DOI:
10.1039/D2RA05239C
(Paper)
RSC Adv., 2022,
12, 28608-28622
Quantum chemical modification of indaceno dithiophene-based small acceptor molecules with enhanced photovoltaic aspects for highly efficient organic solar cells†
Received
21st August 2022
, Accepted 29th September 2022
First published on 7th October 2022
Abstract
In this computational work, with the aim of boosting the ultimate efficiency of organic photovoltaic cells, seven small acceptors (IDST1–IDST7) were proposed by altering the terminal-acceptors of reference molecule IDSTR. The optoelectronic characteristics of the IDSTR and IDST1–IDST7 molecules were investigated using the MPW1PW91/6-31G(d,p) level of theory, and solvent-state computations were examined using time-dependent density functional theory (TD-DFT) simulation. Nearly all the investigated photovoltaic aspects of the newly proposed molecules were found to be better than those of the IDSTR molecule e.g. in comparison to IDSTR, IDST1–IDST7 exhibit a narrower bandgap (Egap), lower first excitation energy (Ex), and a significant red-shift in the absorbance maxima (λmax). According to the findings, IDST3 has the lowest Ex (1.61 eV), the greatest λmax (770 nm), and the shortest Egap (2.09 eV). IDST1–IDST7 molecules have higher electron mobility because their RE of electrons is less than that of IDSTR. Hole mobility of IDST2–IDST7 is higher than that of the reference owing to their lower RE for hole mobility than IDSTR. By coupling with the PTB7-Th donor, the open circuit voltage (VOC) of the investigated acceptor molecules (IDSTR and IDST1–IDST7) was calculated and investigation revealed that IDST4–IDST6 molecules showed higher VOC and fill factor (FF) values than IDSTR molecules. Accordingly, the modified molecules can be seriously evaluated for actual use in the fabrication of OSCs with enhanced photovoltaic and optoelectronic characteristics in light of the findings of this study.
1 Introduction
A lot of work in the areas of novel material research and device technology has led to significant breakthroughs in organic solar cells (OSCs).1,2 Different active layer materials, color customization, being light-weight, their transparency, and their flexibility are just a few of the many benefits of OSCs over silicon solar cells. Attributed to these benefits, OSCs are now considered among the technologies with the most potential for converting solar energy. The donor–acceptor mix in the photo-active layer influences the device performance of OSCs that are bulk heterojunction (BHJ) types.3,4 Fullerene acceptors (FAs) encounter a few significant issues that hinder the device's performance from improving any further: problems with chemical alteration, limited energy level tenability,5 structural instability as well as non-significant UV-visible and near-infrared (NIR) absorption.6,7 Non-fullerene acceptors (NFAs) offer a greater capacity to address these issues than FAs.1,8,9 With comprehensive spectrum coverage, great synthesizing adaptability, minimal voltage loss, and adjustable optoelectronic aspects, NFAs were able to achieve power conversion efficiency (PCE) of over 17%.10–14
It is widely acknowledged that a deeper comprehension of the link between structure and performance is necessary to boost PCEs and the ultimate growth of OSCs. This is due to the fact that slight changes in molecular structures frequently lead to different BHJ variations, including changes in molecular configurations and van der Waals interactions. These variations may have complex effects on exciton/charge behaviors as well as general efficiencies. In order to get a more comprehensive understanding of designing proficient OSCs, a thorough structural modification of small molecule acceptors (SMAs) is still greatly sought.
Several investigations have shown that decreasing the bandgap (Egap) may be achieved by enhancing the electron donating capacity of the donor component by expanding the efficient conjugate part.15 One simple way to accomplish this objective is to extend the core by increasing the fused-rings to extend conjugation in the core that is the donor part of molecule.16,17 However, this method didn't extend the spectrum of absorbance as anticipated, and flaws were found, including low solubility and time-consuming synthesis and purifying stages.18 On the other hand, these issues may be avoided by including heteroatom-based π-bridges into the conjugate system in order to lengthen it through non-covalent forces present within a molecule.19–21
Polymeric donor and NFAs photovoltaic compounds have benefited from the promising consideration of the asymmetric technique.22–24 Consequently, several studies have been reported that include the addition of π-bridges in the molecules in an asymmetric way.25,26 Recent research introduced a single bridge of alkythio-linked thiophene into an extremely crystalline indaceno dithiophene based small molecule (ID-4F). The conjugation was increased and the NFA's Egap was dramatically decreased because of the non-covalent forces (oxygen–sulfur and sulfur–sulfur) and ability to donate electrons in a significant way via these π-bridges. High energy levels and a redshift in the absorption spectrum, improved short circuit current (JSC) as well as 14.3% PCE of the resultant acceptor molecule (IDST-4F) were achieved.25
In the present investigation, indaceno dithiophene and alkythio-linked thiophene bridge-based molecule (IDST-4F) is used as the reference molecule (IDSTR), with the exception that the alkyl side chains have been swapped out with simple methyl groups to save computational costs. The donor core of IDSTR molecule is consist of 4,4,9,9-tetramethyl-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene and 3-methylsulfanyl-thiophene bridge is present on one side of the core of molecule making overall an asymmetric geometry of molecule. In this research, we have regarded both indaceno dithiophene and methylsulfanyl-thiophene units as the donor component of the molecule, as the scientists who designed the bridge that was added to the ID-4F molecule to boost the electron-donating function of the donor core unit. The donor portion of IDSTR is further attached to the molecules' ends by 2-(5,6-difluoro-2-methylene-3-oxo-indan-1-ylidene)-malononitrile units, forming an acceptor–donor–acceptor (A–D–A) type molecule. We have proposed seven new acceptor molecules (IDST1–IDST7) by swapping out the ending acceptor component of the IDSTR molecule with seven different acceptor groups. 1-Dicyanomethylene-6-methyl-2-methylene-3-oxo-indan-5-carboxylic acid methyl ester; compound with methanedione (IDST1), 6-cyano-1-dicyanomethylene-2-methylene-3-oxo-indan-5-carboxylic acid methyl ester (IDST2), 1-dicyanomethylene-2-methylene-3-oxo-indan-5,6-dicarbonitrile (IDST3), 2-(2-methylene-3-oxo-2,3-dihydro-cyclopenta[b]naphthalen-1-ylidene)-malononitrile (IDST4), 2-(2-bromo-6-methylene-7-oxo-6,7-dihydro-1-thia-s-indacen-5-ylidene)-malononitrile (IDST5), 2-(6,7-difluoro-2-methylene-3-oxo-2,3-dihydro-cyclopenta[b]naphthalen-1-ylidene)-malononitrile (IDST6) and 2-(1-chloro-6-oxo-5,6-dihydro-cyclopenta[c]thiophen-4-ylidene)-malononitrile (IDST7) are the new acceptor groups of IDST1–IDST7 molecules respectively as shown in Fig. 1.
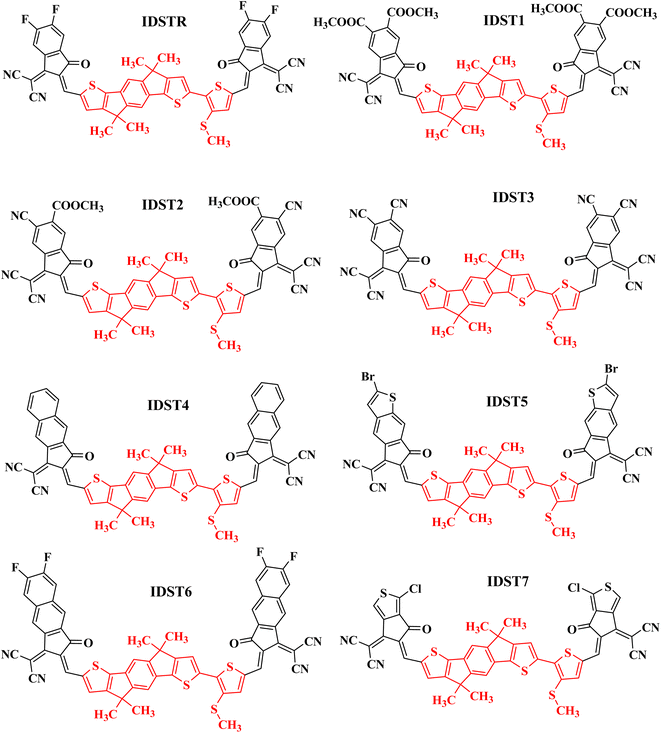 |
| | Fig. 1 ChemDraw structures of IDSTR and IDST1–IDST7 molecules, the red part indicating the donor and black part indicating acceptor site of the molecule. | |
2 Computational approaches
All molecular geometry calculations were done in Gaussian 09,27 and 3D molecular structures were modelled and viewed in GaussView 6.0.16.28 To aid the optimization of the IDSTR, DFT calculations were performed using the B3LYP,29 CAM-B3LYP,30 MPW1PW91,31 and ωB97XD32 functionals in conjunction with the 6-31G(d,p) basis set. Time-dependent density functional theory (TD-DFT)33 computations were employed to forecast IDSTR UV-visible absorption properties after the optimization of geometries and Integral Equation Formalism Polarizable Continuum Model (IEFPCM) model34 was for the investigation of the effect of solvent (chloroform).
To verify the feasibility of the theoretical approach, the maximum absorption (λmax) of IDSTR from four above mentioned functionals was verified with the experimental λmax provided in the literature. When comparing the practical λmax of IDSTR (728 nm)25 to the four computed absorption values i.e. 757 nm, 536 nm, 712 nm and 513 nm correspondingly, we found that the modified Perdew–Wang 1-parameter (MPW1PW91) functional was the closest to the empirical value25 indicating that the combination of this level of theory will be best for computing proposed molecules, as shown in Table 1.
