of 1 using N-iodosuccinimide (NIS) proceeded regioselectively (see 2z1–2z7). In these cases, the reactions were conducted at 0 °C due to the enhanced reactivity of NIS. When lactam-tethered alkenoic acid 1k was subjected to chlorolactonization using N-chlorosuccinimide (NCS), no conversion was observed after 48 h at room temperature. After warming to 60 °C for 1 h, complete conversion was observed. However, both the contra-thermodynamic (5-exo-trig cyclization) and thermodynamic (6-endo-trig cyclization) products were isolated (see 2z8 and 3b, respectively). NOESY experiments (see ESI† for details) revealed that in the case of the 6-endo cyclization product, the syn-diastereomer is the exclusive stereoisomer.
The appreciable stability of the bicycles depicted in Schemes 1 and 2 is commendable since they harbor a benzylic halide, which is typically prone to hydrolysis.23 It is worth emphasizing that molecules containing all-carbon quaternary stereocenters and α-alkoxy quaternary stereocenters such as those depicted in Schemes 1 and 2 have the propensity to mitigate a spectrum of structural diversity issues in medicinal chemistry. Indeed, of the top 120 chiral small-molecule pharmaceuticals by retail sales in the United States in 2018, 13% contained a quaternary stereocenter.24
 | ||
| Scheme 2 Contra-thermodynamic 5-exo-iodolactonization and unselective chlorolactonization of lactam-tethered alkenoic acids. | ||
Extending reactivity trends from one N-heterocycle to another can be daunting and at times foolhardy. Nevertheless, as a testament to the modularity of this methodology, our studies have revealed that the contra-thermodynamic and catalytic halolactonization protocol described herein is not limited to γ-lactam-tethered alkenoic acids. For instance, 2-piperidinone-tethered alkenoic acids19a,f also undergo productive 5-exo cyclization to afford the 6,5-bicycles depicted in Scheme 3 (see 5a–d). These allylic valerolactam acids react significantly faster than the sterically encumbered γ-lactam congeners depicted in Schemes 1 and 2. The rate enhancement in the presence of the NMO catalyst is quite evident given that our previous synthesis of 5a under catalyst-free conditions required 48 h.19f Furthermore, iodolactonization of a morpholinone-tethered alkenoic acid19a proceeds satisfactorily and affords bicycle 5e.
 | ||
| Scheme 3 5-exo-cyclization of piperidinone- and morpholinone-tethered alkenoic acids.19a,f | ||
A widely accepted mechanism for electrophilic halolactonization is that an electrophilic halogen is first transferred from a halogen source to the olefin to form a halonium ion, followed by an intramolecular attack by the carboxylate group. Regarding the mechanistic underpinnings of the N-oxide catalyzed bromolactonization of 1, congruent with Moriyama's insightful findings,15e we surmise that the halogen bonding interaction between the bromine atom in NBS and the axial N-oxide in NMO enhances the electrophilicity of the bromine atom in NBS (Fig. 3, see E), thus, facilitating cleavage of the N–Br bond in NBS. Subsequent attack of the electrophilic bromine on the alkene furnishes bromonium ion F, which is then engaged by the pendant carboxylic acid, giving rise to lactam-lactone 2. It is unclear at this point how the solvent (i.e., DMF) alters the site-selectivity of the reaction.
 | ||
| Fig. 3 Plausible mechanism for N-oxide catalyzed bromolactonization of lactam-tethered alkenoic acids of type 1. | ||
Conclusions
In summary, the site-selective, efficient, and stereocontrolled synthesis of halogenated fused γ-lactone-lactams has been accomplished through the catalytic and contra-thermodynamic halolactonization of readily available allylic lactam acids. The innate tendency of these lactam-tethered-5-aryl-4(E)-pentenoic acids to undergo 6-endo cyclization is overridden in favor of 5-exo cyclization by catalyst- and solvent-controlled reactivity. Several trisubstituted alkenoic acids undergo regioselective and diastereocontrolled halolactonization, leading to the construction of sp3-rich fused lactam-lactones bearing medicinally relevant quaternary and contiguous stereocenters. We anticipate that this efficient strategy would expand the 3D-structural space for the discovery of new lactam-γ-lactones with medicinal value. Computational studies geared toward gaining a better understanding of the role of DMF and NMO in controlling the regioselectivity, are ongoing.Experimental
