
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced degradation of dimethyl phthalate in wastewater via heterogeneous catalytic ozonation process: performances and mechanisms†
Jia Yuan‡
,
Yang Li‡,
Yun Guo and
Zhiwei Wang *
*
State Key Laboratory of Pollution Control and Resource Reuse, Shanghai Institute of Pollution Control and Ecological Security, Tongji Advanced Membrane Technology Center, School of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, China. E-mail: zwwang@tongji.edu.cn
First published on 31st October 2022
Abstract
Ozonation process is a promising yet challenging method for the removal of refractory organic matter due to the sluggish reaction for generating hydroxyl radical (˙OH) at a neutral pH condition. Herein, an efficient heterogeneous catalytic ozonation system using CeO2/Al2O3 catalyst was developed to remove dimethyl phthalate (DMP) from wastewater. Under a neutral condition of pH = 6, this system achieved almost 100% DMP removal within 15 min at an optimized catalyst dosage of 30 g L−1 and the ozone flow rate of 22.5 mg min−1. Moreover, the catalytic ozonation system exhibited a stable degradation performance of DMP in a wider pH range (pH = 5–10). The results of electron paramagnetic resonance (EPR) and quantitative tests confirmed the ultrafast conversion of O3 to ˙OH (0.774 μM min−1) on the surface of CeO2 based ceramic catalyst. The quenching experiments further supported the predominant role of ˙OH in the mineralization of DMP. These results highlight the potential of using the heterogeneous catalytic ozonation system for the efficient removal of refractory organic matter from wastewater.
1. Introduction
Dimethyl phthalate (DMP) is of particular concern since it is a prevalent refractory aromatic pollutant generated in various industries such as plastics, rubber and cosmetics.1–3 Recent studies have highlighted that DMP is a typical environmental endocrine disruptor, which can pose a health risk (e.g., endocrine disorders, reproductive dysfunctions, and aberrations) on humans via the food chains.4–6 Effective approaches are thus needed for removing DMP in water treatment and environmental remediation.To date, numerous technologies such as adsorption,7,8 biological treatment,9,10 and advanced oxidation process11–13 have been widely used to remove refractory organics from wastewater. Among these technologies, the advanced oxidation process (AOP) can effectively mineralize organic matters under ambient conditions with no concentrated waste streams compared to the physical adsorption.14–16 For such process, hydroxyl radical (˙OH) is one of the most effective mediators since it possesses the non-selectivity and high second-order rate constant in the reaction with pollutants.17 However, the generation of ˙OH is highly dependent on the pH value in terms of Fenton reaction,18–20 which is regarded as a knotty problem in the water treatment under the circumneutral pH conditions (e.g., pH 6–9).
With the recent advances in ozonation oxidation, it is now possible to achieve the conversion of O3 to ˙OH at a wide pH range.21 Nevertheless, the ozone has a low solubility in water,22,23 thus leading to a sluggish reaction for producing ˙OH and thus inefficient pollutant removal. For addressing this issue, the catalytic ozonation process has been developed, in which the ozone is supplied to the vicinity of catalyst, accelerating the decomposition of ozone and thereby facilitating the formation of ˙OH. Previous studies have reported the feasible performance of metal oxides (e.g., MgO,24 Co3O4,25 MnO2,26 and CeO2 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27,28) for the application of the catalytic ozonation in water treatment due to their low toxicity. Different from other metal oxides, the reversible reduction of Ce4+ to Ce3+ will effectively tune the storage capacity of oxygen atoms in CeO2,29,30 which may decrease the activation energy barrier of ozone decomposition.
27,28) for the application of the catalytic ozonation in water treatment due to their low toxicity. Different from other metal oxides, the reversible reduction of Ce4+ to Ce3+ will effectively tune the storage capacity of oxygen atoms in CeO2,29,30 which may decrease the activation energy barrier of ozone decomposition.
