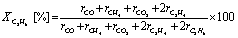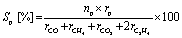DOI:
10.1039/D2RA04401C
(Paper)
RSC Adv., 2022,
12, 24465-24470
Catalytic selective ethane dehydrogenation at low-temperature with low coke formation†
Received
16th July 2022
, Accepted 20th August 2022
First published on 30th August 2022
Abstract
Catalytic ethane dehydrogenation (EDH) was investigated to improve the efficient production of ethylene, an extremely important chemical feedstock. The perovskite oxide YCrO3 was found to be more suitable than earlier reported catalysts because it exhibits greater activity and C2H4 selectivity (94.3%) in the presence of steam at 973 K. This catalyst shows the highest activity than ever under kinetic conditions, and shows very high ethane conversion under integral reaction conditions. Comparison with EDH performance under conditions without steam revealed that steam plays an important role in stabilizing the high activity. Raman spectra of spent catalysts indicated that steam prevents coke formation, which is responsible for deactivating YCrO3. Transmission IR and XPS measurements also revealed a mechanism by which H2O forms surface oxygen species on YCrO3, consequently removing C2H6-derived coke precursors rapidly and inhibiting coke accumulation.
1 Introduction
Ethylene is an extremely important chemical feedstock used by the chemical industry for producing various chemical products such as plastics, synthetic fibres and synthetic rubber.1,2 Currently, global ethylene demand growth is expected to be more than 4% per annum.3 Because of the abundance of cheap ethane from shale gas, the feedstock for ethylene production is shifting from naphtha to ethane.4–6 Currently, the well-known industrial process for ethylene production is cracking: ethane is pyrolysed by a gas-phase radical reaction in the presence of steam. Nevertheless, this process is known to be energy-intensive.7,8 Furthermore, calcination operations to remove coke deposits from the inner wall of the tubular cracking reactor are performed regularly, thereby raising operating costs.9,10 Catalyst coating on tubular surfaces is a promising method to overcome this difficulty related to steam cracking. Several coating techniques have been developed.10–12 Catalysts used in these coated reactors are intended to prevent coke deposition. If the coated catalyst also has the function of selective catalytic dehydrogenation of ethane to ethylene, then the operating temperature can be reduced to aid ethylene production at lower temperatures. However, no good catalyst has been established for this purpose to date. We specifically examined the development of catalysts for highly selective ethane dehydrogenation (EDH) in the presence of steam with less coke formation.
Perovskite-type oxides, with the general formula of ABO3, where A is a rare-earth or an alkali-earth element and where B is typically a transition metal, are promising catalysts for the dehydrogenation of light alkane with steam by virtue of their high hydrothermal stability and redox ability.13–16 The catalytic property of perovskite-type oxides depends on B-site cation characteristics. Few reports have described the dehydrogenation of hydrocarbon under co-feeding of steam on the perovskite-type oxides.17–20 To develop a catalyst with high selectivity and stability for EDH with steam, we specifically examined Cr based perovskite catalyst. This study proposes a novel catalyst of YCrO3 perovskite, which exhibits very high activity, selectivity, and stability in the steam-based dehydrogenation of ethane. X-ray analysis was used to characterise the Cr structure and electronic state. Also, coke formation on YCrO3 was investigated using Raman spectroscopy. The catalyst was found to have high selectivity for ethane dehydrogenation. The reaction mechanism was also elucidated using isotopes.
2 Experimental
2.1. Catalyst preparation
Metal oxide catalysts were prepared by a citric acid complex polymerization method, with a yield per synthesis of 3 g. First, citric acid monohydrate and ethylene glycol (Kanto Chemical Co., Inc.) were dissolved into distilled water using 300 mL PTFE beaker. Then, metal nitrates (Kanto Chemical Co., Inc.) were added to the solution. The molar ratio of total metals: citric acid monohydrate: ethylene glycol was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3. In the case of YCrO3, Y(NO3)3·6H2O of 6.0833 g, Cr(NO3)3·9H2O of 6.4846 g, citric acid monohydrate of 20.3608 g and ethylene glycol of 5.9443 g were used. The obtained solution was stirred in a water bath at ca. 343 K for 16 h and then dried with a hot stirrer to form a powder. The obtained powder was pre-calcined at 673 K for 2 h and calcined at 1123 K for 10 h in static air using an electric muffle furnace (KDF-300Plus, DENKEN-HIGHDENTAL Co., Ltd.). The purity of each chemical used is shown in Table S1 (ESI)†.
