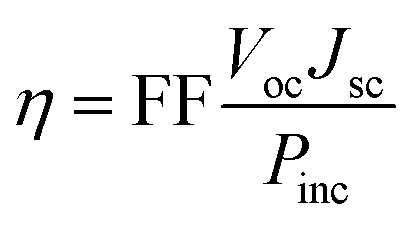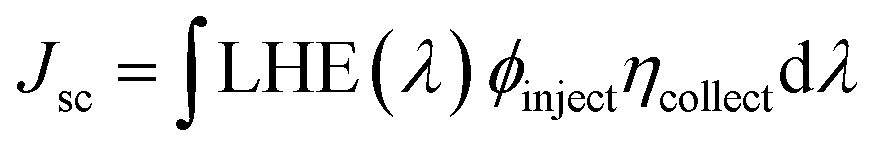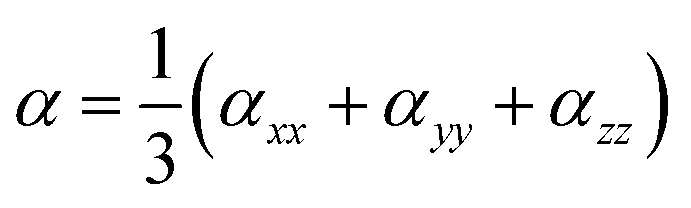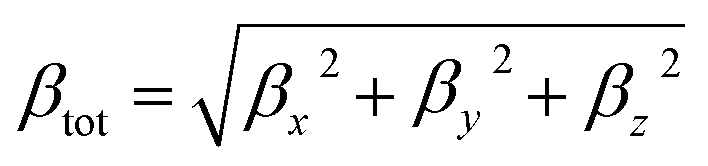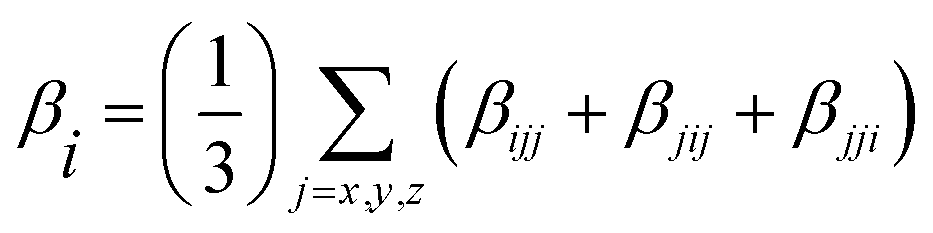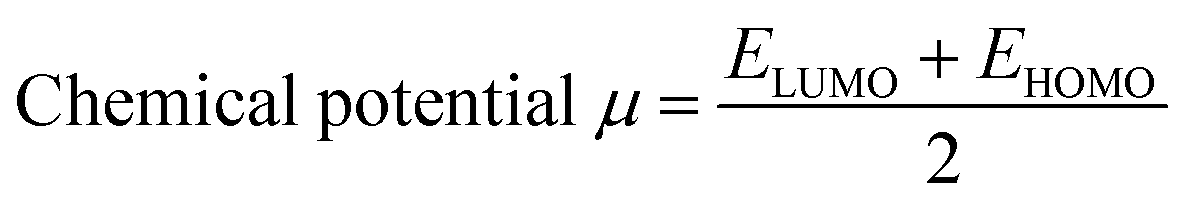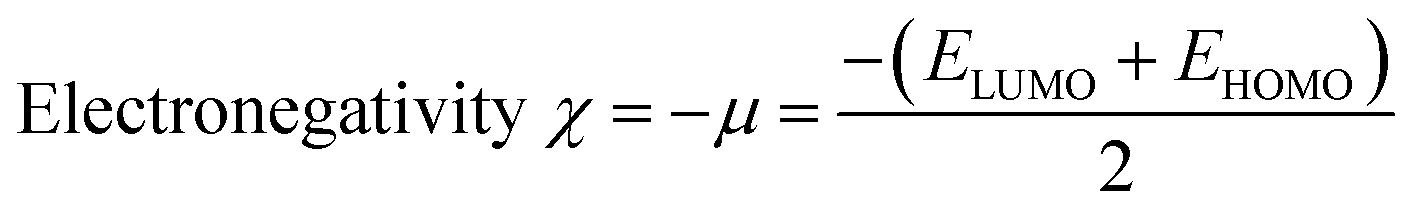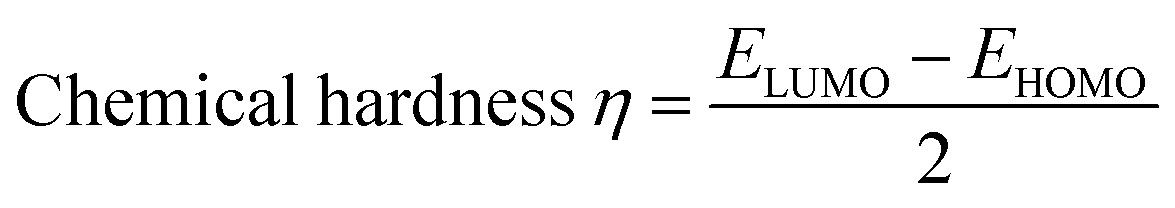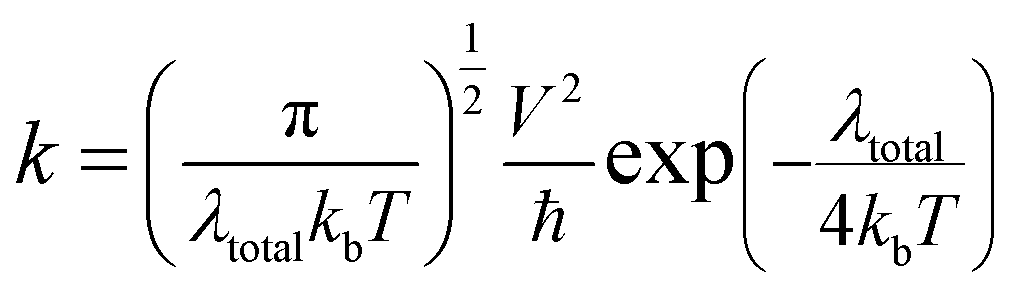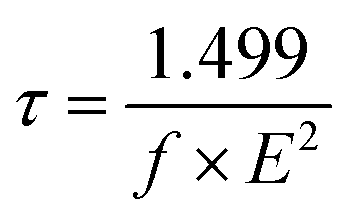DOI:
10.1039/D2RA05297K
(Paper)
RSC Adv., 2022,
12, 30626-30638
New organic dye-sensitized solar cells based on the D–A–π–A structure for efficient DSSCs: DFT/TD-DFT investigations
Received
23rd August 2022
, Accepted 10th October 2022
First published on 26th October 2022
Abstract
Global energy consumption has increased due to population growth and economic development. Solar energy is one of the most important renewable energy sources for human consumption. In this research, four novel organic dyes (D2–D5) of the D–A–π–A structure based on triphenylamine (TPA) were studied theoretically using DFT and TD-DFT techniques for future usage as dye-sensitized solar cells (DSSCs). The effects of modifying the π-spacer of the reference molecule D1 on the structural, electronic, photovoltaic, and optical characteristics of the D2–D5 dyes were studied in detail. D2–D5 exhibited band gaps (Egap) in the range from 1.89 to 2.10 eV with λabs in the range of 508 to 563 nm. The results obtained show that modifying the π-spacer of the dye D1 increased its hole injection and reinforced the intramolecular charge-transfer (ICT) impact, which resulted in a red-shifted ICT absorption with a greater molar extinction coefficient. The theoretically calculated open-circuit voltage (Voc) values ranged from 0.69 to 1.06 eV, while the light-harvesting efficiency (LHE) values varied from 0.95 to 0.99. Indeed, this theoretical research could guide chemists to synthesize effective dyes for DSSCs.