All experiments involving air and moisture sensitive reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle SiliaplateTM glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or KMnO4 stain. Unless otherwise indicated, 1H, 13C, and DEPT-135 NMR, and NOESY spectra were acquired using CDCl3 solvent at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electronspray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx). Brine solutions are saturated solutions of aqueous sodium chloride. Representative GC-MS traces are provided.General procedure A: bromolactonization of lactam acid 1
To an oven-dried 10 mL screw-cap vial, equipped with a stir bar, was added lactam-tethered alkenoic acid 1 (1.0 mmol), dissolved in DMF (1 mL). Then, NBS (196 mg, 1.1 mmol, 1.1 equiv.) and N-methylmorpholine oxide (5.9 mg, 5 mol%) were added. The reaction mixture was stirred at room temperature until TLC and GC-MS showed full conversion. The reaction mixture was then diluted with DCM (20 mL) and quenched with 10% aqueous sodium sulfite (10 mL). The layers were separated and the aqueous layer was extracted once with DCM. The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo to give the desired lactam-lactone, which was purified by flash chromatography on silica.General procedure B: iodolactonization of lactam acid 1
To an oven-dried 10 mL screw-cap vial equipped with a stir bar was added lactam-tethered alkenoic acid 1 (1.0 mmol), dissolved in DMF (1 mL). NIS (247.5 mg, 1.1 mmol, 1.1 equiv.) and N-methylmorpholine oxide (5.9 mg, 5 mol%) were then added. The reaction mixture was stirred at 0 °C until TLC and GC-MS showed full conversion. The reaction mixture was then diluted with DCM (20 mL) and quenched with 10% aqueous sodium sulfite (10 mL). The layers were separated and the aqueous layer was extracted once with DCM. The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo to give the desired lactam-lactone, which was purified by flash chromatography on silica.:30). Yellowish oil. Yield = 404.6 mg, 85%, 95:5 dr (anti:syn). 1H NMR (400 MHz, CDCl3) δ 7.46–7.20 (m, 15H), 5.33 (s, 1H), 4.67 (s, 1H), 3.40–3.29 (m, 2H), 1.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.2, 174.6, 138.9, 135.1, 132.0, 128.7, 128.6, 128.4, 128.1, 127.4, 127.0, 126.5, 125.6, 124.1, 89.0, 71.5, 58.5, 52.3, 44.4, 21.0. FTIR (KBr): 2965.4, 1727.5, 1696.3, 1604.9, 1511.0, 1448.5, 1414.7, 1384.9, 1357.4, 1298.7, 1247.5, 1179.3, 1135.9, 1031.8, 905.8, 839.0. HRMS-EI+ (m/z): calc for C26H22BrNO3 [M]+ 475.0783, found 475.0789.Note: All other lactam-bromolactones depicted in Scheme 1 were prepared as described above. Spectroscopic data can be found in the ESI.
:50). Pale-yellowish oil. Yield = 457.8 mg, 94%, 95:5 dr (anti:syn). 1H NMR (400 MHz, CDCl3) δ 7.40–7.28 (m, 8H), 7.09–7.07 (m, 2H), 5.68 (dddd, J = 17.5, 10.2, 7.5, 4.6 Hz, 1H), 5.33–5.19 (m, 2H), 5.08 (s, 1H), 4.56–4.50 (m, 2H), 3.17–3.12 (m, 2H), 2.94 (d, 1H), 1.64 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.3, 171.9, 138.7, 137.5, 131.4, 130.3, 129.1, 128.8, 128.1, 125.5, 119.6, 88.0, 68.5, 52.8, 45.1, 43.6, 39.5, 23.7. HRMS-EI+ (m/z): calc for C23H22INO3 [M]+ 487.0644, found 487.0649.Note: All other lactam-iodolactones depicted in Scheme 2 were prepared as described above. Spectroscopic data can be found in the ESI.
Author contributions
C. B. – investigation, data curation, methodology; M. J. R. –investigation, data curation, validation; T. K. B. – conceptualization, project administration, supervision, investigation, data curation, methodology, writing – original draft, internal funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University for financial support through startup funds. We thank the Office of Undergraduate Studies and Provost DenBeste for research fellowships to C. B. The school of graduate studies is thanked for research fellowships to T. K. B. and M. J. R. We are grateful to Mr Daniel Hall and Miss Lisa Stowe for timely ordering and delivery of chemicals and supplies during challenging times. Miss Cindy White is thanked for help with instrument and acquisition of 2D NMR data.Notes and references
- (a) D. J. Greenblatt and R. I. Shader, N. Engl. J. Med., 1978, 299, 1342–1344 CrossRef PubMed; (b) I. A. Volchegorskii and E. A. Trenina, Bull. Exp. Biol. Med., 2006, 142, 73–75 CrossRef PubMed; (c) K. Gillard, H. B. Miller and M. S. Blackledge, Chem. Biol. Drug Des., 2018, 92, 1822–1829 CrossRef CAS PubMed.