Herein, we developed a CeO2 based catalytic ozonation system to facilitate the removal of DMP. The quantitative measurements of the active species confirmed that the steady state concentration of ˙OH was significantly higher than that of the ozonation system. The quenching experiments clarified the predominant contribution of ˙OH to the DMP removal. The optimized system can achieve ∼100% degradation efficiency of DMP and the total organic carbon (TOC) removal rate could achieve 44.16% under a neutral condition. Overall, this study provides insight into the application of catalytic ozonation for wastewater treatment.
2. Materials and methods
2.1. Chemical reagents
Dimethyl phthalate (C10H10O4, DMP, >99.0%), 5,5-dimethyl-1-pyrroline-N-oxide (C6H11NO, DMPO, >97.0%), potassium iodide (KI, >99%), dimethyl sulfoxide (DMSO, >99.7%) and acetylacetone (C5H8O2, >99%) were provided by Macklin (Shanghai). Sodium hydroxide (NaOH, >96.0%), tert-butanol (TBA, >99.5%), formaldehyde (HCHO, >99.5%) and chromatographic grade methanol (CH3OH, >99.9%) were purchased from Aladdin (Shanghai). Concentrated sulfuric acid (H2SO4, 98 wt%) was supplied by Sinopharm. Ultrapure water (>18.2 MΩ cm) used in this study was prepared from a Millipore Milli-Q water system. High-purity oxygen (O2, ≥99.9%) and the catalyst were provided by Chunyu Special Gas Company (Shanghai) and Shanmei Shuimei (Beijing), respectively. All chemicals applied in the experiments were at least of reagent grade and used as received without further purification.2.2. Experimental apparatus
As illustrated in Fig. 1, ozonation and catalytic ozonation experiments were both conducted in a plexiglass column reactor (internal diameter of 10 cm and effective volume of 1 L) with a titanium aerator at the bottom. In the batch test, O3 was generated from pure oxygen via the laboratory ozone generator (COM-AD-01, Germany) and continuously bubbled into the reactor containing 70 mg L−1 DMP solution. The catalyst was added into the reactor before introducing the steady ozone flow in the catalytic ozonation experiment. The real-time ozone concentration could be adjusted by the gas valve and measured by the ozone concentration detector (3S-J5000, China). The exhaust gas (excess ozone) was absorbed in a bottle filled with KI solution for preventing air pollution. The initial pH of DMP solution was 5.6. The H2SO4 and NaOH solutions (100 mM) were used to adjust the pH of solution. Water samples were collected at pre-determined intervals during the reaction and filtered with the 0.45 μm polyethersulfone filters. The degradation experiments were repeated twice under the same conditions.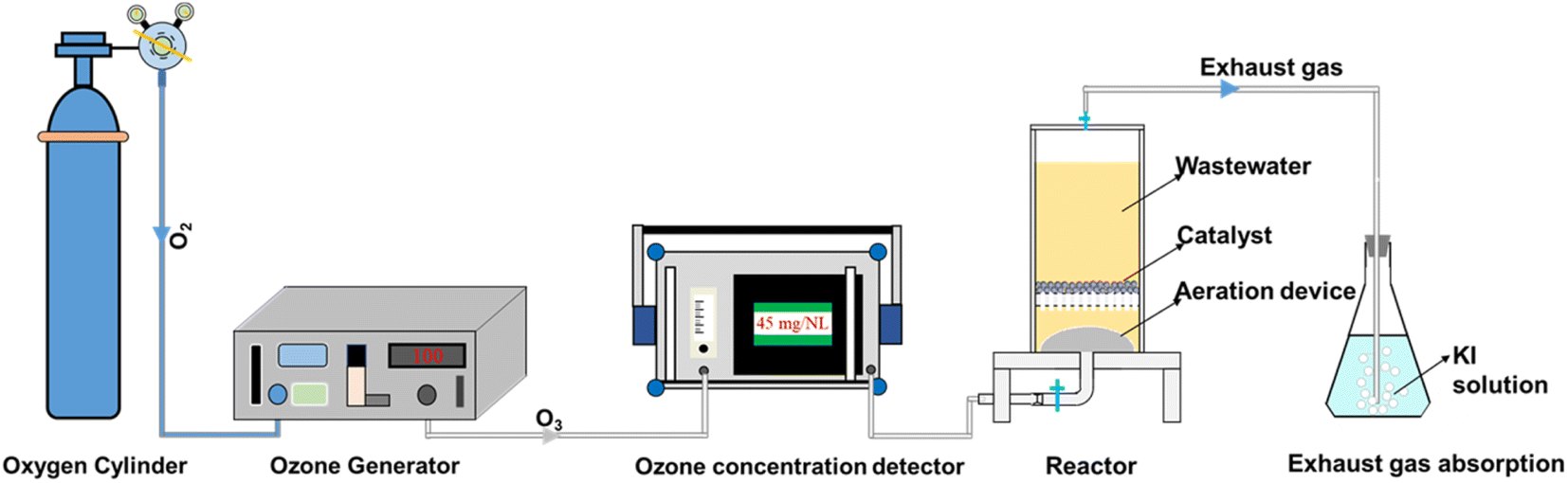 | ||
| Fig. 1 Experimental apparatus and the catalytic ozonation system. | ||
2.3. Characterization of the catalyst
The surface morphology and element composition of the catalyst were investigated by Field Emission Scanning Electron Microscopy (FESEM, ZEISS Gemini 300, Germany) equipped with EDS spectroscopy (OXFORD Xplore). X-ray diffraction (XRD, Bruker D8 Advance, Germany) was used to characterize the crystal structure of the catalyst. Elemental valence of the substance on the surface of catalyst was analyzed by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, America).2.4. Water quality analysis
The high-performance liquid chromatography (HPLC, Agilent 1200, Australia) equipped with an Agilent C18 column (150 mm × 4.6 mm, 5 μm) and an Agilent 1200 ultraviolet detector (VWD) was employed to quantify the concentration of DMP in water samples. Before the quantification analysis, the withdrawn water samples were filtered by a 0.45 μm polyethersulfone filter for removing solid impurities. In the testing process, the mobile phase was a 50/50 (v/v) methanol–water mixture with a constant flow rate of 1.2 mL min−1. The injection volume and test temperature were kept at 20 μL and 40 °C, respectively. The wavelength of the Agilent 1200 ultraviolet detector (VWD) was 274 nm.31 TOC of the water sample was measured by the total organic carbon analyzer (TOC-L, Shimadzu, Japan). The identification of reactive oxygen species (ROS) was conducted by electron paramagnetic resonance spectroscopy (EPR, Bruker EMX X-plus, Germany), using DMPO as spin-trapping agent. The test was carried out under the following conditions: microwave frequency = 9.854 GHz, modulation frequency = 100 kHz, sweep width = 100 G, modulation amplitude = 1 G, center field = 3500 G, static field = 3480.00 G, and microwave power = 20 MW. The quantification of hydroxyl radicals (˙OH) was determined by the Hantzsch reaction method, and the wavelength of UV-vis spectrophotometer was kept at 412 nm.32,33 The inductively coupled plasma optical emission spectrometer (ICP-OES, Agilent 5110, Australia) was used to characterize the concentration of dissolved metal ions after a certain reaction time. The pH of the reaction solution was measured by the pH meter (pH-2s, Hach, USA).3. Results and discussion