Table 1 Maximum absorption of IDSTR using four different functionals and experimental λmax of IDSTR
| Molecule |
B3LYP |
CAM-B3LYP |
MPW1PW91 |
wB97XD |
Exp. λmax |
| IDSTR |
757 nm |
536 nm |
712 nm |
513 nm |
728 nm |
The molecules' UV-visible spectra were generated with the help of Origin 6.0.35 Transition density matrix (TDM) measurements were transformed into visualizations of movement and interaction of excitons using Multiwfn software.36 PyMOlyze 1.1 (ref. 37) was employed to create the density of states (DOS) graphs.38 Marcus theory can be used to examine the extent of charge mobility, both intermolecularly and intramolecularly, by analyzing the reorganization energy (RE).39,40 Recent efforts of ours, however, have centred on the intramolecular charge transfer (ICT). RE is the sum of two reorganizational forces, one external and one internal. Rapid fluctuations in the outer environment and polarisation alterations during charge transport are in external RE,41 whereas the internal RE mainly depends on molecular structural variation. External RE cannot support our calculations, hence for the sake of our research, we only concentrate on internal RE. Using the given eqn (1) and (2), the RE for the mobility of hole (λ+) and electron (λ−) was calculated.
| | |
λ+ = [E0+ − E0] + [E+0 − E+]
| (1) |
| | |
λ− = [E0− − E0] + [E−0 − E−]
| (2) |
E+0 and
E−0 are the cation and anion ground state energies calculated from ground state optimized neutral molecule.
E+ and
E− are the energies of cation and anion computed from cation and anion optimized models.
E0+ and
E0− and are the neutral molecules' energies, that are calculated utilizing optimized cation and anion geometries.
E0 is the ground state energy of molecule neutrally optimized.
42
3 Results and discussion
3.1. Structural optimization
Optoelectronic characteristics are strongly influenced by molecular structure.43 The reference molecule (IDSTR) and the recently proposed molecules (IDST1–IDST7) have been optimized using the DFT functional that was picked as the best fit as represented in Fig. 2. An effective charge transfer is possible due to the molecules' extended conjugation, which extends from the center to the ending regions of the molecule showing that charge transport in these structures is robust due to the π-electrons delocalization. The dihedral angles (θ°) and the bond length (Lb) were calculated to evaluate the level of conjugation as well as planarity of IDSTR and IDST1–IDST7 molecules (Table 2). Double and single carbon bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C) have lengths of 1.34 Å and 1.54 Å, respectively. All the compounds investigated (IDSTR and IDST1–IDST7) had a Lb of around 1.41 Å, it demonstrates increased charge-transfer properties and permits the conjugation-mediated delocalization of π-electrons. All of the molecules that were investigated have θ° that fall within the range of 0.480°–0.669°. This indicates that all of the molecules have a planar geometry, as there is no twist in between end-capped acceptor and donor core, and there are also no groups that cause a hindrance.
C and C–C) have lengths of 1.34 Å and 1.54 Å, respectively. All the compounds investigated (IDSTR and IDST1–IDST7) had a Lb of around 1.41 Å, it demonstrates increased charge-transfer properties and permits the conjugation-mediated delocalization of π-electrons. All of the molecules that were investigated have θ° that fall within the range of 0.480°–0.669°. This indicates that all of the molecules have a planar geometry, as there is no twist in between end-capped acceptor and donor core, and there are also no groups that cause a hindrance.
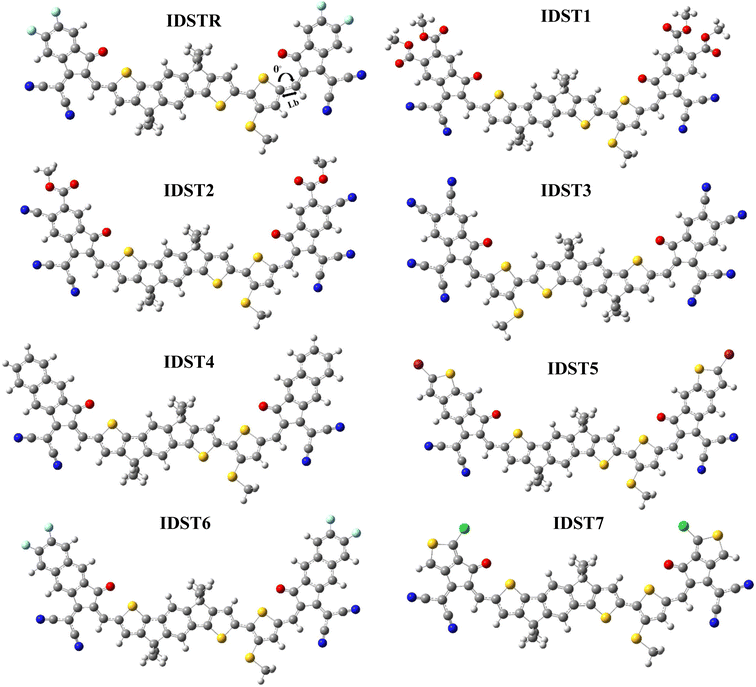 |
| | Fig. 2 Optimized structures of IDSTR and IDST1–IDST7 molecules. | |
Table 2 Computationally estimated Lb and dihedral angles (θ°) of IDSTR and IDST1–IDST7 molecules
| Molecules |
Lb (Å) |
θ° |
| IDSTR |
1.41 |
0.620 |
| IDST1 |
1.41 |
0.480 |
| IDST2 |
1.41 |
0.644 |
| IDST3 |
1.41 |
0.607 |
| IDST4 |
1.41 |
0.584 |
| IDST5 |
1.41 |
0.669 |
| IDST6 |
1.41 |
0.618 |
| IDST7 |
1.41 |
0.015 |
3.2. Frontier molecular orbitals (FMOs) evaluation
Analysis of the FMOs is an essential scientific aspect for determining how well molecules facilitate charge movement and dispersion of electron density.40 It is also useful in understanding the dispersion of charge facilitation and transmission in OSCs. Excitation takes place and electrons transmit from the valence to the conduction band.73 The transmission of charges, light absorption, and electronic characteristics of chromophores are significantly influenced by their HOMO and LUMO energy. OSCs as well as other photovoltaic systems can be identified by the energy gap (Egap), which demonstrates the necessity of exciton dissociation.44 A smaller bandgap is indicative of a higher performing OSCs. Building functional PV devices requires the use of molecules with the lowest possible Egap. Strong polarizability, poor kinetic stability, as well as great chemical reactivity are all characteristics of molecules with small Egap.45 Energy levels of HOMO, LUMO for IDSTR and IDST1–IDST7 HOMO as well as their Egap are examined at select functionals to evaluate the influence of various end group acceptors on the photophysical characteristics of the analyzed molecules and their numerical data are shown in Table 3.
Table 3 HOMO and LUMO energies, Egap, ionization potential (IP) and electron affinity (EA) of IDSTR and IDST1–IDST7 molecules
| Molecules |
HOMO (eV) |
LUMO (eV) |
Egap (eV) |
IP (eV) |
EA (eV) |
| IDSTR |
−5.75 |
−3.53 |
2.22 |
6.39 |
2.89 |
| IDST1 |
−5.72 |
−3.53 |
2.19 |
6.34 |
2.92 |
| IDST2 |
−5.90 |
−3.78 |
2.12 |
6.52 |
3.15 |
| IDST3 |
−6.12 |
−4.03 |
2.09 |
6.85 |
3.33 |
| IDST4 |
−5.59 |
−3.40 |
2.19 |
6.20 |
2.78 |
| IDST5 |
−5.64 |
−3.43 |
2.21 |
6.25 |
2.83 |
| IDST6 |
−5.69 |
−3.52 |
2.16 |
6.30 |
2.91 |
| IDST7 |
−5.73 |
−3.54 |
2.19 |
6.36 |
2.91 |
Fig. 3 displays HOMO and LUMO of every molecule under research with its corresponding Egap. In the ground state, the majority of the charge is located on the center region of the molecule, which is the donor section. However, in the LUMO state, the charge potential shifts to the molecules' acceptor sites, which are located at their peripheral ends. It has been shown that the methylsulfanyl-thiophene bridge enhances the efficiency of charge transfer from the electron donating core to the electron pulling acceptors at that particular location. This strategy indicates the sequential charge transfer competencies of donor core to the acceptor part from HOMO to LUMO. Furthermore, the efficient conjugation that is enabling the effective charge transfer capabilities owing to the planarity of the molecular geometries is shown by the charge transfer from the electron-rich donor portion to the acceptor part of the compound in the excited state. The IDSTR molecule's FMOs; HOMO and LUMO energies were measured to be −5.75 eV and −3.53 eV, respectively, and this molecule had a Egap of 2.22 eV, which is higher than any of our recently proposed compounds. The IDST3 molecule had the least HOMO energy (−6.12 eV) of any of the others, indicating that its HOMO is the stable of all the compounds under study. These IDST3 molecules may be more stable because they have pairs of highly electronegative cyano groups in their peripheral acceptor units that has participated considerably in the charge density dispersal. Moreover, IDST2 possesses the next most stable HOMO after IDST3, due to its comparatively low HOMO energy of −5.90 eV. The existence of both cyano and ester groups in the terminal sites of the IDST2 molecule contributes to its stability. The decreasing patters of HOMO and LUMO energies for IDSTR and IDST1–IDST7 molecules is IDST4 > IDST5 > IDST6 > IDST1 > IDST7 > IDSTR > IDST2 > IDST3 and IDST4 > IDST5 > IDST6 > IDST1 = IDSTR > IDST7 > IDST2 > IDST3 respectively. IDST1–IDST7 molecules have smaller Egap than IDSTR. The Egap of the IDST3 and IDST2 is the smallest and second-smallest among all other recently proposed molecules, respectively, because of their strong peripheral acceptor site, which has contributed sufficiently to improve molecules' ability to transfer charges and resultantly the bandgap is decreased. The descending trend of Egap for studied molecules is IDSTR > IDST5 > IDST1 = IDST4 = IDST7 > IDST6 > IDST2 > IDST3.
