
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of W-modified CeO2/ZrO2 catalysts for selective catalytic reduction of NO with NH3†
Chenglong Lia,
Zhitao Han *a,
Yuqing Hua,
Tingjun Liua and
Xinxiang Panab
*a,
Yuqing Hua,
Tingjun Liua and
Xinxiang Panab
aMarine Engineering College, Dalian Maritime University, No.1, Linghai Road, Dalian 116026, China. E-mail: hanzt@dlmu.edu.cn
bSchool of Electronic and Information Technology, Guangdong Ocean University, Zhanjiang 524088, China
First published on 27th September 2022
Abstract
In this paper, a series of tungsten–zirconium mixed binary oxides (denoted as WmZrOx) were synthesized via co-precipitation as supports to prepare Ce0.4/WmZrOx catalysts through an impregnation method. The promoting effect of W doping in ZrO2 on selective catalytic reduction (SCR) performance of Ce0.4/ZrO2 catalysts was investigated. The results demonstrated that addition of W in ZrO2 could remarkably enhance the catalytic performance of Ce0.4/ZrO2 catalysts in a broad temperature range. Especially when the W/Zr molar ratio was 0.1, the Ce0.4/W0.1ZrOx catalyst exhibited the widest active temperature window of 226–446 °C (NOx conversion rate > 80%) and its N2 selectivity was almost 100% in the temperature of 150–450 °C. Moreover, the Ce0.4/W0.1ZrOx catalyst also exhibited good SO2 tolerance, which could maintain more than 94% of NOx conversion efficiency after being exposed to a 100 ppm SO2 atmosphere for 18 h. Various characterization results manifested that a proper amount of W doping in ZrO2 was not only beneficial to enlarge the specific surface area of the catalyst, but also inhibited the growth of fluorite structure CeO2, which were in favor of CeO2 dispersion on the support. The presence of W was conducive to the growth of a stable tetragonal phase crystal of ZrO2 support, and a part of W and Zr combined to form W–Zr–Ox solid super acid. Both of them resulted in abundant Lewis acid sites and Brønsted acid sites, enhancing the total surface acidity, thus significantly improving NH3 species adsorption on the surface of the Ce0.4/W0.1ZrOx catalyst. Furthermore, the promoting effect of adding W on SCR performance was also related to the improved redox capability, higher Ce3+/(Ce3+ + Ce4+) ratio and abundant surface chemisorbed oxygen species. The in situ DRIFTS results indicated that nitrate species adsorbed on the surface of the Ce0.4/W0.1ZrOx catalyst could react with NH3 due to the activation of W. Therefore, the reaction pathway over the Ce0.4/W0.1ZrOx catalyst followed both Eley–Rideal (E–R) and Langmuir–Hinshelwood (L–H) mechanisms at 250 °C.
1. Introduction
Selective catalytic reduction of nitrogen oxides with NH3 (NH3-SCR) has been widely employed for NOx abatement applications in stationary and mobile sources.1,2 During the past decades, V2O5–WO3 (or MoO3)/TiO2 had been considered the most pervasive and efficient SCR catalysts.3 However, these catalysts still suffer from some inevitable shortcomings in practical application, such as a narrow operation temperature window (300–400 °C), toxicity of vanadium pentoxide and low N2 selectivity at high temperatures.4 Given these disadvantages, great efforts have been made to develop environmentally friendly catalysts with a wide temperature window and high N2 selectivity.In recent years, some non-toxic SCR catalysts such as MnOx, Fe2O3, CuO and CeO2, have been extensively investigated in order to substitute vanadium-based catalysts.5–8 Among them, cerium-based NH3-SCR catalysts have attracted a lot of researchers' interest due to their high oxygen storage/release capacity and remarkable redox properties, which are significant to the oxidation of NOx and the acceleration of NH3-SCR reactions.9 However, pure CeO2 catalysts exhibit poor thermal stability and are easy to sinter at high temperature. In addition, the high active surface oxygen of a pure CeO2 catalyst results in NH3 oxidation on the catalyst surface, especially at high temperature, leading to a decrease in SCR activity.8,10 It is generally believed that acid sites are beneficial to suppress NH3 oxidation and promoting NH3 adsorption on the catalyst surface. Therefore, it should be feasible to enhance acid sites to improve NOx conversion and N2 selectivity of a CeO2 catalyst.11,12
Zirconia (ZrO2) is an acid-based amphoteric oxide with excellent redox capability and high refractory property. Previous studies reported that the addition of ZrO2 to CeO2 led to an improvement on thermal stability and oxygen storage capacity.13 ZrO2-supported CeO2 catalysts exhibited good oxygen storage capacity and highly refractory property at the same time. It can utilize the large surface area of ZrO2 to promote the dispersion of CeO2 on catalyst surface. Previous studies showed that CeO2/ZrO2 catalysts possessed excellent NH3-SCR activity at medium temperature.14,15 Nonetheless, the low-temperature activity and SO2 tolerance of CeO2/ZrO2 catalyst are still not very satisfactory, which hinders their industrial application.
As an important additive in traditional V-based catalysts, WO3 has been recognized as an excellent “chemical” and “structural” promoter to improve SCR performance obviously.16 Previous studies have shown that the addition of WO3 could enhance the adsorption and activation of NH3 by increasing the surface acidity of the catalysts, which was beneficial to the improvement of NH3-SCR activity.17,18 Recently, Fang et al. prepared WO3/Ce0.65Zr0.35O2 catalyst by co-precipitation and impregnation method, it could obtain an excellent NH3-SCR performance at 250–450 °C.19 Väliheikki et al. have proven that the WO3/Ce0.85Zr0.15O2 catalyst exhibited high SO2 and H2O resistance in the temperature range of 300–500 °C.20 In these studies, WO3 was usually used as a surface modifier to modify the catalyst surface. However, there are few reports about the incorporation of W into ZrO2 to form binary metal oxide support for NH3-SCR. Chen et al. reported that, the addition of W in ZrO2 could enhance the total acidity and redox properties by forming W–Zr–Ox, which would greatly promote the SCR performance.21,22 The authors considered that W–Zr–Ox solid super acid could be used as SCR support with a high surface area, which might enhance the catalytic activity of Ce/Zr catalysts greatly.
In this work, we focused on the effects of W doping in ZrO2 on SCR performance of Ce/ZrO2 catalyst. A series of Ce/WZrOx catalysts were prepared by successive co-precipitation and impregnation methods. Catalytic performance tests showed that Ce/WZrOx catalysts exhibited a much higher NOx removal efficiency than that of Ce/ZrO2 catalyst. Further, the effects of W doping in ZrO2 were investigated in detail by using N2 physisorption, XRD, Raman, SEM, TEM, XPS, H2-TPR, NH3-TPD and in situ DRIFTS. Finally, the possible reaction mechanisms were also discussed to gain insights into the effect of WZrOx solid super acid support on SCR reaction pathways.
2. Experimental
2.1 Catalyst preparation
A series of tungsten–zirconium oxides with various molar ratios of W/Zr were prepared by using the co-precipitation method. The typical synthesis process was as follows: a proper amount of Zr(NO3)4·5H2O and (NH4)10H2(W2O7)6·xH2O were dissolved in deionized water. Then the mixed solution was heated to 40 °C and held for 2 h under continuous magnetic stirring. Next, ammonia solution (25 wt%) was added dropwise to the above solution with vigorous stirring to adjust the solution pH to 10. The obtained precipitate was naturally cooled down to room temperature for 5 h and then filtered, and washed with deionized water until pH changed little. Afterwards, the precipitate was washed with anhydrous ethanol, and dried at 80 °C overnight. The collected solid was calcined at 550 °C in air for 3 h, and finally grounded into a fine powder. The prepared tungsten–zirconium mixed oxides were denoted as WmZrOx, where m represented the molar ratio of W/Zr (m = 0.025, 0.05, 0.1, 0.2). Pristine ZrO2 was also prepared for reference by using the precipitation method.Both Ce0.4/ZrO2 and Ce0.4/WmZrOx catalysts were prepared by the impregnation method, where 0.4 represented the molar ratio of Ce/Zr. Firstly, a certain amount of Ce(NO3)3·6H2O was dissolved in deionized water. Then a desired amount of ZrO2 or WmZrOx powder was impregnated in the solution with strong stirring for 0.5 h. Next, the mixture continued to be stirred sufficiently at 80 °C in a water bath to evaporate the solvent. The solid was dried at 100 °C for 12 h, and calcined at 500 °C for 3 h in air. Finally, all catalysts were crushed and sieved to 40–60 mesh for testing.