:3:3. In the case of YCrO3, Y(NO3)3·6H2O of 6.0833 g, Cr(NO3)3·9H2O of 6.4846 g, citric acid monohydrate of 20.3608 g and ethylene glycol of 5.9443 g were used. The obtained solution was stirred in a water bath at ca. 343 K for 16 h and then dried with a hot stirrer to form a powder. The obtained powder was pre-calcined at 673 K for 2 h and calcined at 1123 K for 10 h in static air using an electric muffle furnace (KDF-300Plus, DENKEN-HIGHDENTAL Co., Ltd.). The purity of each chemical used is shown in Table S1 (ESI)†.
2.2. Activity tests
Catalytic activity tests were conducted in a fixed-bed flow-type reactor at atmospheric pressure. The sieved catalyst in the 425–825 μm size range was mixed with SiC (ca. 390 mg) and was charged into a quartz tube (i.d.: 4 mm, o.d.: 6 mm). Activity tests were conducted under atmospheric pressure with steam, designated as “wet condition tests”, and without steam, designated as “dry condition tests”. The reaction gas compositions for the wet condition test and the dry condition test were, respectively, C2H6:H2O:N2 = 1.0:0.50:5.5 and C2H6:N2 = 1.0:6.0. The total gas flow rate was 127 mL min−1. The catalyst bed was heated to 973 K in the N2 flow before introducing the reaction gas. The outlet gases were analyzed using an online GC-FID (GC-8A; Shimadzu Corp.) equipped with a Porapak Q packed column and a methanizer with Ru/Al2O3 catalyst. The ethane conversion (XC2H6) and the selectivity to each product (Sp) were calculated as shown below.
In these equations, r and n respectively denote the flow rate and the number of carbon atoms of each gas. Subscript p signifies a specific product (CO, CH4, CO2, and C2H4).
2.3. Steady-state isotopic transient kinetic analysis: SSITKA
The behaviour of the lattice oxygen in metal oxide catalysts during the reaction was elucidated using steady-state isotopic transient kinetic analysis (SSITKA) with H218O. The experimental protocol is shown in Fig. S1† (ESI). First, the wet condition test was conducted using the reaction gas containing H218O (H216O:H218O = 3:2) for 1 h to replace the lattice oxygen with 18O2−. At this time, the reaction gas composition was C2H6:H2O:Ar:N2 = 1.0:0.50:1.1:4.4 at a total flow rate of 127 mL min−1. Then, to remove physisorbed H218O, the catalyst bed was purged with N2 for 1.5 h. After N2 purge, the wet condition test with only H216O was performed successively. In the test with only H216O, if H218O is detected in the outlet gas, it can be interpreted that the EDH reaction is driven by the Mars-van Krevelen (MvK) mechanism.20,21 Using a quadrupole mass spectrometer (QGA; Hiden Analytical Ltd.), H218O (m/z = 19) and Ar (m/z = 40) in the outlet gas were detected.
2.4. Catalyst characterisation
The crystal structure of the catalyst was confirmed by powder X-ray diffraction (XRD) measurement using an X-ray diffractometer (Smart Lab-III; Rigaku Corp.) with Cu Kα radiation at 40 kV and 40 mA. The BET surface area of the catalyst was calculated from the N2 adsorption isotherm at 77 K (Gemini VII 2390a; Micromeritic Instrument Corp.). Observation of the particle morphology and the distribution of elements were conducted using a scanning electron microscope (SEM; S-3000N, HITACHI, Ltd.) and a transmission electron microscope with an energy-dispersive X-ray spectrometer (TEM-EDX; JEM-2100, JEOL, Ltd.).
The electronic state of Cr in YCrO3 was evaluated by in situ X-ray absorption fine structure (XAFS) measurement at the BL14B2 beamline of SPring-8 in Japan (ID: 2021B1050). The XAFS measurements were taken in transmission mode using a Si (111) crystal monochromator. To prepare the measurement sample, YCrO3 powder mixed with BN was pressed into a 7 mmɸ disk. In situ measurements were taken according to the protocol shown in Fig. S2 (ESI).† First, the sample disk was heated to 973 K in the N2 flow. Then, XAFS spectra were measured under the reaction gas (C2H6:H2O:N2 = 1.0:0.50:5.5), steam (H2O:N2 = 0.50:6.5), and ethane (C2H6:N2 = 1.0:6.0) atmospheres in this order. Inlet gas was supplied at a 127 mL min−1 total flow rate in this experiment. X-ray absorption near-edge structure (XANES) spectra were analyzed using software (xTunes ver. 1.3 Build 20200228; Science & Technology Inst., Co.).
Coke formation on the spent YCrO3 catalyst was confirmed using Raman spectroscopy (NRS-4500; Jasco Corp.). The spectra in the range of 100–3000 cm−1 were measured using a 532 nm laser.