3.1. Characterization of the catalyst
The morphology of CeO2/Al2O3 catalyst is shown in Fig. 2. A large number of irregular rough structures could be observed on the surface of the catalyst. This composite structure could endow the catalyst with a larger specific surface area, which may provide large amounts of active sites for catalytic ozonation. The elemental mapping analysis (Fig. 2c–e) demonstrated that Al, O and Ce were the main elements of catalysts. The BET surface area and pore volume of the catalyst is 169.57 m2 g−1 and 0.44 cm3 g−1, respectively (Fig. 2f). The rough surface and porous structure could provide a number of active sites for the catalyst. Moreover, XRD tests (Fig. 2g) illustrated that the CeO2/Al2O3 catalyst had characteristic diffraction peaks located at 52.9°, 63.0° and 76.3°, which correspond to CeO2 based on JCPDS standard cards (card numbers: 00-078-0484). Besides, the peaks of 38.4°, 40.1° and 70.5° are associated with Al2O3 by referring to JCPDS standard cards (card numbers: 00-001-1305). The existing forms of Al and Ce on the surface of the catalyst were Al2O3 and CeO2, respectively. The XPS analysis revealed that the corresponding binding energy of Al 2p was 74.00 eV, which is consistent with the standard data (73.40 eV), indicating that the Al element was mainly in the form of +3 valence. Fig. 2i shows that the Ce element state on the surface of the catalyst is mainly in the form of +4 valence (with binding energy of Ce 3d3/2 and Ce 3d5/2), which is in line with the standard data of CeO2.34 The results of XPS are consistent with the XRD analysis.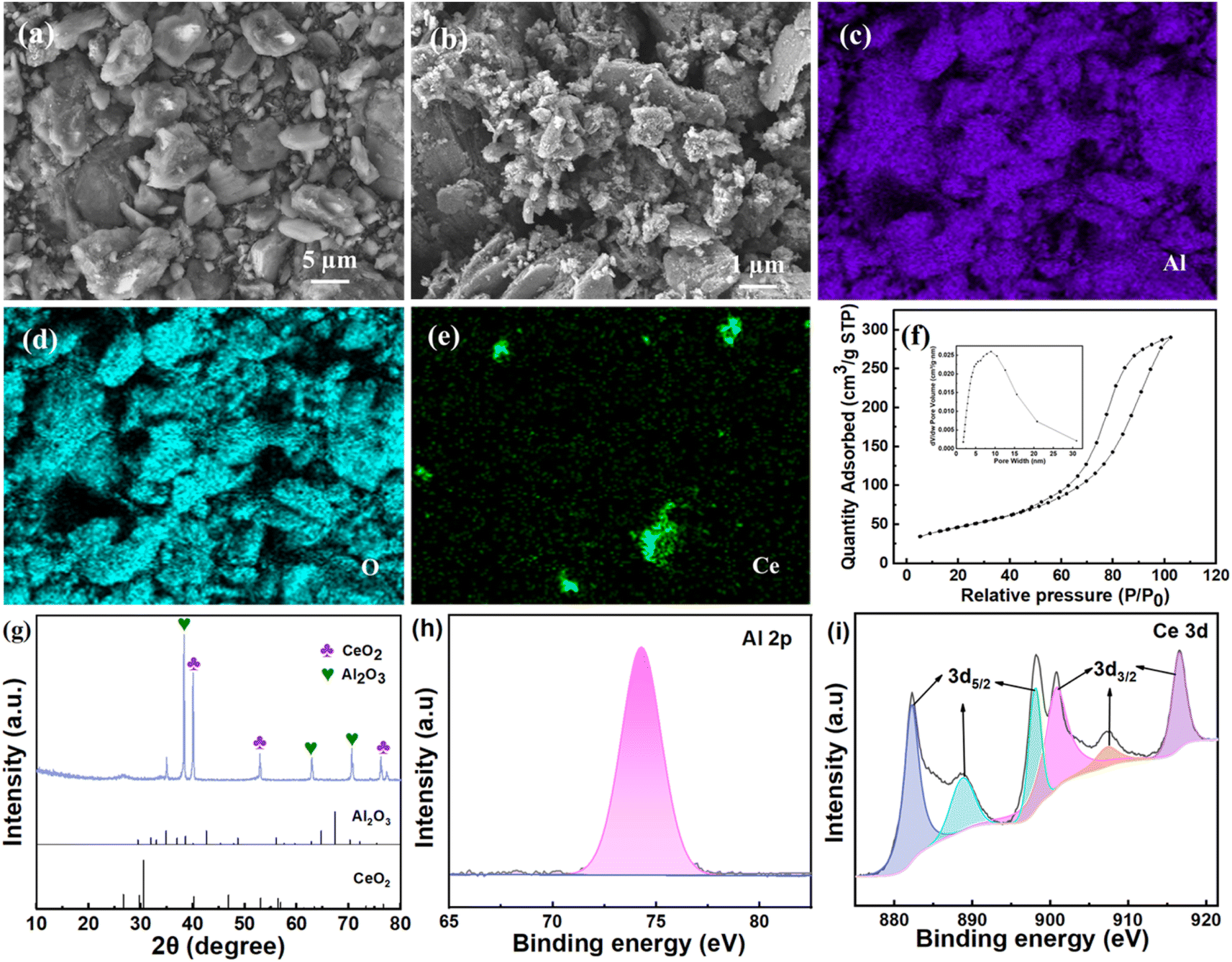 | ||
| Fig. 2 SEM images, elemental mappings, BET test, XRD patterns and XPS tests of the CeO2/Al2O3 catalyst. SEM image at 1000× magnification (a) and 8000× magnification (b); EDS mapping of Al (c), O (d) and Ce (e); BET test (f); XRD patterns (g); XPS tests (h) and (i). | ||
3.2. Optimization of catalytic reaction conditions
According to the previous reports, the performance of the catalytic ozonation system to degrade pollutant might be affected by catalyst dosage, ozone dosage and initial pH.35–37 As shown in Fig. 3a, the DMP removal shows a slight increase with the increase of catalyst dosage. The system can achieve ∼100% DMP removal after 20 min. The catalyst dosage exerts a negligible influence on the apparent rate constant (kapp) of DMP degradation (Fig. 3b). It is worth noting that catalyst dosage has a significant effect on the mineralization of DMP.35 For instance, the TOC removal increased from 26.28% at the dosage of 0 g L−1 to 45.09% at the dosage of 50 g L−1 (Fig. 3c). Furthermore, the catalytic ozonation system achieved the optimized performance for the mineralization of DMP when the dosage of catalyst was 30 g L−1.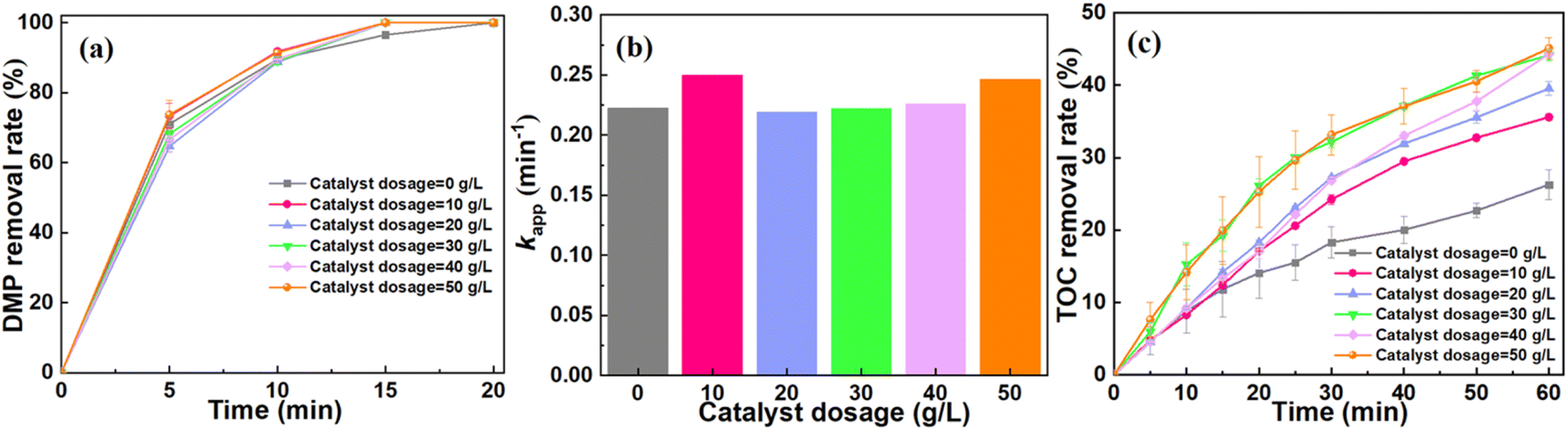 | ||
| Fig. 3 Effect of catalyst dosage on DMP removal efficiency. (a) The DMP removal rate under the conditions of different catalyst dosages; (b) the kapp of DMP degradation at different catalyst dosages; (c) the removal rate of TOC at different catalyst dosages. Experimental conditions: ozone dosage = 22.5 mg min−1, pH = 6, [DMP]0 = 70 mg L−1. | ||
Based on these results, we hypothesized that the degradation of DMP highly depended on the direct oxidation process of O3. During this process, the structure of DMP might be changed when the DMP molecules were oxidized by the dissolved ozone. Since ozone has low solubility in water, the by-product of the DMP oxidation such as formic acid and malonic acid will be difficult to remove.1 For the mineralization of DMP, it might be attributed to the indirect oxidation process. The catalyst enhanced the conversion of O3 to ROS, facilitating the mineralization of DMP.