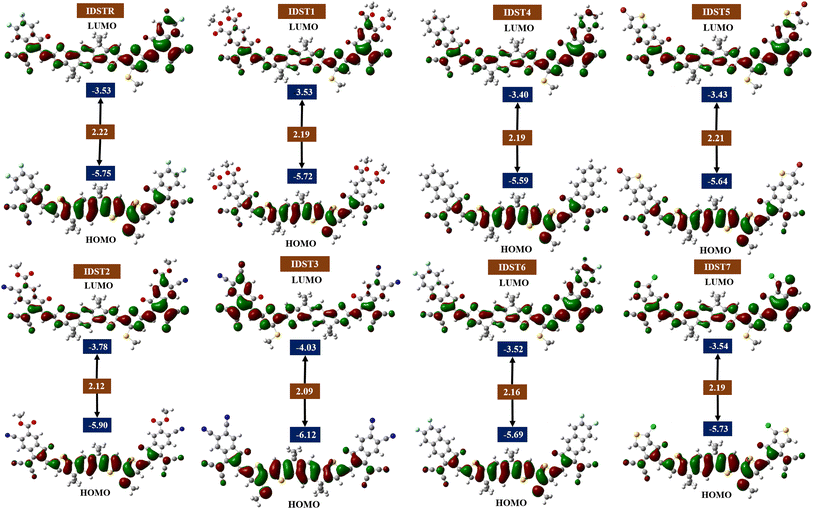 |
| | Fig. 3 FMOs of IDSTR and IDST1–IDST7 molecules. | |
3.3. Ionization potential and electron affinity
A number of variables, including IP and EA, can influence illustrate charge transfer efficacy. Energy released during an electron addition to a molecule is denoted as EA, whereas the energy input necessary for the donating an electron is indicated by IP.46 Molecules with high IPs and EAs have stable HOMO energy states, making it more difficult to remove electrons, while those with low IPs and EAs have the capacity to release electron efficiently. The stabilization of the HOMO via electron withdrawing moieties is the cause of molecules with high IP and EA, while the destabilization of the HOMO by electron donating groups is the cause of molecules with low IP and EA.47 In the current study, IP and EA were calculated using eqn (3) and (4).48
Table 3 contains the IP and EA estimates for the compounds IDSTR and IDST1–IDST7. The HOMO (−5.59 eV) of IDST4 is the most destabilized among all hence possesses smallest IP (6.20 eV). IDST4, with a stabilized HOMO (−6.12 eV), has the highest EA (3.33 eV). Because IDST5 has the second-least stable HOMO (−5.64 eV), it has the second-lowest IP (6.25 eV). IDST2 has the second most stable HOMO (−5.90 eV), which contributes to its second greatest EA (3.15 eV).
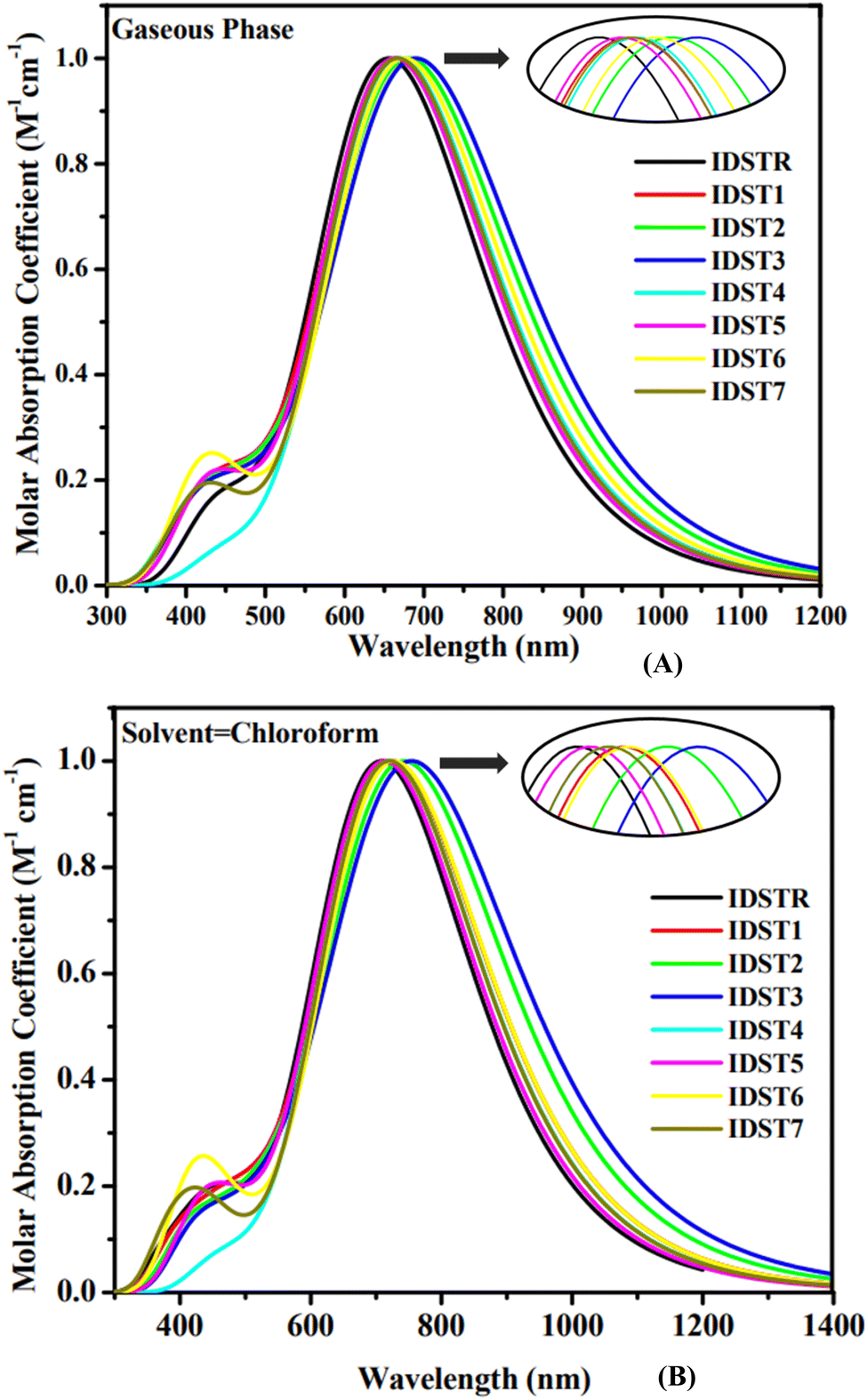
3.4. Absorption spectrum
Quantum absorption spectra can be used to make reasonable estimate of the electrical specifications of chromophores. Tables 4 and 5 show the predicted results for the optical analysis of IDSTR and IDST1–IDST7 molecules in the studied gaseous and solvent (chloroform) phases, respectively, of using MPW1PW91/6-31G(d,p) level of theory. The range of absorption spectrum for all investigated compounds (IDSTR and IDST1–IDST7) in gas phase is from around 350–1200 nm and in chloroform (CHCl3) solvent is from around 350 to 1400 nm as shown in Fig. 4. The λmax of IDSTR in the former phase is 659 nm whereas its λmax in CHCl3 solvent is 712 nm. IDST1–IDST7 molecules have a redshift in their λmax compared to IDSTR in both the gas phase and the CHCl3 solvent, with a range of 666–701 nm and 717–770 nm, respectively. Their UV-visible spectra show that all of the modified molecules exhibited a red shift shift in their λmax in regards to the IDSTR molecule in both evaluated phases, with the IDST1–IDST7 molecules exhibiting a shift of 7 nm to 42 nm and 5 nm to 58 nm, respectively. Better light absorption results from the molecules' planar structure, which promotes greater conjugation within the structure. All investigated molecules exhibited a bathochromic switch when in CHCl3 solvent, as opposed to gaseous form, on the basis of the general fact that the polar solvent stabilize the polar excited state.49 The shorter Egap of all the molecules resulted in a longer wavelength due to the inverse connection of energy and wavelength.50 The IDSTR and IDST1–IDST7 molecules' rising λmax trend in the gas phase and in the CHCl3 is IDSTR < IDST5 < IDST7 < IDST4 = IDST1 < IDST6 < IDST2 < IDST3 and IDSTR < IDST5 < IDST7 < IDST4 < IDST6 < IDST1 < IDST2 < IDST3, respectively. The outcomes reveal that IDST3 has the highest λmax in gas and solvent phases, at 701 nm and 770 nm, respectively, which may be due to the pairs of electronegative cyano group presence in the peripheral acceptor unit.
Table 4 Computed λmax, excitation energy (Ex), oscillator strength (f) and assignment of IDSTR and IDST1–IDST7 molecules in gaseous phase
| Molecules |
Computed λmax (nm) |
Ex (eV) |
f |
Assignment |
| IDSTR |
659 |
1.88 |
2.34 |
H–L (+98%) |
| IDST1 |
672 |
1.84 |
2.37 |
H–L (+99%) |
| IDST2 |
690 |
1.79 |
2.30 |
H–L (+99%) |
| IDST3 |
701 |
1.76 |
2.25 |
H–L (+98%) |
| IDST4 |
672 |
1.84 |
2.53 |
H–L (+99%) |
| IDST5 |
666 |
1.86 |
2.59 |
H–L (+98%) |
| IDST6 |
678 |
1.82 |
2.54 |
H–L (+98%) |
| IDST7 |
670 |
1.85 |
2.43 |
H–L (+97%) |
Table 5 Computed λmax, excitation energy (Ex), oscillator strength (f) and assignment of IDSTR and IDST1–IDST7 molecules in CHCl3 solvent
| Molecules |
Exp. λmax (nm) |
Computed λmax (nm) |
Ex (eV) |
f |
Assignment |
| IDSTR |
∼728 |
712 |
1.74 |
2.58 |
H–L (+98%) |
| IDST1 |
— |
732 |
1.69 |
2.57 |
H–L (+99%) |
| IDST2 |
— |
753 |
1.65 |
2.50 |
H–L (+98%) |
| IDST3 |
— |
770 |
1.61 |
2.42 |
H–L (+97%) |
| IDST4 |
— |
724 |
1.71 |
2.78 |
H–L (+99%) |
| IDST5 |
— |
717 |
1.73 |
2.81 |
H–L (+99%) |
| IDST6 |
— |
731 |
1.70 |
2.79 |
H–L (+98%) |
| IDST7 |
— |
723 |
1.71 |
2.71 |
H–L (+98%) |
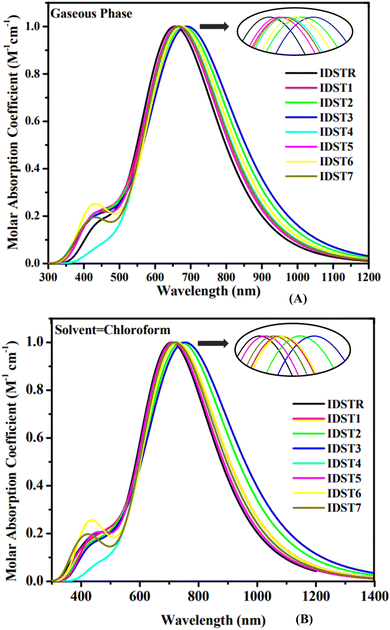 |
| | Fig. 4 Absorption spectra of IDSTR and IDST1–IDST7 in (A) gaseous phase and (B) CHCl3 solvent. | |
The optical properties of organic photovoltaic devices and the amount of radiation produced by electrical stimulation between two levels of energy are both highly dependent on a dimensionless quantity termed oscillator strength (f).51 Energy needed to make a likely transition is called excitation energy (Ex),52 as a matter of fact, improving ICT functionality can be achieved by expanding the absorption spectrum, decreasing the Ex, or raising the f.53 Compared to IDSTR, IDST1–IDST7 molecules have lower Ex while IDST4–IDST7 molecules have higher f, in consequence of this, they might have more efficient ICT.