Introduction
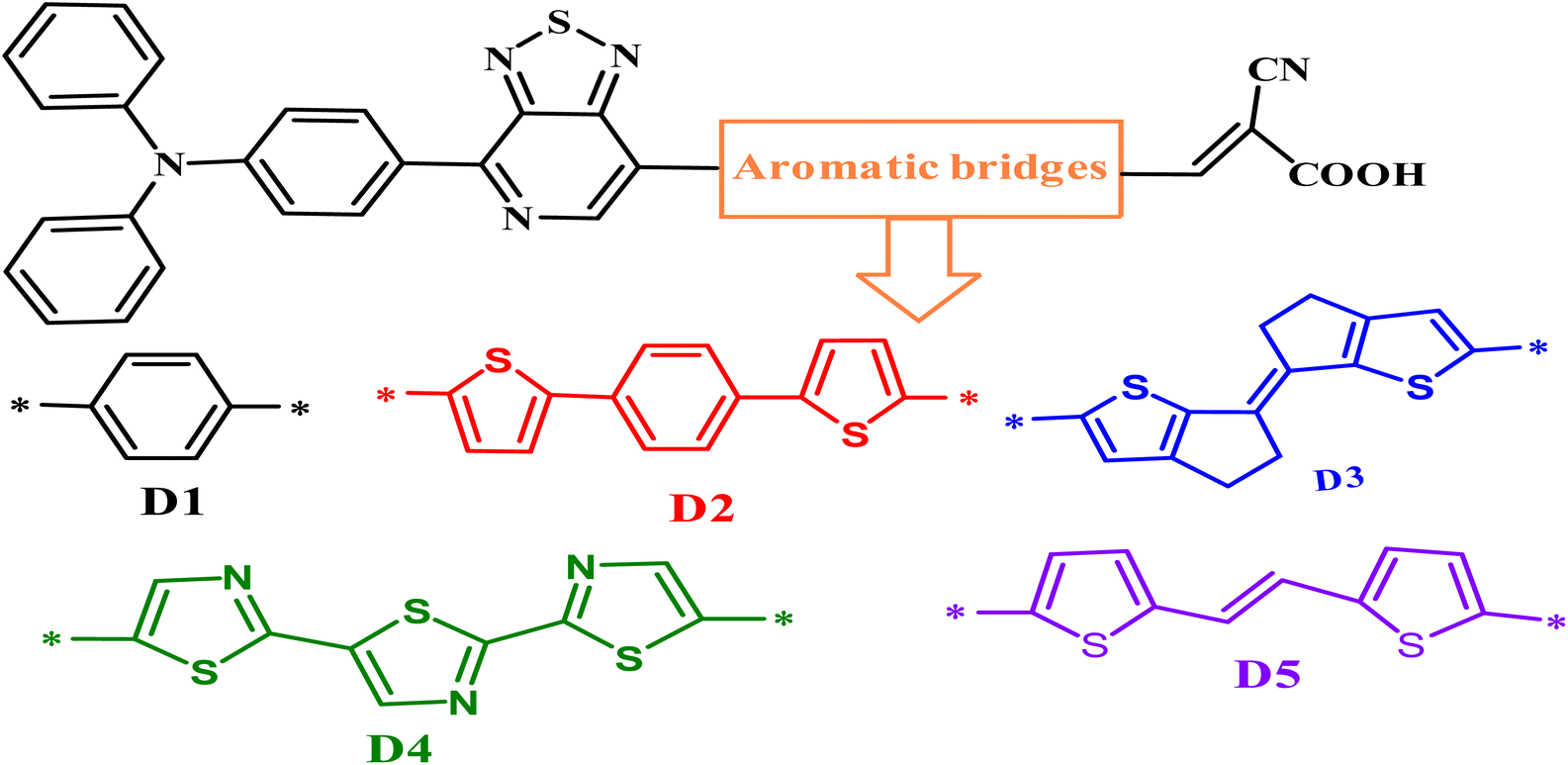
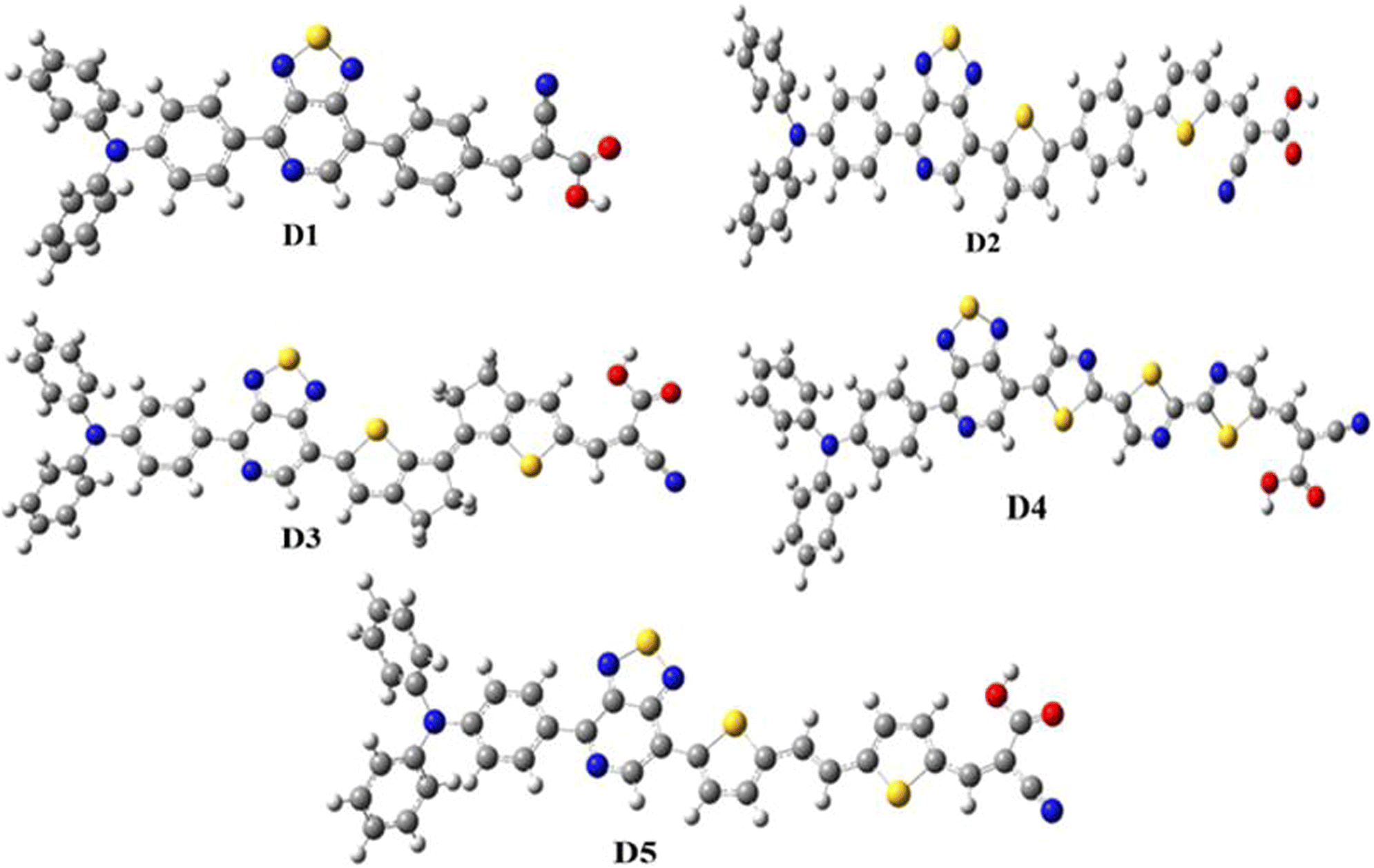
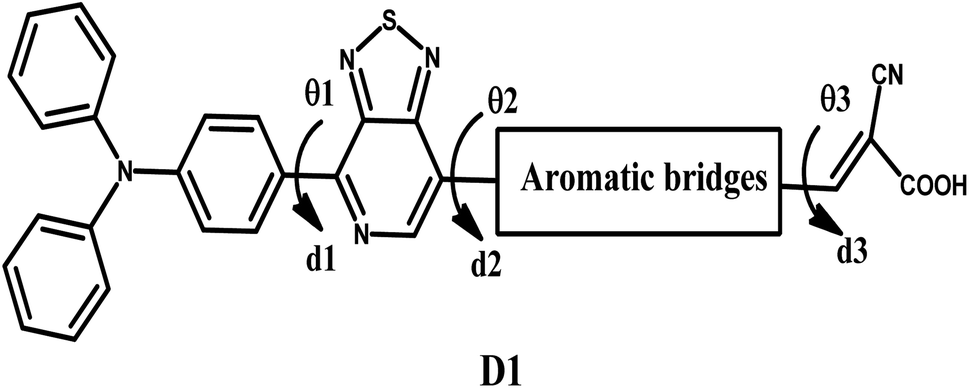
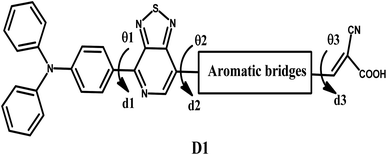
Global energy consumption has expanded rapidly due to economic and population development but has been accompanied by the increasing consumption of non-renewable energy sources and increased environmental pollution. Therefore, resorting to renewable and low-cost energy sources that can help reduce environmental pollution emissions has become the most appropriate option for future energy needs.1–3 Recently, dye-sensitized solar cells (DSSCs) (or Grätzel cells) have been widely used as a new alternative to meet the growing energy demand for safe environmentally friendly, natural, and economical resources,4–6 allowing the opportunity for converting solar energy into electricity with an efficiency of more than 13%.7 The engineering design of DSSCs is based on using photosensitive materials in solar cells; generally, a DSSC includes four main components: the photoanode,8 the cathode,9 the electrolyte, and the sensitizer.10 The photoanode of a DSSC consists of a thin layer of nanocrystalline metal oxide (e.g. titanium dioxide (TiO2) or zinc oxide (ZnO)) deposited on a glass substrate.11 Natural or artificial sensitizers are critical components of DSSCs and play a fundamental role in extending their absorption spectra.12 In this sense, different organic, organometallic, and natural dyes can be utilized as sensitizers.13 The electrolyte of a DSSC consists of a redox couple designated as I−/I3− (iodide/triiodide), closed by a counter-electrode typically composed of platinum (cathode).14 In general, most used organic dyes for DSSCs are artificial sensitizers that possess the donor–π-spacer–acceptor (D–π–A) molecular architecture, which is intended to provide intramolecular charge transport (ICT) from the donor unit (D) to the acceptor group (A) via the π-spacer.15–17 Other potential