2.2 Catalyst activity test
The SCR activity tests of these prepared catalysts were carried out in a fixed-bed quartz reactor (I.D. 6 mm) at atmospheric pressure with a catalyst dosage of 0.5 mL (40–60 mesh). SCR activity measurements were operated in a temperature range of 150–450 °C. The simulated gas consisted of 500 ppm NO, 500 ppm NH3, 5 vol% O2, 100 ppm SO2 (when used) and N2 as balance gas with a total flow rate of 500 mL min−1. The corresponding gas hourly space velocity (GHSV) was 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The outlet concentrations of NO, NO2, NH3 and N2O were monitored by an FTIR spectrometer (Antaris IGS, ThermoFisher Scientific) equipped with a heated low-volume multiple-path gas cell (2 m) and an MCT detector cooled by liquid nitrogen. Here NOx referred to the sum of NO and NO2. NOx conversion efficiency and N2 selectivity were calculated as follows:
000 h−1. The outlet concentrations of NO, NO2, NH3 and N2O were monitored by an FTIR spectrometer (Antaris IGS, ThermoFisher Scientific) equipped with a heated low-volume multiple-path gas cell (2 m) and an MCT detector cooled by liquid nitrogen. Here NOx referred to the sum of NO and NO2. NOx conversion efficiency and N2 selectivity were calculated as follows:
 | (1) |
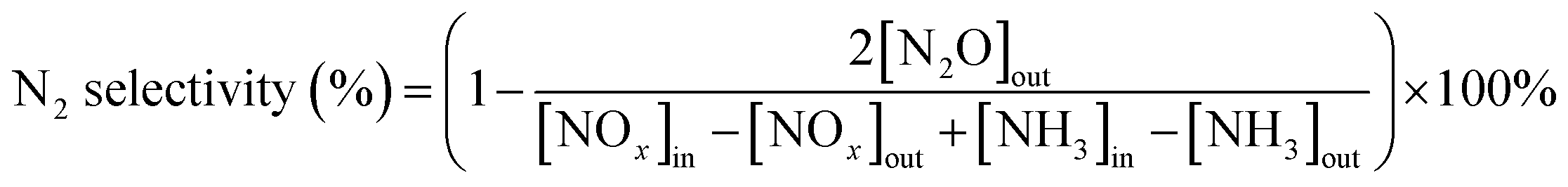 | (2) |
2.3 Catalyst characterization
The textural properties of the prepared samples were measured using N2 physisorption (ASAP 2020 PLUS, Micromeritics). The powder X-ray diffraction (XRD) patterns were performed on a diffractometer (TTRAX III, Rigaku, Japan) with a Cu-Kα radiation source (λ = 0.15406 nm) under 40 kV and 30 Ma. The Raman spectra of samples were carried out at a Raman Spectrometer (RM2000, Renishaw), using an Ar ion laser (532 nm) as the excitation source. The morphology of the samples was characterized by scanning electron microscopic (SEM, Tescan Mira4). The transmission electron microscopic (TEM) images were performed on FEI Talos F200X and the chemical analysis was obtained by energy dispersive X-ray spectrometer (EDS, Oxford Ultim Max65). X-ray photoelectron spectroscopy (XPS) measurement was obtained on a surface analysis photoelectron spectrometer (ESCALAB 250Xi, ThermoFisher Scientific) using Al Kα as a radiation source. Temperature programmed reduction with H2 (H2-TPR) experiments were operated on a chemisorption analyzer (Autochem II 2920, Micromeritics). Temperature programmed desorption of NH3 (NH3-TPD) experiments were operated on a chemisorption analyzer (Autochem II 2920, Micromeritics). In situ DRIFTS measurements were carried out by an FTIR spectrometer (Nicolet iS50, ThermoFisher Scientific) equipped with an MCT/A detector. The spectral resolution was 4 cm−1 with co-addiction 64 scans.3. Results and discussion
3.1 SCR performance
The catalytic performance of the prepared catalysts for NH3-SCR of NOx in the temperature range of 150–450 °C was tested, and the results were displayed in Fig. 1. It could be seen from Fig. 1(a) that W-doped ZrO2 supports imposed significant impacts on SCR catalytic activities of Ce0.4/ZrO2 catalysts. Without W doping, Ce0.4/ZrO2 catalyst showed rather poor SCR activity in the whole temperature region with the maximum NOx conversion of only about 56% at 370 °C, which was in accordance with our previous study.15 In contrast, Ce0.4/WmZrOx catalysts exhibited much better catalytic activity in the test temperature range. With the increase of W/Zr molar ratio from 0.025 to 0.1, the promotional effect of W on SCR activity was observed over W-containing catalysts with dramatically increasing NOx conversion and broadened operation temperature windows. However, further increasing W/Zr molar ratio to 0.2, SCR performance of Ce0.4/W0.2ZrOx catalysts deteriorated obviously in the whole operating temperature, and NOx conversion efficiency was only 41% at 226 °C. After all, Ce0.4/W0.1ZrOx catalyst possessed the largest active temperature window (NOx conversion rate > 80%) of 226–446 °C under GHSV of 60000 h−1. Fig. 1(b) showed the N2 selectivity of Ce0.4/ZrO2 and Ce0.4/WmZrOx catalysts. It could be seen that N2 selectivity over Ce0.4/ZrO2 catalyst began to decline slowly when the reaction temperature was above 375 °C, and reduced to 95% at 450 °C. In contrast, all Ce0.4/WmZrOx catalysts exhibited superior N2 selectivity. It was close to 100% in the whole temperature range. The above results demonstrated that the doping of W in ZrO2 supports could remarkably improve NH3-SCR performance of Ce/Zr catalysts. Since the comprehensive performance of Ce0.4/W0.1ZrOx catalyst was obviously better than other catalysts, comparative investigations between Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts were conducted to elucidate the effect of W–Zr binary metal oxide support on NH3-SCR performance.
 | ||
| Fig. 1 SCR performance test results of prepared catalysts: (a) NOx conversion and (b) N2 selectivity of Ce0.4/ZrO2 and Ce0.4/WmZrOx catalysts. (c) SO2 resistance test over Ce0.4/W0.1ZrOx catalyst at 300 °C. (Reaction conditions: 0.5 mL catalyst, [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, [SO2] = 100 ppm (when used), balance with N2, total flow rate = 500 mL min−1 and GHSV = 60000 h−1). | ||
It is well known that the common catalysts (vanadium-based) would gradually sinter and the catalytic performance decreased seriously after the SCR reactions. Therefore, the thermostability of catalyst was an important factor that must be considered in the practical application. To investigate the thermostability, Ce0.4/W0.1ZrOx catalyst was cycled two times SCR reactions (as shown in Fig. S1†). It could be seen that there was no significant difference in the catalytic performance between two cycles. In addition, XRD, H2-TPR and NH3-TPD techniques were used for the fresh Ce0.4/W0.1ZrOx and the used Ce0.4/W0.1ZrOx (2nd cycle) catalysts to investigate the effect of SCR reaction process on the structure, redox and surface acidity over Ce0.4/W0.1ZrOx catalyst (as shown in Fig. S2–S4†). These results demonstrated that the Ce0.4/W0.1ZrOx catalysts structure, redox and surface acidity were not significantly different before and after the SCR reactions. In other words, Ce0.4/W0.1ZrOx catalyst exhibited excellent thermostability and its catalytic performance remained high even after treatment at high temperature.
Considered that the flue gas usually contained a certain concentration of SO2 in practical cases, which would impose a significant impact on the deactivation of NH3-SCR catalysts. Hence, the effect of SO2 on NOx conversion over Ce0.4/W0.1ZrOx catalyst as a function of time was carried out at 300 °C, and the result was shown in Fig. 1(c). As 100 ppm SO2 was introduced in the feeding gas, NOx conversion efficiency of Ce0.4/W0.1ZrOx catalyst began to decrease slowly, and reduced to 94% within the first 1 h. After stopping SO2 injection, NOx conversion kept still stable at ∼94%. The result indicated that Ce0.4/W0.1ZrOx catalyst had an excellent tolerance to SO2 at 300 °C, and the slight deactivation due to SO2 poisoning was not irreversible.
