DOI:
10.1039/D2RA04974K
(Paper)
RSC Adv., 2022,
12, 25565-25569
Evaluating the effects of OH-groups on the Ni surface on low-temperature steam reforming in an electric field†
Received
9th August 2022
, Accepted 2nd September 2022
First published on 7th September 2022
Abstract
The effect of OH-groups on the surface of a Ni catalyst for low-temperature (473 K) steam reforming of methane in an electric field (EF) was investigated. Ni-doped YSZ (Zr0.65Y0.05Ni0.3O2) was chosen as a highly active catalyst for this purpose. The effects on catalyst activity of adding hydrogen and steam in the pre-treatment were assessed with and without EF. When an EF was applied, activity increased irrespective of the electronic state of Ni, whereas the metallic Ni state was necessary for activity without EF. Furthermore, the highest activity with EF was observed for the pre-treatment with a mixture of H2 and H2O. Investigation of the superiority using XPS measurements showed an increase in the amount of Ni(OH)2, OH groups and H2O near the surface after the activity test, which are regarded as the reaction sites with EF. This finding suggests that a pre-treatment with steam increases the surface OH groups and Ni(OH)2 on the Ni catalyst, and enhances surface proton conduction, thereby improving the activity.
1. Introduction
With the anticipated expansion of hydrogen utilisation, there are high hopes for innovation in hydrogen production technology. Steam reforming of methane at high temperatures, which has been the mainstream of hydrogen production to date, has been industrialised on a large scale worldwide.1 Steam reforming of methane proceeds as in eqn (1) below, followed by a water gas shift reaction as in eqn (2), so that the overall reaction is expressed as in eqn (3).| | |
CH4 + H2O → CO + 3H2, ΔH2980 = 206.1 kJ mol−1
| (1) |
| | |
CO + H2O → CO2 + H2, ΔH2980 = −41.2 kJ mol−1
| (2) |
| | |
CH4 + 2H2O → CO2 + 4H2, ΔH2980 = 164.9 kJ mol−1
| (3) |
The steam reforming reaction, a highly endothermic reaction, has been reported as only being able to proceed at temperatures higher than 1000 K using Ni-based catalysts.1,2 Under such severe conditions, catalyst deactivation and carbon deposition are likely to occur, which poses a challenge. Also such a process requires a large scale operation. A methane steam reforming process that can proceed at lower temperatures with a small scale is highly anticipated.
We investigated the catalytic reaction in an electric field (denoted ER), in which the catalytic performance is improved by application of a direct current to the catalyst bed.3–6 The reaction mechanism in ER was investigated; results showed that the proton conducts on the CeO2 by the Grotthuss mechanism and collides with methane, which promotes the activation of methane.5–7
Consequently, the reaction mechanism of methane steam reforming in ER differs from the conventional catalytic reaction by heating (denoted SR): in ER, proton conduction at the surface (surface protonics) is a crucially important factor for high activity in methane steam reforming under the application of EF. For this study, yttria stabilised zirconia (YSZ),8,9 which exhibits surface proton conductivity, was used as a catalyst support and was doped with Ni, which is the most suitable active metal for steam reforming.10 Active metals are known to be highly dispersible and loaded by reduction treatment after doping.11,12 For methane steam reforming at low temperatures, we investigated utilisation of the surface protonics on the catalyst, and investigated the effects on ER activity attributable to changes in the protonics when the amount of hydroxyl groups and adsorbed water on the catalyst are controlled by pre-treatment.
2. Experimental
2.1 Catalyst preparation
For this experiment, we prepared Ni-doped YSZ: Zr0.65Y0.05Ni0.3O2 as catalysts using a complex polymerization method, which had shown the best ER activity in our earlier work with hydrogen reduction pre-treatment (H2/Ar = 60/60), 873 K for 1 h. After ethylene glycol and citric acid were dissolved with distilled water, we added each nitrate: ZrO(NO3)2 2H2O, Y(NO3)3 6H2O, and Ni(NO3)2 6H2O (Kanto Chemical Co. Inc). This mixture was heated at 343 K for about 20 h with stirring. Subsequently, the water is dried up by stirring on a hot stirrer to make the powdery mixture. After the powder was calcined at 673 K for 2 h with the ramping rate of 5 K min−1, it was calcined at 1123 K for 10 h with the ramping rate of 10 K min−1.