In situ transmission IR measurements were taken to observe the surface species over YCrO3 catalyst using a Fourier transform infrared spectrometer (FT/IR 6200; Jasco Corp.) with an MCT detector and a CaF2 window. The YCrO3 catalyst was pressed and shaped into a 20 mmɸ disk. Regarding the pretreatment, the YCrO3 disk was heated at 993 K for 15 min under Ar flow. Then the temperature was changed to 973 K. The background (BKG) spectra were measured under inert Ar (Dry atmosphere: 100 mL min−1 Ar) or Ar + H2O (Wet atmosphere: 95 mL min−1 Ar + 5 mL min−1 H2O). After BKG spectra were recorded, C2H6 was dosed into the IR cell. Then the spectra were recorded again. The amounts of C2H6 per pulse were, respectively 0.1 mL in the dry atmosphere and 0.5 mL in the wet atmosphere.
To evaluate the electronic states and atomic ratio for the surface elements of YCrO3 catalysts, XPS measurements were taken (Versa Probe II; Ulvac Phi Inc.) using Al Kɑ X-rays. Binding energies for each orbital were calibrated using C 1s (285.0 eV). Catalysts treated under the wet and dry atmospheres with the same condition to the activity tests were moved to the sample chamber using a transfer vessel to avoid exposure to air. The ratio of surface oxygen species was estimated from the area values of deconvoluted peaks.
3 Results and discussion
3.1. Catalytic performance of YCrO3 perovskite-type oxide for EDH in presence of steam
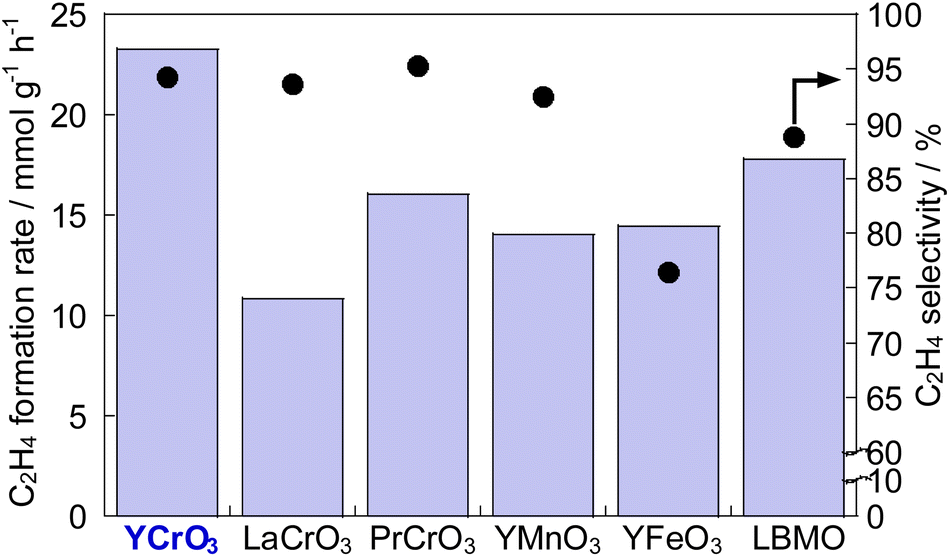
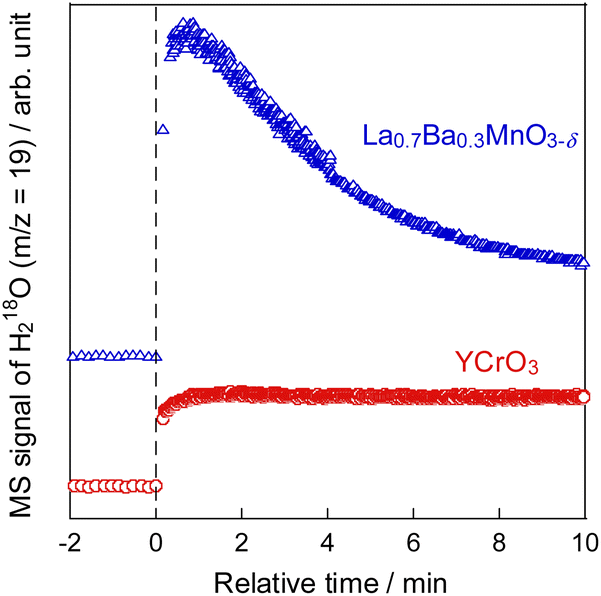
To develop catalysts with high activity and high ethylene selectivity for EDH in the presence of steam, the catalytic performance of Cr-based and Y-based perovskite oxides whose XRD patterns and SEM images are portrayed in Figs S3–S5† was investigated. The ethylene formation rate and selectivity at 60 min with time on stream are presented in Fig. 1 and Fig. S6 (ESI).