3.2 Structural and textural characteristics
 | ||
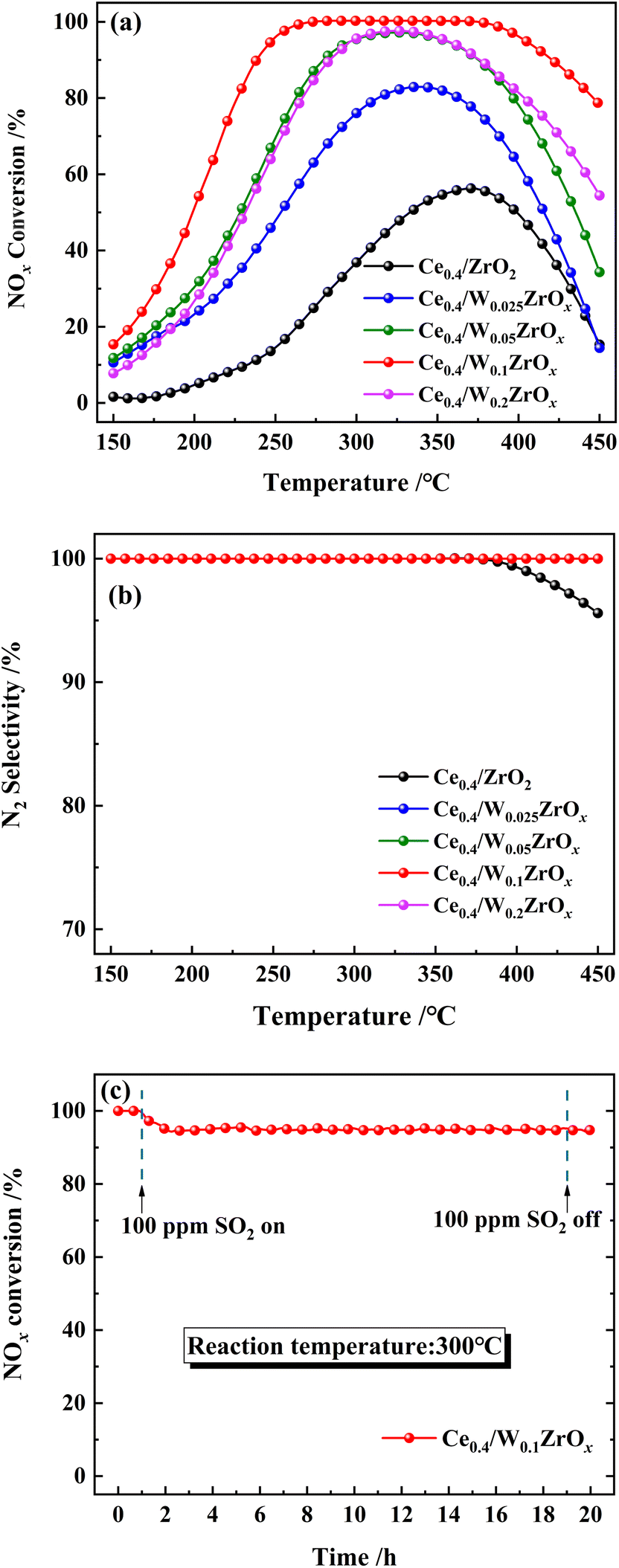
| Fig. 2 N2 adsorption–desorption isotherms of prepared catalysts. | ||
| Catalysts | SBET (m2 g−1) | Pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| Ce0.4/ZrO2 | 46.1 | 11.2 | 0.14 |
| Ce0.4/W0.025ZrOx | 42.9 | 9.1 | 0.11 |
| Ce0.4/W0.05ZrOx | 51.7 | 8.1 | 0.10 |
| Ce0.4/W0.1ZrOx | 57.9 | 6.5 | 0.09 |
| Ce0.4/W0.2ZrOx | 29.4 | 9.7 | 0.07 |
 | ||
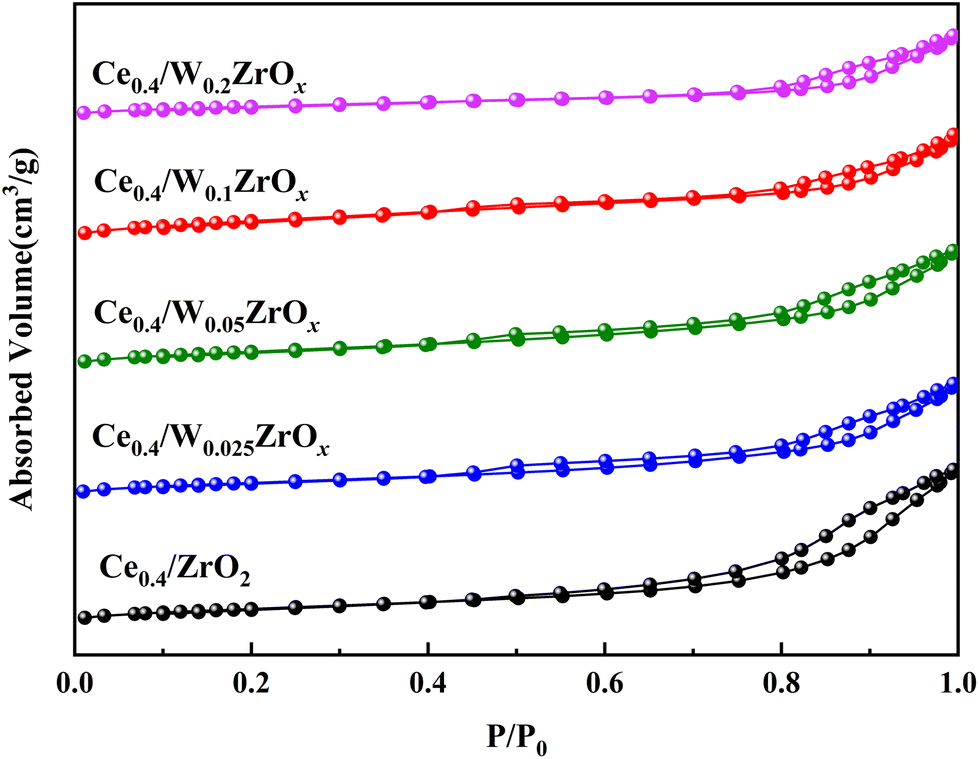
| Fig. 3 (a) XRD patterns and (b) their enlarged views of ZrO2, W0.1ZrOx, Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts. | ||
| Samples | 2θ/° | Cell parameter/Å | Cell volume/Å3 | Crystallite size/Å | |
|---|---|---|---|---|---|
| a = b | c | ||||
| ZrO2 | 50.24 | 3.600 | 5.168 | 67.01 | 79 |
| W0.1ZrOx | 50.43 | 3.601 | 5.150 | 66.79 | 34 |
| Ce0.4/ZrO2 | 50.11 | 3.599 | 5.157 | 66.84 | 85 |
| Ce0.4/W0.1ZrOx | 50.34 | 3.602 | 5.146 | 66.79 | 45 |
After impregnation of CeO2, the crystal structures of ZrO2 and W0.1ZrOx in Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts were the same as to their single supports, as shown in Fig. 3(a). Some diffraction peaks located at 28.6, 33.1, 47.4 and 56.4° could be identified over Ce0.4/ZrO2 catalyst, which was attributed to (111), (200), (220) and (311) planes of fluorite structure CeO2 (PDF-ICDD 34-0394). As to Ce0.4/W0.1ZrOx catalyst, the characteristic peaks corresponding to the crystalline phases of CeO2 could also be detected, but the peak intensities became much weaker compared to those of Ce0.4/ZrO2 catalyst. This phenomenon indicated that the existence of W species could suppress the formation of fluorite structure CeO2, leading to a decrease in the crystallite size. It was beneficial to obtain a highly-dispersed state of ceria oxide active species over W0.1ZrOx support, thus enhancing NH3-SCR activity. Besides, the difference in BET surface area between Ce0.4/ZrO2 and W-containing samples could be interpreted by the crystal phase. From the XRD result, it could be seen that the introduction of W could inhibit the ZrO2 phase transformation from a tetragonal phase to a denser monoclinic phase.29,30 Moreover, an appropriate amount of W led to high dispersion of active species on catalyst surface. It was conducive to the increase of surface area for W-containing samples.
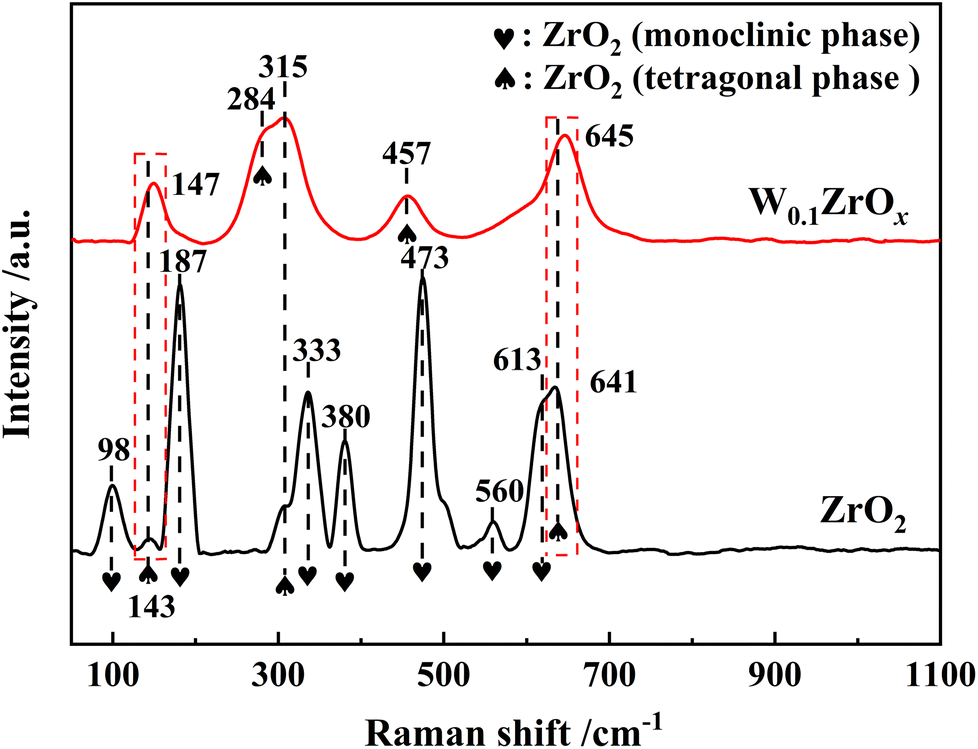
Raman characterization results were presented in Fig. 4. For pristine ZrO2 sample, the band at 98, 187, 333, 380, 473, 560 and 613 cm−1 were assigned to the Raman-active modes for monoclinic phase of ZrO2, and other bands at 143, 315 and 641 cm−1 were assigned to the tetragonal ZrO2.13 As to W0.1ZrOx sample, the peaks at 147, 284, 315, 457 and 645 cm−1 were typically characteristic peaks of tetragonal ZrO2, and no Raman bands corresponding to the characteristic peaks of monoclinic phase were detected.15 Note that, two Raman bands corresponded to tetragonal phase over W0.1ZrOx sample had been shifted to 147 and 645 cm−1 respectively, which might be attributed to a strong interaction between W and Zr in the form of W–Zr-Ox solid super acid. This result was well in accordance with the above XRD results.
