DOI:
10.1039/D2RA04890F
(Paper)
RSC Adv., 2022,
12, 31317-31325
Vacancy defect engineered BiVO4 with low-index surfaces for photocatalytic application: a first principles study†
Received
5th August 2022
, Accepted 18th October 2022
First published on 1st November 2022
Abstract
BiVO4 has been widely investigated as a photocatalyst material for water splitting due to its outstanding photocatalytic properties. In order to further improve its photocatalytic efficiency, it is necessary to conduct an in-depth study of improvement strategies, such as defect engineering. By focusing on the (001) and (011) surfaces, we carried out a systematic theoretical research on pristine and defective systems, including Bi, V and O vacancies. Based on density functional theory (DFT), the electronic properties, band alignments and Gibbs free energy of pristine and defective BiVO4 have been analyzed. The electronic structures of the (001) and (011) surfaces show different band gaps, and O vacancies make the BiVO4 become an n-type semiconductor, while Bi and V vacancies tend to form a p-type semiconductor. Moreover, the band edge positions indicate that holes are indeed easily accumulated on the (011) surface while electrons tend to accumulate on (001). However, the (011) surface with Bi and V vacancies does not have enough oxidation potential to oxidize water. The reaction free energy shows that O and Bi vacancies could lower the overpotential to some extent.
1. Introduction
Photocatalytic water splitting into H2 or O2 has received increasing attention because of its enormous potential to provide a green and renewable way to handle the energy crisis and solve environmental problems.1–3 Since the discovery of the first photocatalyst a series of photocatalysts, such as Fe2O3, TiO2, WO3, SrTiO3 and BiVO4, have been investigated for water splitting.4–8 Among them, BiVO4 has been identified as one of the most promising photocatalysts for water splitting due to its suitable band gap and nontoxic, low-cost and stable nature.9–12 The valence band maximum (VBM) of individual BiVO4 is well below the redox potential of water, causing an outstanding O2 production ability, while its conduction band minimum (CBM) is less positive but very close to the H2 evolution potential.13 Moreover, BiVO4 exhibits a large electron–hole separation yield because of the local polarization caused by the distortion of VO4 tetrahedra.14 However, the rapid recombination of carriers in this photocatalyst results in a main bottleneck for photocatalytic efficiency, which limits the further application of BiVO4 in this field.15
In order to further improve the photocatalytic efficiency of BiVO4, a series of methods have been researched, including morphology control, elemental doping, composite structure and defect engineering.16–21 Recent research shows that defect formation can increase the charge separation efficiency and significantly improve the oxygen evolution reaction (OER) performance of BiVO4 for water splitting.22–27 The most common type of defect in BiVO4 is oxygen vacancies, which can enhance the visible light absorption and promote the charge transfer. Therefore, the photocatalytic efficiency can be improved.28–30 However, in some cases the oxygen vacancies might be counterproductive for photocatalytic performance by acting as electron–hole recombination centers.31 Furthermore, when the Bi vacancy exists, a higher charge diffusion coefficient can be obtained. And the photocurrent density of this system increases remarkably, even higher than that of previously reported O vacancy engineered BiVO4 under the same experimental conditions.32 But defects are not always beneficial to improve the photocatalytic performance. V vacancy defect can induce additional states and act as the recombination centers, which cause the decrease of carrier lifetime and photocurrent.33
The modulation mechanism of defects has been extensively researched in recent years.34,35 In BiVO4, the defects affect the electronic structure significantly and thereby the OER performance can be modulated.36 However, their exact influence on OER is not fully understood due to studies are still limited and inconclusive.37,38 Moreover, for a catalytic process, the reaction occurs on the surface of photocatalyst. Nevertheless most of researches are mainly concentrated on the electronic properties of bulk system with defects.37–40 Therefore, it is necessary to conduct in-depth study of the defects under specific surface.
It has been confirmed that the BiVO4 has the corner-cut truncated bipyramidal morphology. The (001) surface is the most stable,27,41–43 while (011) and (101) comprise the majority of surface area. And the (001), (011), (101) comprise more than 99.3% of the surface area.44 Moreover, (011) and (101) can be considered equivalent due to these two surfaces have similar surface energies and morphology.45 Therefore, in this work, we focus on the (001) and (011) surfaces. The effect of O vacancy, Bi vacancy and V vacancy engineered BiVO4 with representative surface have been investigated based on density functional theory (DFT). We focus on the electronic structure, band edge position and Gibbs free energy of pristine and defective BiVO4. The electronic structure of BiVO4 has been investigated by calculating the partial density of states (DOS). The band edge positions are the focus of analyzing the photocatalytic mechanism. The photocatalytic activities of pristine and defective BiVO4 are studied via analysis of the Gibbs free energy. Results show that Bi vacancy might be an effective mean with the lowest overpotential in our calculated systems. And the V vacancy could enhance the overpotential, thus it should be avoided in the experiment.
2. Computational details
All the structural optimization and static calculations are based on DFT, as implemented in the Vienna ab initio simulation package (VASP).46,47 In the calculation, the projector-augmented wave (PAW) method is selected and the generalized gradient approximation (GGA) functional of Perdew, Burke and Ernzerhof (PBE) is used to describe exchange and correlation potentials,48,49 here the standard PAW potentials have been chosen. The 5d106s26p3 of Bi, 3p63d44s1 of V, and 2s22p4 of O are treated as the valence electrons. The convergence criterion for energy is 10−7 eV for zero-point energy (ZPE) and 10−5 for other calculations, 0.01 eV Å−1 is selected for the convergence criterion of force. And a cut-off kinetic energy is set to 400 eV for plane wave functions. For BiVO4 unit cell and surface system, the Monkhorst–Pack k-point grids setting are 7 × 7 × 5 and 4 × 4 × 1 to the first Brillouin zones, respectively. Also U3d = 2.7 eV has been used on V atom to correct the self-interaction error.11 For the surface system, a vacuum region of 20 Å is added in order to avoid the interactions between layers. And the solvent effect has been considered for the Gibbs free energy calculations as implemented in VASPsol,50 the water was selected as solvent here.