† YCrO3 catalyst showed the highest activity (23.2 mmol g−1 h−1 C2H4 formation rate) among them and high ethylene selectivity (94.3%). The better performance of YCrO3 is thanks to the high selectivity for ethylene on the surface of Cr-based perovskite and a relatively high specific surface area (SSA) among these perovskites (details in ESI Table S2†). Also, the reason for the high activity of YCrO3 may be due to a large amount of stable Cr3+ species on its surface. Although there have been various discussions on the oxidation state of Cr species for non-oxidative dehydrogenation of ethane, depending on the catalyst material, several findings have been reported that Cr3+ species are good active sites.22–25 The Cr cation in YCrO3 is trivalent to maintain the electroneutrality of its perovskite, and as shown in the XPS results (Fig. S7 in ESI†), it remained hardly changed in the reaction atmosphere. Therefore, we considered that these stable Cr3+ species functioned as active sites and led to the high ethane dehydrogenation activity. Then, additional comparison was conducted with La0.7Ba0.3MnO3−δ (LBMO) catalyst reported showing high performance for EDH in presence of H2O by Saito et al.20 The catalytic performance of YCrO3 is higher than that over LBMO (17.8 mmol g−1 h−1 C2H4 formation rate, 88.8% ethylene selectivity) in earlier research. These results demonstrate YCrO3 as a noteworthy catalyst that shows very high activity, selectivity for EDH with steam. To investigate the mechanism of EDH on YCrO3, SSITKA measurements using YCrO3 and LBMO were conducted. The results are presented in Fig. 2. In the measurements, H218O was not detected on YCrO3, although H218O formation was observed on LBMO because EDH proceeds via MvK mechanism over LBMO.20 Furthermore, in situ XAFS measurement for Cr K-edge (Fig. S8 in ESI†) revealed that the oxidation state of Cr in YCrO3 is stable during EDH reaction. These results suggest that non-oxidative dehydrogenation of ethane proceeds on YCrO3.
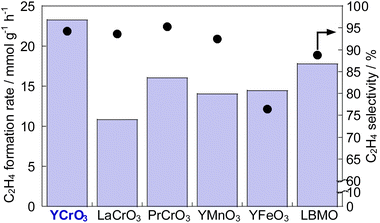 |
| | Fig. 1 C2H4 formation rate and C2H4 selectivity at 973 K over Cr-based and Y-based perovskites, and La0.7Ba0.3MnO3−δ (LBMO). | |
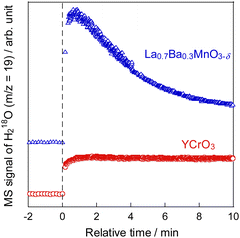 |
| | Fig. 2 Time course of H218O formation in SSITKA at 973 K over YCrO3 and La0.7Ba0.3MnO3−δ. | |
3.2. Role of steam on EDH over YCrO3
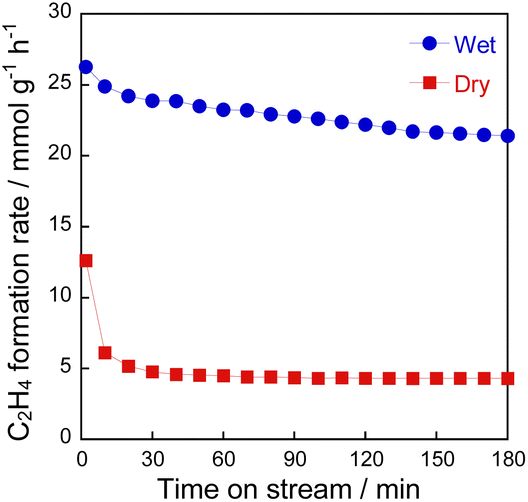
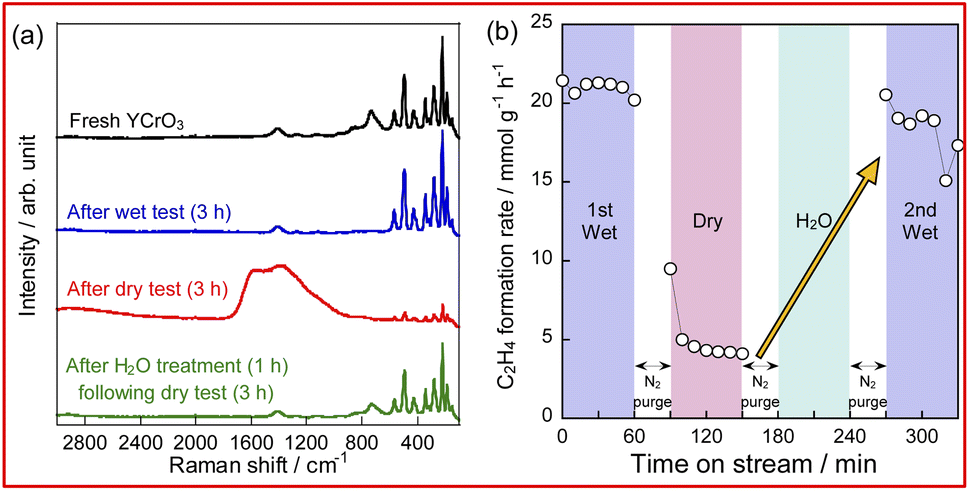