2.2 Activity test
We conducted catalyst activity tests using a fixed-bed flow-type reactor. Catalysts of 80 mg (355–500 μm grain diameter) were filled in a quartz tube (8.0 mm o.d., 6.0 mm i.d). The catalyst bed height was about 2.0 mm. Before the activity tests, we conducted various pre-treatments at a furnace temperature of 873 K for 1 h with the ramping rate of 10 K min−1. The components of pre-treatment gas were H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:Ar = x:(60−x): 60 (x = 0, 12, 30, 48, 60), total flow: 120 SCCM or inert Ar only (total flow: 60 SCCM). The catalyst bed temperature was measured using a thermocouple inserted into the catalyst bed. The furnace temperature was controlled at 473 K when applying EF and at 543–673 K when not applying EF (because no activity was observed at 473 K without EF). We inserted two stainless steel electrodes (2.0 mm o.d.) into the reactor and attached them to both the top and the bottom of the catalyst bed. We applied direct current (DC; 3–9 mA) to the catalyst bed. The response voltage was recorded using a digital phosphor oscilloscope (TDS 2001C; Tektronix Inc). The reaction gas components were CH4:H2O:Ar = 1:2:7 (120 SCCM total flow). The production gas, CH4, CO, and CO2, was analysed using a gas chromatograph (GC-2014; Shimadzu Corp.) with a flame ionization detector (FID) using a methanizer (Ru/Al2O3) and H2 was analysed by a TCD gas chromatograph. The reaction rate was calculated using the amount of CO and CO2 produced (rCO + CO2). The CH4 conversion and CO2 selectivity were calculated using equations shown in ESI.†
:H2O:Ar = x:(60−x): 60 (x = 0, 12, 30, 48, 60), total flow: 120 SCCM or inert Ar only (total flow: 60 SCCM). The catalyst bed temperature was measured using a thermocouple inserted into the catalyst bed. The furnace temperature was controlled at 473 K when applying EF and at 543–673 K when not applying EF (because no activity was observed at 473 K without EF). We inserted two stainless steel electrodes (2.0 mm o.d.) into the reactor and attached them to both the top and the bottom of the catalyst bed. We applied direct current (DC; 3–9 mA) to the catalyst bed. The response voltage was recorded using a digital phosphor oscilloscope (TDS 2001C; Tektronix Inc). The reaction gas components were CH4:H2O:Ar = 1:2:7 (120 SCCM total flow). The production gas, CH4, CO, and CO2, was analysed using a gas chromatograph (GC-2014; Shimadzu Corp.) with a flame ionization detector (FID) using a methanizer (Ru/Al2O3) and H2 was analysed by a TCD gas chromatograph. The reaction rate was calculated using the amount of CO and CO2 produced (rCO + CO2). The CH4 conversion and CO2 selectivity were calculated using equations shown in ESI.†
2.3 Characterisation
In situ transmission XANES measurements were taken to estimate the electronic state of Ni in the bulk material at different pre-treatments. Catalysts of 17.05 mg and 121.5 mg of BN were mixed for 2 h in an agate mortar as a suitable quantity for XANES measurements. The mixture was placed in a moulding machine and was pressed at 60 kN to form a 10 mm ϕ disc. The disc was placed in an in situ cell, which was then installed in the XAFS measurement system at the beamline BL14B2 or BL01B1 of SPring-8 in Hyogo, Japan. The cell was pre-treated with a gas flow of H2:H2O:He = (30−x):x:90 (x = 0, 15, 30, total flow rate 120 SCCM) at a cell temperature of 873 K and a holding time of 1 h. After the pre-treatment, the temperature was controlled to be 473 K for evaluation with following gas flow; CH4:H2O:He = 1:2:7. As standard samples, Ni foil, Ni(OH)2, and NiO were used.
An XPS system (PHI5000 VersaProbe II; Ulvac-Phi Inc.) was used to estimate the state near the surface after each pre-treatment and after the ER test. About 80 mg of each catalyst was first pre-treated with H2:H2O:Ar = (60−x):x:60 (x = 0, 30, 60, total flow: 120 SCCM) at a furnace temperature of 873 K for 1 h, using the reactor described in Section 2.2. The temperature was then controlled to be 473 K. The total flow rate of the reaction gas was 120 SCCM, with reaction gas of CH4:H2O:Ar = 1:2:7. Current of 9 mA was applied to the catalyst bed for 30 min. After each pre-treatment and after application of the EF, the reaction tube was placed in a glove bag to avoid exposure to the open air. It was then placed in a transfer vessel under high purity Ar conditions. The transfer vessel was placed in an XPS apparatus. We obtained the peak of C 1s, O 1s, and Ni 2p2/3 from the measurements. The peaks attributed to C–C and C–H bonds in the C 1s orbital were charge-corrected to 284.8 eV. As Ni standards, Ni foil, Ni(OH)2, and NiO were used.
