DOI:
10.1039/D2RA04684A
(Paper)
RSC Adv., 2022,
12, 27267-27274
Effect of the dangling aromatic ring on neutral luminescent bis(phosphine) Cu(I)/Ag(I) complexes with the asymmetric pyridyl-tetrazolate ligands†
Received
27th July 2022
, Accepted 13th September 2022
First published on 26th September 2022
Abstract
A series of neutral luminescent bis(phosphine) Cu(I) complexes of pyridyl-tetrazolate ligands (L1–L3) with the general formula [CuI(Ln)(P^P)] (1–6) were synthesized, which have been well characterized by IR, UV/vis, CV, 1H NMR and 31P NMR. For comparison, an Ag(I) complex [AgI(L2)(PPh3)2] (7) was also synthesized. The crystal structures of 2 and 7 have been further determined by X-ray crystallography. All these Cu(I) compounds show bright luminescence in the solid state with photoluminescence quantum yields (PLQYs) in the range of 25.8% to 85.0%. More interestingly, the Cu(I) complexes bearing an additional dangling aromatic ring on the diimine ligands exhibit enhanced luminescent performance in various solutions and their PLQYs are significantly higher than those of related Cu(I) complexes without steric protection. Compared with 1, the Cu(I) complexes with an additional dangling tetrazole moiety show a significant solvatochromic effect, which is uncommon for luminescent Cu(I) complexes. Moreover, [CuI(L2)(PPh3)2] (2) was further designed as an OLED material, which showed a high external quantum efficiency of 7.7%.
Introduction
Luminescent Cu(I) complexes have gained increasing interest for their application in organic light emitting devices (OLEDs),1 as they are environmentally benign and economically viable compared with noble metal complexes of Ru(II), Pt(II), Os(II) and Ir(III).2–6 In addition, they possess interesting photophysical and photochemical properties associated with the closed-shell d10 electronic configuration, such as thermally activated delayed fluorescence (TADF), which is attributed to the moderate spin–orbit coupling (ξCu = 857 cm−1) and the raised non-radiative deactivating ligand-field state.7 Since the highly luminescent mononuclear Cu(I) complex, [Cu(dmp)(POP)]+ (where dmp = 2,9-dimethyl-1,10-phenanthroline, POP = bis[2-(diphenylphosphino)phenyl]ether) was reported by McMillin's group,8 a large number of luminescent Cu(I) complexes have been reported so far.9 Among these mononuclear Cu(I) complexes, the distorted tetrahedrally coordinated heteroleptic complexes [CuI(N^N)(P^P)]+/0 (N^N = various diimine ligands and P^P = diphosphine ligands) have been intensively studied, mainly because their emission properties are readily tuned by the systematic modification of electronic and steric properties of the diimine and diphosphine ligands.10 Accordingly, some luminescent heteroleptic Cu(I) complexes were designed as LED materials with a high external quantum efficiency (EQE ˃ 15%).11 Compared with the traditionally well-established diimine ligands, such as the 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) and their derivatives, Cu complexes of tetrazole (tz)-containing ligands, e.g. 5-(2-pyridyl)tetrazolate (L1), were relatively less studied. However, these ligands possess some obvious advantages: (1) tz has a stronger coordination ability to Cu(I) centers than the pyridyl ring, due to its soft base property; (2) the ligand can be readily protonated/deprotonated due to the mesomeric structure of tz rings; (3) neutral Cu(I) complexes are readily formed, which enhance the post-processability for their application as an OLED material; (4) non-radiative decay through C–H vibrational quenching can be reduced by replacing the pyridine (py) ring with a tz unit; (5) the asymmetric coordination environment will improve their stability in the excited states.12 A typical example is the well-studied neutral heteroleptic Cu(I) complex [CuI(L1)(P^P)] (L1 = 5-(2-pyridyl)tetrazolate).13 As revealed by DFT study, the HOMO is mainly confined to Cu(I) and the LUMO is located primarily on the pyridine ring and to a less extent on the tz ring. The MLCT absorption band mainly involves the metal d orbital and π* orbital of the pyridine ring. Thus, the M–N(py) bond is weakened on the excited state, while the M–N(tz) bond remains almost unchanged. Recent study on the TADF properties of [CuI(L1)(POP)] in the solid state have found an interesting direct intersystem crossing process.14 These unique properties have aroused our interest for the in-depth study of luminescent Cu(I) complexes containing this class of ligands.
Although [CuI(L1)(PPh3)2] and its related complexes exhibit excellent luminescent properties in the solid state, their fluid luminescent properties are usually very poor, mainly due to the geometrical distortion from tetrahedron to square plane on the excited state. One of the traditional strategies to improve the luminescent properties of Cu(I) complexes is to employ the sterically demanding a,a′-disubstituted diimine ligands, which effectively suppress the deformation of excited states. However, both the a,a′-positions in these diimine ligands should be occupied by bulky functional groups to suppress the excited geometric distortion, while the existence of a bulky group on only one of the a,a′-positions is not enough.15 Nevertheless, the charge-neutrality of resultant Cu(I) complexes is not easy to maintain, if the bulky alkyl groups are added on the a,a′-positions of these pyridyl tetrazolate ligands. In order to maintain the charge neutrality of Cu(I) complexes and improve their emission properties simultaneously, an additional aromatic ring was introduced on the 5-position of 5-(2-pyridyl)tetrazolate (L1) (Fig. 1). Accordingly, a series of Cu(I) complexes, [CuI(Ln)(P^P)] (2–6, Fig. 2) containing tz ligands (L1–L3) have been prepared. It is noted that these new Cu(I) complexes exhibit excellent luminescence not only in the solid state, but also in solution, including polar protic solvents. These Cu(I) complexes have no steric protection on the a,a′-positions, and exhibit enhanced solution-state luminescence. These compounds represent a class of neutral mononuclear Cu(I) complexes with the interesting TADF behaviour. To provide more insight on their luminescence, analogous Ag complex [Ag(L2)(PPh3)2] (7) was also synthesized as a comparison. Moreover, 2 has been further used to fabricate light-emitting diodes (OLEDs).