 | ||
| Fig. 4 Raman results of ZrO2 and W0.1ZrOx samples. | ||
 | ||
| Fig. 5 SEM images of ZrO2 (a), W0.1ZrOx (b), Ce0.4/ZrO2 (c) and Ce0.4/W0.1ZrOx (d). | ||
 | ||
| Fig. 6 XPS spectra of (a) Ce 3d, (b) O 1s, (c) Zr 3d and (d) W 4f of prepared catalysts. | ||
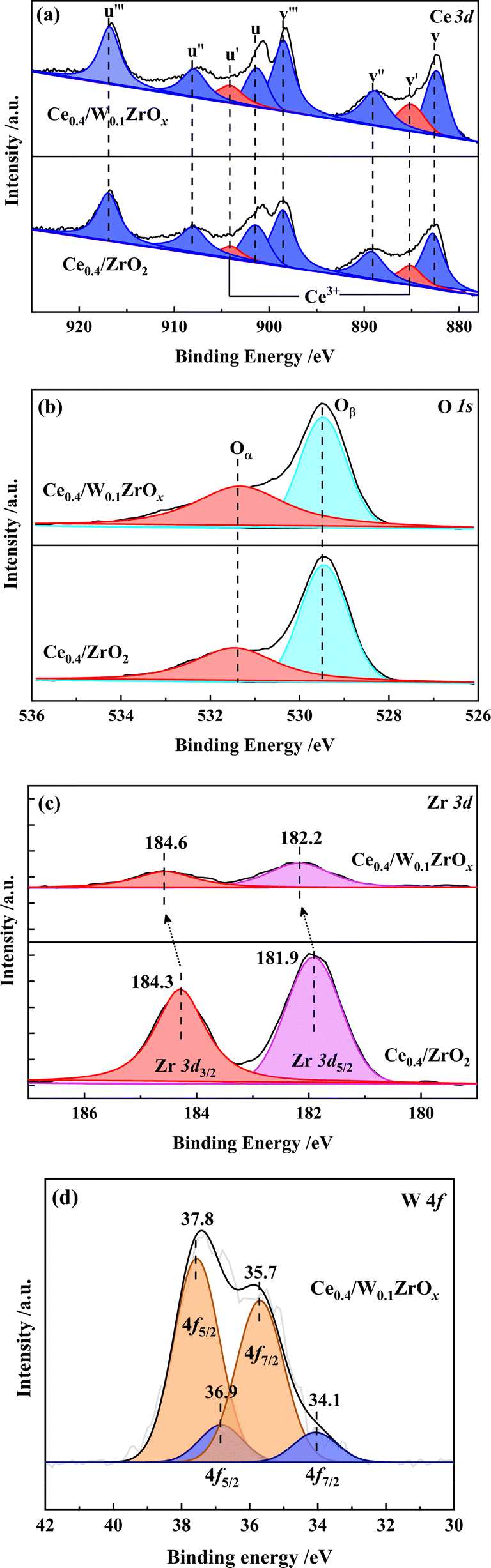
As shown in Fig. 6(a), the XPS spectra of Ce were fitted into 8 sub-peaks, in which two sub-bands marked in red represent 3d104f1 state of Ce3+, and the other ones marked in blue correspond to 3d104f0 state of Ce4+.32,33 The Ce3+/(Ce3+ + Ce4+) ratios were calculated as the integral areas of the corresponding curves, and the results were listed in Table 3. Compared to Ce0.4/ZrO2 catalyst, the ratio of Ce3+/(Ce3+ + Ce4+) at the surface of Ce0.4/W0.1ZrOx catalyst increased from 12.6% to 16.5%. The increase of Ce3+ content might be due to the interaction between cerium and the neighboring W atoms.16 Since the existence of Ce3+ species could induce a charge imbalance and unsaturated chemical bonds on the catalyst surface, it was conducive to improve redox properties and surface active oxygen content.32,33 As there were abundant Ce3+ species at the surface of Ce0.4/W0.1ZrOx catalyst, it was reasonable to obtain an enhancement effect on NO oxidation into NO2, thus facilitating the fast SCR reaction in denitrification process.
| Samples | Surface atom concentration (%) | The relative molar ratio (%) | ||||
|---|---|---|---|---|---|---|
| Ce | Zr | O | W | Ce3+/(Ce3+ + Ce4+) | Oα/(Oα + Oβ) | |
| Ce0.4/ZrO2 | 17.9 | 6.8 | 75.3 | — | 12.6 | 41.7 |
| Ce0.4/W0.1ZrOx | 21.6 | 1.6 | 75.9 | 0.9 | 16.5 | 51.6 |
The O 1s XPS information of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts was presented in Fig. 6(b). Two kinds of surface oxygen species were identified by performing a peak-fitting deconvolution. The peaks at a lower binding energy of 529.0–531.0 eV were assigned to surface lattice oxygen (donated as Oβ), and the peaks at a higher binding energy of 531.0–533.0 eV were attributed to the surface chemisorbed oxygen (donated as Oα).7 Previous studies pointed out that, surface chemisorbed oxygen (Oα) was highly active in NO oxidation and NH3 activation process due to its higher mobility than lattice oxygen (Oβ).34 The Oα/(Oα + Oβ) ratios of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts were calculated by the area integral of Oα and Oβ curves. As shown in Table 3, the Oα/(Oα + Oβ) ratio in Ce0.4/ZrO2 catalyst (12.6%) was much lower than that of Ce0.4/W0.1ZrOx catalyst (16.5%). It was possible that the addition of W species resulted in the formation of low-valence state metal cations, thus producing a great deal of oxygen vacancies, charge unbalance and unsaturated chemical bonds on the surface of Ce0.4/W0.1ZrOx catalyst.17,18 This was also in favor of boosting NO oxidation to NO2, promoting SCR reactions proceeding through a ‘fast SCR’ route.
Fig. 6(c) presented the Zr 3d XPS spectra of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts. There were two peaks at binding energy of 184.0–185.0 eV (Zr 3d3/2) and 181.5–182.5 eV (Zr 3d5/2), which corresponded to Zr4+ species.35 Apparently, the peak intensities of Zr4+ for Ce0.4/ZrO2 catalyst were much higher than those for Ce0.4/W0.1ZrOx catalyst. Moreover, the peaks of Zr4+ for Ce0.4/ZrO2 catalyst had been shifted to higher binding energy values. It was possibly due to the introduction of W, which resulted in W–Zr–Ox solid super acid at catalyst surface, arising a change in the electron density and lattice spacing of ZrO2. The results were in accordance with the XRD and Raman results.
Fig. 6(d) presented the XPS spectra of W 4f over Ce0.4/W0.1ZrOx catalyst. Spectrum deconvoluted into two doublets showed two chemical states of W on the surface of Ce0.4/W0.1ZrOx catalyst. The spectral peaks at 35.7 and 37.8 eV corresponded to W 4f7/2 and W 4f5/2 respectively, which were attributed to W6+ state. Doublet with relatively low intensity (peaks at 34.1 and 36.9 eV) corresponded to W5+ state.36