- (a) C. Saturnino, B. Fusco, P. Saturnino, G. D. E. Martino, F. Rocco and J.-C. Lancelot, Biol. Pharm. Bull., 2000, 23, 654–656 CrossRef CAS PubMed; (b) J. Wei, X. Pan, Z. Pei, W. Wang, W. Qiu, Z. Shi and G. Xiao, J. Trauma Acute Care Surg., 2012, 73, 654–660 CrossRef CAS PubMed.
- (a) R. M. Trend, Y. K. Ramtohul, E. M. Ferreira and B. Stoltz, Angew. Chem., Int. Ed., 2003, 42, 2892 CrossRef CAS PubMed; (b) X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (c) W. Yang, S. Wang, Q. Zhang, Q. Liu and X.-X. Xu, Chem. Commun., 2015, 51, 661 RSC; (d) X.-M. Xie and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3767 CrossRef CAS PubMed; (e) H. Shigehisa, M. Hayashi, H. Ohkawa, T. Suzuki, H. Okayasu, M. Mukai, A. Yamazaki, R. Kawai, H. Kikuchi, Y. Satoh, A. Fukuyama and K. Hiroya, J. Am. Chem. Soc., 2016, 138, 10597 CrossRef CAS PubMed; (f) Y.-J. Zhang, T. Abe, T. Tanaka, C.-R. Yang and I. Kouno, J. Nat. Prod., 2001, 64, 1527 CrossRef CAS PubMed; (g) J. J. Beck and S.-C. Chou, J. Nat. Prod., 2007, 70, 891 CrossRef CAS PubMed; (h) Y. Wache, M. Aguedo, J. M. Nicaud and J. M. Belin, Appl. Microbiol. Biotechnol., 2003, 61, 393 CrossRef CAS PubMed; (i) A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef PubMed; (j) M. I. Konaklieva and B. J. Plotkin, Mini-Rev. Med. Chem., 2005, 5, 73 CrossRef PubMed; (k) I. Collins, J. Chem. Soc., Perkin Trans., 1998, 1, 1869–1888 RSC.
- (a) S. Rashid, B. A. Bhat and G. Mehta, Org. Lett., 2015, 17, 3604–3607 CrossRef PubMed; (b) J. Cao and P. Perlmutter, Org. Lett., 2013, 15, 4327–4329 CrossRef PubMed.
- (a) L. I. Llarrull, S. A. Testero, J. F. Fisher and S. Mobashery, Curr. Opin. Microbiol., 2010, 13, 551–557 CrossRef PubMed; (b) J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC; (c) A. Lepikhina, O. Bakulina, D. Dar’in and M. Krasavin, RSC Adv., 2016, 6, 83808–83813 RSC; (d) P. Gross and J. A. Zapp, CRC Crit. Rev. Toxicol., 1984, 13, 205–216 CrossRef CAS; (e) F. J. R. Rombouts, G. Tresadern, O. Delgado, C. Martinez Lamenca, M. Van Gool, A. Garcia-Molina, S. A. Alonso de Diego, D. Oehlrich, H. Prokopcova, J. M. Alonso, N. Austin, H. Borghys, S. Van Brandt, M. Surkyn, M. De Cleyn, A. Vos, R. Alexander, G. Macdonald, D. Moechars, H. Gijsen and A. A. Trabanco, J. Med. Chem., 2015, 58, 8216 CrossRef CAS.
- (a) M. Shahid, F. Sobia, A. Singh, A. Malik, H. M. Khan, D. Jonas and P. M. Hawkey, Crit. Rev. Microbiol., 2009, 35, 81–108 CrossRef CAS PubMed; (b) K.-F. Kong, L. Schneper and K. Mathee, APMIS, 2010, 118, 1–36 CrossRef CAS; (c) G. Kapoor, S. Saigal and A. Elongavan, J. Anaesthesiol., Clin. Pharmacol., 2017, 33, 300–305 CrossRef CAS.
- (a) R. D. Marco, A. Bedini, S. Spampinato, L. Comellini, J. Zhao, R. Artali and L. Gentilucci, J. Med. Chem., 2018, 61, 5751–5757 CrossRef PubMed; (b) S. M. Rawls, W. Robinson, S. Patel and A. Baron, Neuropharmacology, 2008, 55, 865 CrossRef CAS.
- E. J. Velthuisen, B. A. Johns, D. P. Temelkoff, K. W. Brown and S. C. Danehower, Eur. J. Med. Chem., 2016, 117, 99–112 CrossRef CAS PubMed.