3.5. Light-harvesting efficiency (LHE)
Every component of SCs must be able to generate a charge after the absorption of light, known as LHE.54 Short-circuit current generation is significantly affected by LHE, which than effect the device's solar efficacy.55 Using eqn (5), the LHE of every chemical studied was computed.In above equation, ηλ is the LHE and the IDSTR and IDST1–IDST7 LHE values have been computed, and they are shown in Table 6. Compared to IDSTR, the LHE of IDST4–IDST7 molecules is higher because their f is higher, as LHE is directly related to f. Due to IDST5's greater oscillator strength, LHE is at its ultimate level. Consistent with the findings, the LHE of IDST5–IDST7 molecules is significantly affected by the ending acceptor moieties.
Table 6 f, and LHE of IDSTR and IDST1–IDST7
| Molecules |
f |
LHE |
| IDSTR |
2.58 |
0.9973 |
| IDST1 |
2.57 |
0.9973 |
| IDST2 |
2.51 |
0.9969 |
| IDST3 |
2.42 |
0.9961 |
| IDST4 |
2.79 |
0.9983 |
| IDST5 |
2.82 |
0.9985 |
| IDST6 |
2.80 |
0.9984 |
| IDST7 |
2.71 |
0.9980 |
3.6. Dipole moment
The dipole moment (D) has a significant role in determining solubility as well as crystallinity. These characteristics have a crucial role in defining polarization processes in the necessary solvent for efficient organic photovoltaic devices.56 Consistent transfer of charge is enabled by the planar and ordered morphologies of molecules with strong dipole moments, which lead to close molecular packing, improved crystallinity, as well as higher solubility in polar solvents. For better charge conduction, molecules with a larger D tend to be more crystallized and dissolve more easily in polar solvents.42 Molecules with no dipole moment are often insoluble in polar organic solvents like chloroform. Hence the connection between them is not universal, every molecule does have a different chemical structure, which affects its charge carrier as well as solubility properties.57 As per given numerical values in Table 7, the raising order of D for IDSTR and IDST1–IDST7 molecules is same in gas phase and CHCl3 solvent i.e., IDSTR < IDST7 < IDST2 < IDST6 < IDST5 < IDST1 < IDST4 < IDST3. All modified molecules exhibit superior crystallinity and solubility than IDSTR due to their greater D values. The highest D value was found in IDST3, that may be attributed to the presence of cyano groups at both of its ends.
Table 7 Dipole moment of IDSTR and IDST1–IDST7 molecules in gas phase and CHCl3 solvent
| Molecules |
D (gas phase) |
D (solvent phase) |
| IDSTR |
2.10 |
2.62 |
| IDST1 |
4.66 |
4.83 |
| IDST2 |
2.39 |
3.35 |
| IDST3 |
11.32 |
13.64 |
| IDST4 |
6.65 |
7.95 |
| IDST5 |
3.74 |
4.54 |
| IDST6 |
2.78 |
3.52 |
| IDST7 |
2.24 |
2.75 |
3.7. Charge mobility analysis
The reorganization energy (RE) of IDST1–IDST7 and IDSTR molecules were determined at MPW1PW91 functional to analyze the charge transfer from donor unit acceptor unit. The main factor for the creation of effective substances for OSCs is the RE, which quantifies the charge that is transported from donor fragment to acceptor portions of a compound and is linked to the electron and hole mobility.58 The RE has an inverse relationship to the charges mobility i.e., electrons and holes. As a result, lowering the values of the RE will result in a more effective charge transport.59 One of the many factors that affect the RE are the geometric configurations of anions and cations. RE of electron and hole are calculated for all of the investigated compounds using eqn (1) and (2), and the findings are given and shown in Table 8 and Fig. 5. The findings suggest that the improved molecular blend of planar electronic structures is responsible for the enhanced electron and hole mobilities seen in the newly suggested molecules.
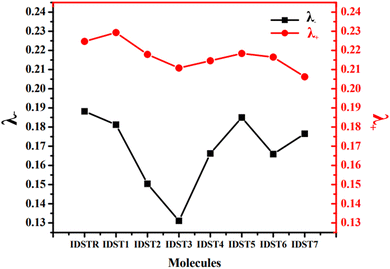
Table 8 RE of electron and hole (λ− and λ+, respectively) for IDSTR and IDST1–IDST7 molecules
| Molecules |
λ− (eV) |
λ+ (eV) |
| IDSTR |
0.1882 |
0.2247 |
| IDST1 |
0.1812 |
0.2293 |
| IDST2 |
0.1504 |
0.2179 |
| IDST3 |
0.1311 |
0.2108 |
| IDST4 |
0.1662 |
0.2146 |
| IDST5 |
0.1850 |
0.2184 |
| IDST6 |
0.1659 |
0.2165 |
| IDST7 |
0.1765 |
0.2062 |
 |
| | Fig. 5 RE plot of IDSTR and IDST1–IDST7 molecules. | |
The RE of the IDSTR molecule is 0.1882 eV for the λ− and 0.2247 eV for the λ+. All the modified molecules have higher electron mobilities than IDSTR because their RE for λ− are smaller than those of the IDSTR molecule. Because of the planar molecule structure, the resulting molecular mix will have a smooth morphology, which improves exciton dissociation and electron mobility. Since the RE of IDST3 was observed to be the smallest (0.1311 eV) of all the molecules examined, this suggests that the molecule's new peripheral acceptors have had a significant impact on increasing its electron transport. The order of λ− RE for the studied molecules is IDST3 < IDST2 < IDST6 < IDST4 < IDST7 < IDST1 < IDST5 < IDSTR.
IDST2–IDST7 molecules have improved mobility of hole than reference as these molecules have smaller λ+ RE value than IDSTR molecule. The drastic drop in the RE of hole for the IDST7 molecule indicates that the terminal acceptor is contributing significantly to the lowering of the RE to promote mobility of hole. IDST7 < IDST3 < IDST4 < IDST6 < IDST2 < IDST5 < IDSTR < IDST1 is the sequence of λ+ for the studied molecules.