To further explore the oxidation performance of catalytic ozone system, the batch tests under different ozone flow rates (7.5 mg min−1, 15 mg min−1, 22.5 mg min−1, 30 mg min−1) were conducted. As shown in Fig. 4a and b, the ultrafast degradation of DMP can be achieved with the increase of ozone flow rate. Specifically, the kapp increased from 0.069 min−1 at the flow rate of 7.5 mg min−1 to 0.295 min−1 at the flow rate of 30 mg min−1. Fig. 4c illustrates the impact of ozone flow rate on the TOC removal. After 60 min, the TOC removal achieved 44.2% when the ozone flow rate increased to 22.5 mg min−1. This result is in agreement with the previous studies, which is related to the enhanced mass transfer and conversion under the sufficient ozone supply.38 The effect of pH on the catalytic ozonation was further investigated. As shown in Fig. S1,† this system exhibited the promising DMP degradation in a wider pH range (pH = 5–10) compared to literature (pH = 7–10).39
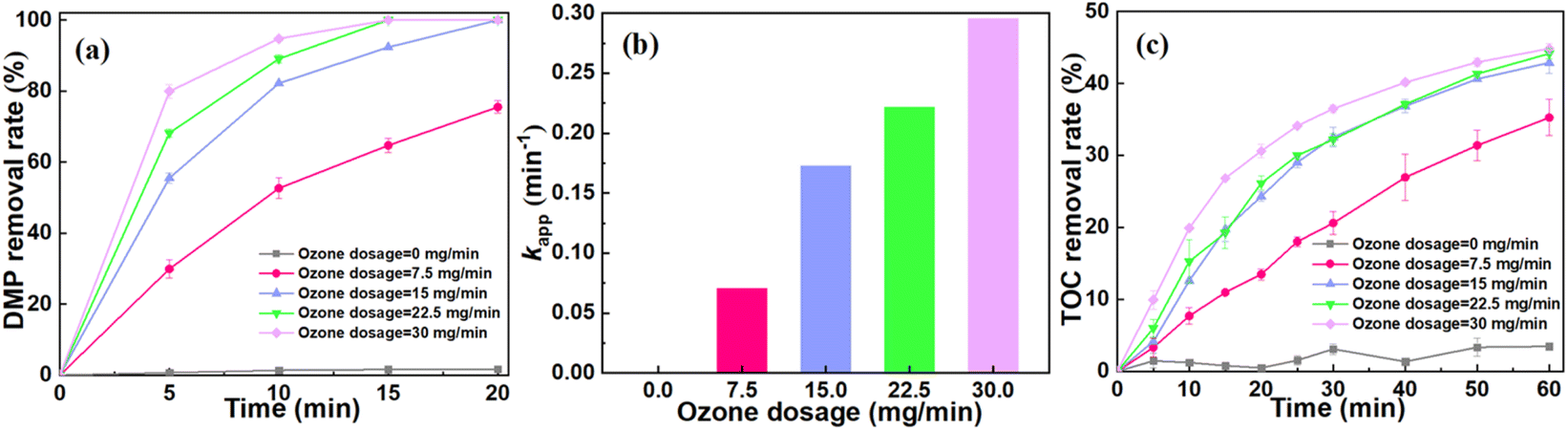 | ||
| Fig. 4 Effect of ozone dosage on DMP removal efficiency. (a) The DMP removal rate under the conditions of different ozone dosages; (b) the kapp of DMP degradation at different ozone dosages; (c) the removal rate of TOC in the system at different ozone dosages. Experimental conditions: catalyst dosage = 30 g L−1, pH = 6, [DMP]0 = 70 mg L−1. | ||
Furthermore, the catalytic ozonation system in this study shows a superior higher DMP degradation efficiency than most catalytic systems documented elsewhere.1,40–42 As shown in Table S1,† the catalytic ozonation system can achieve ∼100% DMP removal at a high concentration (70 mg L−1) with a short hydraulic retention time (within 15 min). The CeO2/Al2O3 catalyst in this study exhibited a better performance in degrading organic matters.
3.3. Determination of the active species
To validate the ROS in the catalytic ozonation system, EPR tests under different quenching reagents (e.g., methanol and TBA) were conducted. As illustrated in Fig. 5a, there was no DMPO-OH characteristic peak in the ozonation system without introducing catalyst, confirming the ineffective conversion of O3 to ˙OH. In contrast to the ozonation system, the characteristic peaks with an intensity ratio of 1:2:1:2:1:2:1 were clearly detected in the catalytic ozonation system, which can be attributed to DMPOX (5,5-dimethyl-1-pyrrolidone-2-oxyl).36,43 The high concentration of ˙OH may facilitate the overoxidation of DMPO-OH, resulting in the formation of DMPOX (Fig. S2†). After the introduction of TBA, the characteristic peaks disappeared. Similarly, there were no characteristic peaks in the methanol-added solution which showed undetected concentration of O2˙− in the catalytic system.44 These results suggested that the ˙OH should be the dominant ROS in the catalytic ozonation system rather than that of O2˙−.