- (a) M. Baiula, P. Galletti, G. Martelli, R. Soldati, L. Belvisi, M. Civera, S. D. Dattoli, S. M. Spampinato and D. Giacomini, J. Med. Chem., 2016, 59, 9721–9742 CrossRef CAS; (b) D. Kuhn, C. Coates, K. Daniel, D. Chen, M. Bhuiyan, A. Kazi, E. Turos and Q. P. Dou, Front. Biosci., 2004, 9, 2605–2617 CrossRef; (c) B. Xing, J. Rao and R. Liu, Mini-Rev. Med. Chem., 2008, 8, 455–471 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- A. G. H. Wee, G.-J. Fan and H. M. Bayirinoba, J. Org. Chem., 2009, 74, 8261–8271 CrossRef CAS.
- A. W. J. Logan, S. J. Sprague, R. W. Foster, L. B. Marx, V. Garzya, M. S. Hallside, A. L. Thompson and J. W. Burton, Org. Lett., 2014, 16, 4078–4081 CrossRef PubMed.
- M. Maier, in Science of Synthesis, ed. E. M. Carreira and J. S. Panek, Thieme, Stuttgart, 2010, vol. 20, pp. 1421−1551 Search PubMed.
- (a) A. N. French, S. Bissmire and T. Wirth, Chem. Soc. Rev., 2004, 33, 354 RSC; (b) H. Fujioka and K. Murai, Heterocycles, 2013, 87, 763 CrossRef; (c) Y. Cheng, W. Yu and Y.-Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC.
- (a) M. Okada, K. Kaneko, M. Yamanaka and S. Shirakawa, Org. Biomol. Chem., 2019, 17, 3747–3751 RSC; (b) R. Kristianslund, J. E. Tungen and T. V. Hansen, Org. Biomol. Chem., 2019, 17, 3079–3092 RSC; (c) T. Chen, T. J. Y. Foo and Y.-Y. Yeung, ACS Catal., 2015, 5, 4751–4755 CrossRef; (d) J. Wong and Y.-Y. Yeung, RSC Adv., 2021, 11, 13564–13570 RSC; (e) K. Moriyama, M. Kuramochi, S. Tsuzuki, K. Fujii and T. Morita, Org. Lett., 2021, 23, 268–273 CrossRef CAS PubMed; (f) J. E. Tungen, R. Kristianslund, A. Vik and T. V. Hansen, J. Org. Chem., 2019, 84, 11373–11381 CrossRef CAS; (g) Y. A. Cheng, T. Chen, C. K. Tan, J. J. Heng and Y.-Y. Yeung, J. Am. Chem. Soc., 2012, 134, 16492–16495 CrossRef CAS PubMed.
- W. J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396 CrossRef CAS.
- (a) F. Freeman, Chem. Rev., 1975, 75, 439–490 CrossRef; (b) K. Murai, A. Nakamura, T. Matsushita, M. Shimura and H. Fujioka, Chem. – Eur. J., 2012, 18, 8448–8453 CrossRef PubMed.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef.
- (a) H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC; (b) T. K. Beng and A. Moreno, New J. Chem., 2020, 44, 4257–4261 RSC; (c) T. K. Beng, M. Bauder, M. J. Rodriguez and A. Moreno, New J. Chem., 2018, 42, 16451–16455 RSC; (d) K. Hovenkotter, H. Braunstein, S. Langevin and T. K. Beng, Org. Biomol. Chem., 2017, 15, 1217–1221 RSC; (e) T. K. Beng and A. Moreno, RSC Adv., 2020, 10, 8805–8809 RSC; (f) J. Garcia, J. Eichwald, J. Zesiger and T. K. Beng, RSC Adv., 2022, 12, 309–318 RSC.
- (a) T. K. Beng, J. Fessenden, K. Quigley, J. Eichwald, and J. Zesiger, New J. Chem., revisions requested Search PubMed; (b) T. K. Beng, M. Rodriguez and C. Borg, RSC Adv., 2022, 12, 17617–17620 RSC.
- T. K. Beng and R. E. Gawley, J. Am. Chem. Soc., 2010, 132, 12216 CrossRef CAS.
- A. Calcaterra, L. Mangiardi, G. D. Monache, D. Quaglio, S. Balducci, S. Berardozzi, A. Iazzetti, R. Franzini, B. Botta and F. Ghirga, Molecules, 2020, 25, 414 CrossRef.
- C. K. Tan, L. Zhou and Y. Y. Yeung, Org. Lett., 2011, 13, 2738–2741 CrossRef PubMed.
- (a) M. C. Carreno, J. L. G. Ruano, G. Sanz, M. A. Toledo and A. Urbano, Tetrahedron Lett., 1996, 37, 4081–4084 CrossRef; (b) K. Lu, J. Chu, H. M. Wang, X. L. Fu, D. W. Quan, H. X. Ding, Q. W. Yao and P. Yu, Tetrahedron Lett., 2013, 54, 6345–6348 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d2ra04177d |
| This journal is © The Royal Society of Chemistry 2022 |