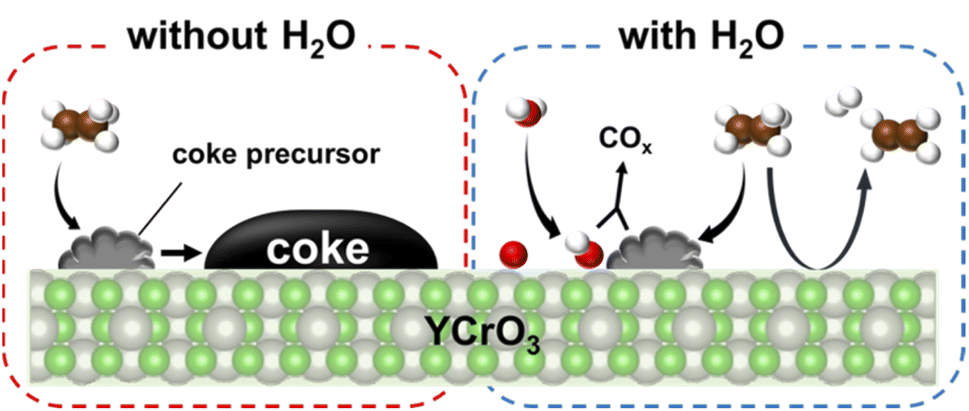
To confirm the influence of coexisting H2O on the reaction over YCrO3 catalyst, the wet condition and the dry condition tests (details in the ESI†) were conducted for 180 min. The comparison of ethylene formation rate with time course under each condition is depicted in Fig. 3. Although the activity dropped to the same level as the gas-phase reaction within 30 min in the dry condition, high activity was maintained for 180 min in the wet condition. As shown in Figs S5(b) and S9–S13,† the structure of YCrO3 after the activity tests were the same as that of the fresh one, indicating that the crystal structure is stable in both reaction atmospheres. Despite not being the MvK mechanism in which H2O plays a key role as the media of release and regeneration of lattice oxygen, results showed that coexisting steam positively affected the catalytic performance over YCrO3. Then, we specifically examined coke formation, which is one cause for the deactivation of catalysts during ethane dehydrogenation. Fig. 4a presents Raman spectra of fresh and spent YCrO3 catalyst. After the dry condition test, the notable band with peaks at around 1350 and 1600 cm−1 appeared. The band was deconvoluted as shown in Fig. S14 (ESI).† The deconvoluted peaks can be assigned to graphitic carbon (G, D1 and D2 band) and amorphous carbon (D3 band),26 indicating that these carbon species were deposited on YCrO3 during EDH without steam. These bands of carbon species disappeared completely by H2O treatment following the dry condition test. Fig. 4b presents results of the dry–wet condition switching test with H2O treatment. The activity dropped by switching from wet to dry conditions. Then, by conducting H2O treatment for 60 min, the activity was restored to the same level as the first wet condition test. Because the behaviour of the activity corresponds to that of Raman peaks, the main cause of deactivation under the dry condition is regarded as coke formation on YCrO3 surface. Therefore, the role of coexisting steam is the prevention of coke deposition, leading to high stability of the activity over YCrO3.