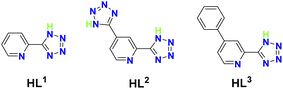 |
| | Fig. 1 The structures of HL1–HL3. | |
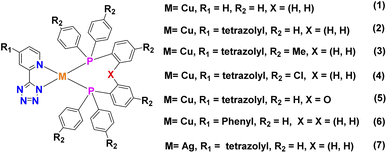 |
| | Fig. 2 Structures of neutral Cu(I)/Ag(I) complexes 1–7 containing various tz ligands. | |
Results and discussion
The ligands HL1, HL2 and HL3 were synthesized by similar procedures via [3+2] cycloaddition reactions of organonitriles and N3−.17 Treatment of [CuI(MeCN)4](ClO4) with phosphine ligands and various asymmetric diimine ligands afforded the neutral Cu(I) complexes 1–6 with the general formula [CuI(Ln)(P^P)] (n = 1–3), which were isolated as yellow micro-crystalline solids with moderate yields. In the similar condition, treatment of HL2 with AgNO3 afforded the colourless mononuclear neutral complex [Ag(L2)(PPh3)2] (7). All these complexes have been characterized by IR, UV/vis, 1H NMR and 31P NMR spectra, satisfactory elemental analysis. The IR spectra of these compounds show strong stretches at ∼1618 and 1435 cm−1, which are assigned to v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and v(NN) stretching bands from tz moieties and comparable with most related tz compounds.9d,16 Moreover, a v(N–H) stretch at ∼3125 cm−1 is found in 2–5. The absence of stretching band of the anion (ClO4−) proves that these compounds are neutral in the solid state. The 1H NMR spectra show that there are three well distinguished sharp proton signals in the normal range, which are consistent with their diamagnetic d10 electronic configurations. These complexes have also been characterized by 31P{1H} NMR spectroscopy and broadening of their 31P signals were observed, which is similar to those reported in related Cu(I) and Ag(I) complexes (Fig. S3–S7†).9d,10b,21
N) and v(NN) stretching bands from tz moieties and comparable with most related tz compounds.9d,16 Moreover, a v(N–H) stretch at ∼3125 cm−1 is found in 2–5. The absence of stretching band of the anion (ClO4−) proves that these compounds are neutral in the solid state. The 1H NMR spectra show that there are three well distinguished sharp proton signals in the normal range, which are consistent with their diamagnetic d10 electronic configurations. These complexes have also been characterized by 31P{1H} NMR spectroscopy and broadening of their 31P signals were observed, which is similar to those reported in related Cu(I) and Ag(I) complexes (Fig. S3–S7†).9d,10b,21
Crystal structures of 2 and 7
The crystal structures of 2 and 7 have been determined by X-ray crystallography, and selected bond parameters are summarized in Table 1. As shown in Fig. 3, the two compounds are isostructural, and the metal centres are coordinated by a bidentate diimine ligand and two PPh3 ligands in a distorted tetrahedron. In 2, the Cu1–N2 bond length (2.049(3) Å) is shorter than that of Cu1–N1 (2.160(3) Å), indicating that the coordinated tz moiety is deprotonated and forms the stronger back-bonding interaction with Cu atom. In the structurally similar complex [Cu(N^N)(POP)]+ (N^N = 2-(2-tert-butyl-2H-tetrazol-5-yl)pyridine),16 the Cu–N(py) and Cu–N(tz) bond lengths are essentially identical, since both heterocycles are neutral. Compared with pyridine ring, the deprotonated tz moiety has stronger coordination ability and higher electron density, resulting in an asymmetric coordination environment. Compared with [CuI(N^N)(PPh3)2]+ with symmetric diimine ligands, the Cu–N(tz) bond length is also shorter. The shortening of Cu–N bond distance leads also to a slightly smaller P–Cu–P bite angle (122.48(4)°). Similar result is also found in 7, where the Ag–N6 and Ag–N7 bond distances are 2.308(6) and 2.513(6) Å, respectively and the P–Ag–P bond angle is 128.27(7)°. Although 2 and 7 are isostructural, the N-M-N bite angle of 2 (79.16(12)°) is significantly larger than the corresponding angle (69.84(19)°) in 7. The pyridyl ring is co-planar with the deprotonated tz ring with small dihedral angles of 1.8 and 2.8°, for 2 and 7 respectively. It is noted that the dangling tz ring and the coordinating pyridyl tetrazole ligands have dihedral angles of 9.0 and 7.9° respectively. Extensive weak interactions, such as π–π stacking interactions of aromatic rings and H-bondings via NH⋯N of adjacent Cu(I) complexes, have been found in their solid structures. (Fig. S9 and S10†)
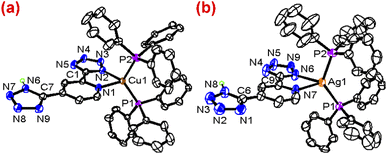 |
| | Fig. 3 The ORTEP drawing of structures for 2 (a) and 7 (b). | |
Table 1 Selected bond parameters (Å, °) for 2 and 7
| 2 |
7 |
| Cu1–N2 |
2.049(3) |
Ag1–N6 |
2.308(6) |
| Cu1–N1 |
2.160(3) |
Ag1–N7 |
2.513(6) |
| Cu1–P1 |
2.235(2) |
Ag1–P1 |
2.419(2) |
| Cu1–P2 |
2.265(2) |
Ag1–P2 |
2.471(2) |
| N2–Cu1–N1 |
79.16(12) |
N6–Ag1–N7 |
69.84(19) |
| P1–Cu1–P2 |
122.48(4) |
P1–Ag1–P2 |
128.27(7) |
Electronic absorption and emission properties