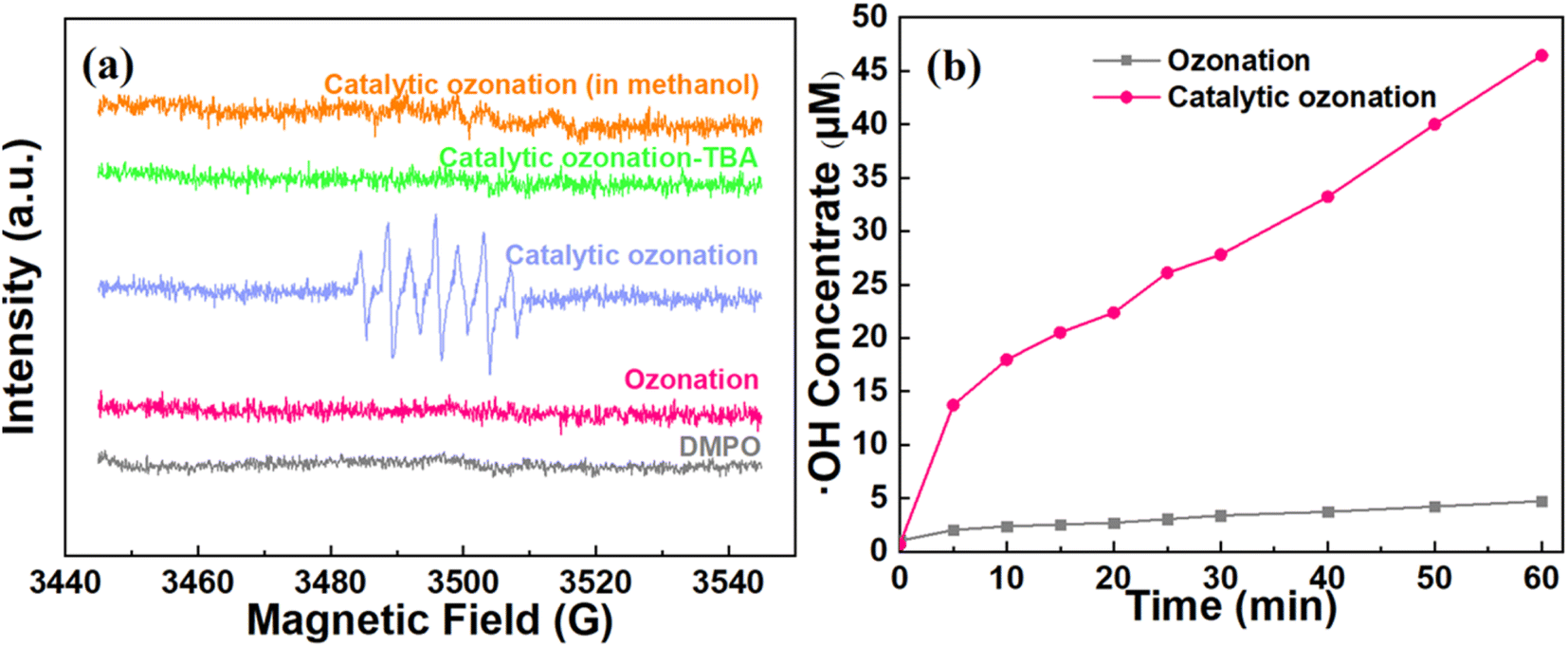 | ||
| Fig. 5 Qualitative and quantitative tests of the active species in the system. (a) DMPO spin-trapping EPR spectra for the catalytic ozonation system. Experimental conditions: catalyst dosage = 30 g L−1, ozone dosage = 22.5 mg min−1, [DMPO]0 = 0.22 M, DMPO dosage = 20 μL, sample volume = 20 μL. (b) Quantitative test for ˙OH concentrations generated in different systems. Experimental conditions: catalyst dosage = 30 g L−1, ozone dosage = 22.5 mg min−1, pH = 6. | ||
Since the ˙OH possesses a strong oxidation ability towards organic matters in the wastewater (oxidation–reduction potential = 2.80 V),45 the quantitative measurements were used to analyze the concentration of ˙OH in the ozonation and catalytic ozonation systems (Text S1†). Fig. 5b shows that the ˙OH concentration in the catalytic ozonation system (46.44 μM) is significantly higher than that of the ozonation system (4.75 μM) at 60 min. It confirmed the ultrafast conversion of O3 to ˙OH (0.774 μM min−1) on the surface of CeO2 based ceramic catalyst compared to that in ozonation system (0.08 μM min−1). These results supported that the system could achieve an efficient heterogeneous catalytic ozonation for the ultrafast conversion of O3 to ˙OH.
3.4. Quenching experiments
For further clarifying the contribution of ˙OH to the degradation of DMP, the quenching experiments were conducted. The tert-butanol (TBA) was used as the scavenger (k˙OH, TBA = 3.8–7.6 × 108 M−1 s−1),36 and the reaction rate constant between ˙OH and TBA is much higher than that with bicarbonate (k˙OH, bicarbonate = 8.5 × 106 M−1 s−1).1 Therefore, in this quenching experiment, we chose TBA as the scavenger of ˙OH. The DMP removal rate was inhibited in both systems after the TBA was introduced into the system (Fig. 6a and b). For example, the DMP removal was ∼100% in both ozonation and catalytic ozonation systems, respectively, after 60 min oxidation reaction. Furthermore, the catalytic ozonation system still exhibited a considerable performance for removing DMP during the quenching experiments. This may be attributed to the fact that TBA can predominantly react with the free ˙OH in bulk solution rather than the ˙OH generated on the surface of catalyst.12,46 These results indicated that the interfacial oxidation on the catalyst surface was a predominant process to remove DMP.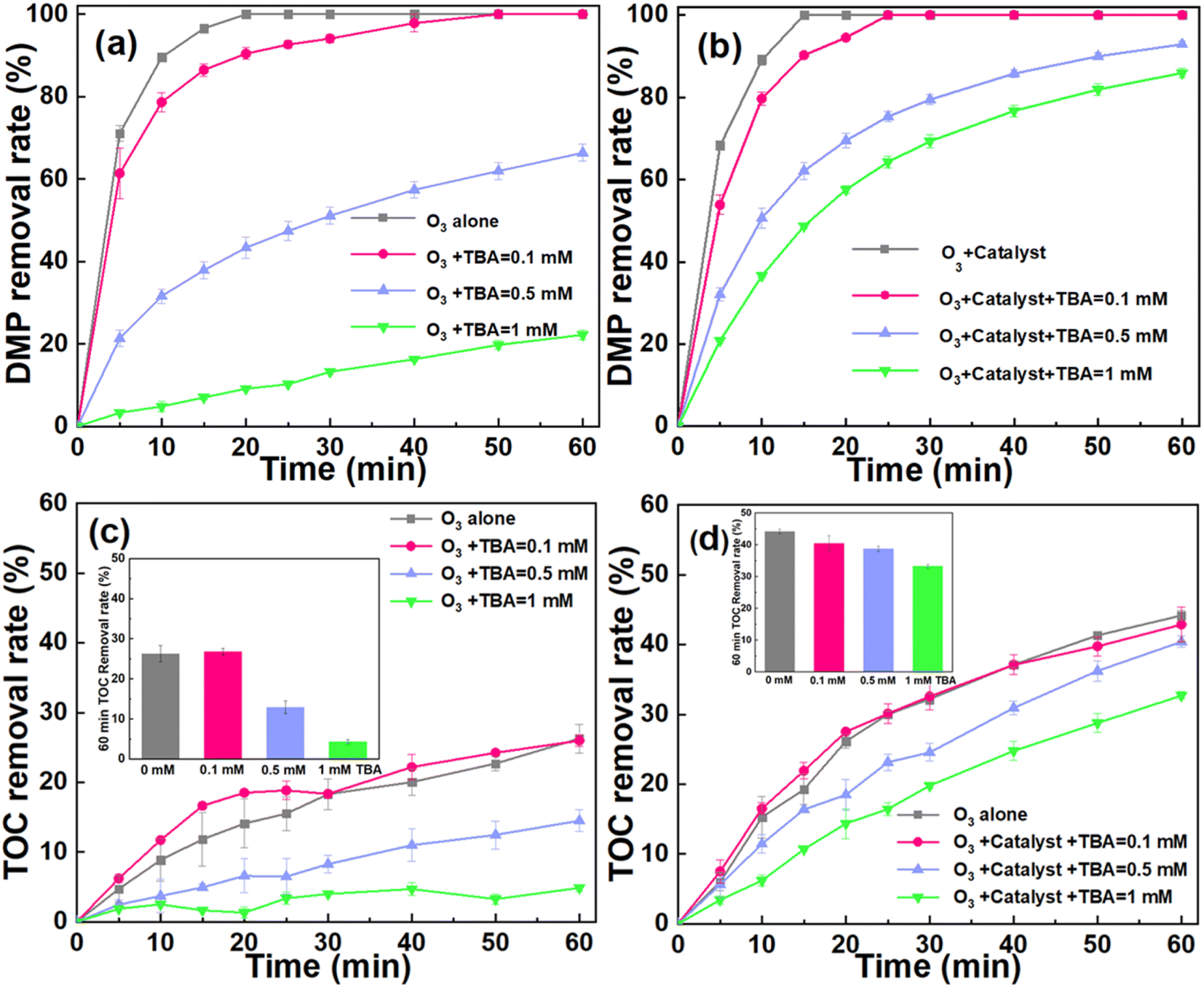 | ||
| Fig. 6 The effect of adding TBA on the DMP degradation in ozonation and catalytic ozonation systems. (a) DMP removal rate in ozonation system (a) and catalytic ozonation system (b). TOC removal rate in ozonation system (c) and catalytic ozonation system (d). Experimental conditions: catalyst dosage = 30 g L−1, ozone dosage = 22.5 mg min−1, pH = 6, [DMP]0 = 70 mg L−1. | ||