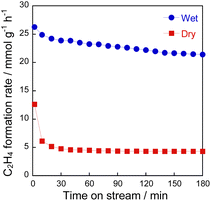 |
| | Fig. 3 Time course of the C2H4 formation rate at 973 K over YCrO3 under wet and dry atmospheres. | |
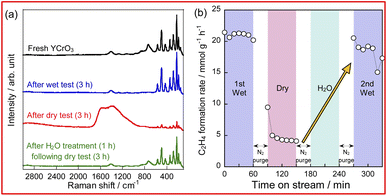 |
| | Fig. 4 Raman spectra of fresh and spent YCrO3 (a) and time course of C2H4 formation rate during activity tests with H2O treatment (b). | |
3.3. Mechanism of coke removal by steam on YCrO3
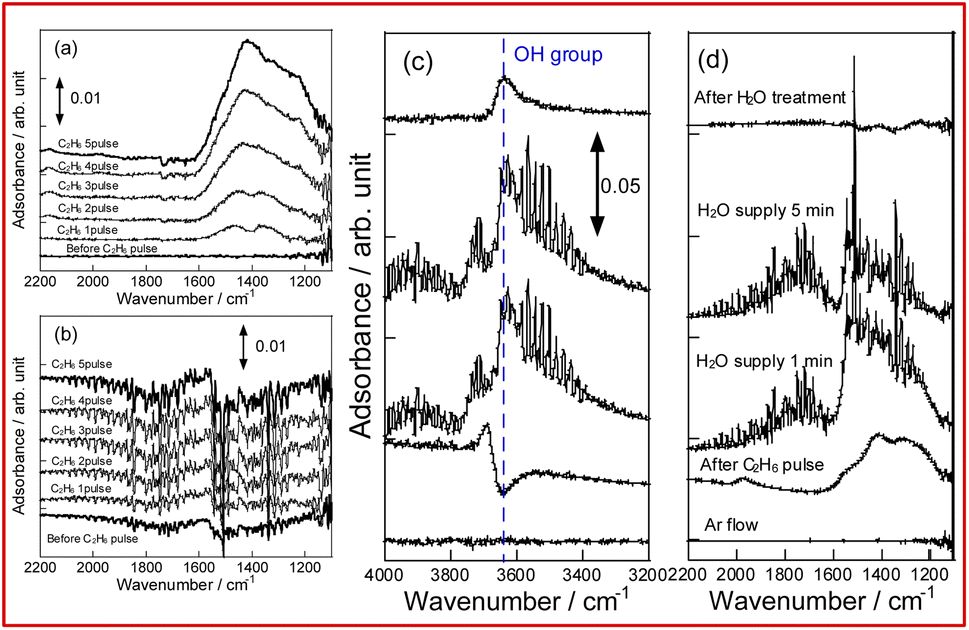
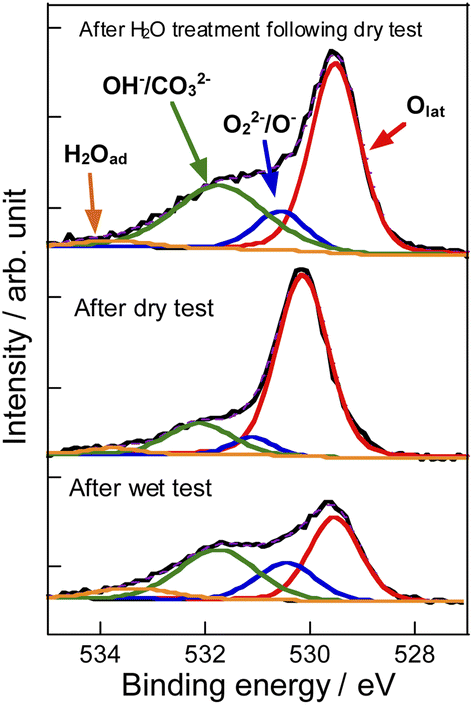
To evaluate the mechanism of preventing coke formation by steam, in situ transmission IR measurements were conducted. Fig. 5 shows the IR spectra (1100–2200 cm−1) of YCrO3 supplying C2H6 under the dry and wet atmosphere. Details of the measurement protocol are described in the ESI.† In the dry atmosphere, a notable absorption band at the 1200–1600 cm−1 region was observed after the first dose of C2H6 (0.1 mL). Then, as the ethane was pulsed further, the band broadened and increased in intensity (Fig. 5a). Details of the deconvoluted spectra and assignments for these bands are presented in Fig. S15 (ESI) and Table S3 (ESI).† These changes of IR spectra indicate that aliphatic and aromatic hydrocarbon species accumulate on YCrO3 surface in the dry C2H6 atmosphere. These species are generally regarded as coke precursors that eventually form the coke via further dehydrogenation and polymerization.27–32 In contrast, despite having five times as much C2H6 supplied per pulse, no band attributed to the coke precursors was observed under the wet atmosphere. Furthermore, an atmosphere-switching test was conducted: beforehand, coke precursors were accumulated on YCrO3 by C2H6 supply under the dry atmosphere, then dosing steam into the IR cell and recording the spectra with the time course. As shown in Fig. 5c and d, the bands of coke precursors gradually attenuated and disappeared, indicating that steam removes coke precursors from the YCrO3 surface. Furthermore, the negative peak at 3639 cm−1, which is attributed to OH group, appeared after the C2H6 pulse. The negative peak of OH group changed to positive after the H2O treatment, indicating that the OH group consumed because of the contact with C2H6 was regenerated. The electronic state of surface oxygen species for YCrO3 was investigated using XPS measurements. The O 1s spectra, which were deconvoluted into four peaks, of spent YCrO3 after various treatments, are depicted in Fig. 6. The peaks at the highest and lowest binding energy are assigned respectively to lattice oxygen O2− (denoted as Olat) and the adsorbed H2O (denoted as H2Oad). The two intermediate peaks are attributed to surface oxygen species (denoted as O−/O22− and OH−/CO32−).33–35 Binding energies of these peaks and the ratio of surface oxygen species are presented in Table 1. The surface oxygen species changed drastically depending on the treatment applied to YCrO3. The O 1s spectrum after the wet condition test showed a higher ratio of surface oxygen species than that after the dry condition test. Then, conducting H2O treatment of YCrO3 after the dry condition test, the ratio recovered to a high value. These results demonstrate that surface oxygen species such as OH and O−/O22− consumed because of the contact with C2H6 are regenerated by steam, which corresponds well with results of in situ IR measurements. From findings obtained using in situ IR and XPS, it was inferred that the surface oxygen species formed on YCrO3 by coexisting steam rapidly remove the coke precursors before they accumulate on the active sites, as portrayed in Fig. 7.