The UV/vis spectra of these compounds in CH2Cl2 solution and the ligands HL1–HL3 in MeOH solution have been collected at room temperature. As shown in Fig. S1,† these ligands exhibit the strong ligand-centred π–π* absorption bands <330 nm. The overlaid absorption spectra of 1–7 are shown in Fig. 4. Apart from the intense ligand-centred (LC) π–π* transitions of phosphine and tz ligands in CH2Cl2 solutions with molar extinction coefficients on the order of 104 M−1 cm−1 in the range of 220–330 nm, 1–6 also show moderate absorption bands with molar extinction coefficients on the order of 103 M−1 cm−1 from 330 to 400 nm tailing down off 450 nm. Compared with related cationic complexes [CuI(N^N)(PPh3)2]+, these absorption bands are obviously blue-shifted, in agreement with the raising of π*-orbitals of diimine ligands. Since neither phosphine ligands nor tz ligands show absorption bands in these ranges, these absorption bands are mainly assigned to metal-to-ligand charge transfer (MLCT) of {d(Cu) → π*(tz)}, possibly mixed with somewhat contributions of inter-ligand charge transfer. As suggested by TD-DFT studies, the lowest-lying transitions of 1–6 are assigned as the 1MLCT/1LLCT charge-transfer (Cu/PPh3-N^N) transitions, mixed with some intra-ligand (1IL) transitions of the anionic chelated ligands. These assignments are further supported by the dependence of the absorption energy on the π-accepting ability of phosphine ligands, with the order of P(PhMe)3 < POP < PPh3 < P(C6H4Cl)3 for compounds 2–5 containing the same diimine ligand L2. The increase of the π-accepting ability of phosphine ligands leads to the blue-shifted of the lowest-energy absorption bands. The assignment of the lowest energy absorption bands of these Cu(I) complexes are also supported by the dependence of the absorption energy on the electronic nature of diimine ligands, which is in line with the order of expected π* orbital energy level [L2 < L1 < L3] for the complexes 1, 2 and 6 bearing the same PPh3 ligand. For Ag(I) complex 7, the corresponding MLCT absorption band is not observed due to the lower-lying d orbital of the Ag+ ion.
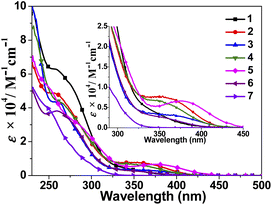 |
| | Fig. 4 The UV/vis spectra of 1–7 in CH2Cl2 solution. | |
The emission properties of these complexes have been investigated in CH2Cl2 solution at room temperature (Table 2, Fig. 5b). All these Cu(I) complexes exhibited yellow to orange emissions (539–595 nm) with photoluminescence quantum yields (PLQYs) up to 4.7% upon excitation with λex = 380 nm. Although the PLQYs of these compounds in solution are lower than that of [CuI(dmp)(PPh3)2]+ (dmp = 2,9-dimethylphenanthroline),18 they are significantly higher than most of the Cu(I) complexes [Cu(N^N)(PPh3)2]+ without the sterically-protected diimine ligands.18,19 All these emission spectra are broad and structureless (with ∼110 nm of the full width at half-maximum), consistent with the MLCT assignment with the large structural distortion on the excited state. The emission maxima of 2–5 with the same L2 ligand are in the range of 589 to 600 nm and red-shifted as compared with 1. In contrast to the observed trend in the lowest energy absorption, the emission energies of 2–5 varied with the substituents on phosphine ligands, which is in the trend of 2 (600 nm, PPh3) > 3 (595 nm, P(PhMe)3) > 4 (590 nm, P(PhCl)3) > 5 (589 nm, POP), as reflected from the overlaid emission spectra of 1–6 (Fig. 5b). Similar observation is also recently reported in our luminescent Cu(I) complexes containing various phosphine ligands.10b
Table 2 Photophysical data for 1–7
| |
Medium |
Emissiona λ/nm (τ0/μs) |
ϕem (×103) |
| Excitation at 380 nm in CH2Cl2 solution and 355 nm for glassy state at 77 K. Ref. 13. |
| 1 |
CH2Cl2 (298) |
571 (0.48) |
2 |
| Solid (298)b |
512 (20.6) |
850 |
| Glass (77) |
534 (230.47) |
|
| 2 |
CH2Cl2 (298) |
600 (0.52) |
31 |
| Solid (298) |
567 (1.65) |
258 |
| Glass (77) |
578 (112.10) |
|
| 3 |
CH2Cl2 (298) |
595 (0.65) |
12 |
| Solid (298) |
544 (8.23) |
393 |
| Glass (77) |
569 (75.43) |
|
| 4 |
CH2Cl2 (298) |
590 (0.72) |
47 |
| Solid (298) |
548 (2.31) |
403 |
| Glass (77) |
543 (15.42) |
|
| 5 |
CH2Cl2 (298) |
589 (0.07) |
0.3 |
| Solid (298) |
546 (8.72) |
628 |
| Glass (77) |
568 (102.50) |
|
| 6 |
CH2Cl2 (298) |
539 (2.02) |
5.2 |
| Solid (298) |
524 (6.69) |
486 |
| Glass (77) |
528 (263.39) |
|
| 7 |
CH2Cl2 (298) |
438 (<10 ns) |
1 |
| Solid (298) |
528 (15.78) |
66 |
| Glass (77) |
517 (208.28) |
|
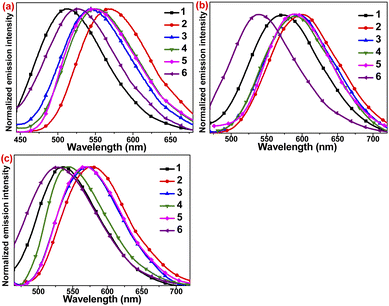 |
| | Fig. 5 (a) Emission spectra Cu(I) complexes 1–6 in the solid state at room temperature (b) in CH2Cl2 at room temperature and (c) in low-temperature 77 K glassy media EtOH–MeOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) (λex = 355 nm). :1, v/v) (λex = 355 nm). | |
The luminescence of 2–4 in CH2Cl2 is significantly enhanced as compared with 1. It is noted that although 1 exhibits the excellent PLQY of ∼85% in the solid state, its PLQY is only 0.2% in degassed CH2Cl2 solution. On the introduction of an additional aromatic ring into the pyridyl ring, the PLQYs in CH2Cl2 solution increase from 1.2% to 4.7%. It is noted that most Cu(I) complexes are nearly non-emissive or poorly emissive without the bulky substituents on the a,a′-positions of diimine ligands.18 The emission properties of these luminescent Cu(I) complexes reported herein are attributed to the dangling aromatic rings on the pyridyl ring, which enhance the π-conjugation system. Moreover, 5 with a POP ligand is nearly non-emissive with PLQY ∼0.03% and its lifetime (τ = 0.07 μs) is also much shorter than the other Cu(I) complexes in the range of 0.48 to 2.02 μs. Considering the spectroscopic studies on the related Cu(I) complexes,19 as well as the microsecond emission lifetime and tunable emission, these emissions are ascribed to 3MLCT {d(Cu) → π*[N^N]} probably admixed with somewhat 3LLCT {π(phosphine) → π*[N^N] excited states. All the emission maxima of Cu(I) complexes in solutions are relatively red-shifted as compared with those in solid state, which result from to deformation of excited states due to lack of steric protection. On the contrary, the Ag(I) complex 7 shows a blue emission at λem = 438 nm with lifetime less than 10 ns in CH2Cl2 solution (Fig. S11a†). Thus, it is tentatively assigned to metal-perturbed ligand-centred π–π* transitions, which are similarly observed in structurally related mononuclear Ag(I) complexes.20