3. Results and discussion
3.1 Bulk geometric optimization
In our calculations, the unit cell of BiVO4 has been optimized at first, and here the crystal cell and shape are allowed to be changed. Assuming c is the longest axis, the optimized lattice parameters are a = 5.17 Å, b = 5.16 Å, c = 11.76 Å, α = 89.999°, β = 90.003°, γ = 90.145°. The photocatalyst BiVO4 exits in monoclinic scheelite (ms-) and tetragonal scheelite (ts-) phases. In the process of optimization based on DFT, the ms-BiVO4 (a = 5.194 Å, b = 5.09 Å, c = 11.667 Å, α = β = 90°, γ = 90.4°) would spontaneously transform to ts-BiVO4 (a = b = 5.147 Å, c = 11.722 Å, α = β = γ = 90°) when crystal cell and shape are allowed to be changed,41,51 therefore, the BiVO4 might tend to ts-BiVO4 in our calculation, and the lattice constants slightly expand by less than 1% compared with ts-BiVO4. Due to ms-BiVO4 and ts-BiVO4 have a similar structure, both ms-BiVO4 and ts-BiVO4 can be used for calculating photocatalytic performance, which would not change the important results and conclusions.52
3.2 Surface geometric structure
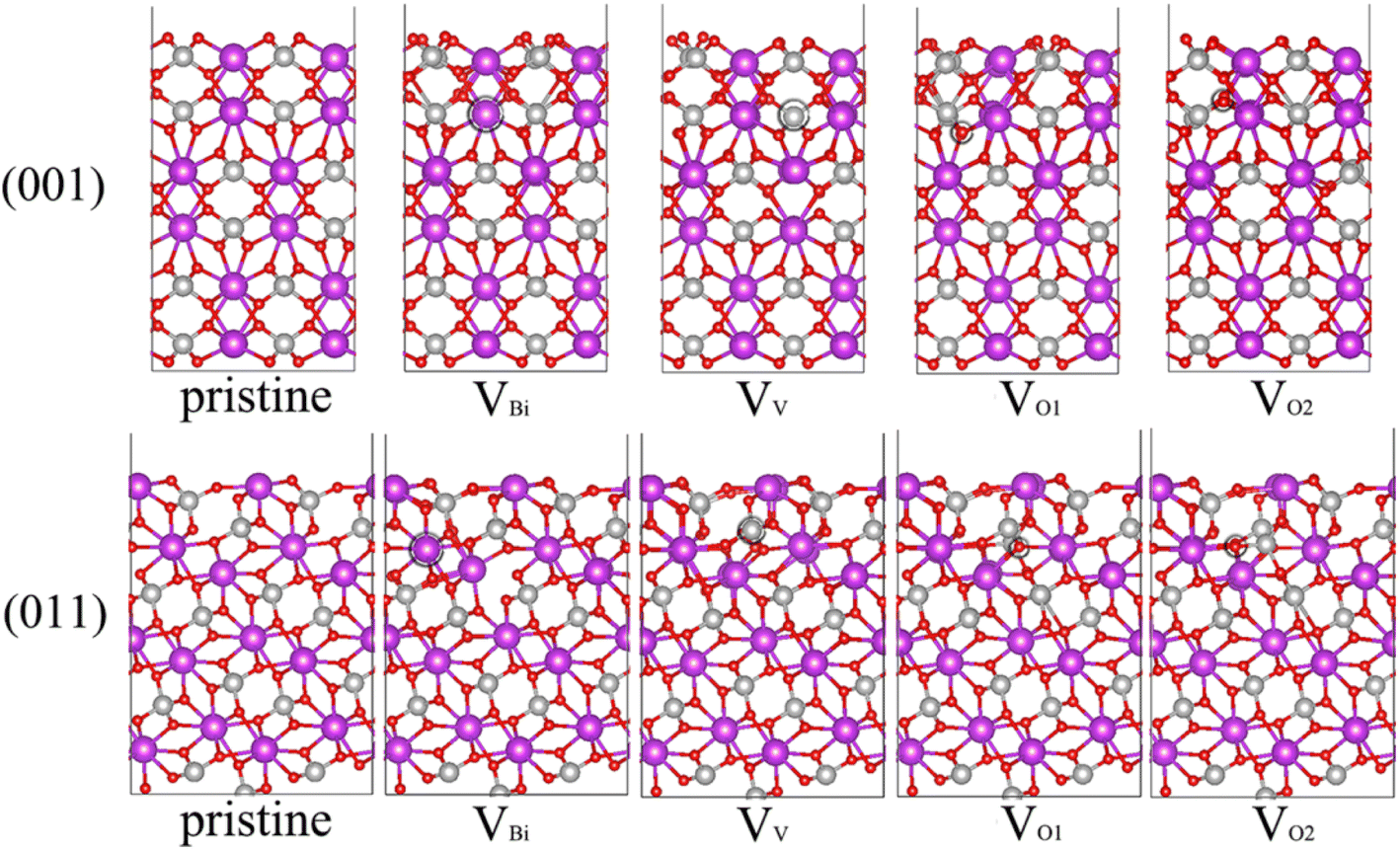
Due to the (001), (011) and (101) surfaces cover most area of BiVO4 and (011) and (101) can be considered equivalent,44,45 thus we focus on (001) and (011) surfaces. In some previous studies, the b is set as the longest axis. In this case, the corresponding surfaces should define (010), (110) and (011). Moreover, there are some studies rotating the BiVO4 90° around the longest axis c compared our bulk BiVO4, thus (011) should define (101) in these existent studies. For one BiVO4 unit cell, it contains one kind of Bi, one kind of V and two kinds of O. Therefore, four possible kinds of vacancy defects of BiVO4 are examined, namely, Bi vacancy engineered BiVO4 (VBi), V vacancy engineered BiVO4 (VV), the first kind of O vacancy engineered BiVO4 (VO1) and the second kind of O vacancy engineered BiVO4 (VO2), also the pristine BiVO4 with (001) and (011) surface are considered here. The unit cell of bulk BiVO4 has been cleaved to obtain the (001) and (011) surfaces, then the two slabs has been adjusted to make the thickness of them larger than 15 Å. And the BiVO4 (001) and (011) models are constructed by 2 × 2 and 2 × 1 corresponding slabs, respectively. The side and top views of optimized geometric structures are shown in Fig. 1 and S1,† respectively. The vacancy concentration of (001) is 4.2% for VBi and VV, 1.0% for VO1 and VO2, while that of (011) is 3.6% for VBi, 3.3%for VV, 0.9% for VO1 and VO2. After carefully checking the structures of defective BiVO4, it can be seen that the obvious distortion has been formed near the defect, which might modulate the electronic properties and affect the photocatalytic performance of BiVO4 significantly.