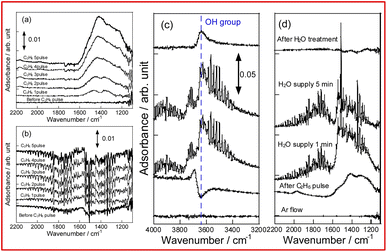 |
| | Fig. 5 In situ IR spectra for YCrO3 under dry atmosphere (a), wet atmosphere (b), and during removal of coke precursors by steam (c and d). | |
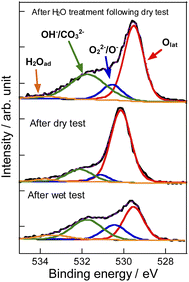 |
| | Fig. 6 Deconvoluted XP spectra of O 1s region for YCrO3 after dry test, wet test, and H2O treatment following the dry test. | |
Table 1 Binding energy and the ratio of surface oxygen species for O 1s spectra of YCrO3 after various treatments
| Treatment |
Binding energy/eV |
Ratio of surface oxygen species/lattice oxygen |
| Olat |
O−/O22− |
OH−/CO32− |
H2Oad |
| Wet test |
529.5 |
530.5 |
531.8 |
533.4 |
1.38 |
| Dry test |
530.2 |
531.1 |
532.1 |
533.8 |
0.29 |
| H2O treatment |
529.5 |
530.6 |
531.7 |
533.8 |
0.82 |
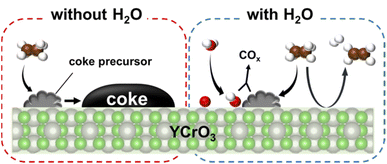 |
| | Fig. 7 Schematic image showing the reaction mechanism. | |
4 Conclusion
To develop catalysts with high EDH performance at 973 K in the presence of steam, Cr-based and Y-based perovskite oxides were investigated. Results show that YCrO3 had the highest activity and highest C2H4 selectivity of the catalysts tested. Comparison of the activities in wet and dry conditions indicates that the presence of steam contributes to the stability of the high EDH performance of YCrO3. Raman spectra of the spent catalysts show that coke was formed on the catalyst surface under dry conditions, demonstrating that H2O contributed to the inhibition of coke formation and the removal of the formed coke. The mechanism by which H2O prevents coke formation was investigated using in situ transmission IR spectroscopy and XPS. In a dry atmosphere, an absorption band of a ‘coke precursor’ was observed after the C2H6 pulse. In a wet atmosphere, no such band appeared, indicating that steam hindered the coke precursor formation. Furthermore, the coke precursor bands which appeared under the dry atmosphere decreased and disappeared with the time course when steam was supplied, confirming that the coke on the YCrO3 surface was removed by H2O. The XPS measurements showed that the ratio of surface oxygen species on YCrO3 under dry conditions (O−/O22− and OH−/CO32−) were reduced compared to those after other treatments, indicating that the coexistence of H2O allows the formation of surface oxygen species and rapid removal of coke precursors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We appreciate Dr Tetsuo Homma and Dr Takeshi Watanabe (Japan Synchrotron Radiation Research Institute, SPring-8) for their great supports in XAFS measurements in BL14B2. We also appreciate Prof. S. Yamazoe (Tokyo Metropolitan Univ.) for his great support in XAFS analyses. We appreciate Mr Hiroshi Sampei and Mr Taku Matsuda (Waseda Univ.) for giving great support for TEM and SEM measurement. The TEM, EDX and SEM studies of this work were the results of using equipment (TEM and EDX: JEM-2100; JEOL, SEM: S-3000N; HITACHI) in the Material Characterization Central Laboratory shared on the MEXT Project for promoting public usage of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMX04405022. We also appreciate Mr Takahiro Gotoh (Material Characterization Central Laboratory, Waseda Univ.) for his support on the transmission electron diffraction of the TEM measurements.
Notes and references
- H. Saito and Y. Sekine, RSC Adv., 2020, 10, 21427–21453 RSC.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3(9), 2130–2149 CrossRef CAS.
- Deloitte, The Future of Petrochemicals: Growth Surrounded by Uncertainty, 2019 Search PubMed.
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS PubMed.