In the solid state, these complexes show intense broad green (512 to 567 nm) unstructured photoluminescence, typically observed in luminescent Cu(I) complexes (Fig. 5a and Table 2). The lifetimes are in the range of 1.65 to 20.6 μs. Moreover, the PLQYs in solid state for 2–6 ranging between 25.8% and 62.8%, which are lower than the reported PLQY of 1 (85%), possibly due to the non-radiative decay through C–H/N–H vibrational quenching of the dangling aromatic rings. The enhanced solid emissions of these Cu(I) complexes are attributed to the decrease in the excited state distortion of Cu(I) complexes and non-radiative decay in the solid state. Since the rigidochromic effect impedes the structural distortion in the excited state, the solid-state emissions for most of these Cu(I) complexes are blue-shifted as compared with their emissions in solution. In contrast to the solution emissions, the solid-state emissions show a different trend on the electronic properties of phosphine ligands and diimine ligands. The deviation can be explained by the presence of various factors such as inter- and inner-molecular π-stacking in the solid state, as previously reported in the solid-state emissions of related Cu(I) complexes.9d,10b In contrast, the emission maxima of complexes 2–5 with the same L2 ligand range from 544 to 567 nm and are red-shifted compared with that of 1. Upon excitation at λex = 380 nm, 7 exhibits a green emission peaking at 528 nm. Based on the long decay times, large Stokes shifts, and spectroscopic studies of the related Ag(I) complexes,20 the emission of 7 in the solid state is assigned as intra-ligand 3LC phosphorescence. (Fig. S11b†)
In 77 K EtOH–MeOH (4:1, v/v) glassy medium, all these complexes display intense luminescence (Fig. 5c and Table 2). Compared with their emissions in solution, the emissions in the glassy state are generally blue-shifted and longer-lived. This is attributed to the rigidochromic effect, which reduces the structural distortion on the excited state and is also commonly observed in MLCT emitters. However, compared with the emission spectra in the solid state at room temperature, the emission spectra at 77 K are red-shifted, which are normally observed for [CuI(N^N)(P^P)]+ complexes due to the presence of TADF behaviour.8 The longer excited state lifetimes in the glassy medium at 77 K further support the assignment of triplet excited state. These emissions are also tentatively assigned to 3MLCT [d(Cu) → π*(N^N)] excited state. The emission band of 7 in low-temperature glassy medium is highly structured with a vibrational progression spacing of ∼1200 cm−1. The significantly different low-temperature emission properties of 7 is suggestive of a different emission origin, which is ascribed to the 3LC excited state of the L2 ligand. (Fig. S11c†) The emission maximum of 7 is very close to those of the Cu(I) complexes of L2 in glassy state. Thus, there may be contributions from 3LC excited state to the photoluminescence of Cu(I) complexes at low temperature.
Electronic structure calculations
Density functional theory (DFT) calculations of the complexes were performed by using the Gaussian 09 program to determine the nature of frontier orbitals. The lowest energy triplet excited states of 2 and 7 were computed and the two singly occupied molecular orbitals (SOMOs) were identified. Contour plots of the two SOMO and the spin densities of the complexes were shown in Fig. 6. Hydrogen atoms in the structures were omitted for clarity. For the Cu(I) complex 2, the lower energy SOMO of the complex consist of dπ(Cu) with contributions by lone pairs of P (from PPh3) and N (from L2), while the higher one consist of the π* orbital of L2 ligand, which suggests the triplet excited state of the complex is MLCT [Cu → L2] in nature, with some mixing of LLCT [PPh3 → L2]. For the Ag(I) complex 7, the two SOMOs consist of π and π* orbital of the L2 ligand, which suggests the triplet excited state of the complex is ligand-centred (IL) in nature.
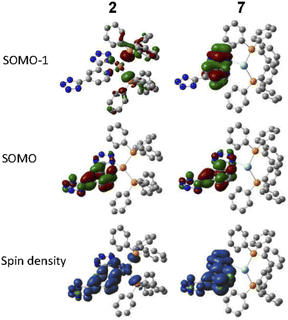 |
| | Fig. 6 Frontier molecular orbitals for the triplet state of complexes 2 and 7 calculated at the B3LYP/6-31+G(d)/LANL2DZ level of theory. | |
Solvatochromic effect
Many Cu(I) complexes exhibit low stability in various solutions, especially in their excited states, which limit their applications as probes/sensors for O2, pH, and various ions. Herein, the asymmetric ligands were used instead of bipyridine and phenanthroline diimine ligands. These asymmetric diimine ligands may provide obvious advantages over the symmetric one. In the ground state, the electron-rich tz moiety acts as a soft-base, which could form stronger bonds with Cu(I) ions. In the excited state, the MLCT transition could be attributed to the Cu(I) centre and pyridyl group of the ligands, while the tz moiety is not involved significantly due to its high lying π* orbital and effectively stabilize the excited state of these Cu(I) complexes. Moreover, the L2 in complexes 2–5 have an additional protonated tz moiety, whose pKa value (∼4.5) is comparable with the carboxylic acid. Thus, their emissions may be sensitive towards the pH variation.