Other characterizations including XRD, BET, etc. are presented in ESI.†
3. Results and discussion
3.1. Catalytic activity
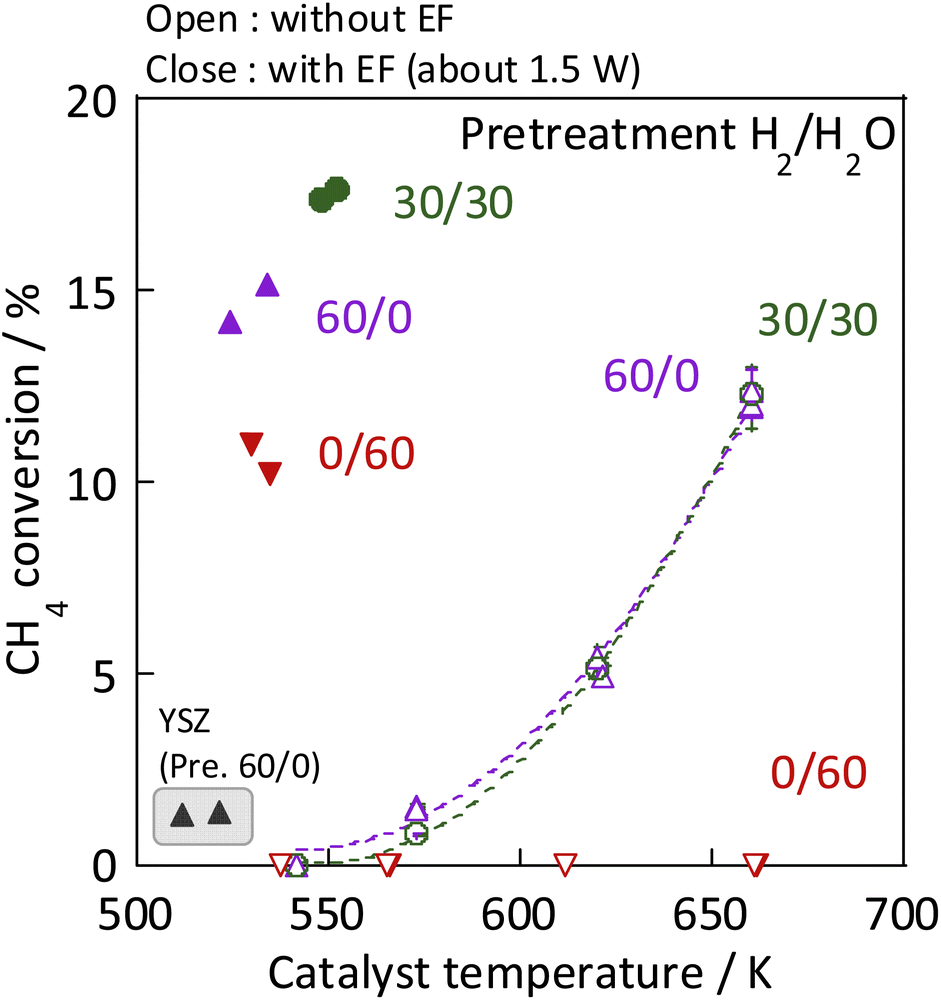
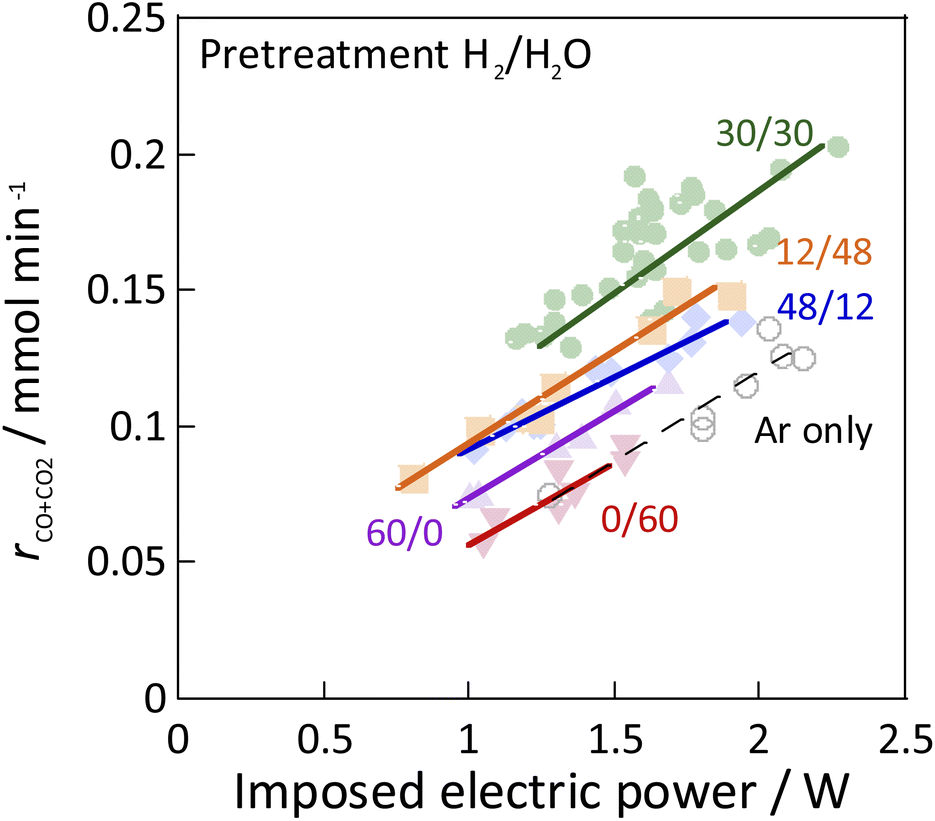
As the catalyst, Ni, which is known to be highly active for steam reforming of methane, was chosen: YSZ catalysts doped with it were prepared. The catalytic activity of methane steam reforming over conventional heated catalysis (SR) and low-temperature catalysis with an applied electric field (ER) were compared after reductive deposition of Ni as the active site. Prepared Ni-YSZ catalyst showed high activity compared to other Ni catalysts in EF, so the correlation between surface structure and activity was investigated under various pre-treatment conditions with steam and/or hydrogen for this catalyst. Three pre-treatment conditions were used: only H2O, H2O + H2 mixture, and H2. The results are presented in Fig. 1 (and Table S1 in ESI†). The catalyst which was pre-treated with steam only (i.e. H2:H2O = 0:60) showed no SR activity, even at 673 K, because the Ni was regarded as oxidised by the steam. The Ni0 state is generally known to be necessary for the conventional catalytic reaction by heating, which proceeds by the Langmuir–Hinshelwood mechanism.10,13,14 In the steam-only pre-treatment, Ni is regarded as oxidised, therefore is inactive. However, the low-temperature catalytic reaction with the application of EF showed sufficient activity with only a steam pre-treatment, which indicates that the reaction in ER proceeds by a different reaction mechanism than in SR, even on oxidized Ni. Furthermore, to confirm the role of the presence of Ni, ER activity was measured using pre-reduced YSZ without Ni doping, which showed much smaller ER activity than Ni-YSZ. These phenomena are not electrode-dependent, and the same results can be obtained with carbon rod electrodes and other electrodes, regardless of