As illustrated in Fig. 6c and d, the TOC removal was suppressed in both systems during the TBA quenching experiments. For instance, the TOC removal of the ozonation and catalytic ozonation systems were 26.28% and 44.16% at 60 min, respectively. After introducing TBA (1 mM), the TOC removal decreased to 4.39% in the ozonation system and 32.37% in the catalytic ozonation system. The quenching of ˙OH greatly reduces the catalytic system's ability to mineralize organic pollutants. Moreover, ozone was unable to mineralize TBA (Fig. S4†). Therefore, the addition of TBA had no effect on the removal of TOC. These results further confirmed that the ˙OH generated on the surface of catalyst played the dominant role in the oxidation of DMP.
3.5. Stability of catalyst and the effect of leached ions
The ICP test was conducted to measure the stability of the catalyst after 60 min reaction. The result showed that the aluminum ion concentration was 0.76 mg L−1. Meanwhile, the cerium ion was undetectable. These results suggested that the CeO2 based catalyst possessed the durability during the catalytic ozonation process. Considering that the aluminum ions can be leached from catalyst, the effect of aluminum ions on the degradation of DMP was further explored.47 Fig. 7a and b illustrates the DMP and TOC removal among the ozonation system, ozonation system introduced by aluminum ions (0.76 mg L−1), and catalytic ozonation system. It is worth noting that the DMP and TOC removal of the ozone system introduced by aluminum ions were similar to that of the ozone system. Thus, the leached aluminum ions had a negligible effect on DMP degradation in catalytic ozonation system. Therefore, aluminum oxide was used as the substrate while the loaded cerium may contribute to the catalytic ozonation process.11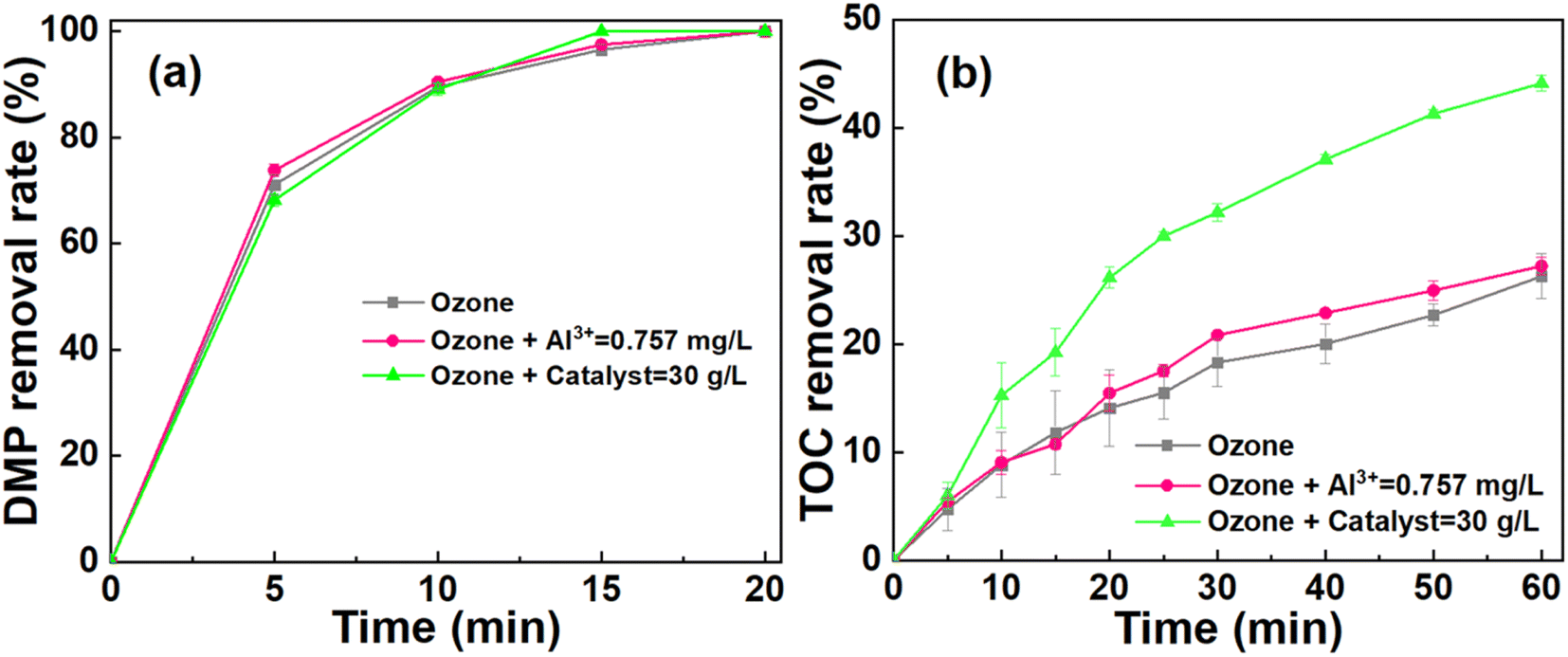 | ||
| Fig. 7 The effect of dissolved ions on DMP degradation. (a) The effect on DMP removal rate. (b) The effect on TOC removal rate. Experimental conditions: catalyst dosage = 30 g L−1, ozone dosage = 22.5 mg min−1, pH = 6, [DMP]0 = 70 mg L−1, [Al3+]0 = 0.757 mg L−1. | ||
3.6. Mechanisms of DMP degradation in the catalytic system
The results of batch tests and quenching experiments confirmed the proposed ozone oxidation process, which could be divided into two pathways of direct ozonation and ˙OH-mediated indirect oxidation. It should be noted that the ozone could act as the oxidant and directly oxidize a small amount of DMP to CO2 and H2O. In this study, the ozonation system can achieve ∼100% DMP removal after 20 min and 26.28% TOC removal rate without catalyst (Fig. 3a and c). Part of DMP was oxidized and mineralized. However, this process is limited by the low solubility and low redox potential (oxidation–reduction potential of 2.07 V) of ozone.48 Compared to the direct ozonation process, the ˙OH-mediated indirect oxidation was more important for DMP degradation due to the stronger oxidative ability of ˙OH (oxidation–reduction potential of 2.80 V).49 In Section 3.5 about the effect of leached ions, aluminum was proved to have no promoting effect in catalytic ozonation process. Therefore, the cerium oxide is the active material. As shown in Fig. 8, the electron transfer process through redox couples of Ce4+ and Ce3+ in the CeO2 could enhance ozone decomposition and ˙OH formation due to the high oxygen storage capacity and oxygen release capacity of CeO2.50,51 Via quenching experiments, the interfacial ˙OH (˙OHI) was verified to govern the DMP removal and mineralization while free ˙OH (˙OHF) made a less significant contribution to the catalytic process. As such, the interfacial ˙OH generated by the CeO2 active layer played an important role in the DMP degradation.