structures of artificial sensitizers, including D–A–π–A,18–20 D–D–π–A,21,22 (D–π–A)2,23,24 D–π–A–A,25,26 and A–π–D–π–A,27,28 have also been established and gained significant importance due to their high efficacy,29 but the most common structure is (D–π–A) because it facilitates electron movement, which improves the efficiency of dyes and allows them to be used in DSSCs.30–32 Since the efficiency of a DSSC cell is highly dependent on the photosensitive dye used in the cell, the following points can be used when designing the cell to improve and raise the efficiency of DSSCs: (a) it must include the most appropriate π-spacers between the electron donor and the acceptor; (b) it is important to have a broad and strong absorption band capable of covering practically all the visible and near-infrared spectral region (NIR); (c) appropriate redox and oxidation potentials of the compound should be investigated to ensure regeneration; (d) a high electron connection is necessary between the molecules and conduction band of the semiconductor (TiO2) to accelerate the transfer of electrons from the dye to TiO2.33–35 It is essential to study the electronic and optoelectronic characteristics of sensitizers for DSSC devices.36 Also, the highest occupied molecular orbital (HOMO) level of the dye being examined should be less than the redox couple energy level (iodide/triiodide I−/I3−) for the dye to be successfully regenerated.37 In addition to allowing the electron to be injected into the anode quickly, the lowest occupied molecular orbital (LUMO) should be above the conduction band (CB) of TiO2.38 Recent research introduced an electron auxiliary acceptor into the conjugated system between the electron-donor moiety and π-linker and used this in the design of a new architecture known as the D–A–π–A system,39–42 where sensitizers are coupled with auxiliary acceptor units, such as quinoxaline,43–46 benzothiadiazole,47–49 thieno[3,4-c]-pyrrole-4,6-dione,50,51 benzotriazole,52–54 