the material of the electrode. Therefore, it was found that Ni has an important role in the activity in ER, and that YSZ only is not sufficient. Furthermore, from Fig. 2, the ER activity was in the order of H2:H2O = 30:30 > 12:48 > 48:12 > 60:0 > 0:60 as the composition ratio of the pre-treatment. The simultaneous supply of hydrogen and steam was found to have a higher ER activity than the hydrogen reduction and steam pre-treatments. This difference was observed to persist during the long-term test (ESI Fig. S2†). This result indicates that the difference in ER activity between these different pre-treatment conditions persists for a long time, the difference is not a transitional state phenomenon.
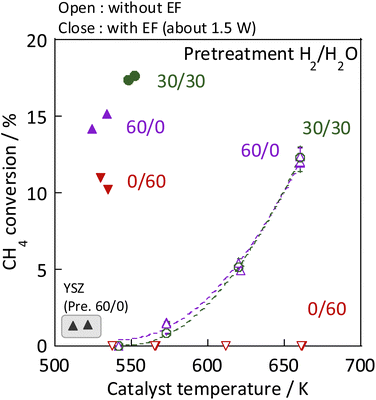 |
| | Fig. 1 Results of activity tests conducted with and without the EF: catalyst, Zr0.65Y0.05Ni0.3O2 and Zr0.95Y0.05O2; catalyst amount, 80 mg; pre-treatment, H2:H2O:Ar = x:(60−x):60 (x = 0, 30, 60); reaction gas flow, CH4:H2O:Ar = 1:2:7; total 120 SCCM; furnace temp.: 473 K with EF, 543–673 K without EF. | |
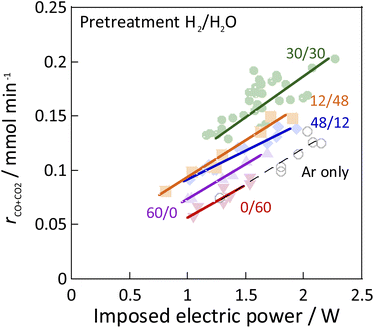 |
| | Fig. 2 Results of activity tests conducted with the EF: catalyst, Zr0.65Y0.05Ni0.3O2; catalyst amount: 80 mg; pre-treatment, H2:H2O:Ar = x:(60−x):60 (x = 0, 12, 30, 48, 60), or Ar only; reaction gas flow, CH4:H2O:Ar = 1:2:7; total 120 SCCM; furnace temp.: 473 K; current: 3–9 mA. | |
3.2. Evaluation of the Ni state by XANES
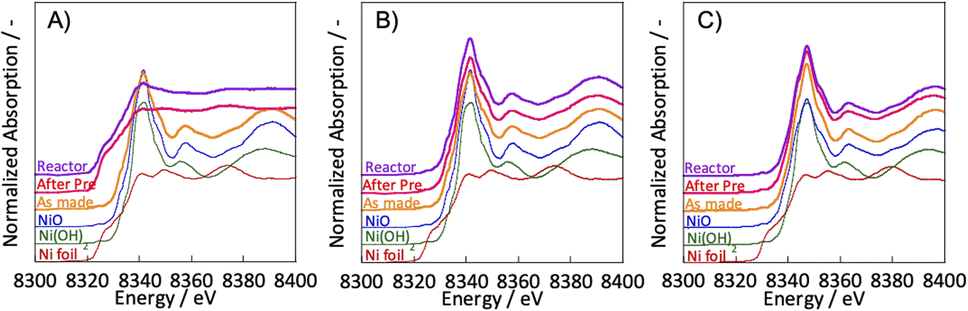
To clarify the differences in activity between SR and ER by the different pre-treatments, the Ni state of the catalyst after pre-treatment was evaluated using in situ XANES. In situ XANES measurements were taken under three pre-treatment conditions: only H2, H2O + H2 mixture, and only H2O. The XANES spectra after preparation, pre-treatment, and in the reaction atmosphere are depicted in Fig. 3. The Ni state of the as-made catalyst was confirmed to be NiO. These