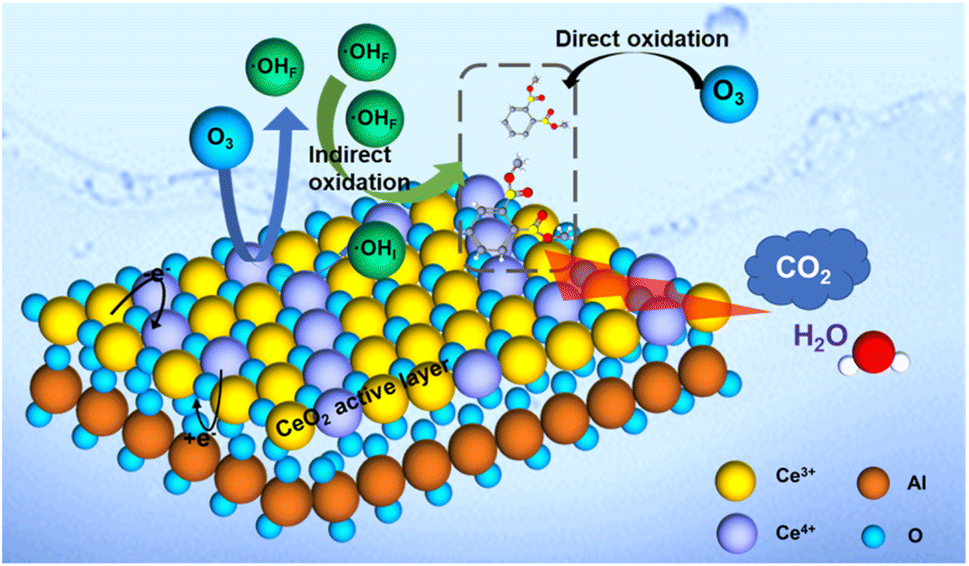 | ||
| Fig. 8 The proposed mechanism of the DMP removal in the catalytic ozonation system. | ||
4. Conclusions
In this work, a catalytic ozonation system was utilized to remove a refractory organic matter (DMP) from wastewater. In a wider pH range of 5–10, the system showed a stable degradation efficiency towards the DMP. Especially in the circumneutral condition of pH = 6, DMP could be completely removed from wastewater within 15 min when 30 g L−1 catalyst dosage and 22.5 mg min−1 ozone flow rate. Meanwhile, the TOC removal of this system could achieve 44.16%. EPR analysis and quantitative tests confirmed the efficient conversion of O3 to ˙OH. The free radicals quenching experiments verified that the interfacial ˙OH generated by the catalyst governed the DMP deterioration in the catalytic ozonation system. These results supported that the CeO2 based catalyst has a desirable potential for improving the heterogeneous catalytic ozonation in water treatment and environmental remediation.Author contributions
Jia Yuan: data curation, software, formal analysis, writing – original draft, writing – review & editing. Yang Li: visualization, supervision, validation, writing – review & editing. Yun Guo: visualization, writing – review & editing, supervision. Zhiwei Wang: conceptualization, supervision, resources, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the National Key Research and Development Program of China (grant 2019YFC0408200) for the financial support of the work.References
- Y. Liu, D. Wu, S. Peng, Y. Feng and Z. Liu, Sep. Purif. Technol., 2019, 209, 588–597 CrossRef CAS.
- X. Pang, N. Skillen, N. Gunaratne, D. W. Rooney and P. K. J. Robertson, J. Hazard. Mater., 2021, 402, 123461 CrossRef CAS PubMed.
- A. Paluselli, V. Fauvelle, F. Galgani and R. Sempere, Environ. Sci. Technol., 2019, 53, 166–175 CrossRef CAS.
- M. Dolatabadi, T. Świergosz and S. Ahmadzadeh, Sci. Total Environ., 2021, 772, 145323 CrossRef CAS.
- H. Zhao, Y. Chen, Q. Peng, Q. Wang and G. Zhao, Appl. Catal., B, 2017, 203, 127–137 CrossRef CAS.
- X. Zhang, M. Feng, R. Qu, H. Liu, L. Wang and Z. Wang, Chem. Eng. J., 2016, 301, 1–11 CrossRef CAS.
- J. Rodríguez, L. Castrillón, E. Marañón, H. Sastre and E. Fernández, Water Res., 2004, 38, 3297–3303 CrossRef.
- A. E. Ameh, O. O. Oyekola and L. F. Petrik, J. Cleaner Prod., 2022, 338, 130571 CrossRef CAS.
- P. Roslev, K. Vorkamp, J. Aarup, K. Frederiksen and P. H. Nielsen, Water Res., 2007, 41, 969–976 CrossRef CAS.
- M. J. Luján-Facundo, M. I. Iborra-Clar, J. A. Mendoza-Roca and M. I. Alcaina-Miranda, J. Cleaner Prod., 2019, 238, 117866 CrossRef.