and diketopyrrolopyrrole.55,56 Indeed, the D–A–π–A system has shown multiple advantages, including reducing electron recombination, minimizing aggregation, adjusting the molecular energy levels, causing a red-shift of the absorption band, and increasing the photovoltaic efficiency and stability;57–59 whereas several organic dyes of triphenylamine-based compounds have been used as electron donors due to the excellent light-harvesting ability of triphenylamine (TPA)-based molecules, their capacity to control the energy levels of molecules, promote charge generation, and facilitate the formation of intermolecular interactions.60–64 For DSSCs, several electron-acceptor materials have been developed and synthesized.65–67 However, cyanoacrylic acid is often employed as an electron-acceptor material due to its high electron-attracting capacity and solid binding to the TiO2 surface, which facilitates electron injection.68 The selection of the π-spacer is critical when designing D–π–A dyes because it plays a crucial function in regulating the HOMO–LUMO energy levels and expanding the absorption range of organic sensitizers.69 Ying et al. synthesized and elaborated a series of materials with the D–A–π–A system as sensitizers.58 Among these sensitizers, PT2 (reference molecule), which we named in this article as D1, demonstrated a good performance of 6.11% (Voc = 0.668 V, Jsc = 12.61 mA cm−2, and FF = 0.74), using TPA as the donor, [1,2,5]-thiadiazolo[3,4-c]-pyridine (PT) as the supplementary acceptor fragment, benzene as the π-bridge, and cyanoacetic acid as the electron-acceptor moiety (Fig. 1). The π-bridge plays an essential role in the rapid transfer of electrons for the DSSC charge; due to this, it should have the following features: easy charging, prolonged π-spacer that diminishes a large part of the light-absorption spectrum, and it should facilitate the absorption capacity.70,71 Hence, choosing a suitable π-bridge is one of the most important steps in building high-performance photovoltaic cells. Several π-bridges have been implemented experimentally and theoretically to fabricate novel sensitizers.72