comparisons of pre-treatments by in situ XANES indicated differences in the bulk Ni state. For the two pre-treatments with steam in the composition of the pre-treatment gas; H2 + H2O mixture and only H2O, the bulk Ni state was confirmed to be NiO. For the hydrogen reduction pre-treatment with only H2, the bulk Ni state was identical to that of Ni foil, i.e., metallic. These results indicate that the ER activity is independent of the bulk Ni state and that oxidised nickel can also be highly active, even in the oxidised state. We have already studied the reaction mechanism in the ER.5,15–17 In the literature, we concluded that the reaction is promoted by the application of an electric field, which causes the proton conduction over the catalyst support to collide with methane at the ‘interface’ between the support and the active metal. The surface protonics changes significantly when the electric potential is switched on and off, which can be confirmed by surface operando analysis using infrared spectroscopy.5 The rotational spectrum of adsorbed water appears at 855 cm−1 only when an electric field is applied. Also, by comparing two TOFs; by the specific surface area of active metal (TOF-s), and by the length of its perimeter (TOF-p), and we found that the activity of the ER was strongly dependent on the TOF-p. As described in the previous section, the reaction in the electric field proceeds regardless the electrode material. Therefore, we believe that ER does not necessarily need the metallic surface of the supported metal to show the catalytic activity, but only the particle interface. Therefore, we considered that ER activity could occur even with NiO rather than Ni in the metallic state.
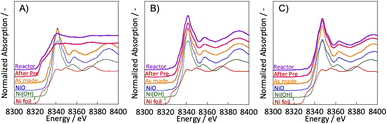 |
| | Fig. 3 Ni K-edge XANES spectra with various pre-treatments: (A) H2:H2O = 30:0 (SCCM); (B) H2:H2O = 15:15; (C) H2:H2O = 0:30; catalyst: Zr0.65Y0.05Ni0.3O2. | |