- D. Wu, Y. Liu, H. He and Y. Zhang, Chemosphere, 2016, 155, 127–134 CrossRef CAS PubMed.
- X. Li, W. Chen, L. Ma, Y. Huang and H. Wang, J. Cleaner Prod., 2019, 222, 174–181 CrossRef CAS.
- X.-K. Zhao, G.-P. Yang, Y.-J. Wang and X.-C. Gao, J. Photochem. Photobiol., A, 2004, 161, 215–220 CrossRef CAS.
- L. Lupa, L. Cocheci, R. Pode and I. Hulka, Sep. Purif. Technol., 2018, 196, 82–95 CrossRef CAS.
- C. Na, Y. Zhang, X. Quan, S. Chen, W. Liu and Y. Zhang, J. Hazard. Mater., 2017, 338, 186–193 CrossRef CAS PubMed.
- G.-P. Yang, X.-K. Zhao, X.-J. Sun and X.-L. Lu, J. Hazard. Mater., 2005, 126, 112–118 CrossRef CAS.
- L. Wang, B. Li, D. D. Dionysiou, B. Chen, J. Yang and J. Li, Environ. Sci. Technol., 2022, 56, 3386–3396 CrossRef CAS.
- Y. Qi, Y. Mei, J. Li, T. Yao, Y. Yang, W. Jia, X. Tong, J. Wu and B. Xin, Chem. Eng. J., 2019, 373, 1158–1167 CrossRef CAS.
- H. Zeng, G. Zhang, Q. Ji, H. Liu, X. Hua, H. Xia, M. Sillanpaa and J. Qu, Environ. Sci. Technol., 2020, 54, 14725–14731 CrossRef CAS.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- B. Zhu, G. Jiang, S. Chen, F. Liu, Y. Wang and C. Zhao, Chem. Eng. J., 2022, 430, 132843 CrossRef CAS.
- M. Izadifard, G. Achari and C. H. Langford, Chemosphere, 2018, 197, 535–540 CrossRef CAS PubMed.
- H. Mohebali, G. Moussavi, M. Karimi and S. Giannakis, Chem. Eng. J., 2022, 434, 134614 CrossRef CAS.
- X. Zhang, T. Shen, Y. Ding and S. Tong, Ozone: Sci. Eng., 2019, 41, 541–550 CrossRef CAS.
- Z. Zhang, H. Ai, M.-L. Fu, Y.-b. Hu, J. Liu, Y. Ji, V. Vasanthakumar and B. Yuan, Chem. Eng. J., 2021, 418, 129461 CrossRef CAS.
- J. Ma, S. Zhang, X. Duan, Y. Wang, D. Wu, J. Pang, X. Wang and S. Wang, Chemosphere, 2021, 267, 129287 CrossRef CAS PubMed.
- S. Xing, X. Lu, J. Liu, L. Zhu, Z. Ma and Y. Wu, Chemosphere, 2016, 144, 7–12 CrossRef CAS.
- J. Akhtar, N. S. Amin and A. Aris, Chem. Eng. J., 2011, 170, 136–144 CrossRef CAS.
- A. Trovarelli and J. Llorca, ACS Catal., 2017, 7, 4716–4735 CrossRef CAS.
- H. Ha, S. Yoon, K. An and H. Y. Kim, ACS Catal., 2018, 8, 11491–11501 CrossRef CAS.
- Y. Nie, C. Hu, J. Qu and X. Zhao, Appl. Catal., B, 2009, 87, 30–36 CrossRef CAS.
- S. Dominguez, P. Ribao, M. J. Rivero and I. Ortiz, Appl. Catal., B, 2015, 178, 165–169 CrossRef CAS.
- T. Nash, Biochem. J., 1953, 55, 416–421 CrossRef CAS PubMed.
- D. A. Al Farraj, A. M. Al-Mohaimeed, R. M. Alkufeidy and N. A. Alkubaisi, Colloid Interface Sci. Commun., 2021, 41, 100375 CrossRef CAS.
- X. Li, W. Chen, L. Ma, H. Wang and J. Fan, Chemosphere, 2018, 195, 336–343 CrossRef CAS.
- Z. Sun, L. Zhao, C. Liu, Y. Zhen and J. Ma, Environ. Sci. Technol., 2019, 53, 10342–10351 CrossRef CAS.
- L. Li, W. Ye, Q. Zhang, F. Sun, P. Lu and X. Li, J. Hazard. Mater., 2009, 170, 411–416 CrossRef CAS PubMed.
- H. Chen and J. Wang, Chemosphere, 2019, 234, 14–24 CrossRef CAS.
- Z. Yan, J. Zhu, X. Hua, D. Liang, D. Dong, Z. Guo, N. Zheng and L. Zhang, J. Cleaner Prod., 2020, 272, 122856 CrossRef CAS.
- Z. Bai, Q. Yang and J. Wang, Environ. Technol., 2017, 38, 2048–2057 CrossRef CAS.
- Y. Tang, Z. Pan and L. Li, J. Colloid Interface Sci., 2017, 508, 196–202 CrossRef CAS PubMed.
- H. Kan, T. Wang, Z. Yang, R. Wu, J. Shen, G. Qu and H. Jia, J. Hazard. Mater., 2020, 383, 121185 CrossRef CAS.
- C. Zhang, D. He, J. Ma and T. D. Waite, Water Res., 2018, 145, 220–230 CrossRef CAS.
- H. Yan, P. Lu, Z. Pan, X. Wang, Q. Zhang and L. Li, J. Mol. Catal. A: Chem., 2013, 377, 57–64 CrossRef CAS.
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- S.-Q. Tian, J.-Y. Qi, Y.-P. Wang, Y.-L. Liu, L. Wang and J. Ma, Water Res., 2021, 193, 116860 CrossRef CAS.
- S. Peng, Y. Feng, Y. Liu and D. Wu, Chemosphere, 2018, 212, 438–446 CrossRef CAS.
- A. Fischbacher, J. von Sonntag, C. von Sonntag and T. C. Schmidt, Environ. Sci. Technol., 2013, 47, 9959–9964 CrossRef CAS PubMed.
- M. Kermani, B. Kakavandi, M. Farzadkia, A. Esrafili, S. F. Jokandan and A. Shahsavani, J. Cleaner Prod., 2018, 192, 597–607 CrossRef CAS.
- C. A. Orge, J. J. M. Órfão, M. F. R. Pereira, A. M. Duarte de Farias, R. C. R. Neto and M. A. Fraga, Appl. Catal., B, 2011, 103, 190–199 CrossRef CAS.
- C. A. Orge, J. J. M. Órfão, M. F. R. Pereira, A. M. Duarte de Farias and M. A. Fraga, Chem. Eng. J., 2012, 200–202, 499–505 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05048j |
| ‡ These two authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2022 |