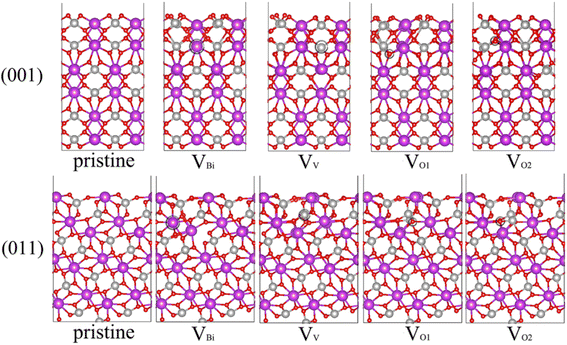 |
| | Fig. 1 Side view of optimized geometric structures of BiVO4 surface. Bi (purple), V (silver) and O (red) atoms are shown in colored spheres. The vacancy site is marked by black circle. | |
3.3 Electronic structures
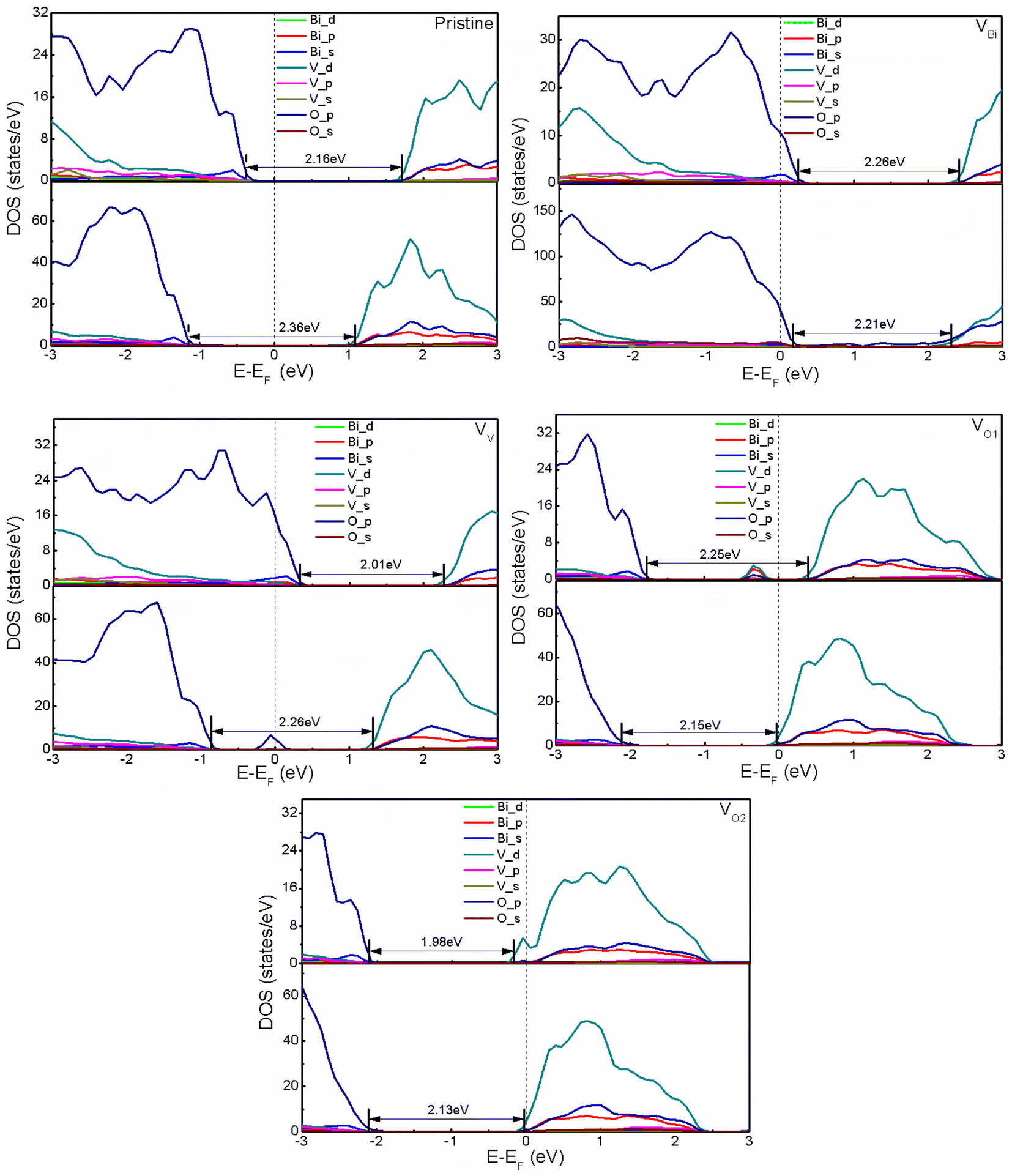
After obtaining the most stable structures, the electronic structure of defective BiVO4 (001) and (011) have been investigated by calculating the partial DOS. And the calculated results are shown in Fig. 2. Clearly, the band gaps of pristine BiVO4 (001) and (011) are 2.16 eV and 2.36 eV, respectively. This result is consistent with the previous GGA + U calculation (2.123/2.083 eV for (001) surface and 2.244/2.250 eV for (011) surface) and LDA + U calculation (2.18 eV for (001) surface and 2.38 eV for (011) surface).45,52 And the Fermi level of pristine BiVO4 (001) is close to the VBM and far from the CBM, while that of pristine BiVO4 (011) is almost in the middle of band gap. Here the VBM is mainly populated by O 2p and the CBM is composed of V 3d in both pristine BiVO4 (001) and (011) systems. For the band edges of defective BiVO4 (001) and (011), also it can be observed VBM and CBM are contributed by O 2p and V 3d, respectively. However, there are still some differences existing in different structures, especially the Fermi level and band gap. It can be seen that VBi and VV tend to make the pristine BiVO4 become a p-type semiconductor, while VO1 and VO2 tend to form a n-type semiconductor. Generically speaking, O vacancy is easy to form in the process of synthesis, thus the BiVO4 is often treated as a n-type semiconductor rather than p-type semiconductor in the experiments. This phenomenon indicates the VBi and VV could introduce holes while VO1 and VO2 would introduce electrons, respectively. Moreover, the band gap, the Fermi level and band edge could be controlled by defects, indicating the oxidation and reduction capacity probably can be modulated according to introducing defects. Notably, a peak appears near the middle of band gap for VV with (011) surface and VO1 with (001) surface. The peak of VV with (011) is very close to the Fermi level, which might be caused by defect states, and it probably acts as the recombination center, which is not good for photocatalysis to some extent.