As expected, the MLCT absorption bands of these Cu(I) complexes showed a dependence on the nature of the solvents. Taking 3 as an example (Fig. 7), the lowest-energy MLCT absorption bands in the range 350–400 nm are slightly dependent on the solvents used, while the LC absorption bands remain almost unchanged in different solvents. In contrast, the emission spectra of 3 show a stronger solvatochromic effect in various solvents. The emission maxima (λem) follow the order of DMF (553 nm) < MeOH (560 nm) < CH3CN (564 nm) < CHCl3 (584 nm) < CH2Cl2 (594 nm). Such a variation trend is mainly attributed to the existence of an acidic N–H bond in the dangling tz moiety, which is readily affected by solvent polarity and H-bonding interaction. In MeOH, the acidic N–H group forms strong H-bonding with solvents, which raises the π* orbital of ligand. Thus a higher-energy emission could be expected, which is supported by the emission observed in basic DMF with higher electron-density. This hypothesis is also supported by the negligible solvatochromic effect for 1 which does not bear a dangling tz unit (Fig. S2†).
 |
| | Fig. 7 The absorption spectra (a) and emission spectra (b) of 3 in various solvents. | |
Electroluminescence performance
As a result of the excellent solid-state luminescent properties of these neutral Cu(I) complexes, OLED containing 2 was fabricated to evaluate their EL properties. A structure of ITO/HAT-CN/TAPC/TCTA/mCP/mCP: PO-T2T: 20 wt% of 2/PO-T2T/LiF/Al was designed. Among them, HAT-CN, TAPC, TCTA, mCP, PO-T2T, and LiF are used as hole injection layer, hole transport layer, electron blocking layer, exciton blocking layer, electron transport layer, and electron injection layer, respectively. The corresponding energy level diagrams and molecular structures of the charge-transport materials are shown in Fig. 9. ITO conductive glass was used as the anode. The ITO conductive glass was treated with UV ozone and then placed in an organic electroluminescence integrated deposition system. Each organic material was formed on the ITO conductive glass by solution spin coating method on a 2500 r/s spin coater in turn. The film thickness was monitored by a quartz oscillating plate, and the light-emitting area was 3 mm × 3 mm. The HOMO and LUMO levels of these Cu(I) complexes were determined using the cyclic voltammetry (CV) and the optical bandgaps are estimated (Fig. S12 and Table S1†). As shown in Fig. 8b, the device containing 2 emits bright green emission peaking at 526 nm, and the emission spectrum of the electroluminescent devices is comparable with the solid-state emission spectrum, indicating that EL emission comes directly from 2. However, the emission maxima in these spectra exhibit a slight blue shift at different voltages, possibly due to the difficulty in managing excitons in a single emission layer. Since the primary mCP and charge transfer material used are non-emissive, an effective combination of holes and electrons occurred in the dopant. The Commission Internationale de L'Eclairage (CIE) coordinates of this device is (0.365, 0.551) (Fig. 9d). The current density–voltage–brightness curves are displayed in Fig. 8c, revealing a turn-on voltage at 2.4 V and a luminance of 1000 cd m−2 for lighting at 3.5 V. For most Cu(I) devices, a 2.4 V turn-on voltage is very low. The low resistance at doping concentration of 20 wt% suggests that 2 is an efficient charge carrier, as high doping concentration usually leads to the concentration quenching effect. The maximum current efficiency was 22.96 cd A−1 at a current density of 0.01 mA cm−2 (Fig. 8d). At the maximum current efficiencies, the maximum external quantum efficiency (EQE) was 7.7% (Fig. 9a), comparable to the EQEs of most Cu(I)-doped OLEDs. The good performance of this device could be attributed to the TADF-type launch.
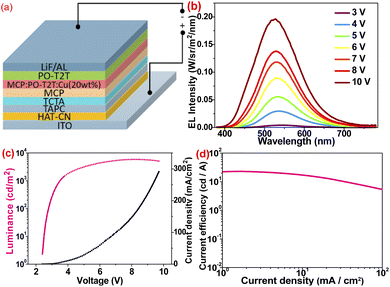 |
| | Fig. 8 (a) The OLED device structure of 2-based device; (b) the EL spectra of 20 wt% doping device under different voltages of 2; (c) current density–voltage–brightness (I–V–B) curves of 2-based device; (d) the dependence of the current efficiency of 2 on the current density. | |
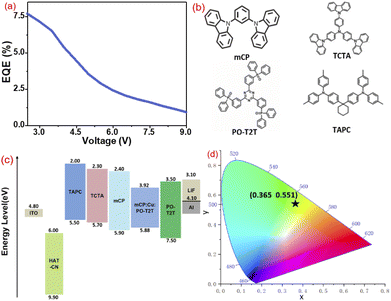 |
| | Fig. 9 (a) EQE–voltage characteristics of 2-based device; (b) the molecular structures of the charge transfer compound in the device; (c) the energy band diagram for 2; (d) CIE diagram of 2-based device. | |
Conclusion
Functionalized pyridyl-tetrazole ligands have been studied for the design of neutral heteroleptic Cu(I)-based compounds with the general formula [Cu(N^N)(P^P)] (1–6). All these Cu(I) complexes exhibit strong luminescence in the solid state. Upon the introduction of a dangling aromatic moisty on the pyridyl ring, the luminescent performance in solutions is significantly enhanced as compared to parent pyridyl-tetrazole complex 1. OLED containing 2 exhibits high EQE and current efficiency. These features suggest that heteroleptic Cu(I) complexes with pyridyl-tetrazole ligands could be used as an economic luminescent material.