3.3 Redox properties
H2-TPR experiments were performed to evaluate the redox properties of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts, and the results were shown in Fig. 7 and Table 4. Two distinctive peaks at 503 and 802 °C could be observed in H2-TPR profiles of Ce0.4/ZrO2 catalyst, corresponding to the reduction of surface Ce4+ species to Ce3+ and bulk Ce4+ to Ce3+.37,38 For Ce0.4/W0.1ZrOx catalyst, there were three broad reduction peaks around 410, 520 and 792 °C, in which the first peak was assigned to the reduction of the surface Ce4+ species to Ce3+, the second one assigned to the reduction of W6+ to W5+, and the third peak assigned to the reduction of bulk Ce4+ to Ce3+.17,18 Compared with Ce0.4/ZrO2 catalyst, the peak corresponding to the reduction of surface Ce4+ to Ce3+ over Ce0.4/W0.1ZrOx catalyst had been shifted to a lower temperature (410 °C). It indicated that the surface Ce4+ species became more reducible after doping W species. Previous study reported that when host oxide (such as CeO2) was reducible, the dopant might donate extra electrons to the host cations.39 In view of this, it was very possible that W as dopant would donate electrons to adjacent Ce4+ species, resulting in a strong interaction between W and Ce, thus improving the redox properties of Ce0.4/W0.1ZrOx catalyst. Furthermore, H2 consumption amount over Ce0.4/W0.1ZrOx catalyst (2.34 mmol g−1) was much higher than that of Ce0.4/ZrO2 catalyst (1.48 mmol g−1). In other words, addition of W species in Ce0.4/ZrO2 catalyst support could greatly enhance the redox properties, which was an important factor for promoting SCR catalytic activity of Ce0.4/W0.1ZrOx catalyst at low-temperature.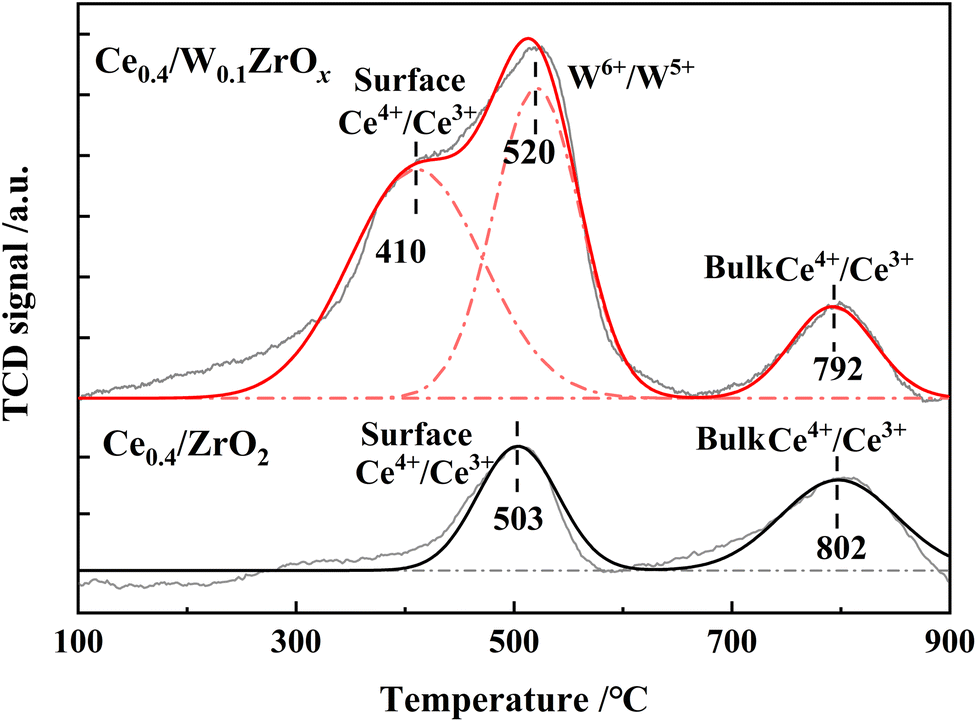 | ||
| Fig. 7 H2-TPR profiles of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts in the range of 100–900 °C. | ||
| Catalysts | Reduction peak temperature (°C) | H2 consumption (mmol g−1) | ||
|---|---|---|---|---|
| Peak 1 | Peak 2 | Peak 3 | ||
| Ce0.4/ZrO2 | 503 | — | 802 | 1.48 |
| Ce0.4/W0.1ZrOx | 410 | 520 | 792 | 2.34 |
3.4 Surface acidity
Surface acidity of NH3-SCR catalysts was one more critical factor in denitrification reaction. NH3-TPD experiment was performed to probe the number of acid sites in Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts, and the results were presented in Fig. 8. The quantitative analysis results of total surface acidities were listed in Table 5. It could be seen from Fig. 8 that NH3-TPD profiles of both Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts exhibited three desorption peaks, which were labeled as α, β and γ, respectively. The peak α was attributed to weak acid sites, the peak β was assigned to medium acid sites, and the peak γ was ascribed to strong acid sites.21,40 As shown in Fig. 8, there were only slight differences in the peak positions of weak and medium acid sites between these two catalysts. However, the peak position of strong acid sites over Ce0.4/W0.1ZrOx catalyst shifted to a much lower temperature compared to Ce0.4/ZrO2 catalyst. It could be ascribed to the formation of W–Zr–Ox solid super acid at the surface of Ce0.4/W0.1ZrOx catalyst.21 As shown in Table 5, the total acid amounts at the surface of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts were 1.42 and 1.94 mmol g−1, respectively. The results demonstrated that, Ce0.4/W0.1ZrOx catalyst possessed a superior total acidity over Ce0.4/ZrO2 catalyst. The doping W in ZrO2 support effectively improved the surface acidity of Ce0.4/ZrO2 catalyst, which was beneficial to adsorb more NH3 species, thus enhancing SCR performance.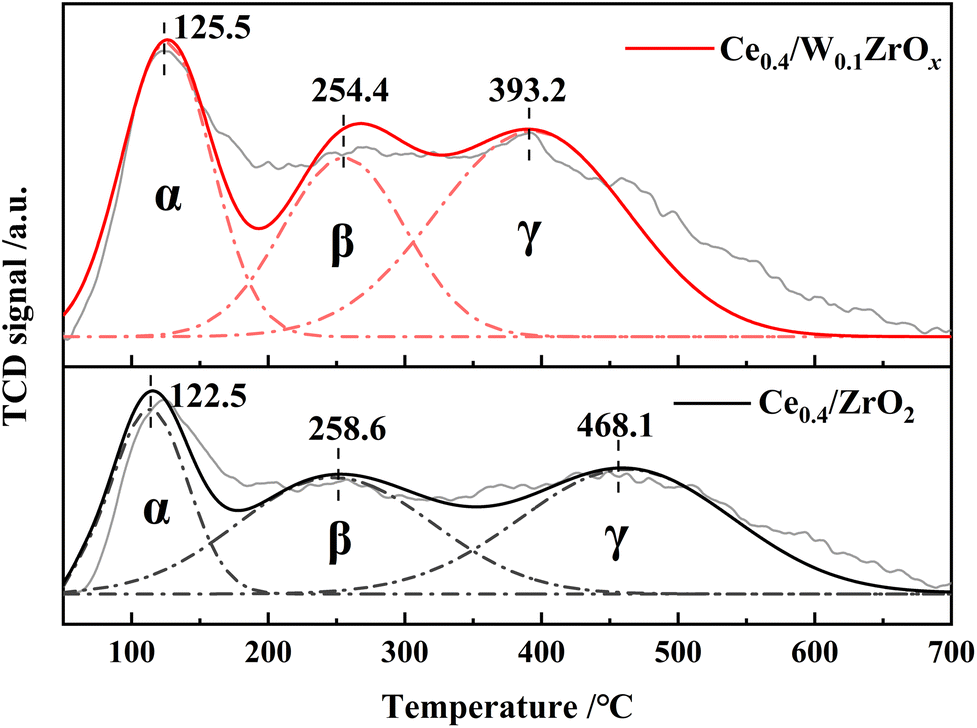 | ||
| Fig. 8 NH3-TPD curves of Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts in the range of 50–700 °C. | ||