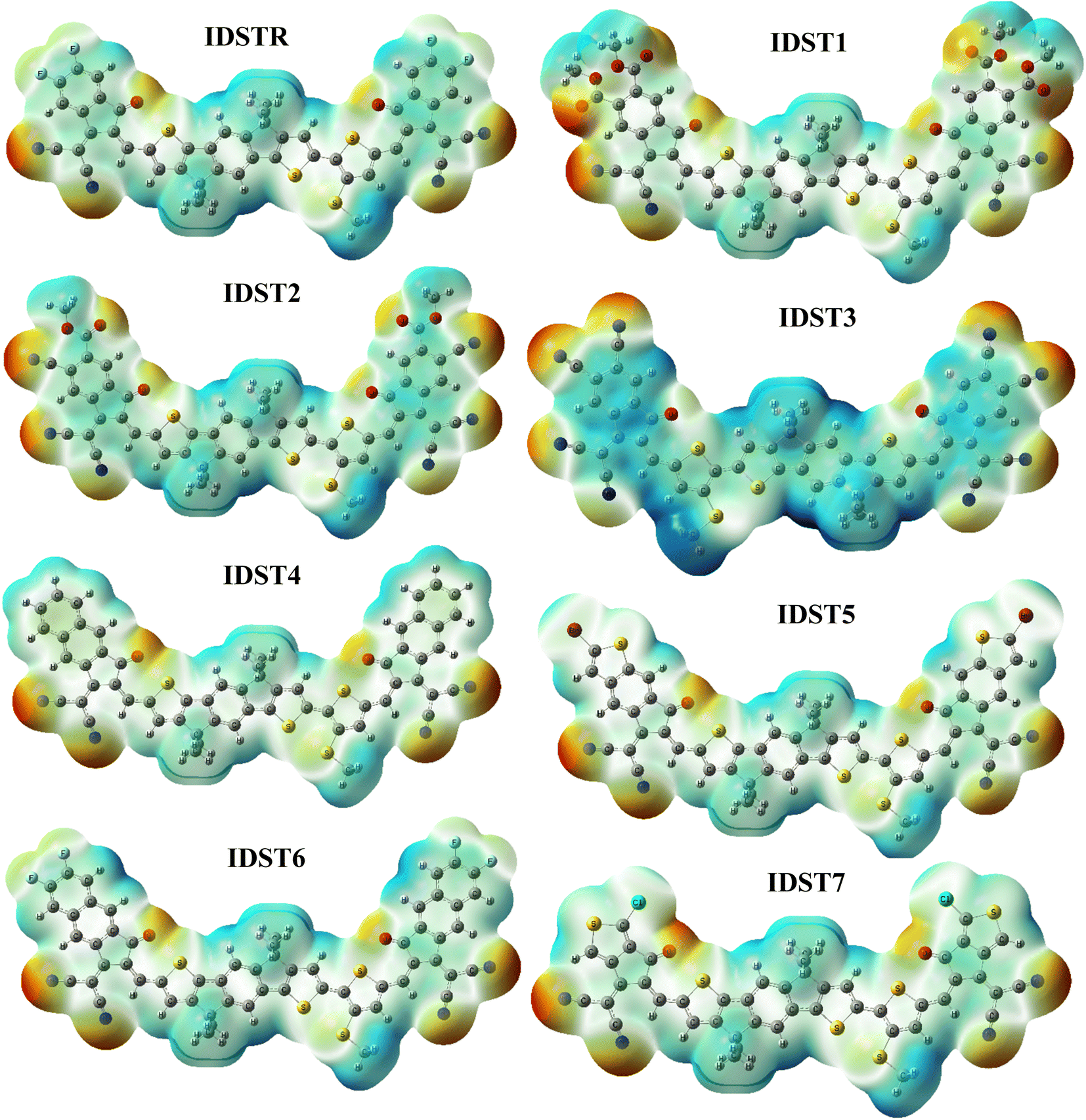
3.8. Electrostatic potential (ESP)
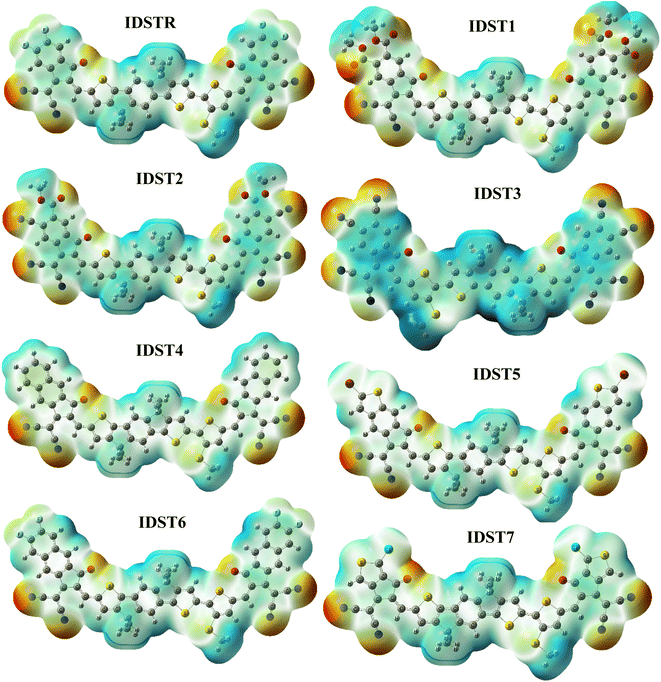
ESP highlights the existence of electrons as well as distribution of charges on the molecule three dimensionally (3D).60 On our carefully investigated compounds, ESP analysis was done in order to forecast the reactive sites of a molecular structure.61 The 3D configuration of lone-pairs, electrons, and electronegative materials, which are conveniently exposed to nucleophilic activity are shown on ESP map. On the ESP mapping, red indicates an area with a high concentration of electrons, green indicates a neutral region, and blue indicates a region with a lower density of electrons.
Fig. 6 displays the IDSTR and IDST1–IDST7 colorful ESP maps. An abundance of electron concentration can be seen over the oxygen as well as nitrogen atoms at the outer acceptor areas of the molecules, which shows up as dark red spots on ESP layouts.
 |
| | Fig. 6 ESP maps of IDSTR and IDST1–IDST7 molecules. | |
Donor regions with thiophene rings, benzene rings and methyl groups are indicated as blue on the ESP maps, indicating an extreme deficiency of electrons at those locations.
3.9. Density of states (DOS) analysis
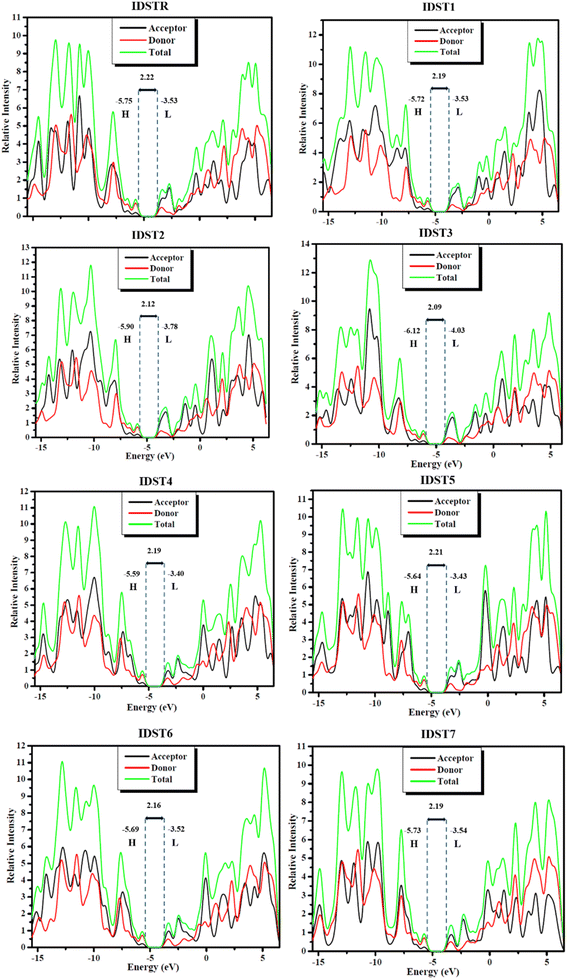
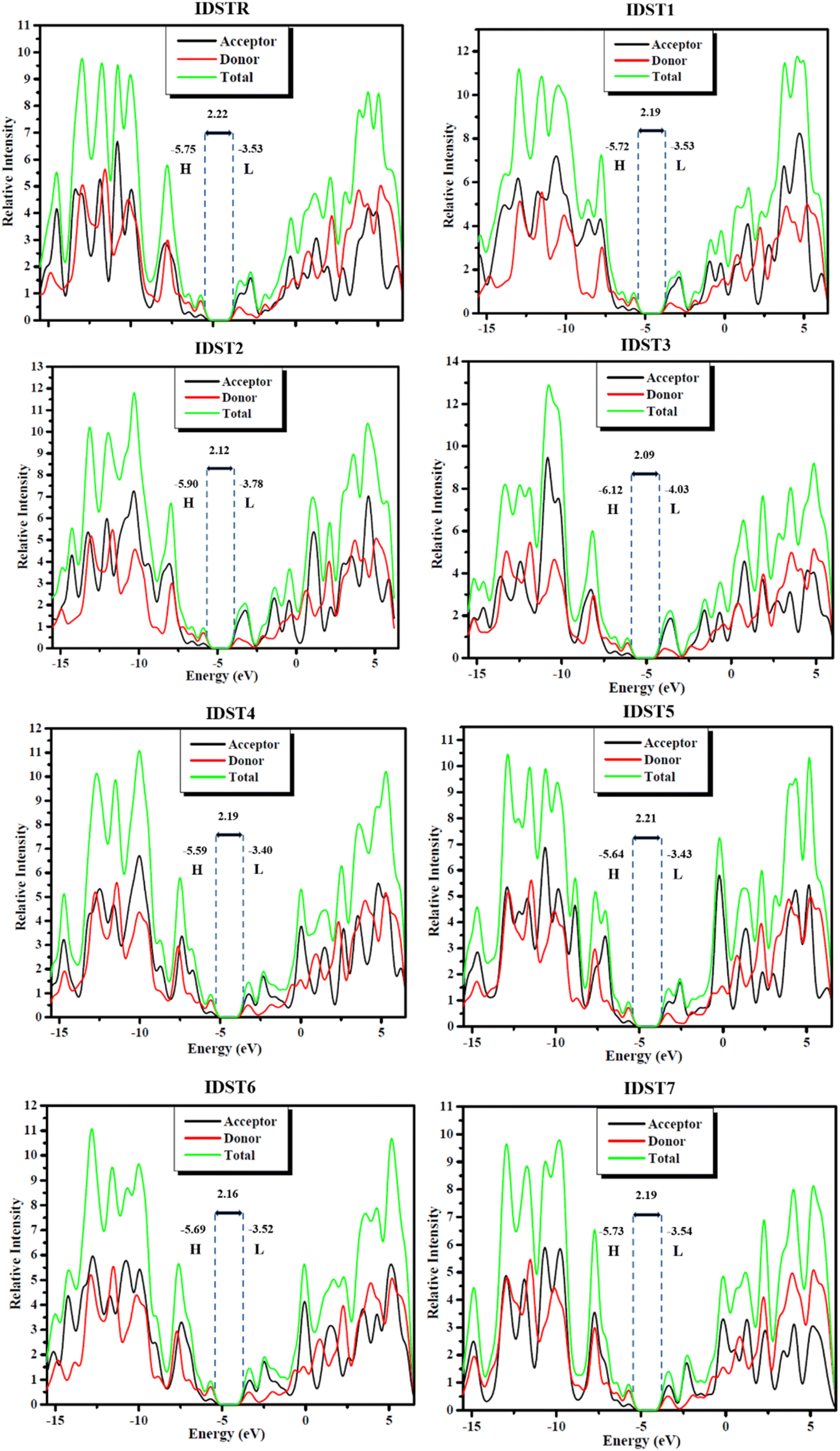
It is essential to carry out DOS analyses to gain a deeper comprehension of the activities of every molecule's units i.e., donor, acceptor. The entire capabilities of compounds are confirmed by their corresponding DOS, which are either partial or total DOS.61 DOS estimation is crucial in establishing the arrangement of frontier molecular orbitals (FMOs) with respect to Mulliken charge density.62 All compounds under study had their DOS calculated by employing MPW1PW91/6-31G(d,p) level, and plots of peaks were generated in PyMOlyze 1.1. The energy (represented by the x-axis) and relative intensity (represented by the y-axis) are shown graphically in DOS plots. The HOMO energies are represented by peaks on the left of the center plane (the bandgap) in the graphs, while the LUMO energies are represented by peaks on the right. Each molecule was subdivided into its respective donor and acceptor halves for investigation of the individual unit contribution to the FMOs. In the DOS plots of IDSTR and IDST1–IDST7, donor and acceptor participation is shown by red and black lines, while overall contribution to FMO growth by the moieties is shown by a green line as seen in Fig. 7, and the participation information is included in Table 9. DOS studies showed that all compounds (IDSTR, IDST1–IDST7) had similar donor and acceptor contributions patterns, with the donor fragment being more significant in HOMO and the acceptor portions being more significant in LUMO. This clearly demonstrates the flow of charge from donor unit to acceptor units of molecule efficiently by consecutive conjugation which may increase the OSCs' ultimate efficiency. These findings corroborate with the aforementioned FMOs of IDSTR and IDST1–IDST7, as demonstrated in Fig. 3. The data show that the HOMO is dominated by the donor central part of the molecule that is rich with electrons, whereas the charge is flowing from the donor central region to the electron-withdrawing terminal units of molecules in LUMO pointing to the molecules' planar geometry, which allow for such efficient charge transfer.