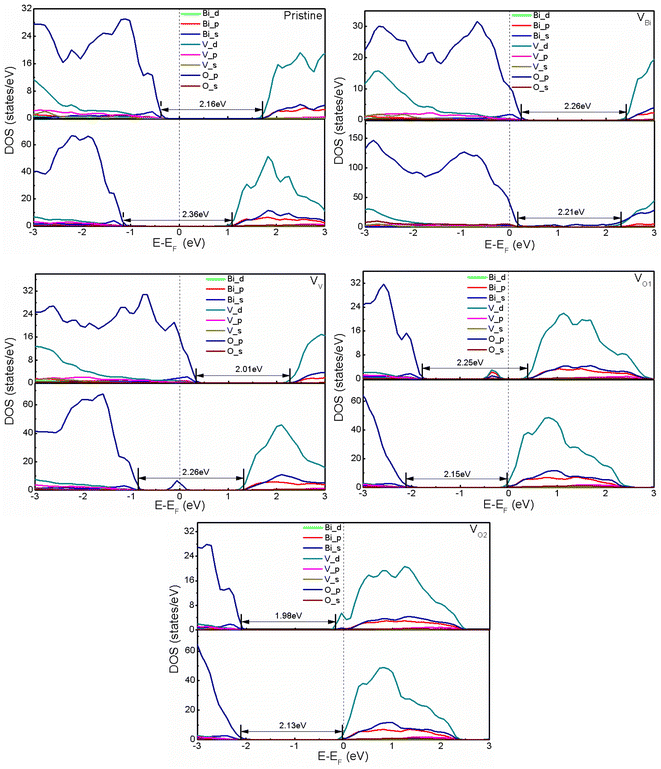 |
| | Fig. 2 The calculated DOS of pristine and defective BiVO4. The upper part and bottom part represent (001) and (011), respectively. The Fermi level is set to zero. | |
3.4 Band alignments
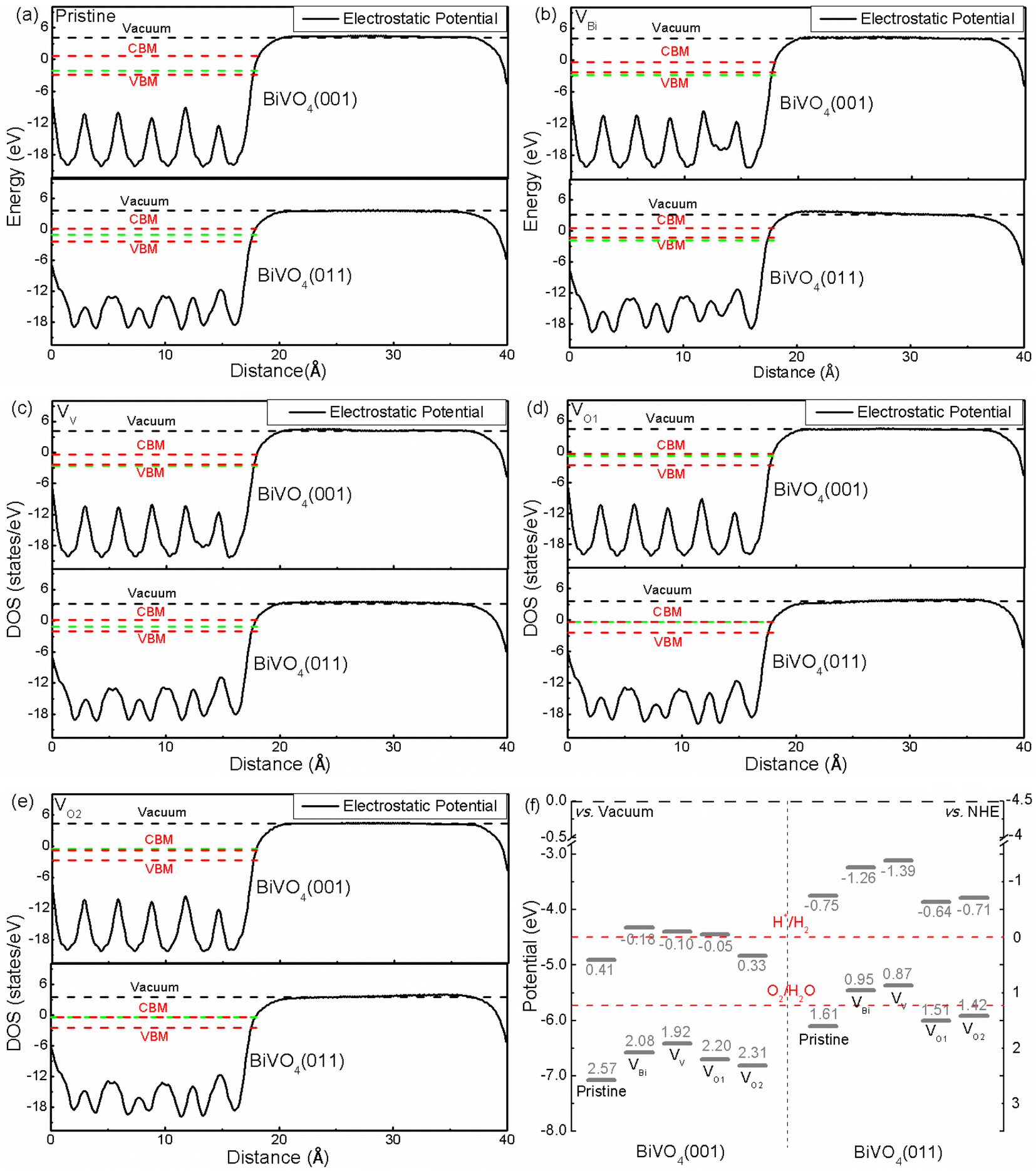
The band edge position is an important role for photocatalytic application. Hence the band edge position of BiVO4 has been analyzed based on macroscopic averaging method,53 where the electrostatic potential has been chosen as a reference in order to obtain the band edge. The relative positions between the Fermi level and CBM/VBM are obtained according to their individual supercell. The results are shown in Fig. 3(a)–(e). Clearly, the electrostatic potentials of different facets are quite different. And the difference valued between CBM/VBM and vacuum level is 4.91/7.07 eV for pristine BiVO4 (001), and 3.75/6.11 eV for pristine BiVO4 (011), respectively, meaning the CBM and VBM of pristine BiVO4 (011) is higher than that of pristine BiVO4 (001). Therefore, the photogenerated electrons on the (011) would migrate to the (001) and the photogenerated holes could transfer from (001) to (011). This is consistent with the experimental results, where the researchers obtained direct evidence that holes are indeed easily accumulated on the (011) surface and electrons tend to accumulate on (001).54 Although the average potentials of two facets are dramatically changed, similar behaviors could be found in VBi, VV, VO1 and VO2, where the CBM and VBM of (011) are still higher that of (001). And the charge separation could be promoted near the crystal boundary of these two facets. Under the light irradiation, the photogenerated electrons would migrate to BiVO4 (001) from BiVO4 (011), and the photogenerated holes would transfer from BiVO4 (001) to BiVO4 (011) at the same time. Therefore, the BiVO4 (001) and BiVO4 (011) could be the reduction site and oxidation site, respectively. This result is consistent with the previous experimental results. Moreover, it can be inferred that the VBi, VV, VO1 and VO2 cannot change this situation due to CBM and VBM of BiVO4 (011) are still higher than BiVO4 (001) after introducing these defects.