| Catalysts | Peak center temperature (°C) | Total acidity (mmol g−1) | ||
|---|---|---|---|---|
| Peak α | Peak β | Peak γ | ||
| Ce0.4/ZrO2 | 122 | 258 | 468 | 1.42 |
| Ce0.4/W0.1ZrOx | 125 | 254 | 393 | 1.94 |
3.5 In situ DRIFTS
Fig. 9 showed the in situ DRIFTS spectra of NH3 adsorption over Ce0.4/ZrO2 and Ce0.4/W0.1ZrOx catalysts at different temperatures (100–350 °C). For Ce0.4/ZrO2 catalyst, after NH3 adsorption, several bands were detected in the range of 1000–1800 cm−1. The bands peaked at 1542 cm−1 and 1152 cm−1 were assigned to asymmetric and symmetric N–H bending vibrations of N–H bonds in coordinated NH3 linked to Lewis acid sites.41,42 The band peaked at 1358 cm−1 could be ascribed to the amide (–NH2) species.43 Obviously, there was no obvious band corresponding to Brønsted acid sites in the in situ DRIFS spectra of NH3 adsorption over Ce0.4/ZrO2 catalyst.
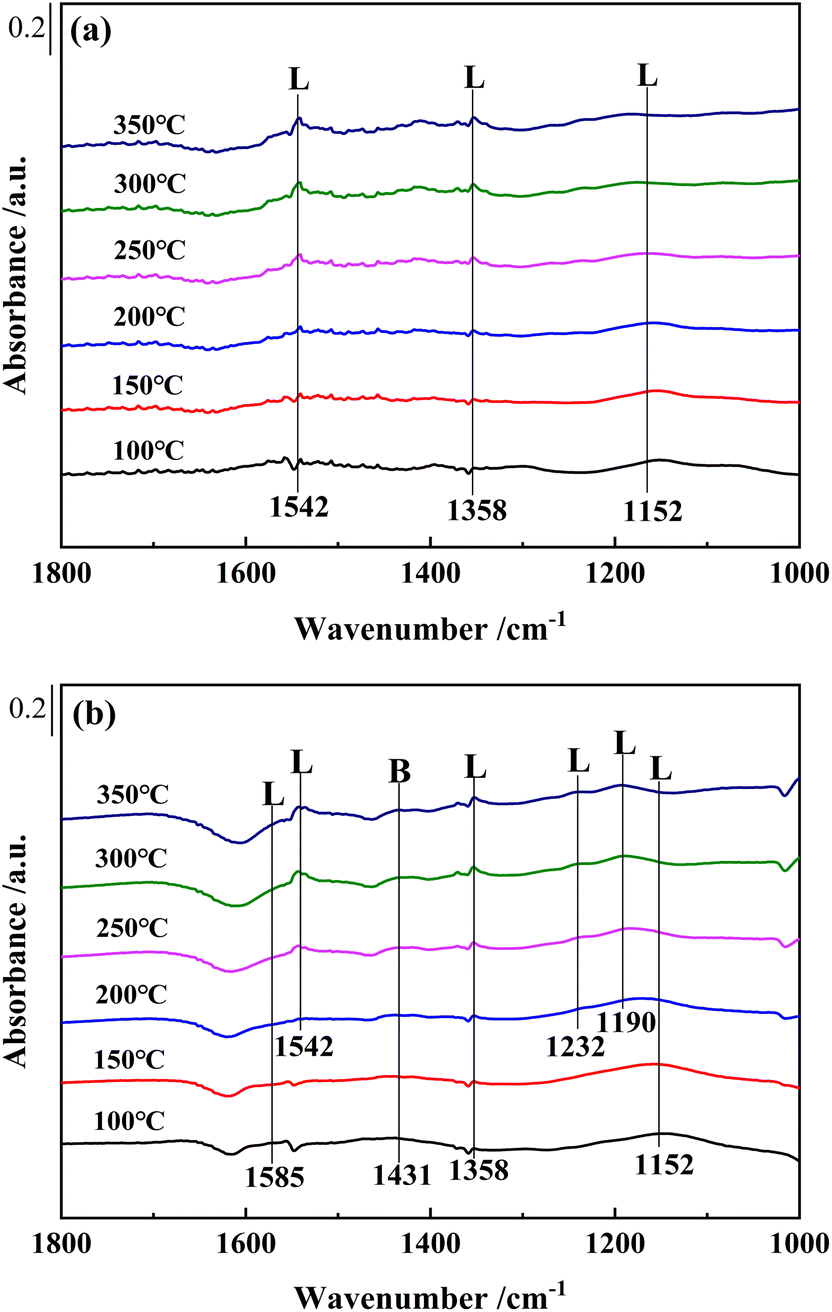 | ||
| Fig. 9 In situ DRIFTS spectra of (a) Ce0.4/ZrO2 and (b) Ce0.4/W0.1ZrOx catalysts at different temperatures. Condition: [NH3] = 500 ppm and N2 as balance gas. | ||
For Ce0.4/W0.1ZrOx catalyst, the in situ DRIFTS spectra of NH3 adsorption over Ce0.4/W0.1ZrOx catalyst were quite different from those for Ce0.4/ZrO2 catalyst. The NH3 species adsorbed on Ce0.4/W0.1ZrOx catalyst surface (1152 and 1542 cm−1) were attributed to coordinated NH3 on Lewis acid sites. But several new bands could also be detected: the bands peaked at 1585 cm−1 and 1190, 1232 cm−1 were assigned to asymmetric and symmetric bending vibrations of N–H bonds in coordinated NH3 linked to Lewis acid sites, and the band peaked at 1431 cm−1 was attribute to NH4+ species on Brønsted acid sites.24,42,44 Compared to Ce0.4/ZrO2 catalyst, much more NH3 could be adsorbed on the surface of Ce0.4/W0.1ZrOx catalyst, which was in accordance with NH3-TPD results. This result suggested that the introduction of W species tremendously increased the amount of both Brønsted acid sites and Lewis acid sites on catalyst surface, thus significantly improving the adsorption of NH3 species, which played a key role in NH3-SCR process.45
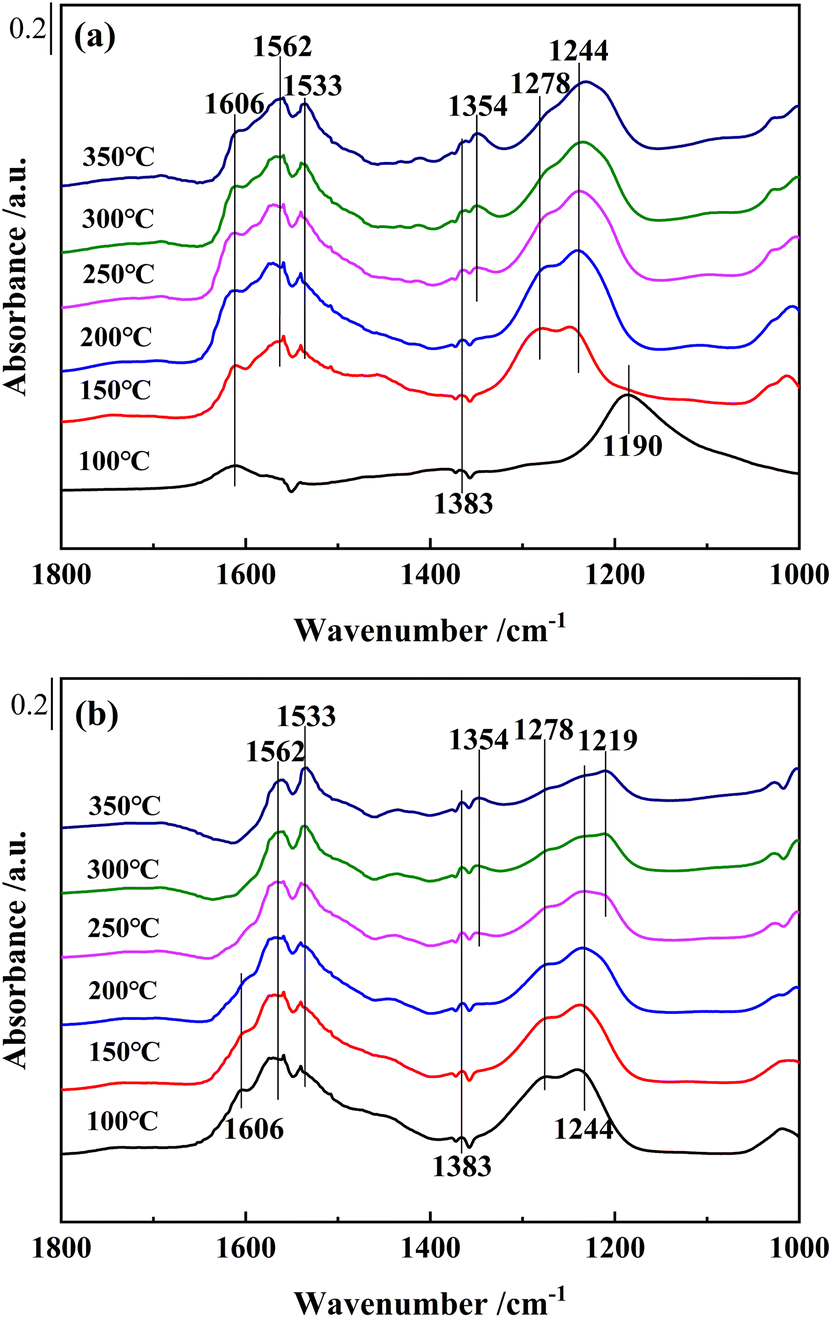 | ||
| Fig. 10 In situ DRIFTS spectra of (a) Ce0.4/ZrO2 and (b) Ce0.4/W0.1ZrOx catalysts at different temperatures. Condition: [NO] = 500 ppm, [O2] = 5 vol% and N2 as balance gas. | ||
As shown in Fig. 10(b), for Ce0.4/W0.1ZrOx catalyst, several bands, bidentate nitrates (1244, 1533 and 1562 cm−1), monodentate nitrate (1278 cm−1) and bridged nitrate (1219 cm−1), could also be detected after NO + O2 adsorption, which could be assigned to adsorbed NOx species.48,49 The bands peaked at 1354, 1383 cm−1 and 1606 cm−1 were attributed to M-NO2 nitro compounds and gaseous NO2 molecules. Compared to in situ DRIFTS spectra of NO + O2 co-adsorption over Ce0.4/ZrO2 catalysts, it was worth noting that the band intensity of adsorbed NOx species on the surface of Ce0.4/W0.1ZrOx catalyst was significantly weaker. Moreover, with the increase of reaction temperature, the band intensities of adsorbed NOx species on the surface of Ce0.4/W0.1ZrOx catalyst became weaker gradually. The above results indicated that the introduction of W species not only resulted in more Brønsted acid sites and Lewis acid sites formed on the surface of Ce0.4/W0.1ZrOx catalyst, but also reduced the thermal stability of the inactive nitrate species, leaving more active sites available for the adsorption of NH3 species. It was conducive to improving SCR performance.
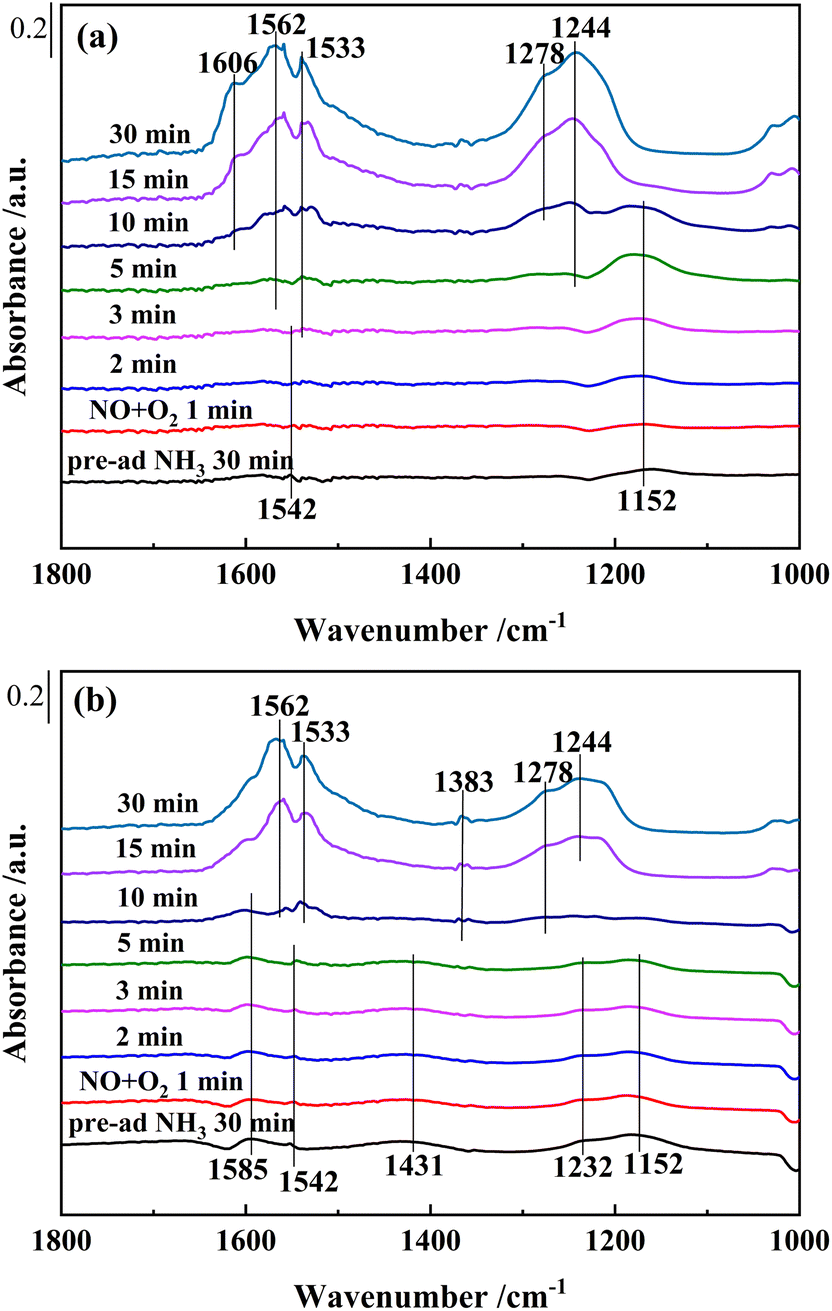 | ||
| Fig. 11 In situ DRIFTS of reactions between pre-adsorbed NH3 species and NO + O2 over (a) Ce0.4/ZrO2 and (b) Ce0.4/W0.1ZrOx catalysts at 250 °C. | ||