 |
| | Fig. 7 DOS plots of IDSTR and IDST1–IDST7 molecules. | |
Table 9 Participation of donor and acceptor units in FMOs of studied molecules
| Molecules |
|
Donor (%) |
Acceptor (%) |
| IDSTR |
HOMO |
77.9 |
22.1 |
| LUMO |
38.3 |
61.7 |
| IDST1 |
HOMO |
77.2 |
22.8 |
| LUMO |
36.3 |
63.7 |
| IDST2 |
HOMO |
76.5 |
23.5 |
| LUMO |
34.2 |
65.8 |
| IDST3 |
HOMO |
76.1 |
23.9 |
| LUMO |
33.6 |
66.4 |
| IDST4 |
HOMO |
76.5 |
23.5 |
| LUMO |
37.5 |
62.5 |
| IDST5 |
HOMO |
77.0 |
23.0 |
| LUMO |
39.0 |
61.0 |
| IDST6 |
HOMO |
76.3 |
23.6 |
| LUMO |
37.2 |
62.8 |
| IDST7 |
HOMO |
76.5 |
23.5 |
| LUMO |
39.3 |
60.7 |
3.10. Transition density matrix (TDM) and exciton binding energy (Eb)
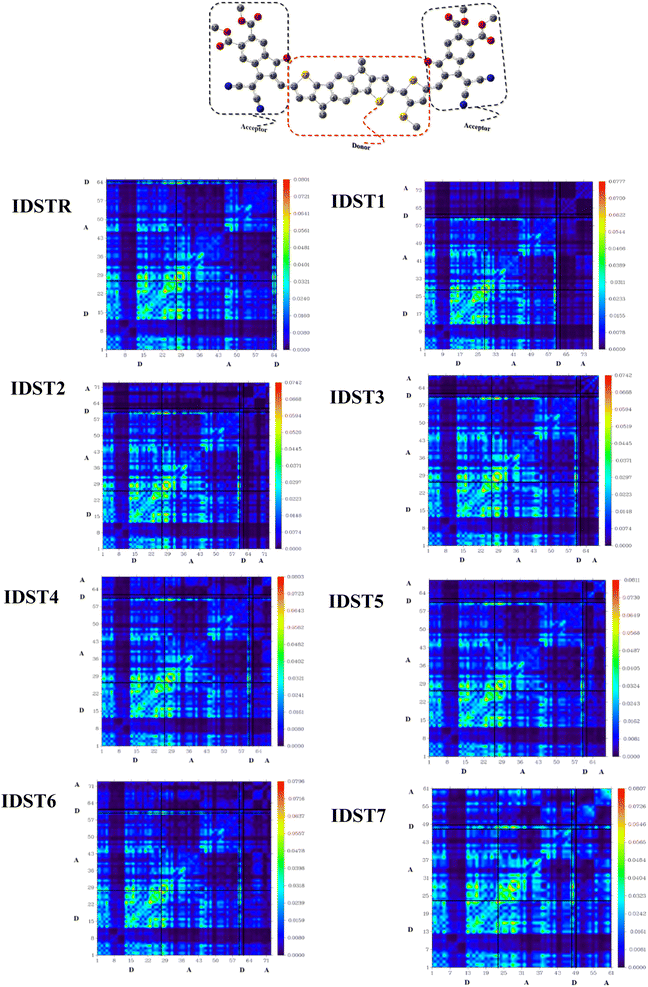
TDM analysis is mandatory to provide an accurate forecast of exciton movement between donor and acceptor zones at specific places in conjugated molecular geometry.63 This approach has the potential to investigate a number of different types of charge phenomena, including prediction of charge transmission, primary charge sites, and positioning of exciton movement during absorption and emission in the excited state.64 TDM plots are usually generated to analyze electronic characteristics, such as the resonance effects and the degree of delocalization, and to get insight into the charge mobility inside a molecule. Due to the negligible impact in charge transport, hydrogen's participation is often disregarded in TDM analysis.74,75 Numbers of atoms in compounds other than hydrogen are shown along the left y-axis and the lower x-axis, respectively. The charge density coefficient is represented by the multicolored bar on the x-axis of the right side, whose colors go from blue to red. As can be seen in Fig. 8, the electronic potential is existing in both the regions (acceptor and the donor) of all of the molecules we looked at (IDSTR and IDST1–IDST7). Consistency in electron density travels from donor to acceptor in a diagonal and off-diagonal pattern, with the diagonal being the more prevalent. The scheme depicted the smooth transport of electronic potential from the electron rich donor part to the acceptor component of the molecule by sequential conjugation, suggesting effective charge dispersion.
 |
| | Fig. 8 TDM plots of IDSTR and IDST1–IDST7 molecules. | |
The interaction coefficient (IC) for all studied molecules is computed and given in Table 10, as well as their increasing pattern is IDST5 < IDST4 < IDST6 < IDST1 < IDSTR < IDST2 < IDST3 < IDST7. The IC of IDST5 is small which indicate that electrons can easily be transferred from the donor and acceptor sites of this molecule. This is due to the fact that increased charge mobility is predicted by a smaller IC of molecule.57
Table 10 Egap, Eb (gas phase), Eb (CHCl3 solvent), Interaction Coefficient (IC) of IDSTR and IDST1–IDST7 molecules
| Molecules |
Egap (eV) |
Eb (eV) |
Eb (eV) |
IC |
| Gaseous |
Solvent |
| IDSTR |
2.22 |
0.34 |
0.48 |
0.69670 |
| IDST1 |
2.19 |
0.35 |
0.50 |
0.69639 |
| IDST2 |
2.12 |
0.32 |
0.48 |
0.69673 |
| IDST3 |
2.09 |
0.34 |
0.48 |
0.69712 |
| IDST4 |
2.19 |
0.35 |
0.48 |
0.69591 |
| IDST5 |
2.21 |
0.35 |
0.49 |
0.69574 |
| IDST6 |
2.16 |
0.33 |
0.47 |
0.69619 |
| IDST7 |
2.19 |
0.34 |
0.48 |
0.69748 |
The Eb is also an important consideration, that can be used to evaluate the potential for the separation of electron–hole (exciton), the performance as well as the electrical characteristics of OSCs.65 Eb is the value that is used to calculate the coulombic interactions that take place between electron–hole pair.66 When the electron–hole coulombic interaction is small, as indicated by a reduced Eb, and vice versa. Eb was estimated using eqn (6) in this study and the results are given in Table 10.
The calculated values show that in the gas phase, IDST2 and IDST6 have the lower Eb value (at 0.32 eV and 0.36 eV respectively) compared to the IDSTR molecule, IDST3 and IDST7 are close to the reference Eb, and IDST2 has the lowest Eb value overall. Eb of IDST6 (0.47 eV) is smallest in CHCl3 solvent. Since these molecules have lower Eb values, it is simple for exciton to diffuse into free charges, making them interesting candidates for higher charge densities.
3.11. Performance of device
An essential aspect of understanding the operation of any photovoltaic device is measuring its open circuit voltage (VOC), which can done to assess its photovoltaic performance.67 VOC accurately reflects the maximum voltage that an optical device is capable of providing at zero voltage input.68 Factors like ambient light, charge transmission, temperature of photovoltaic system, etc. all have a significant impact on VOC.69 The maximal voltage is achieved when the donor material's HOMO is associated to the acceptor material's LUMO. In order to achieve the increased VOC values, the donor molecule must have a lower HOMO, whereas the acceptor molecule must have a greater LUMO level. A rise in VOC leads to an increment in fill factor, that is the foundation for the higher PCE of solar devices. In this investigation, we coupled the HOMO of a well-known polymeric donor (PTB7-Th) with the LUMO of the recently proposed non-fullerene acceptors (IDSTR, IDST1–IDST7), to produce the maximum VOC feasible. PTB7-Th, with its HOMO and LUMO of −5.20 eV and −3.60 eV respectively, is a robust donor, as shown by the existing literature. The eqn (7) was used in this study to statistically estimate VOC.| |
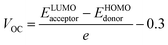 | (7) |
In the preceding equation, e stands for the molecular charge, which is 1, and a factor of intersurface charge, which has a typical value of 0.30. Fig. 9 shows the theoretically predicted VOC for IDSTR and IDST1–IDST7 in connection with PTB7-Th, and Table 11 displays the corresponding empirically predicted values. The findings show that IDST4–IDST6 molecules have a larger VOC than the IDSTR molecules, which have a VOC of 1.37 eV. The ascending order of VOC for IDSTR and IDST1–IDST7 is IDST3 < IDST2 < IDST7 < IDSTR = IDST1 < IDST6 < IDST5 < IDST4. The IDST4 was shown to have the greatest VOC value in this study, suggesting its potential for usage in boosting OSCs' PCE.