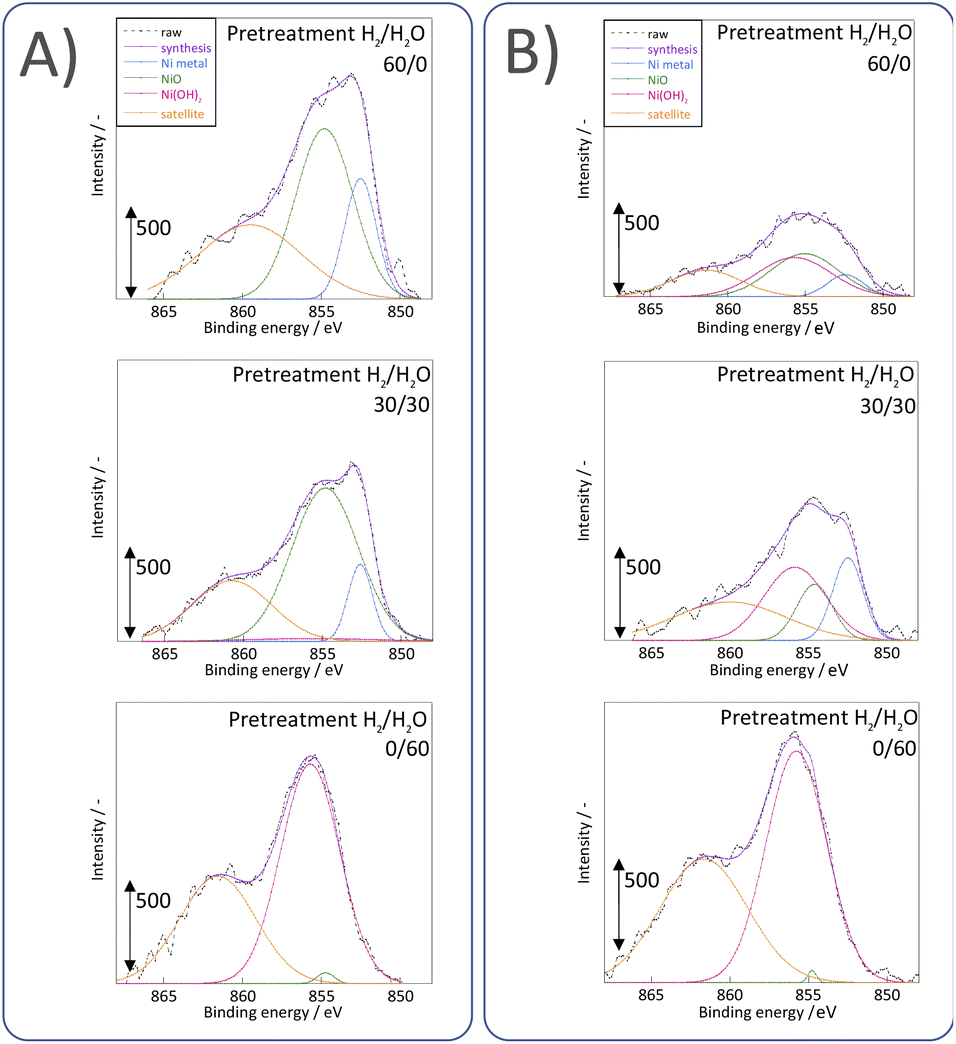
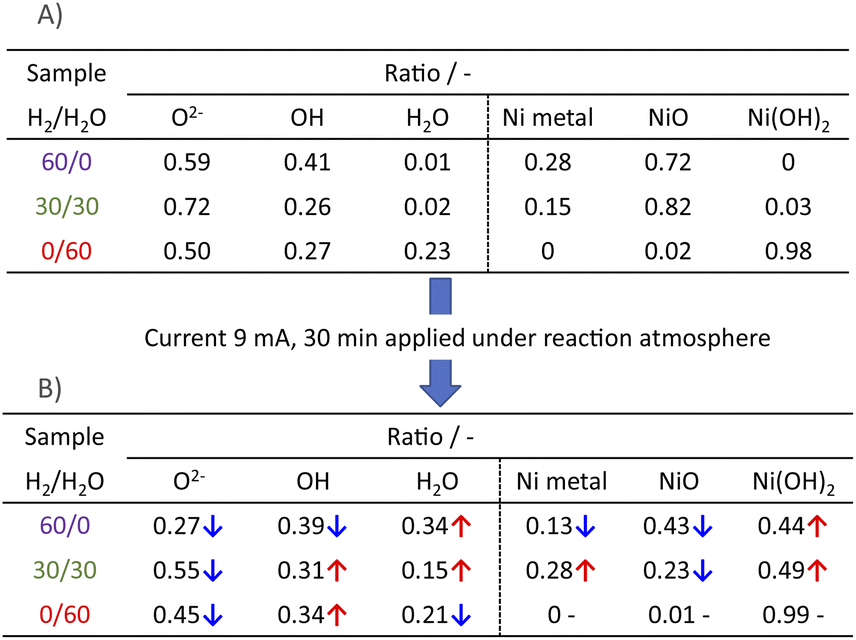
3.3 Evaluation of the Ni state near the surface
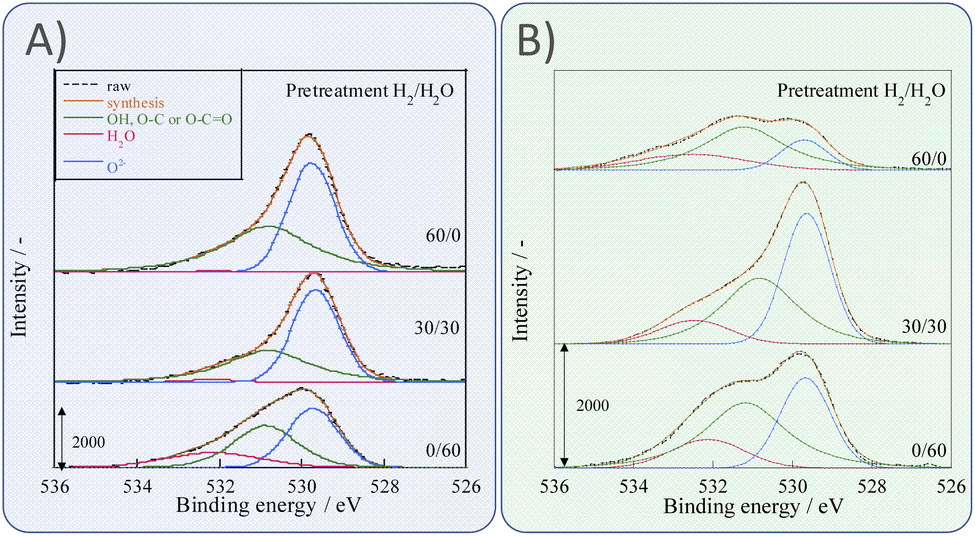
Next, we observed the surface states of Ni for these catalysts after each pre-treatment by XPS because the state of the surface Ni is regarded as dominating the catalytic activity. The Ni and O spectra were obtained for the three pre-treatment conditions; only H2, H2O + H2 mixture, and only H2O, respectively, after pre-treatment and after ER testing. Fig. 4 and 5 portray the XPS spectra. Table 1 presents the Ni and O composition ratios. The ratio of Ni(OH)2, OH and H2O at the surface increased only under the pre-treatment condition of H2:H2O = 30:30, where hydrogen and steam were supplied simultaneously. The ratio of any of them decreased under other conditions. The increase in Ni(OH)2, the OH and H2O ratio were then compared with the results of the ER test, as described in Section 3.1. The surface OH group and adsorbed H2O mediate the Grotthuss mechanism, one of the proton migration mechanisms. Under pre-treatment conditions in which hydrogen and steam were supplied simultaneously (H2:H2O = 30:30), the ratio of Ni(OH)2 increased, and it also plays a role on surface proton conduction. We examined the catalytic activity of Ni(OH)2 without catalyst support in the EF, and we confirmed that the Ni(OH)2 showed catalytic activity on steam reforming of methane in the EF (see ESI Fig. S3†), so the role of surface protonics on Ni(OH)2 has confirmed experimentally.
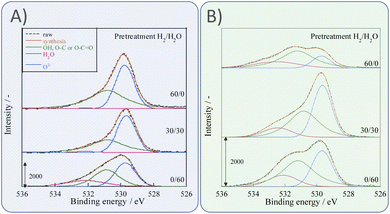 |
| | Fig. 4 XPS spectra for O 1s after various pre-treatments followed by reaction (A) without application of EF and (B) after the application of the EF of 9 mA. | |
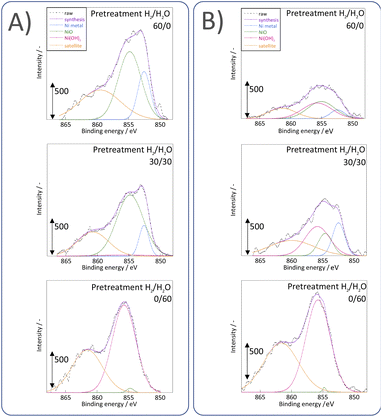 |
| | Fig. 5 XPS spectra for Ni 2p3/2 after various pre-treatments followed by reaction (A) without application of EF and (B) after the application of the EF of 9 mA. | |