Experimental section
Materials and methods
Triphenylphosphine (PPh3), tri(p-tolyl)phosphine [P(PhMe)3], tri(4-chlorophenyl)phosphine [P(PhCl)3], bis{2-(diphenylphosphanyl)phenyl} ether (POP), were purchased from Acros Organic Chemical Company. 2-(1H-tetrazol-5-yl)pyridine (HL1), 2,4-di(1H-tetrazol-5-yl)pyridine (HL2), 4-phenyl-2-(1H-tetrazol-5-yl)pyridine (HL3) were synthesized by the similar procedures via [3 + 2] cycloaddition reactions of organonitriles and N3−.17 [Cu(MeCN)4](ClO4)21 and [CuI(L1)(PPh3)2] (1)13 were synthesized according to literature procedures. The commercially available reagents and solvents of analytical grade were used without further purification.
Synthesis and characterization
[CuI(L2)(PPh3)2] (2). [Cu(MeCN)4]ClO4 (50 mg, 0.153 mmol), PPh3 (80.3 mg, 0.31 mmol) were added to CH3CN (15 mL) and the mixture was stirred at room temperature for 1 h under nitrogen atmosphere. Then, HL2 (32.9 mg, 0.15 mmol) was added and was further stirred for 1 h. After removal of solvent under reduced pressure, the residue was purified by column chromatography on silica gel eluted with acetone/dichloromethane (v:v, 1:4). The bright yellow layer was collected. The light yellow microcrystalline solid was obtained and was further purified by slow evaporation of a mixed CH2Cl2/MeOH solvent of 2. Yield: 105 mg, 64.0%. Elemental analysis for C43H34CuN9P2: calcd C 64.37, H 4.27, N 15.71%; found C 64.33, H 4.15, N 15.74%, selected IR (KBr, cm−1): v(N–H) 3139, v(NN) 1435, v(CN) 1619. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1):228 (71110), 256sh (48870), 366sh (7000). 1H NMR (400 MHz, CDCl3): 9.41 (s, 1H, L2-H), 9.12 (d, J = 5.4 Hz, 1H, L2-H), 8.18 (dd, J = 5.4, 1.7 Hz, 1H, L2-H), 7.38–7.30 (m, 6H, PPh3-H), 7.23–7.08 (m, 24H, PPh3-H); 31P{1H} NMR (162 MHz, CDCl3): δ: 0.68 (s, PPh3).
{CuI(L2)[P(PhMe)3)2]} (3). The synthetic procedure of 3 is similar to that of 2 except that P(PhMe)3 (93.07 mg, 0.306 mmol) was used instead of PPh3. Yield: 104 mg, 52.8%. Elemental analysis for C49H46CuN9P2: calcd C 66.39, H 5.23, N 14.22%; found C 66.54, H 5.28, N 14.43%. Selected IR (KBr, cm−1): v(N–H) 3128, v(NN) 1436, v(CN) 1621. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1): 230 (102300), 263sh (42980), 371sh (2900). 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.3, 1H, L2-H), 7.76 (d, J = 5.4 Hz, 1H, L2-H), 7.56 (dd, J = 11.8, 8.1 Hz, 10H), 7.32 (s, 1H), 7.26 (d, J = 2.6 Hz, 6H), 7.12 (d, J = 16.5 Hz, 8H), 1.62 (s, 18H, –CH3). 31P{1H} NMR (162 MHz, CDCl3): δ 29.41 (s, P(PhMe)3).
{CuI(L2)[P(PhCl)3)2]} (4). The synthetic procedure of 4 is similar to that of 2 except that P(PhCl)3 (112.0 mg, 0.307 mmol) was used instead of PPh3. Yield: 124 mg, 55.3%. Elemental analysis for C43H28Cl6CuN9P2: calcd C 51.19, H 2.80, N 12.49%; found C 52.23, H 2.68, N 12.54%. Selected IR (KBr, cm−1): v(N–H) 3130, v(NN) 1435, v(CN) 1620. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1):232 (88390), 264sh (43000), 357sh (5600). 1H NMR (400 MHz, CDCl3): 10.66 (s, 1H, L2-H), 8.25 (d, J = 8.0 Hz, 1H, L2-H), 7.84 (d, J = 8.0 Hz, 1H, L2-H), 7.72–7.29 (m, 4H, PPh3-H), 7.13–7.01 (m, 10H, PPh3-H), 6.93–6.80 (m, 10H, PPh3-H). 31P{1H} NMR (162 MHz, CDCl3): δ −2.01 (s, P(PhCl)3).
[CuI(L2)(POP)] (5). The synthetic procedure of 5 is similar to that of 2 except that POP (82.33 mg, 0.153 mmol) was used instead of PPh3. Yield: 87 mg, 50.9%. Elemental analysis for C43H32CuN9OP2: calcd C 63.27, H 3.95, N 15.44%; found C 63.56, H 3.76, N 15.54%. Selected IR (KBr, cm−1): v(N–H) 3132, v(NN) 1435, v(CN) 1622. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1): 228 (76630), 254sh (49280), 290sh (27892), 380 (6503). 1H NMR (400 MHz, CDCl3): 9.83 (s, 1H, L2-H), 8.10 (d, J = 5.4 Hz, 1H, L2-H), 8.05 (d, J = 5.4 Hz, 1H, L2-H), 7.40–7.66 (m, 4H, POP-H), 7.35–7.25 (m, 4H, POP-H), 7.22–7.11 (m, 8H, POP-H), 7.06–6.98 (m, 4H, POP-H), 6.98–6.90 (m, 4H, POP-H), 6.80–6.66 (m, 4H, POP-H). 31P{1H} NMR (162 MHz, CDCl3): δ −12.98 (s, POP).