As shown in Fig. 11(b), after saturated adsorption of NH3 for 30 min, several bands appeared in the spectra over Ce0.4/W0.1ZrOx catalyst. The bands peaked at 1152, 1232, 1542 and 1585 cm−1 on Lewis acid sites were attributed to NH3 species, while the band peaked at 1431 cm−1 on Brønsted acid sites was assigned to NH4+.44 After introduction of NO + O2, all bands belonging to NH3 species on Lewis acid sites and Brønsted acid sites decreased obviously in intensity. It could be seen that these NH3 species had been completely substituted by nitrate species after 10 min. This result indicated that both coordinated NH3 and NH4+ species on Ce0.4/W0.1ZrOx catalyst surface could act as reducing agents to reduce NOx. Furthermore, the coordinated NH3 species over Ce0.4/W0.1ZrOx catalyst played a dominant role in SCR reactions, and the NH4+ species was also involved in SCR reactions. As the doping of W to Ce0.4/ZrO2 catalyst resulted in more coordinated NH3 and ionic NH4+, both of them led to the improvement of NH3-SCR performance.21,24
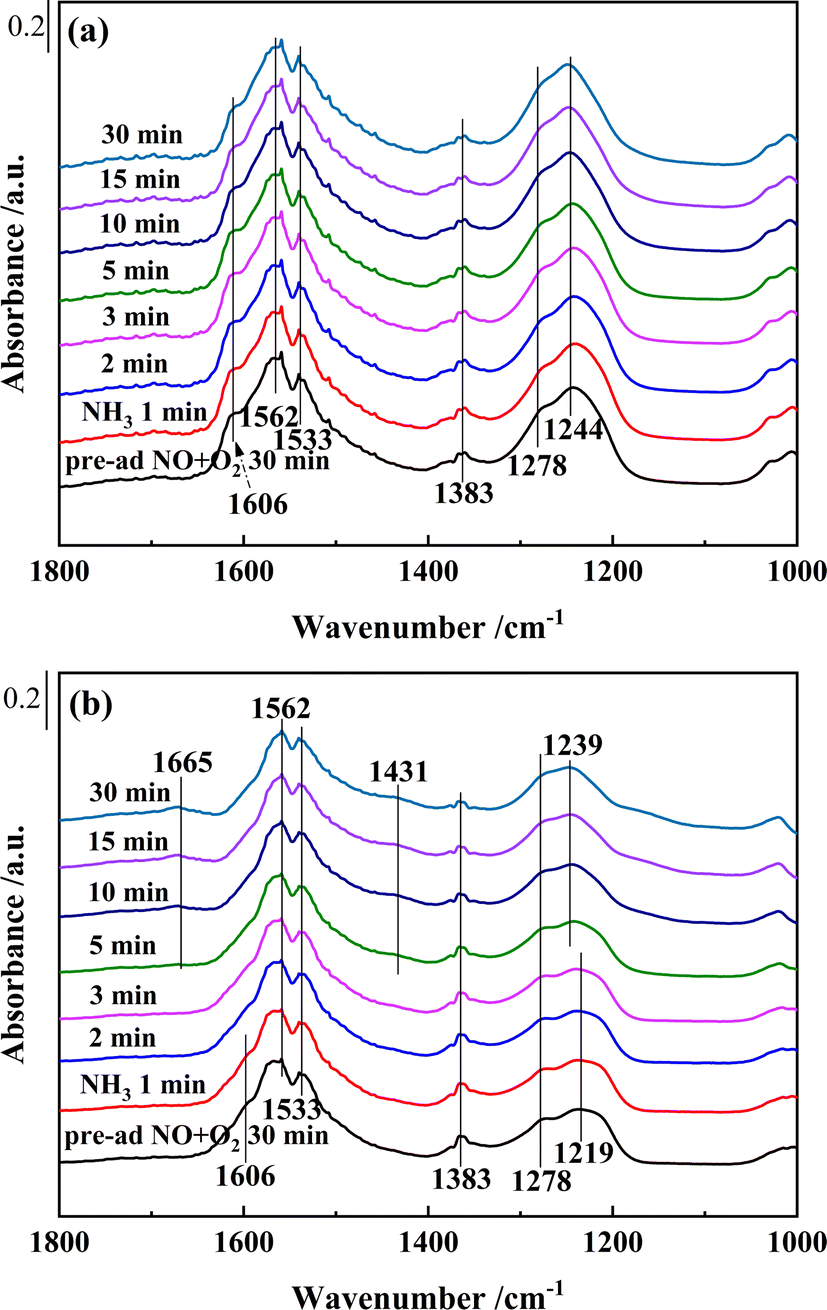 | ||
| Fig. 12 In situ DRIFTS of reactions between pre-adsorbed NO + O2 species and NH3 over (a) Ce0.4/ZrO2 and (b) Ce0.4/W0.1ZrOx catalysts at 250 °C. | ||
As shown in Fig. 12(a), after saturated pre-adsorption of NO + O2 on the surface of Ce0.4/ZrO2 catalyst, several bands, monodentate nitrate (1278 cm−1), bidentate nitrates (1244, 1533 and 1562 cm−1), M–NO2 nitro compounds (1383 cm−1) and gaseous NO2 molecules (1606 cm−1), could be detected. After switching to NH3, the bands corresponding to NOx species hardly changed in 30 min, this result showed that the pre-adsorbed NOx species on the surface of Ce0.4/ZrO2 catalyst hardly reacted with NH3. In the meanwhile, with the injection of NH3, no characteristic bands of NH3 species appeared, which might be due to the occupation of active sites by stable nitrate/nitrite species. Thus, the poor NH3-SCR activity of Ce0.4/ZrO2 catalyst might be related to the suppressive effect of nitrate/nitrite species on active sites.
Fig. 12(b) showed that, after pre-adsorbed with NO + O2 on Ce0.4/W0.1ZrOx catalyst surface, bridged nitrate (1219 cm−1), monodentate nitrate (1278 cm−1) and bidentate nitrates (1533 and 1562 cm−1) were formed. Meanwhile, the bands corresponding to M–NO2 (1383 cm−1) and gaseous NO2 molecules (1606 cm−1) also appeared. After switching to NH3, the intensities of the bands (1278, 1383, 1533 and 1562 cm−1) assigned to adsorbed NOx species decreased gradually, while other bands peaked at 1606 and 1219 cm−1 disappeared in 1 and 3 min, respectively. From then on, Ce0.4/W0.1ZrOx catalyst surface was mainly covered by adsorbed NH3 species, which were in the form of coordinated NH3 (1239 and 1665 cm−1) bonded to Lewis acid sites and NH4+ species (1431 cm−1) bonded to Brønsted acid sites. This result showed that the adsorption of nitrate species on Ce0.4/W0.1ZrOx catalyst surface could react with NH3. Though the addition of W might inhibit the adsorption of nitrate species on Ce0.4/W0.1ZrOx catalyst surface (see Fig. 10), the reactions between adsorbed nitrate species and NH3 could still play an important role in NH3-SCR of NOx.
As to Ce0.4/W0.1ZrOx catalyst, both Eley-Rideal (E–R) and Langmuir–Hinshelwood (L–H) mechanisms had been followed during NH3-SCR reactions at 250 °C. Moreover, E–R rather than L–H mechanism was the dominant reaction pathway. The coordinated NH3 species were considered the most important intermediates in E–R mechanism. Abundant Lewis acid sites had been formed on the surface of Ce0.4/W0.1ZrOx catalyst due to the introduction of W, which promoted the generation of coordinated NH3 species. Different from Ce0.4/ZrO2 catalyst, ionic NH4+ species on Brønsted acid sites had been formed on the surface of Ce0.4/W0.1ZrOx catalyst, which could further react with NOx, thus providing a supplementary pathway for N2 formation. As to L–H mechanism, the addition of W species favored the activation of adsorbed NOx species, especially bridged nitrates and adsorbed NO2, promoting the reactions between adsorbed NOx species and NH3.