- Z. Kapsalyamova and S. Paltsev, Energy Econ., 2020, 92, 104984 CrossRef.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- T. Ren, M. K. Patel and K. Blok, Energy, 2018, 33, 817–833 Search PubMed.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- C. Zhao, C. Liu and Q. Xu, Ind. Eng. Chem. Res., 2010, 49, 5765–5774 CrossRef CAS.
- C. M. Schietekat, S. A. Sarris, P. A. Reyniers, L. B. Kool, W. Peng, P. Lucas, K. M. Van Geem and G. B. Marin, Ind. Eng. Chem. Res., 2015, 54, 9525–9535 CrossRef CAS.
- BASF, CAMOL™ catalytic coatings for steam cracker furnace tubes, BASF, Canada, 2012 Search PubMed.
- S. H. Symoens, N. Olahova, A. E. M. Gandarillas, H. Karimi, M. R. Djokic, M. Reyniers, G. B. Marin and K. M. Van Geem, Ind. Eng. Chem. Res., 2018, 57, 16117 CrossRef CAS.
- M. Misono, Stud. Surf. Sci. Catal., 2013, 176, 67–95 CrossRef.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- Y. Sugiura, D. Mukai, Y. Murai, S. Tochiya and Y. Sekine, Int. J. Hydrogen Energy, 2013, 38, 7822–7829 CrossRef CAS.
- D. Mukai, S. Tochiya, Y. Murai, M. Imori, T. Hashimoto, Y. Sugiura and Y. Sekine, Appl. Catal., A, 2013, 453, 60–70 CrossRef CAS.
- R. Watanabe, M. Tsujioka and C. Fukuhara, Catal. Lett., 2016, 146, 2458–2467 CrossRef CAS.
- R. Watanabe, Y. Hondo, K. Mukawa, C. Fukuhara, E. Kikuchi and Y. Sekine, J. Mol. Catal. A: Chem., 2013, 377, 74–84 CrossRef CAS.
- R. Watanabe, M. Ikushima, K. Mukawa, F. Sumomozawa, S. Ogo and Y. Sekine, Front. Chem., 2013, 1(21), 1–11 CAS.
- H. Saito, H. Seki, Y. Hosono, T. Higo, J. G. Seo, S. Maeda, K. Hashimoto, S. Ogo and Y. Sekine, J. Phys. Chem. C, 2019, 123, 26272–26281 CrossRef CAS.
- Y. Hosono, H. Saito, T. Higo, K. Watanabe, K. Ito, H. Tsuneki, S. Maeda, K. Hashimoto and Y. Sekine, J. Phys. Chem. C, 2021, 125, 11411–11418 CrossRef CAS.
- T. V. M. Rao, E. M. Zahidi and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 159–165 CrossRef CAS.
- V. Z. Fridman, R. Xing and M. Severance, Appl. Catal., A, 2016, 523, 39–53 CrossRef CAS.
- U. Olsbye, A. Virnovskaia, O. Prytz, S. J. Tinnemans and B. M. Weckhuysen, Catal. Lett., 2005, 103, 143–148 CrossRef CAS.
- H. H. Shin and S. McIntosh, ACS Catal., 2015, 5, 95–103 CrossRef CAS.
- Y. Sekine and H. Fujimoto, ISIJ Int., 2019, 59, 1437–1439 CrossRef CAS.
- S. Srihiranpullop and P. Praserthdam, Catal. Today, 2004, 93–95, 723–727 CrossRef CAS.
- L. Carlos, E. L. Jablonski, J. M. Parera, F. Roger and L. Frederic, Ind. Eng. Chem. Res., 1992, 31, 1017–1021 CrossRef.
- L. Emdadi, L. Mahoney, I. C. Lee, A. C. Leff, W. Wu, D. Liu, C. K. Nguyen and D. T. Tran, Appl. Catal., A, 2020, 595, 117510 CrossRef CAS.
- J. V. lbarra, C. Royo, A. Monzón and J. Santamaría, Vib. Spectrosc., 1995, 9, 191–196 CrossRef.
- Y. He and Y. He, Catal. Today, 2002, 74, 45–51 CrossRef CAS.
- H. Zhang, S. Shao, R. Xiao, D. Shen and J. Zeng, Appl. Catal., A, 2001, 212, 83–96 CrossRef.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- C. Zhang, C. Wang, W. Hua, Y. Guo, G. Lu, S. Gil and A. Giroir-Fendler, Appl. Catal., B, 2016, 186, 173–183 CrossRef CAS.
- K. Chu, F. Liu, J. Zhu, H. Fu, H. Zhu, Y. Zhu, Y. Zhang, F. Lai and T. Liu, Adv. Energy Mater., 2021, 11, 2003799 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hideaki Tsunekia,
Shun Maeda
a,
Hideaki Tsunekia,
Shun Maeda