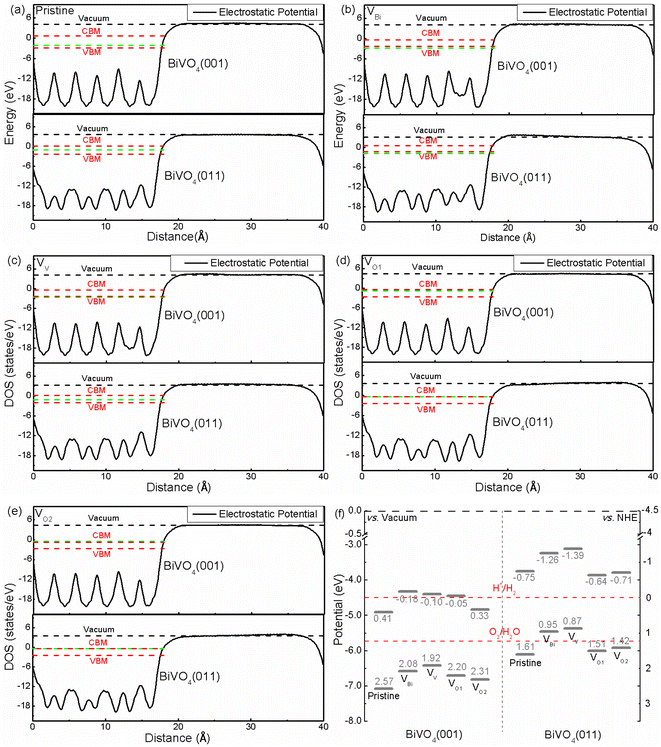 |
| | Fig. 3 (a)–(e) The relative positions of electrostatic potential of pristine and defective BiVO4, (f) band edge potentials for BiVO4 (001) and (011). The green dash line represents the Fermi level. | |
Considering the relationship between potentials of normal hydrogen electrode (NHE) and vacuum level (Evacuum = −ENHE − 4.5 eV), the band edge positions related to H+/H2 level and O2/H2O level are plotted, as shown in Fig. 3(f). We can see that the band edge positions are totally different. For (011) surface, the VBM of VBi and VV are located above the corresponding O2/H2O potential, indicating the biased voltage is necessary for VBi and VV to produce O2. As for (001) surface, the VBM of pristine BiVO4, VBi, VV, VO1 and VO2 are below the O2/H2O level, meaning these systems have ability to produce O2 without biased voltage. Totally speaking, for (011) surface, VBi and VV might not be a good way for photocatalytic application due to that the biased voltage must be added.
3.5 Overpotential
In order to further investigate the photocatalytic activity, the OER performance of pristine BiVO4, VBi, VV, VO1 and VO2 with (001) and (011) surfaces have been analyzed according to overpotential. Here the computational hydrogen electrode (CHE) model has been adopted.55 Generally speaking, there are four steps in the OER process, and each step contains one electron transfer. For the first step, the H2O could be dissociated at the Bi site under the influence of photogenerated hole, and then a proton would be released and the OH radical would be formed. The second step is the reaction that OH radical releases another a proton and forms the O with the interaction of photogenerated hole. Then, the generated O would be combined with adjacent H2O, forming the OOH radical and releasing a proton. At last, the OOH radical would further release a proton and form the O2, then the O2 leaves the surface. The optimal OER reaction path could be described as:| | |
H2O + * ⇌ OH* + H+ + e−
| (1) |
| | |
O* + H2O ⇌ OOH* + H+ + e−
| (3) |
where * stands for the reaction site of photocatalyst, and H*, OH*, O* and OOH* refer to adsorbed intermediates in the OER process. The decisive role for overpotential (η) is determined by the largest Gibbs free energy change (ΔG) among four reaction steps:| | |
η = −max[|ΔGOH*|, |ΔGO* − ΔGOH*|, |ΔGOOH* − ΔGO*|, |4.92 − ΔGOOH*|]/e − 1.23
| (5) |
Without the biased voltage, the ΔG can be obtained by calculating the difference of Gibbs free energy between product and reactant:
| | |
ΔG = ΔE + ΔEZPE − TΔS
| (6) |
in which the Δ
E refers to adsorption energy, Δ
EZPE and Δ
S stand for ZPE and entropy at the specific temperature
T, respectively. The relationship of Gibbs free energy in the CHE model meet the conditions:
| | |
G(H+) + G(e−) = 1/2G(H2)
| (7) |
| | |
G(H+) + G(OH−) = G(H2O)
| (8) |
| | |
2G(H2) + G(O2) − 2G(H2O) = 4.92 eV
| (9) |
In our calculation, the free energy of O2 is obtained by eqn (9) rather than DFT because of the large error for calculating O2 in VASP program. The calculated free energy of H2, O2 and H2O are listed in Table S1.†
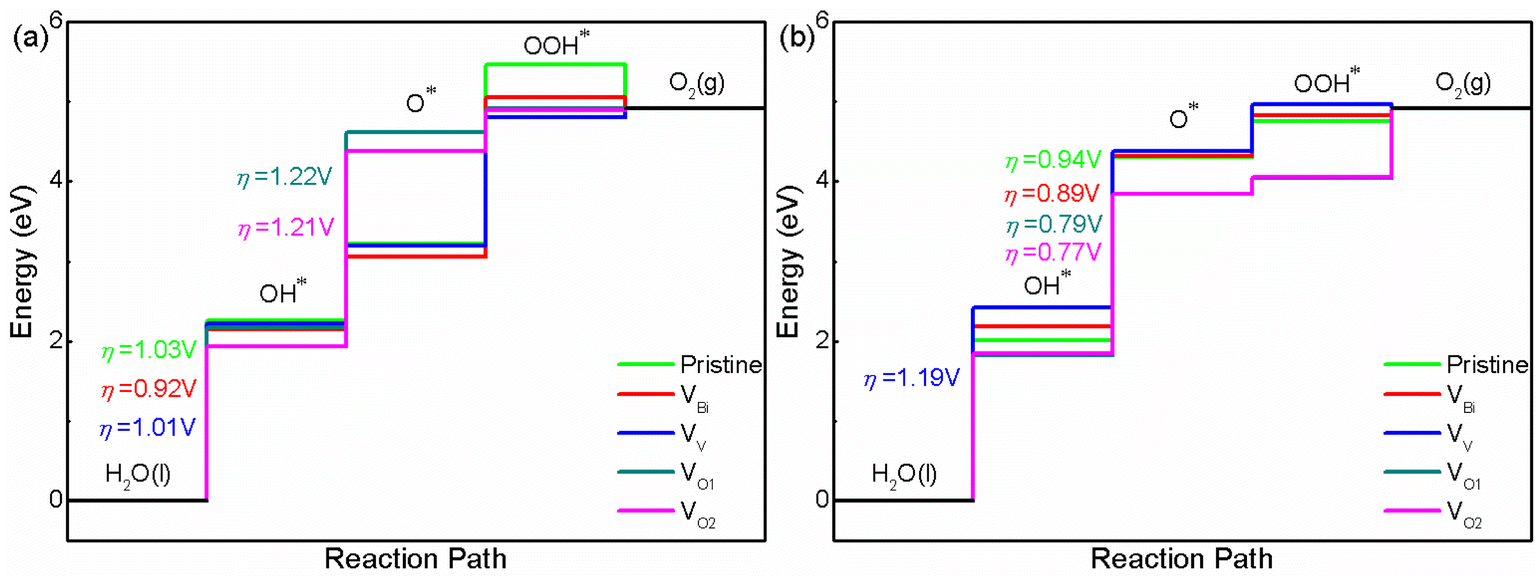
After obtaining the free energy of H2, O2 and H2O, the adsorbed intermediates have been investigated. The structures of adsorbed intermediates (OH*, O* and OOH*) of pristine BiVO4, VBi, VV, VO1 and VO2 with (001) and (011) surface are plotted in Fig. S2 and S3.† And the calculated total energy, ZPE and entropy for all the structures are shown in Table S2.† For these structures, BiVO4 substrate has been fixed and the adsorbed atom has been optimized at first, then the adsorbed atom and the top layer of substrate are relaxed together. The calculated results about overpotential of pristine and defective BiVO4 are shown in Fig. 4(a) and (b). For all the systems, the limiting step is the first step or the second step. Clearly, VBi, VV, VO1 and VO2 could impact the free energy greatly. Here we focus on pristine BiVO4 at first. It can be found that (001) has a lower overpotential compared with (011). Hence, (011) is more active than (001) for OER. For this model, the overpotential is close to previous calculated