4. Conclusion
In this work, WmZrOx-supported Ce-based catalysts have been prepared, and the effects of W doping in ZrO2 on NH3-SCR performance over Ce0.4/WmZrOx catalysts have been investigated systematically. It was found that various W/Zr molar ratios imposed a distinctive impact on the SCR activity of the prepared Ce0.4/WmZrOx catalysts. Compared to Ce0.4/ZrO2 catalyst, the addition of W in ZrO2 promoted the catalytic performance in a broad temperature range. Especially, Ce0.4/W0.1ZrOx catalyst exhibited the widest active temperature window (NOx conversion rate > 80%) of 226–446 °C and nearly 100% N2 selectivity. It was attributed to the enhanced redox property, W doping would lead to an increase in Ce3+ and Oα contents on the surface of Ce0.4/W0.1ZrOx catalyst. Besides, Ce0.4/W0.1ZrOx catalyst also had good SO2 tolerance, which could maintain more than 94% of NOx conversion efficiency after being exposed to 100 ppm SO2 atmosphere for 18 h. The results showed that introduction of W in ZrO2 resulted in a larger specific surface area, and formed more Brønsted acid sites and Lewis acid sites at the surface of Ce0.4/W0.1ZrOx catalyst, which enhanced the total surface acidity. Moreover, the thermal stability of inactive nitrate species had also been reduced significantly, leaving more active sites available for the adsorption of NH3 species. It was conducive to improving SCR performance. The in situ DRIFTS results indicated that coordinated NH3 and ionic NH4+ species were active intermediates, and bridging nitrates, monodentate nitrates and bidentate nitrates were involved in SCR reactions over Ce0.4/W0.1ZrOx catalyst at 250 °C. Therefore, SCR reactions occurred over Ce0.4/W0.1ZrOx catalyst might follow both Eley–Rideal (E–R) mechanism and Langmuir–Hinshelwood (L–H) mechanism.Author contributions
Chenglong Li: conceptualization, investigation, writing–original draft, review and editing; Zhitao Han: conceptualization, validation, supervision, project administration, funding acquisition, writing, review and editing; Yuqing Hu: formal analysis, investigation, data curation; Tingjun Liu: formal analysis, investigation; Xinxiang Pan: project administration and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by National Natural Science Foundation of China (51779024, 51979045 and 52271356), Natural Science Foundation of Liaoning Province of China (2020MS130), Fundamental Research Funds for the Central Universities (3132019330), and Guangdong Province Natural Resources Project (2022-32).References
- D. Wang, B. Huang, Z. Shi, H. Long, L. Li, Z. Yang and M. Dai, RSC Adv., 2021, 11, 18458–18467 RSC.
- X. Sun, Q. Liu, X. Zhang and S. Liu, RSC Adv., 2021, 11, 22780–22788 RSC.
- S. Xiong, Y. Peng, D. Wang, N. Huang, Q. Zhang, S. Yang, J. Chen and J. Li, Chem. Eng. J., 2020, 387, 123090 CrossRef.
- P. Wang, L. Yao, Y. Pu, L. Yang, X. Jiang and W. Jiang, RSC Adv., 2019, 9, 36658–36663 RSC.
- G. Zhai, Z. Han, X. Wu, H. Du, Y. Gao, S. Yang, L. Song, J. Dong and X. Pan, J. Taiwan Inst. Chem. Eng., 2021, 125, 132–140 CrossRef CAS.
- H. Du, Z. Han, X. Wu, Q. Wang, C. Li, Y. Gao, S. Yang, L. Song, J. Dong and X. Pan, J. Environ. Chem. Eng., 2021, 9, 105653 CrossRef CAS.
- M. Shen, C. Li, J. Wang, L. Xu, W. Wang and J. Wang, RSC Adv., 2015, 5, 35155–35165 RSC.
- Q. Zhang, Z. Song, P. Ning, X. Liu, H. Li and J. Gu, Catal. Commun., 2015, 59, 170–174 CrossRef CAS.
- Z. Ma, D. Weng, X. Wu and Z. Si, J. Environ. Sci., 2012, 24, 1305–1316 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- S. Liu, P. Yao, Q. Lin, S. Xu, M. Pei, J. Wang, H. Xu and Y. Chen, Catal. Today, 2021, 382, 34–41 CrossRef CAS.
- Z. Liu, S. Zhang, J. Li and L. Ma, Appl. Catal., B, 2014, 144, 90–95 CrossRef CAS.
- S. Letichevsky, C. A. Tellez, R. Avillez, M. Silva, M. A. Fraga and L. G. Appel, Appl. Catal., B, 2005, 58, 203–210 CrossRef CAS.
- S. Gao, X. Chen, H. Wang, J. Mo, Z. Wu, Y. Liu and X. Weng, J. Colloid Interface Sci., 2013, 394, 515–521 CrossRef CAS PubMed.
- Z. Han, X. Li, X. Wang, Y. Gao, S. Yang, L. Song, J. Dong and X. Pan, J. Colloid Interface Sci., 2022, 608, 2718–2729 CrossRef CAS.
- S. Zhan, H. Zhang, Y. Zhang, Q. Shi, Y. Li and X. Li, Appl. Catal., B, 2017, 203, 199–209 CrossRef CAS.
- D. W. Kwon and S. C. Hong, Appl. Surf. Sci., 2015, 356, 181–190 CrossRef CAS.
- L. Chen, D. Weng, J. Wang, D. Weng and L. Cao, Chinese. J. Catal., 2018, 39, 1804–1813 CrossRef CAS.
- Z. Fang, B. Yuan, T. Lin, H. Xu, Y. Cao, Z. Shi, M. Gong and Y. Chen, Chem. Eng. Res. Des., 2015, 94, 648–659 CrossRef CAS.
- A. Väliheikki, T. Kolli, M. Huuhtanen, T. Maunula and R. L. Keiski, Top. Catal., 2015, 58, 1002–1011 CrossRef.
- M. Chen, Q. Jin, X. Tao, Y. Pan, S. Gu and Y. Shen, Catal. Today, 2020, 358, 254–262 CrossRef CAS.
- H. Zhu, A. Ramanathan, J. F. Wu and B. Subramaniam, ACS Catal., 2018, 8, 4848–4859 CrossRef CAS.
- K. J. P. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Ding, F. Liu, X. Shi, K. Liu, Z. Lian, L. Xie and H. He, ACS Appl. Mater. Interfaces, 2015, 7, 9497–9506 CrossRef CAS PubMed.
- X. Lu, C. Song, S. Jia, Z. Tong, X. Tang and Y. Teng, Chem. Eng. J., 2015, 260, 776–784 CrossRef CAS.
- B. Thirupathi and P. G. Smirniotis, J. Catal., 2012, 288, 74–83 CrossRef CAS.
- J. Duehansen, S. Boghosian, A. Kustov, P. Fristrup, G. Tsilomelekis, K. Stahl, C. Christensen and R. Fehrmann, J. Catal., 2007, 251, 459–473 CrossRef CAS.
- L. Li, W. Tan, X. Wei, Z. Fan, A. Liu, K. Guo, K. Ma, S. Yu, C. Ge, C. Tang and L. Dong, Catal. Commun., 2018, 114, 10–14 CrossRef CAS.
- P. Bautista, M. Faraldos, M. Yates and A. Bahamonde, Appl. Catal., B, 2007, 71, 254–261 CrossRef CAS.
- N. Li, A. Wang, M. Zheng, X. Wang, R. Cheng and T. Zhang, J. Catal., 2004, 225, 307–315 CrossRef CAS.
- P. Maitarad, H. Jin, D. Zhang, L. Shi and S. Namuangruk, J. Phys. Chem. C, 2014, 118, 9612–9620 CrossRef CAS.
- P. Ning, Z. Song, H. Li, Q. Zhang, X. Liu, J. Zhang, X. Tang and Z. Huang, Appl. Surf. Sci., 2015, 332, 130–137 CrossRef CAS.
- Z. Ma, X. Wu, Z. Si, D. Weng, J. Ma and T. Xu, Appl. Catal., B, 2015, 179, 380–394 CrossRef CAS.
- Y. Peng, C. Liu, X. Zhang and J. Li, Appl. Catal., B, 2013, 140–141, 276–282 CrossRef CAS.
- J. Fan, P. Ning, Z. Song, X. Liu, L. Wang, J. Wang, H. Wang, K. Long and Q. Zhang, Chem. Eng. J., 2018, 334, 855–863 CrossRef CAS.
- V. V. Ganbavle, G. L. Agawane, A. V. Moholkar, J. H. Kim and K. Y. Rajpure, J. Mater. Eng. Perform., 2014, 23, 1204–1213 CrossRef CAS.
- S. Xiong, Y. Liao, H. Dang, F. Qi and S. Yang, RSC Adv., 2015, 5, 27785–27793 RSC.
- X. Qi, L. Han, J. Deng, T. Lan, F. Wang, L. Shi and D. Zhang, Environ. Sci. Technol., 2022, 56, 5840–5848 CrossRef CAS.
- E. W. Mcfarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- L. Li, P. Li, W. Tan, K. Ma, W. Zou, C. Tang and L. Dong, Chinese. J. Catal., 2020, 41, 364–373 CrossRef CAS.
- J. Yang, S. Ren, Y. zhou, Z. Su, L. Yao, J. Cao, L. Jiang, G. Hu, M. Kong, J. Yang and Q. Liu, Chem. Eng. J., 2020, 397, 125–446 Search PubMed.
- Z. Liu, H. Su, J. Li and Y. Li, Catal. Commun., 2015, 65, 51–54 CrossRef CAS.
- G. Qi and R. T. Yang, J. Phys. Chem., 2004, 108, 15738–15747 CrossRef CAS.
- S. Xie, L. Li, L. Jin, Y. Wu, H. Liu, Q. Qin, X. Wei, J. Liu, L. Dong and B. Li, Appl. surf. Sci., 2020, 515, 146014 CrossRef CAS.
- G. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225 CrossRef CAS.
- Z. B. Wu, B. Jiang, Y. Liu and H. Wang, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS PubMed.
- Y. Peng, K. Li and J. Li, Appl. Catal., B, 2013, 140–141, 483–492 CrossRef CAS.
- F. Liu, W. Shan, Z. Lian, L. Xie, W. Yang and H. He, Environ. Sci. Technol., 2013, 3, 2699 CAS.
- X. Yao, Q. Yu, Z. Ji, Y. Lv, Y. Cao, C. Tang, F. Gao, L. Dong and Y. Chen, Appl. Catal., B, 2013, 130–131, 293–304 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CrossRef CAS.
- R. B. Jin, Y. Liu, Y. Wang, W. L. Cen, Z. B. Wu, H. Q. Wang and X. L. Weng, Appl. Catal., B, 2014, 148, 582–588 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04862k |
| This journal is © The Royal Society of Chemistry 2022 |