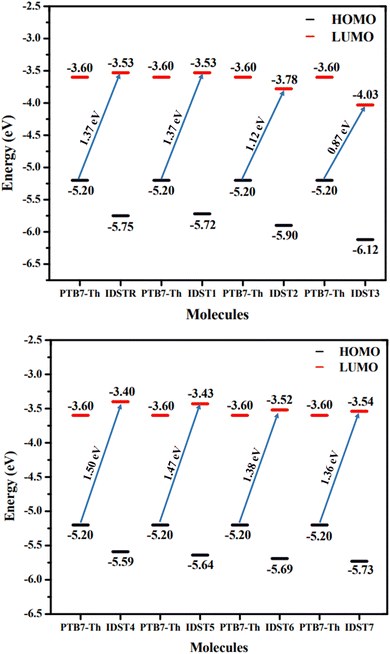 |
| | Fig. 9 Computed VOC of IDSTR and IDST1–IDST7 acceptors with the combination of PTB7-Th. | |
Table 11 VOC, normalized VOC and FF of IDSTR and IDST1–IDST7
| Molecule |
VOC (eV) |
Normalized VOC |
FF |
| IDSTR |
1.37 |
53.00 |
0.9077 |
| IDST1 |
1.37 |
53.00 |
0.9077 |
| IDST2 |
1.12 |
43.33 |
0.8920 |
| IDST3 |
0.87 |
33.66 |
0.8690 |
| IDST4 |
1.50 |
58.02 |
0.9140 |
| IDST5 |
1.47 |
56.87 |
0.9128 |
| IDST6 |
1.38 |
53.38 |
0.9082 |
| IDST7 |
1.36 |
52.61 |
0.9071 |
Among the most crucial factors in calculating a PV system's PCE is its fill factor (FF), since the two are directly connected. This characteristic is significantly influenced by the VOC at the donor–acceptor contact. The FF of all of our investigated compounds was calculated using eqn (8).70
| |
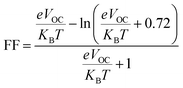 | (8) |
is normalized
VOC and
e is the customary charge and always equals 1.
KB is the Boltzmann constant (8.61733034 × 10
−5 electron volts per kelvin) and
T is the temperature (constant value 300 K).
Table 11 displays the derived normalized
VOC and FF values for
IDSTR and
IDST1–IDST7.
IDSTR has an FF of 0.9077 and a normalized
VOC of 53.00. According to the given results,
IDST4–IDST6 molecules have greater normalized
VOC (58.02, 56.87, 53.38) and FF (0.9140, 0.9128, 0.9082) than
IDSTR.
The power conversion efficiency (PCE), which aggregates all solar device operational factors into a single statistic, is estimated to determine if the solar substance is sufficiently efficient for usage in operational implementations.71 The PCE of a molecule is affected in a direct manner by the VOC, FF, and the short circuit voltage (JSC). This connection is well described by eqn (9).72
| |
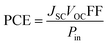 | (9) |
In this work, we hypothetically determine the VOC and FF using the previous equation for the IDSTR and IDST1–IDST7 molecules; however, the JSC has not been computed because of the inadequate facilities associated. The LHE, which is one of the JSC's criteria, has already been analyzed above. Based on the above findings, the PCE of IDST1 and IDST7 molecules is predicted to be very close to that of IDSTR. IDST4, IDST5, and IDST6 molecules have a greater PCE since they have a higher LHE, VOC and FF than IDSTR. The theoretical estimations demonstrate that these non-fullerene acceptors may be useful in practice due to their outstanding optoelectronic characteristics.
4 Conclusion
In this computational approach, seven new indaceno dithiophene based small acceptor molecules (IDST1–IDST7) have been developed of A–D–A type by the alteration of IDSTR molecule. A variety of optoelectronic aspects such as energy levels of HOMO–LUMO with Egap, optical bandgap, EA and IP, transition density matrix, LHE, oscillator strength, absorbance maxima (λmax), binding energy, density of states, RE for the mobility of hole and electron, VOC of IDSTR and IDST1–IDST7 molecules have been investigated in this research by employing MPW1PW91 functional and 6-31G(d,p) basis set. The outcomes revealed that newly altered molecules (IDST1–IDST7) have reduced Egap, higher λmax as well as smaller excitation energy than IDSTR molecule. According to the estimations, IDST3 possesses the shortest Egap (2.09 eV) lowest Ex (1.61 eV), as well as greatest λmax (770 nm). LHE of IDST4–IDST7 is more efficient than IDSTR and among all IDST5 has the maximum LHE (0.9985) due to its highest oscillator strength (2.82). All modified molecules exhibit superior crystallinity and solubility due to their greater dipole moment values (2.75–13.64 D) than the dipole moment of IDSTR (2.62 D). IDST1–IDST7 molecules have higher electron mobilities than IDSTR because their RE for λ− (0.1311–0.1812 eV) are smaller than those of the IDSTR (0.1882 eV) molecule. IDST2–IDST7 molecules have improved mobility of hole than reference as these molecules have smaller λ+ RE (0.2061–0.2184 eV) value than IDSTR molecule (0.2247 eV). By coupling the IDST1–IDST7 acceptors with PTB7-Th donor the VOC was estimated and the results showed that VOC of IDST4 (1.50 eV), IDST5 (1.47 eV) and IDST6 (1.38 eV) is higher than IDSTR VOC (1.37 eV). These results suggest that IDST4, IDST5, and IDST6 are the most promising choices for using these altered molecules in the potential development of OSCs with improved photovoltaic properties.
Author contributions
Ehsan Ullah Rashid: all the authors showed efficient contribution and dedication to their corresponding work in this manuscript and their credit to the manuscript is summarized as: Ehsan Ullah Rashid: investigation, visualisation, validation, writing original draft, interpretation of data. N. M. A. Hadia: conceptualization, revision, visualization, data curation, validation, writing, review, and editing. Omaymah Alaysuy: conceptualization, revision, visualization, data curation, validation, writing, review, and editing. Javed Iqbal: supervision, resources, software, interpretation of data, visualization, data curation, validation, revision, writing, review and editing. M. M. Hessien: methodology, software, interpretation of data, visualization, data curation, validation, writing, review, and editing. Gaber A. M. Mersal: conceptualization, revision, visualization, data curation, validation, writing, review, and editing. Ahmed M. Shawky: formal analysis, validation, visualization, writing, acquisition, interpretation of data. Rana Farhat Mehmood: funding acquisition, data curation, resources, investigation, writing, review, and editing. Muhammad Imran Khan: conceptualization, revision, visualization, data curation, validation, writing, review, and editing. Rasheed Ahmad Khera: methodology, supervision, resources, software, interpretation of data, visualization, data curation, validation, writing, review, and editing.
Conflicts of interest
The authors of this manuscript proclaim no conflicts of interest.
Acknowledgements
The authors greatly appreciate the technical support and the Department of Chemistry, University of Agriculture (UAF), Faisalabad, Pakistan. This work was supported by Taif University Researchers Supporting Project number TURSP-2020/109, Taif University, Taif, Saudi Arabia and authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: (22UQU4331174DSR23). The authors also thankful to Dr Khurshid Ayub, COMSATS University, Islamabad, Abbottabad Campus, Pakistan for additional resources.
References
- G. Zhang, J. Zhao, P. C. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- G. Wang, M. A. Adil, J. Zhang and Z. Wei, Adv. Mater., 2019, 31, 1805089 CrossRef CAS.
- H. Wang, J. Cao, J. Yu, Z. Zhang, R. Geng, L. Yang and W. Tang, J. Mater. Chem. A, 2019, 7, 4313–4333 RSC.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- M. Reyes-Reyes, K. Kim, J. Dewald, R. López-Sandoval, A. Avadhanula, S. Curran and D. L. Carroll, Org. Lett., 2005, 7, 5749–5752 CrossRef CAS PubMed.
- B. A. Collins, J. R. Tumbleston and H. Ade, J. Phys. Chem. Lett., 2011, 2, 3135–3145 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 1–19 CrossRef.
- H. Chen, Y. Zou, H. Liang, T. He, X. Xu, Y. Zhang, Z. Ma, J. Wang, M. Zhang and Q. Li, Sci. China: Chem., 2022, 1–12 Search PubMed.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- X. Wang, H. Chen, A.-R. Ran, L. Luo, P. P. Chan, C. C. Tham, R. T. Chang, S. S. Mannil, C. Y. Cheung and P.-A. Heng, Med. Image Anal., 2020, 63, 101695 CrossRef PubMed.
- T. Liu, R. Ma, Z. Luo, Y. Guo, G. Zhang, Y. Xiao, T. Yang, Y. Chen, G. Li and Y. Yi, Energy Environ. Sci., 2020, 13, 2115–2123 RSC.
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC.
- F. Huang, T. He, M. Li, L. Meng, W. Feng, H. Liang, Y. Zhou, X. Wan, C. Li and G. Long, Chem. Mater., 2022, 34, 6009–6025 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed.
- J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma and W. You, Adv. Mater., 2018, 30, 1704713 CrossRef PubMed.
- S. Dai, T. Li, W. Wang, Y. Xiao, T. K. Lau, Z. Li, K. Liu, X. Lu and X. Zhan, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed.
- S. Dai, Y. Xiao, P. Xue, J. James Rech, K. Liu, Z. Li, X. Lu, W. You and X. Zhan, Chem. Mater., 2018, 30, 5390–5396 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS.
- J. Lee, S. J. Ko, M. Seifrid, H. Lee, C. McDowell, B. R. Luginbuhl, A. Karki, K. Cho, T. Q. Nguyen and G. C. Bazan, Adv. Energy Mater., 2018, 8, 1801209 CrossRef.
- S. Feng, C. e. Zhang, Y. Liu, Z. Bi, Z. Zhang, X. Xu, W. Ma and Z. Bo, Adv. Mater., 2017, 29, 1703527 CrossRef PubMed.
- S. Li, L. Zhan, N. Yao, X. Xia, Z. Chen, W. Yang, C. He, L. Zuo, M. Shi and H. Zhu, Nat. Commun., 2021, 12, 1–11 CrossRef.
- D. Liu, Q. Zhu, C. Gu, J. Wang, M. Qiu, W. Chen, X. Bao, M. Sun and R. Yang, Adv. Mater., 2016, 28, 8490–8498 CrossRef CAS PubMed.
- Q. Guo, J. Lin, H. Liu, X. Dong, X. Guo, L. Ye, Z. Ma, Z. Tang, H. Ade and M. Zhang, Nano Energy, 2020, 74, 104861 CrossRef CAS.