Table 1 Comparison of the ratio from O 1s, Ni 2p3/2 XPS spectra: (A) after pre-treatments without application of EF; (B) with application of the EF of 9 mA; sample, Zr0.65Y0.05Ni0.3O2, pre-treatment, H2:H2O:Ar = x:(60−x):60 (x = 0, 30, 60)
3.4 Discussion
The differences in activity of Ni-doped YSZ catalysts in the methane steam reforming reaction with hydrogen and/or steam as a pre-treatment were investigated for two reactions: heated catalysis (SR) and electric field-applied catalysis (ER).
Results showed that the catalyst after steam treatment, which was not active in SR, was highly active in ER. Results obtained from XANES and XPS showed that Ni was oxidised by the steam treatment in both cases. Therefore, it is clear that ER activity can occur irrespective of the electronic state of Ni, supporting the conventional low-temperature catalytic reaction mechanism by the surface protonics.15–17 Because YSZ is also a proton conductor, it is useful as an interface between the support and the active metal irrespective of its electronic state. The ER activity of Ni-doped YSZ was regarded as occurring in the presence of an interface between the catalyst support and the active metal.5
Furthermore, higher ER activity was observed when hydrogen and steam were mixed in the pre-treatment. The highest activity was obtained when H2 and H2O were mixed in equal amounts. To investigate the factors responsible for this activity order, XPS measurements of the electronic structure of O and Ni near the surface showed that the ratio of Ni(OH)2, OH and H2O increased only after pre-treatment with a mixture of equal amounts of H2 and H2O. This finding leads us to believe that the surface protonics, which is important in ER, was improved because of the increased ratio of OH and H2O on the surface, not only on the catalyst-support but also near Ni, and caused high reaction activity of Ni-doped YSZ.