[CuI(L3)(PPh3)2] (6). The synthetic procedure of 6 is similar to that of 1 except that HL3 (34.13 mg, 0.153 mmol) was used instead of HL1. Yield: 65 mg, 52.4%. Elemental analysis for C48H38CuN5P2: calcd C 71.14, H 4.73, N 8.64%; found C 71.62, H 4.62, N 8.48%. Selected IR (KBr, cm−1): v(NN) 1434, v(CN) 1613. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1): 230 (98420), 260sh (62450), 353sh (3000). 1H NMR (400 MHz, CDCl3): 8.52 (s, 1H, L3-H) 7.91 (d, J = 5.5 Hz, 1H, L3-H), 7.71 (dd, J = 8.0, 1.5 Hz, 2H, L3-H), 7.54–7.45 (m, 4H, L3-H),7.32–7.26 (m, 6H, PPh3), 7.15–7.13 (m, 24H, PPh3-H); 31P{1H} NMR (162 MHz, CDCl3): δ 10.25 (s, PPh3).
[AgI(L2)(PPh3)2] (7). AgBF4 (50 mg, 0.25 mmol), PPh3 (134.0 mg, 0.5 mmol) were added to CH3CN (15 mL) and was stirred at room temperature for 1 h under nitrogen atmosphere. After that, HL2 (54 mg, 0.25 mmol) was added, and the mixture was further stirred for 1 h. After removal of the solvent under reduced pressure, the residue was further purified by slow diffusion of diethyl ether into a CH2Cl2 solution of 7 to give colorless crystals. Yield: 96 mg, 59.4%. Elemental analysis for C43H34AgN9P2: calcd C 61.00, H 4.05, N 14.89%; found C 61.43, H 4.32, N 14.98%. Selected IR (KBr, cm−1): v(N–H) 3135, v(NN) 1436, v(CN) 1618. UV/vis (CH2Cl2): λmax/nm (ε/mol−1 dm3 cm−1): 230 (52327), 252sh (38046). 1H NMR (400 MHz, CDCl3): 9.63 (s, 1H, L2-H), 8.11 (d, J = 5.1 Hz, 1H, L2-H), 8.03 (d, J = 5.4 Hz, 1H, L2-H), 7.42–4.34 (m, 6H, PPh3-H), 7.34–7.29 (m, 8H, PPh3-H), 7.28–7.22 (m, 16H, PPh3-H); 31P{1H} NMR (162 MHz, CDCl3): δ 9.22 (s, PPh3).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21771026), Hubei Provincial Natural Science Foundation of China (2018CFA047) and the Natural Science Foundation of Anhui Province (2008085QD173). Financial support from the Hong Kong Research Grants Council (projects no. 18301418 and no. 18301319) is also acknowledged.
Notes and references
-
(a) Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Mater., 2004, 16, 432–436 CrossRef CAS;
(b) Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203–1208 CrossRef CAS;
(c) Q. Zhang, J. Ding, Y. Cheng, L. Wang, Z. Xie, X. Jing and F. Wang, Adv. Funct. Mater., 2007, 17, 2983–2990 CrossRef CAS;
(d) N. Armaroli, G. Accorsi, M. Holler, O. Moudam, J. F. Nierengarten, Z. Zhou, R. T. Wegh and R. Welter, Adv. Mater., 2006, 18, 1313 CrossRef CAS;
(e) G. Che, Z. Su, W. Li, B. Chu and M. Li, Appl. Phys. Lett., 2006, 89, 103511 CrossRef;
(f) C. W. Hsu, C. C. Lin, M. W. Chung, Y. Chi, G. H. Lee, P. T. Chou, C. H. Chang and P. Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085–12099 CrossRef CAS PubMed.
-
(a) Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC;
(b) E. Holder, B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109–1121 CrossRef CAS;
(c) L. He, J. Qiao, L. Duan, G. Dong, D. Zhang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2009, 19, 2950–2960 CrossRef CAS;
(d) T. Tsuzuki and S. Tokito, Adv. Mater., 2007, 19, 276–280 CrossRef CAS;
(e) C. H. Yang, Y. M. Cheng, Y. Chi, C. J. Hsu, F. C. Fang, K. T. Wong, P. T. Chou, C. H. Chang, M. H. Tsai and C. C. Wu, Angew. Chem., Int. Ed., 2007, 119, 2470–2473 CrossRef.
-
(a) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS;
(b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS;
(c) C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem. - Eur. J., 2010, 16, 233–247 CrossRef CAS;
(d) J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS;
(e) J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
-
(a) J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS;
(b) P. T. Chou and Y. Chi, Eur. J. Inorg. Chem., 2006, 2006, 3319–3332 CrossRef;
(c) J. Breu, C. Kratzer and H. Yersin, J. Am. Chem. Soc., 2000, 122, 2548–2555 CrossRef CAS;
(d) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS;
(e) M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
-
(a) H. Yersin, Top. Curr. Chem., 2004, 241, 1 CrossRef CAS;
(b) Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe, C. Adachi and Y. Kawamura, Appl. Phys. Lett., 2005, 86, 71104–71107 CrossRef;
(c) H. Sasabe, J. I. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003–5007 CrossRef CAS PubMed;
(d) C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
-
(a) F. Yu, C. Shen, T. Zheng, W.-K. Chu, J. Xiang, Y. Luo, C.-C. Ko, Z. Q. Guo and T.-C. Lau, Eur. J. Inorg. Chem., 2016, 2016, 3641–3648 CrossRef CAS;
(b) F. Yu, W.-K. Chu, C. Shen, Y. Luo, J. Xiang, S.-Q. Chen, C.-C. Ko and T.-C. Lau, Eur. J. Inorg. Chem., 2016, 2016, 3892–3899 CrossRef CAS;
(c) C. Shen, F. Yu, W.-K. Chu, J. Xiang, P. Tan, Y. Luo, H. Feng, Z.-Q. Guo, C.-F. Leung and T.-C. Lau, RSC Adv., 2016, 6, 87389–87399 RSC;
(d) L.-J. Luo, Q.-Q. Su, S.-C. Cheng, J. Xiang, W.-L. Man, W.-M. Shu, M.-H. Zeng, S.-M. Yiu, C.-C. Ko and T.-C. Lau, Inorg. Chem., 2020, 59, 4406–4413 CrossRef CAS PubMed;
(e) P.-Y. Ho, S.-C. Cheng, S.-M. Yiu, V. K.-M. Au, J. Xiang, C.-F. Leung and C.-C. Ko, Inorg. Chem., 2019, 58, 11372–11381 CrossRef CAS PubMed.