- J. Lin, Q. Guo, Q. Liu, J. Lv, H. Liang, Y. Wang, L. Zhu, F. Liu, X. Guo and M. Zhang, Chin. J. Chem., 2021, 39, 2685–2691 CrossRef CAS.
- A. Frisch, Gaussian 09W Reference, Wallingford, USA, 2009, p. 25 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, 2009, see also: https://gaussian.com/.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. D. Hack and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 7917–7926 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- L. Deschenes and A. David, https://www.originlab.com, Commercial price, 2000, vol. 595.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. Tenderholt, Stanford University, CA Stanford, 2006.
- S. Alexander and R. Orbach, J. Phys., Lett., 1982, 43, 625–631 CrossRef.
- Z. Shuai, W. Li, J. Ren, Y. Jiang and H. Geng, J. Chem. Phys., 2020, 153, 080902 CrossRef CAS PubMed.
- E. U. Rashid, J. Iqbal, M. I. Khan, Y. A. El-Badry, K. Ayub and R. A. Khera, RSC Adv., 2022, 12, 12321–12334 RSC.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339–2350 CrossRef CAS PubMed.
- M. I. Khan, J. Iqbal, S. J. Akram, Y. A. El-Badry, M. Yaseen and R. A. Khera, J. Mol. Graphics Modell., 2022, 113, 108162 CrossRef CAS PubMed.
- J. Zhou, X. Wan, Y. Liu, G. Long, F. Wang, Z. Li, Y. Zuo, C. Li and Y. Chen, Chem. Mater., 2011, 23, 4666–4668 CrossRef CAS.
- E. U. Rashid, J. Iqbal, R. F. Mehmood, Y. A. El-Badry, S. J. Akram and R. A. Khera, Comput. Theor. Chem., 2022, 1211, 113669 CrossRef.
- E. U. Rashid, N. Hadia, J. Iqbal, R. F. Mehmood, H. Somaily, S. J. Akram, A. M. Shawky, M. I. Khan, S. Noor and R. A. Khera, RSC Adv., 2022, 12, 21801–21820 RSC.
- J. Widmer, M. Tietze, K. Leo and M. Riede, Adv. Funct. Mater., 2013, 23, 5814–5821 CrossRef CAS.
- A. Farhat, R. A. Khera, S. Iqbal and J. Iqbal, Opt. Mater., 2020, 107, 110154 CrossRef CAS.
- H. Sahu and A. N. Panda, Phys. Chem. Chem. Phys., 2014, 16, 8563–8574 RSC.
- U. Azeem, R. A. Khera, A. Naveed, M. Imran, M. A. Assiri, M. Khalid and J. Iqbal, ACS Omega, 2021, 6, 28923–28935 CrossRef CAS PubMed.
- Y. Tian, K. Wang, H. Zhang, X. Wu and C. Zhong, Tetrahedron, 2022, 113, 132756 CrossRef CAS.
- I. Zubair, R. A. Kher, S. J. Akram, Y. A. El-Badry, M. U. Saeed and J. Iqbal, Chem. Phys. Lett., 2022, 793, 139459 CrossRef CAS.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, M. Riaz, S. J. Akram, H. Somaily, A. M. Shawky, M. Raheel, M. I. Khan and E. U. Rashid, J. Phys. Chem. Solids, 2022, 110906 CrossRef CAS.
- M. Waqas, J. Iqbal, R. F. Mehmood, S. J. Akram, A. M. Shawky, M. Raheel, E. U. Rashid and R. A. Khera, J. Mol. Graphics Modell., 2022, 116, 108255 CrossRef CAS PubMed.
- M. Rafiq, R. A. Khera, M. Salim, M. Khalid, K. Ayub and J. Iqbal, Chem. Phys. Lett., 2021, 782, 139018 CrossRef CAS.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, S. J. Akram, Y. A. El-Badry, S. Noor and R. A. Khera, Surf. Interfaces, 2022, 30, 101875 CrossRef CAS.
- A. Sharif, S. Jabeen, S. Iqbal and J. Iqbal, Mater. Sci. Semicond. Process., 2021, 127, 105689 CrossRef CAS.
- M. Rafiq, M. Salim, S. Noreen, R. A. Khera, S. Noor, U. Yaqoob and J. Iqbal, J. Mol. Liq., 2022, 345, 118138 CrossRef CAS.
- M. R. Aslam, R. A. Khera, Y. A. El-Badry, M. Rafiq, A. Naveed, M. T. Shehzad and J. Iqbal, J. Mol. Graphics Modell., 2022, 112, 108146 CrossRef CAS PubMed.
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J.-L. Brédas, J. Am. Chem. Soc., 2002, 124, 7918–7919 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong and B. Gao, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- M. Ans, J. Iqbal, B. Eliasson and K. Ayub, Comput. Mater. Sci., 2019, 159, 150–159 CrossRef CAS.
- C. Fonseca Guerra, J. W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- P. Å. Malmqvist, Int. J. Quantum Chem., 1986, 30, 479–494 CrossRef CAS.
- A. Luzanov, A. Sukhorukov and V. Umanskii, Theor. Exp. Chem., 1976, 10, 354–361 CrossRef.
- P. Gong, P. Guo, Y. Wang, L. Yan, Z. Liang, M. Ding, J. Tong, J. Li and Y. Xia, ACS Appl. Energy Mater., 2021, 4, 9627–9638 CrossRef CAS.
- Y. A. Duan, Y. Geng, H. B. Li, J. L. Jin, Y. Wu and Z. M. Su, J. Comput. Chem., 2013, 34, 1611–1619 CrossRef CAS PubMed.
- U. Mubashar, A. Farhat, R. A. Khera, N. Iqbal, R. Saleem and J. Iqbal, J. Mol. Model., 2021, 27, 1–13 CrossRef PubMed.
- M. Salim, M. Rafiq, R. A. Khera, M. Arshad and J. Iqbal, Sol. Energy, 2022, 233, 31–45 CrossRef CAS.
- I. Zubair, R. A. Khera, A. Naveed, R. A. Shehzad and J. Iqbal, Mater. Sci. Semicond. Process., 2022, 148, 106812 CrossRef CAS.
- J. D. Chen, C. Cui, Y. Q. Li, L. Zhou, Q. D. Ou, C. Li, Y. Li and J. X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- R. Rajeswari, N. Islavath, M. Raghavender and L. Giribabu, Chem. Rec., 2020, 20, 65–88 CrossRef CAS PubMed.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- M. Ans, F. Manzoor, K. Ayub, F. Nawaz and J. Iqbal, J. Mol. Model., 2019, 25, 1–12 CrossRef CAS PubMed.
- M. Ans, M. Paramasivam, K. Ayub, R. Ludwig, M. Zahid, X. Xiao and J. Iqbal, J. Mol. Liq., 2020, 305, 112829 CrossRef CAS.
- M. Ans, K. Ayub, S. Muhammad and J. Iqbal, Comput. Theor. Chem., 2019, 1161, 26–38 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Omaymah Alaysuyc,
Javed Iqbal
*b,
Omaymah Alaysuyc,
Javed Iqbal



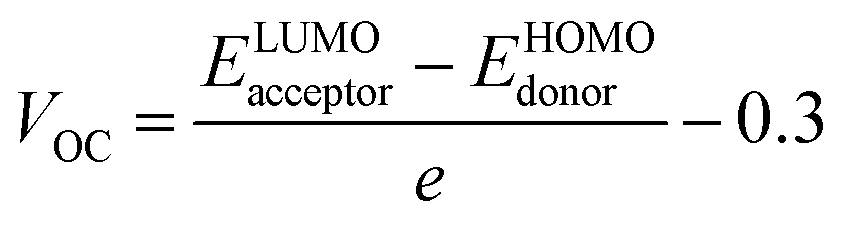

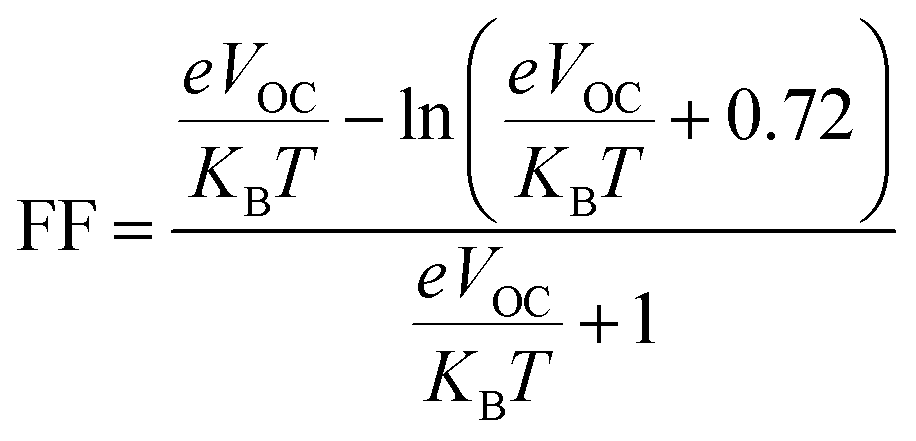
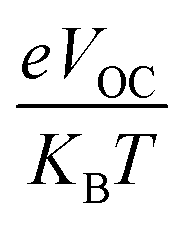 is normalized VOC and e is the customary charge and always equals 1. KB is the Boltzmann constant (8.61733034 × 10−5 electron volts per kelvin) and T is the temperature (constant value 300 K). Table 11 displays the derived normalized VOC and FF values for IDSTR and IDST1–IDST7. IDSTR has an FF of 0.9077 and a normalized VOC of 53.00. According to the given results, IDST4–IDST6 molecules have greater normalized VOC (58.02, 56.87, 53.38) and FF (0.9140, 0.9128, 0.9082) than IDSTR.
is normalized VOC and e is the customary charge and always equals 1. KB is the Boltzmann constant (8.61733034 × 10−5 electron volts per kelvin) and T is the temperature (constant value 300 K). Table 11 displays the derived normalized VOC and FF values for IDSTR and IDST1–IDST7. IDSTR has an FF of 0.9077 and a normalized VOC of 53.00. According to the given results, IDST4–IDST6 molecules have greater normalized VOC (58.02, 56.87, 53.38) and FF (0.9140, 0.9128, 0.9082) than IDSTR.