studies, such as PBE0 with implicit solvent model calculation (1.2 V for (001) and 0.9 V for (011) surface) and GGA calculation (1.42 V for (001) and 1.14 V for (011) surface).44,56 Then we turn to defective BiVO4. For (001), it can be seen that the VBi, VV, VO1 and VO2 could lower the free energy for forming OH* and OOH*. These two steps consist of one H removal, which is more efficient in all of the reaction steps happening on a surface. This phenomenon might be because the defects introduce additional holes or electrons near the Fermi level (the VBi and VV introduce holes while VO1 and VO2 introduce electrons). These carriers could promote the charge transfer and lower the free energy. In the process of forming O*, VBi, VV show a lower free energy compared with pristine BiVO4, while VO1 and VO2 show a higher free energy. This phenomenon might due to the pure O tend to be combined with holes rather than electrons for this surface, in these system, the VBi, VV introduce additional holes while VO1 and VO2 introduce electrons. Totally speaking, the VO1 and VO2 reduce the photocatalytic activity due to a large ΔG, while VV only lower the overpotential slightly and the difference could be negligible, thus VV probably do not affect the photocatalytic performance for this surface. And VBi could improve the photocatalytic performance to some extent. Then we turn to OER on the (011) surface, and here an analogous set of calculations was performed. The results show that the pristine (011) surface is the more efficient than (001), and the second step becomes the potential limiting step. With respect to the pristine surface, the free energy of defective BiVO4 with (011) surface show a different variation trend compared with (001). It can be seen that the VO1 and VO2 lower the free energy while VBi and VV enhance the free energy for forming OH*, O* and OOH*. This phenomenon is completely different compared with (001), indicating the OH, O and OOH radicals might tend to be combined with electrons rather than holes for this surface. For (011) surface, we can see that the OER efficiency is reduced when the VV is introduced, while it can be improved when the VBi, VO1 and VO2 is introduced. The overpotential of VO1 and VO2 shows a similar behavior with some previous studies,57,58 where the O vacancies enhance the overpotential for (001). However, there are also some previous studies showing a different situation, where the O vacancies lower the overpotential for (001) and enhance the overpotential for (011).44 This phenomenon probably due to the different location of O vacancies, thus it can be inferred that O vacancies is an effective but uncontrollable method for OER. In our calculated systems, the VV is not beneficial to enhance OER performance while VBi would contribute to improving OER performance. The calculated results is consistent with the previous experimental results.32,33 Therefore, the VV should be avoided in the experiment. And the VBi should be adopted to improve the OER performance.
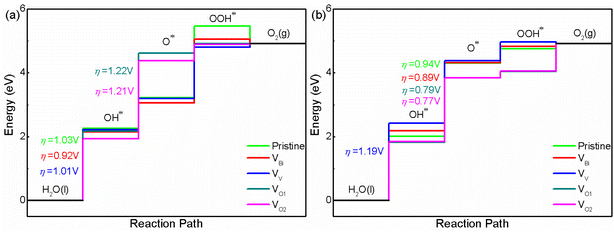 |
| | Fig. 4 The calculated OER free energy of (a) BiVO4 (001) and (b) BiVO4 (011). | |
3.6 Origins of free energy change
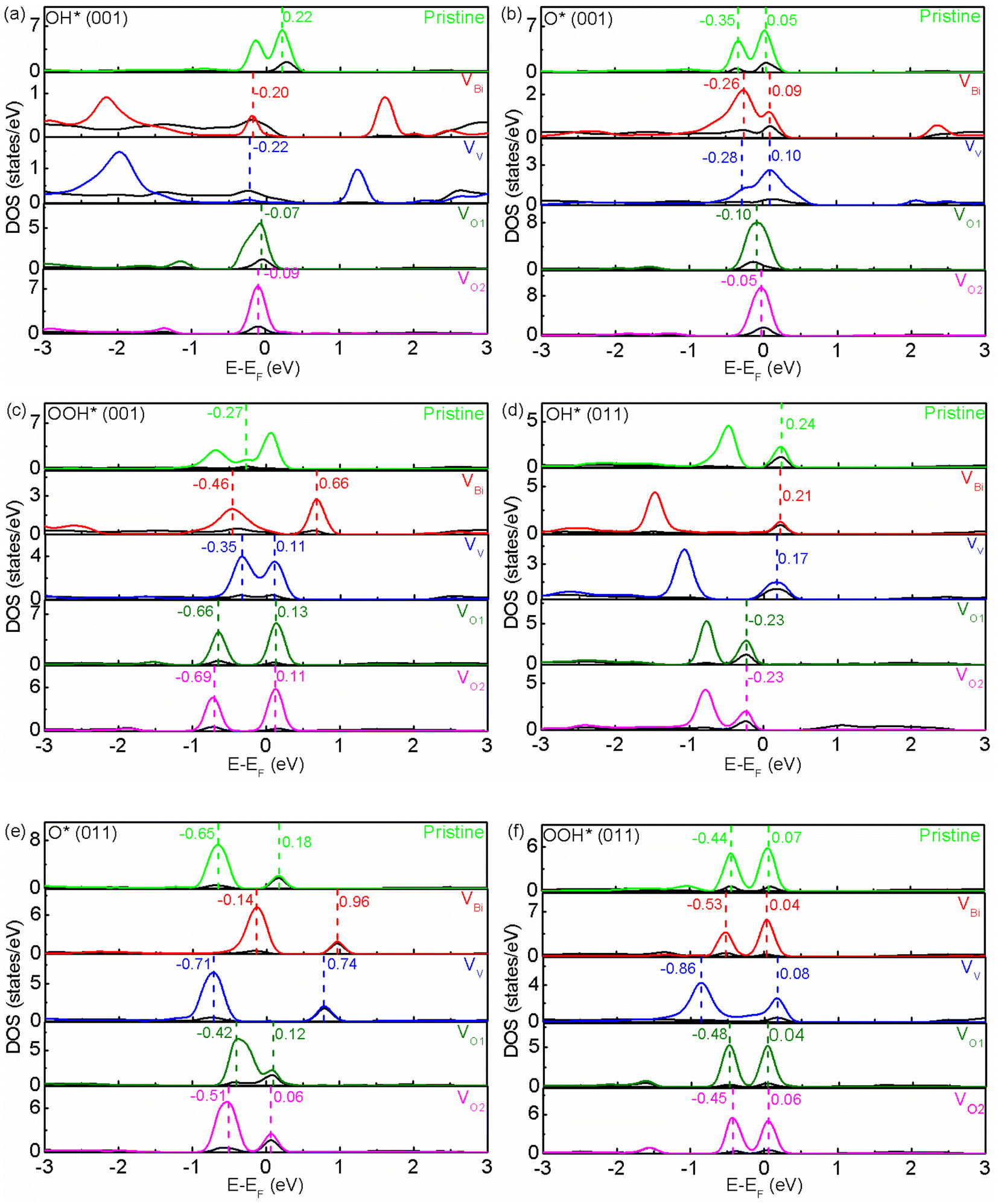
In order to reveal the origins of free energy change, the partial DOS of active sites and adsorbate have been examined. In a chemical reactions, the frontier molecular orbitals in the proximity of Fermi level play an important role.59 As show in Fig. 5(a)–(f), it can be seen there are two or one hybridized states between active site and adsorbate for O* and OOH*. Totally speaking, two hybridized states near the Fermi level would have a lower free energy. For (001), the pristine BiVO4, VBi and VV have a much lower free energy compared with VO1 and VO2 for O*, and pristine BiVO4 has a much higher free energy compared with VBi, VV, VO1 and VO2 for OOH*. Moreover, the free energy would increase as the distance between two hybridized states become greater. This phenomenon might due to a larger distance between two hybridized states increases the difficulty in transferring charge and weaken the adsorption. And one hybridized state makes the charge