-
(a) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS;
(b) Y. R. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS.
-
(a) D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS;
(b) S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwich and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
-
(a) J.-L. Chen, Z.-H. Guo, Y.-S. Luo, L. Qiu, L.-H. He, S.-J. Liu, H.-R. Wen and J.-Y. Wang, New J. Chem., 2016, 40, 5325–5332 RSC;
(b) X. F. Liu, R. F. Li, L. F. Ma, X. Feng and Y. Q. Ding, New J. Chem., 2016, 40, 619–625 RSC;
(c) F. S. Wu, J. Li, H. B. Tong, Z. Y. Li, C. Adachi, A. Langlois, P. D. Harvey, L. Liu, W.-Y. Wong, W.-K. Wong and X. J. Zhu, J. Mater. Chem. C, 2015, 3, 138–146 RSC;
(d) J. Xiang, S.-C. Cheng, X.-X. Jin, Q.-Q. Su, X. Zhou, W.-K. Chu, C.-F. Leung and C.-C. Ko, Dalton Trans., 2019, 48, 741–750 RSC.
-
(a) J. H. Min, Q. S. Zhang, W. Sun, Y. X. Cheng and L. X. Wang, Dalton Trans., 2011, 40, 686–693 RSC;
(b) X.-X. Jin, T. Li, D.-P. Shi, L.-J. Luo, Q.-Q. Su, J. Xiang, H.-B. Xu, C.-F. Leung and M.-H. Zeng, New J. Chem., 2020, 44, 13393–13400 RSC;
(c) L.-L. Hu, C. Shen, W.-K. Chu, J. Xiang, F. Yu, G. Xiang, Y. Nie, C.-L. Kwok, C.-F. Leung and C.-C. Ko, Polyhedron, 2017, 127, 203–211 CrossRef CAS;
(d) X.-L. Chen, R. M. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang and C.-Z. Lu, Chem. Mater., 2013, 25, 3910–3920 CrossRef CAS;
(e) J. Xiang, Y.-G. Yin and P. Mei, Inorg. Chem. Commun., 2007, 10, 1168–1171 CrossRef CAS.
-
(a) G. B. Che, Z. S. Su, W. L. Li, B. Chu, M. T. Li, Z. Z. Hu and Z. Q. Zhang, Appl. Phys. Lett., 2006, 89, 103511 CrossRef;
(b) Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203–1208 CrossRef CAS;
(c) S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshinoa and M. Osawa, J. Mater. Chem. C, 2013, 1, 542–551 RSC.
- X.-L. Ding, L. Shen, L.-Y. Zou, M.-S. Ma and A.-M. Ren, MOLECULAR PHYSICS, 2018, 116, 898–909 CrossRef CAS.
- L. Bergmann, J. Friedrichs, M. Mydlak, T. Baumann, M. Nieger and S. Bräse, Chem. Commun., 2013, 49, 6501–6503 RSC.
- L. Bergmann, G. J. Hedley, T. Baumann, S. Bräse and I. D. W. Samuel, Sci. Adv., 2016, 2, e1500889 CrossRef PubMed.
-
(a) S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Prescimone, G. Longo, A. Pertegás, M. Sessolob and H. J. Bolink, Dalton Trans., 2014, 43, 16593–16596 RSC;
(b) S. Keller, A. Pertegás, G. Longo, L. Martínez, J. Cerdá, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Ortí and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857–3871 RSC.
- C. Femoni, S. Muzzioli, A. Palazzi, S. Stagni, S. Zacchini, F. Monti, G. Accorsi, M. Bolognesi, N. Armaroli, M. Massi, G. Valenti and M. Marcaccio, Dalton Trans., 2013, 42, 997–1010 RSC.
-
(a) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed;
(b) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed;
(c) J. Xiang and Y. Luo, Inorg. Chem. Commun., 2013, 37, 12–16 CrossRef CAS;
(d) J. Xiang, Y. Luo, L.-L. Zhao, C.-H. Wang and J.-S. Wu, Inorg. Chem. Commun., 2013, 31, 23–28 CrossRef CAS.
- C. E. A. Palmer and D. R. McMillin, Inorg. Chem., 1987, 26, 3837–3840 CrossRef CAS.
- R. A. Rader, D. R. McMillin, M. T. Buckner, T. G. Matthews, D. J. Casadonte, R. K. Lengel, S. B. Whittaker, L. M. Darmon and F. E. Lytle, J. Am. Chem. Soc., 1981, 103, 5906–5912 CrossRef CAS.
-
(a) K. Matsumoto, T. Shindo, N. Mukasa, T. Tsukuda and T. Tsubomura, Inorg. Chem., 2010, 49, 805–814 CrossRef CAS PubMed;
(b) A. Kaeser, O. Moudam, G. Accorsi, I. Séguy, J. Navarro, A. Belbakra, C. Duhayon, N. Armaroli, B. Delavaux-Nicot and J. Nierengarten, Eur. J. Inorg. Chem., 2014, 2014, 1345–1355 CrossRef CAS;
(c) J. Chen, T. Teng, L. J. Kang, X. L. Chen, X. Y. Wu, R. M. Yu and C. Z. Lu, Inorg. Chem., 2016, 55, 9528–9536 CrossRef CAS;
(d) A. Kaeser, B. Delavaux-Nicot, C. Duhayon, Y. Coppel and J. Nierengarten, Inorg. Chem., 2013, 52, 14343–14354 CrossRef CAS.
- J. V. Hanna, R. D. Hart, P. C. Healy, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 2321–2326 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental section, Fig. S1–S12, Table S1 and S2. CCDC 2178439 and 2178440. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04684a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d and
Jing Xiang
*d and
Jing Xiang