4. Conclusion
To carry out steam reforming of methane efficiently at low temperatures for hydrogen production, the catalytic activity of a Ni-doped YSZ catalyst was investigated by application of a DC electric field to the catalyst. The catalyst after steam treatment showed no activity at any temperature by conventional heated catalysis. However, the catalysts with a DC electric field were found to be highly active after the steam treatment, even at low temperatures. Both XANES and XPS showed that, in both cases, Ni was oxidised by the steam treatment. Therefore, the steam reforming activity in the EF proceeds at lower temperatures by virtue of the surface protonics, independent of the electronic state of Ni. Furthermore, results demonstrated that pre-treatment with hydrogen and coexisting steam caused higher performance in the EF because such pre-treatment increased the ratio of Ni(OH)2 on the catalyst. We consider that the increase in the ratio of OH and H2O on the surface near Ni as well as on the support is also effective for low-temperature catalytic reactions through surface protonics, which results in high reaction activity.
Conflicts of interest
The authors have no conflict to declare.
Acknowledgements
We appreciate Dr Tetsuo Homma and Dr Takeshi Watanabe (Japan Synchrotron Radiation Research Institute, SPring-8) for their great support with XAFS measurements in BL14B2. We also appreciate Prof. S. Yamazoe (Tokyo Metropolitan Univ.) for his great support with XAFS analyses.
References
- M. A. Nieva, M. M. Villaverde, A. Monzón, T. F. Garetto and A. J. Marchi, Chem. Eng. J., 2014, 235, 158–166 CrossRef CAS.
- Y. Sekine, M. Haraguchi, M. Tomioka, M. Matsukata and E. Kikuchi, J. Phys. Chem. A, 2010, 114(11), 3824–3833 CrossRef CAS.
- Y. Sekine, M. Haraguchi, M. Matsukata and E. Kikuchi, Catal. Today, 2011, 171(1), 116–125 CrossRef CAS.
- K. Oshima, T. Shinagawa, M. Haraguchi and Y. Sekine, Int. J. Hydrogen Energy, 2013, 38(7), 3003–3011 CrossRef CAS.
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 38007 CrossRef CAS PubMed.
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS.
- A. Takahashi, R. Inagaki, M. Torimoto, Y. Hisai, T. Matsuda, Q. Ma, J. G. Seo, T. Higo, H. Tsuneki, S. Ogo, T. Nordy and Y. Sekine, RSC Adv., 2020, 10, 14487–14492 RSC.
- S. Miyoshi, Y. Akao, N. Kuwata, J. Kawamura, Y. Oyama, T. Yagi and S. Yamaguchi, Chem. Mater., 2014, 26, 5194–5200 CrossRef CAS.
- S. Ø. Stub, E. Vøllestad and T. Norby, J. Phys. Chem. C, 2017, 121(23), 12817–12825 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1984, 85, 31–43 CrossRef CAS.
- S. Li, C. Zhang, Z. Huang, G. Wu and G. Gong, Chem. Commun., 2013, 49, 4226–4228 RSC.
- J. D. A. Bellido, E. Y. Tanabe and E. M. Assaf, Appl. Catal., B, 2009, 90, 485–488 CrossRef CAS.
- J. L. Rogers, M. C. Mangerella, A. D. D'Amico, J. R. Gallagher, M. R. Dutzer, E. Stavitski, J. T. Miller and C. Sievers, ACS Catal., 2016, 6, 5873–5886 CrossRef CAS.
- V. M. Shinde and G. Madras, RSC Adv., 2014, 4, 4817–4826 RSC.
- N. Nakano, M. Torimoto, H. Sampei, R. Yamashita, R. Yamano, K. Saegusa, A. Motomura, K. Nagakawa, H. Tsuneki, S. Ogo and Y. Sekine, RSC Adv., 2022, 12, 9036–9043 RSC.
- Y. Hisai, Q. Ma, T. Qureishy, T. Watanabe, T. Higo, T. Norby and Y. Sekine, Chem. Commun., 2021, 57, 5737–5749 RSC.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J.-G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ayako Takahashia,
Jun Sasakia,
Takuma Higo
a,
Ayako Takahashia,
Jun Sasakia,
Takuma Higo