transfer more difficultly and further weaken the adsorption. Similar behavior also can be found in (011) although the exact locations of hybridized states are different. For OH*, there is one hybridized state between active site and adsorbate. And it can be seen the free energy is lower when the hybridized state is below the Fermi level. Moreover, it can be found the free energy is related to the distance between the Fermi level and the hybridized state. When the hybridized state is below the Fermi level, the free energy decrease with a smaller distance. However, when the hybridized state is above the Fermi level, the free energy is unexpectedly enhanced with a smaller distance. This result is difficult to explain and ought to be investigated in more detail in the future. We guess this phenomenon is due to positive charges have already accumulated on the active site, which would promote holes transfer and inhibit electrons transfer between active site and adsorbate. Therefore, the free energy could be modulated. The Bader charge analysis for active site has been shown in Fig. S4,†60 it is clearly that positive charges have accumulated on the active site. After inducing defects, the charge distribution on this site has a slight, not obvious change. According to the results of hybridized states and Bader charge, it can be inferred defects mainly affect the hybridized states between active site and adsorbate, thus the free energy for each step can be adjusted and the overpotential can be changed.
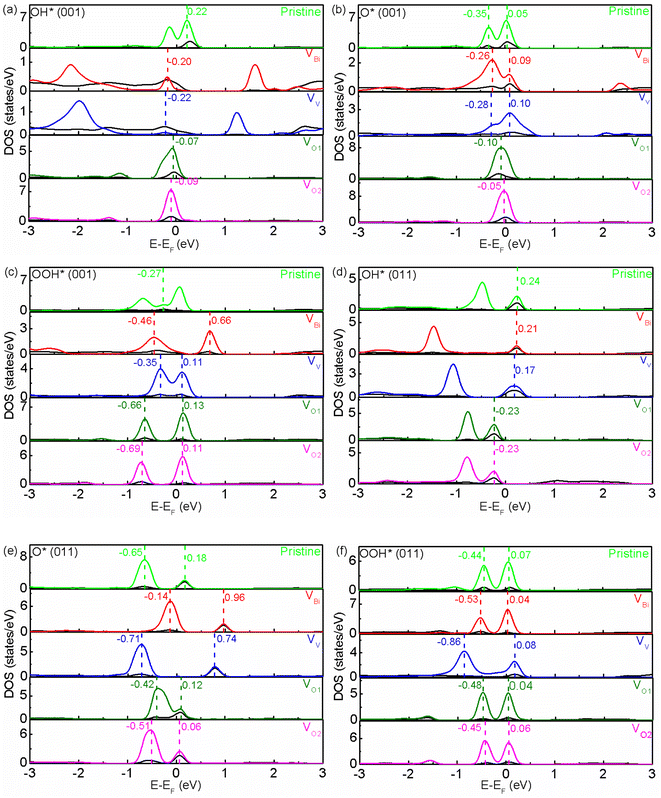 |
| | Fig. 5 (a)–(f) The partial DOS of pristine and defective BiVO4. The black and multicoloured line represent the partial DOS of active site and adsorbate, respectively. | |
4. Conclusions
In summary, we have carried out a comprehensive periodic density functional theory (DFT) simulations for the defective BiVO4 with (001) and (011) surfaces to improve its photocatalytic performance. It is found that defects have a great effect on BiVO4. VV with (011) surface and VO1 with (001) surface create defect states near the middle of band gap, which might not be good for photocatalysis to some extent. The band edge position indicates that VBi and VV with (011) cannot produce O2 without biased voltage. According to modeling all of the reaction intermediates for different water oxidation mechanisms, we have shown that the most favorable photocatalytic process on BiVO4 is the (011) surface. The defects could change the overpotential greatly, and VBi, VO1 and VO2 exhibit the best photocatalytic activity due to its lower overpotential in our calculated systems. Moreover, the defects have a great effect on hybridized state between active site and adsorbate. By controlling the exposed surface facet and the vacancy content, OER performance can be improved, which is important for the design of novel photocatalyst.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Funding for this research was provided by National Natural Science Foundation of China (NSFC) (11674396).
References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- J. Eichhorn, C. Kastl, J. K. Cooper, D. Ziegler, A. M. Schwartzberg, I. D. Sharp and F. M. Toma, Nat. Commun., 2018, 9, 2597 CrossRef.
- O. F. S. Khasawneh and P. Palaniandy, Environ. Technol. Innovation, 2021, 21, 101230 CrossRef.
- W. Zhang, H. He, H. Li, L. Duan, L. Zu, Y. Zhai, W. Li, L. Wang, H. Fu and D. Zhao, Adv. Energy Mater., 2021, 11, 2003003 Search PubMed.
- V. Dutta, S. Sharma, P. Raizada, V. K. Thakur, A. A. P. Khan, V. Saini, A. M. Asiri and P. Singh, J. Environ. Chem. Eng., 2021, 9, 105018 CrossRef CAS.
- S. Patial, V. Hasija, P. Raizada, P. Singh, A. A. P. K. Singh and A. M. Asiri, J. Environ. Chem. Eng., 2020, 8, 103791 CrossRef CAS.
- H. L. Tan, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2017, 5, 16498–16521 RSC.
- S. C. Wang, P. Chen, Y. Bai, J. H. Yun, G. Liu and L. Z. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- Y. C. Qiu, W. Liu, W. Chen, G. M. Zhou, P. C. Hsu, R. F. Zhang, Z. Liang, S. S. Fan, Y. G. Zhang and Y. Cui, Sci. Adv., 2016, 2, 1501764 CrossRef.
- T. W. Kim, Y. Ping, G. A. Galli and K. S. Choi, Nat. Commun., 2015, 6, 8769 CrossRef CAS.
- L. Zhou, C. Q. Zhao, B. Giri, P. Allen, X. W. Xu, H. Joshi, Y. Y. Fan, L. V. Titova and P. M. Rao, Nano Lett., 2016, 16, 3463–3474 CrossRef CAS PubMed.
- C.-K. Huang, T. Wu, C.-W. Huang, C.-Y. Lai, M.-Y. Wu and Y.-W. Lin, Appl. Surf. Sci., 2017, 399, 10–19 CrossRef CAS.
- Z. Zhao, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2011, 13, 4746–4753 RSC.
- Y. Chen, T. Shi, P. Liu, X. Ma, L. Shui and C. Shang, J. Mater. Chem. A, 2018, 6, 19167–19175 RSC.
- M. Huang, C. Li, L. Zhang, Q. Chen, Z. Zhen, Z. Li and H. Zhu, Adv. Energy Mater., 2018, 8, 1802198 CrossRef.
- J. Wang, L. Xu, T. Wang, R. Li, Y. Zhang, J. Zhang and T. Peng, Adv. Energy Mater., 2021, 11, 2003575 CrossRef CAS.
- J. Safaei, H. Ullah, N. A. Mohamed, M. F. M. Noh, M. F. Soh and A. A. Tahir, Appl. Catal., B, 2018, 234, 296–310 CrossRef CAS.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS.
- J. Mao, Z. Gu, Y. Yu, H. Liu, J. Qu and X. An, ACS Appl. Energy Mater., 2021, 4, 2543–2551 CrossRef CAS.
- R. Guo, A. Yan, J. Xu, B. Xu, T. Li, X. Liu, T. Yi and S. Luo, J. Alloys Compd., 2020, 817, 153246–153257 CrossRef CAS.
- X. Lu, K. Ye, S. Zhang, J. Zhang, J. Yang, Y. Huang and H. Ji, Chem. Eng. J., 2022, 428, 131027 CrossRef CAS.
- M. A. Gaikwad, U. P. Suryawanshi, U. V. Ghorpade, J. S. Jang, M. P. Suryawanshi and J. H. Kim, Small, 2022, 18, 2105084 CrossRef CAS.
- S. Liu, R. Gao, R. Zhang, Z. Wang, X. Liu, T. Nakajima, X. Zhang, Y. Su and L. Wang, Appl. Catal., B, 2021, 298, 120610 CrossRef CAS.
- Z. Kang, X. Lv, Z. Sun, S. Wang, Y. Zheng and X. Tao, Chem. Eng. J., 2021, 421, 129819 CrossRef CAS.
- M. Lamers, S. Fiechter, D. Friedrich, F. F. Abdi and R. Krol, J. Mater. Chem. A, 2018, 6, 18694–18700 RSC.
- S. M. Thalluri, S. Hernández, S. Bensaida, G. Saraccoa and N. Russo, Appl. Catal., B, 2016, 180, 630–636 CrossRef CAS.
- S. Wang, P. Chen, Y. Bai, J. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- X. Zhao, J. Hu, X. Yao, S. Chen and Z. Chen, ACS Appl. Energy Mater., 2018, 1, 3410–3419 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed., 2018, 57, 2248–2252 CrossRef CAS PubMed.
- S. Wang, T. He, P. Chen, A. Du, K. Ostrikov, W. Huang and L. Wang, Adv. Mater., 2020, 32, 2001385 CrossRef CAS.
- Y. Lu, Y. Yang, X. Fan, Y. Li, D. Zhou, B. Cai, L. Wang, K. Fan and K. Zhang, Adv. Mater., 2022, 34, 2108178 CrossRef CAS.
- T. Tran-Phu, Z. Fusco, I. D. Bernardo, J. Lipton-Duffin, C. Y. Toe, R. Daiyan, T. Gengenbach, C. Lin, R. Bo, H. T. Nguyen, G. M. J. Barca, T. Wu, H. Chen, R. Amal and A. Tricoli, Chem. Mater., 2021, 33, 3553–3565 CrossRef CAS.
- S. Lardhi, L. Cavallo and M. Harb, J. Phys. Chem. C, 2018, 122, 18204–18211 CrossRef CAS.
- N. Österbacka, F. Ambrosio and J. Wiktor, J. Phys. Chem. C, 2022, 126, 2960–2970 CrossRef.
- W. Yin, S. Wei, M. M. Al-Jassim, J. Turner and Y. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155102 CrossRef.
- H. Ullah, A. A. Tahir and T. K. Mallick, Appl. Catal., B, 2018, 224, 895–903 CrossRef CAS.
- M. D. Bhatt and J. Y. Lee, J. Electroanal. Chem., 2018, 828, 97–101 CrossRef CAS.
- T. Liu, M. Cui and M. Dupuis, J. Phys. Chem. C, 2020, 124, 23038–23044 CrossRef CAS.
- J. Zhang, X. Chen, M. Deng, H. Shen, H. Li and J. Ding, Phys. Chem. Chem. Phys., 2020, 22, 25297–25305 RSC.
- N. Österbacka and J. Wiktor, J. Phys. Chem. C, 2021, 125, 1200–1207 CrossRef.
- D. Wang, H. Jiang, X. Zong, Q. Xu, Y. Ma, G. Li and C. Li, Chem.–Eur. J., 2011, 17, 1275–1282 CrossRef CAS PubMed.
- J. Yang, D. Wang, X. Zhou and C. Li, Chem.–Eur. J., 2013, 19, 1320–1326 CrossRef CAS PubMed.
- P. Nikačević, F. S. Hegner, J. R. Galán-Mascarós and N. López, ACS Catal., 2021, 11, 13416–13422 CrossRef.
- J. Hu, W. Chen, X. Zhao, H. Su and Z. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 5475–5484 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed.
- P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef.
- I. Laraib, M. A. Carneiro and A. Janotti, J. Phys. Chem. C, 2019, 123, 26752–26757 CrossRef CAS.
- J. Shi, W. Zhang and Q. Gu, J. Phys. Chem. C, 2022, 126, 9541–9550 CrossRef CAS.
- C. G. Van de Walle and R. M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5621 CrossRef CAS.
- Y. Zhao, R. Li, L. Mu and C. Li, Cryst. Growth Des., 2017, 17, 2923–2928 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- P. Li, X. Chen, H. He, X. Zhou, Y. Zhou and Z. Zou, Adv. Mater., 2018, 30, 1703119 CrossRef.
- A. Massaro, A. Pecoraro, S. Hernandez, G. Talarico, A. B. Munoz-García and M. Pavone, Mol. Catal., 2022, 517, 112036 CrossRef CAS.
- J. Hu, X. Zhao, W. Chen, H. Su and Z. Chen, J. Phys. Chem. C, 2017, 121, 18702–18709 CrossRef CAS.
- K. Fukui, Science, 1982, 218, 747–754 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Top view of optimized geometric structures of BiVO4 surface. Free energy of H2, O2 and H2O. Structures of the intermediates in the OER processes. Total energy and zero-point energy (ZPE) correction of adsorbates. Bader charge of pristine and defective BiVO4. See DOI: https://doi.org/10.1039/d2ra04890f |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*