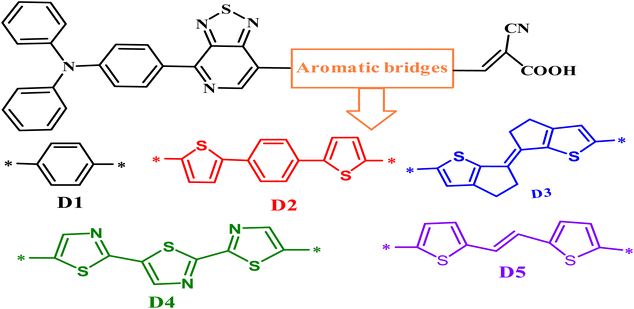 |
| | Fig. 1 Chemical structures of the proposed molecules in this work, Di (i = 1–5). | |
PT2 was used to investigate the efficiency of D–A–π–A structural molecules of thiophene derivatives as the π-bridge (Fig. 1) by using theoretical DFT and TD-DFT methods to explain the influences of the π-spacer compounds D2–D5. By finding the critical theoretical elements, a high current charge density (Jsc), open-circuit voltage (Voc), light-harvesting efficiency (LHE), reorganization energy (λtotal), electronic injection driving force (ΔGinject), and driving force of regeneration (ΔGreg) were determined. Furthermore, other parameters, such as the energy levels, frontier molecular orbitals (FMOs), natural bond orbital (NBO), nonlinear optical properties (NLO), density of states (DOS), chemical indices, and absorption spectra were also considered. These theoretical parameters help to elucidate the relationship between the molecular structure and performance, which aids selecting better dyes.
Computational section
Several computational parameters can influence the functioning of DSSCs, including the power conversion efficiency (η), and this parameter depends on several variables, including the values of the Voc, Jsc, fill factor (FF), and the incident solar power (Pinc):73| |
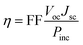 | (1) |
The parameter Voc can be evaluated by the analytical relation according to eqn (2):74
where
ELUMO indicates the dye's energy level, and
ECB designates the energy of the CB of TiO
2 (
ECB = −4.0 eV).
75 In a DSSC,
Jsc can also be determined by the following formula:
76| |
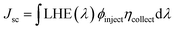 | (3) |
where LHE(
λ) signifies the light-harvesting efficiency at a specific wavelength,
Φinject represents the electron injection efficiency, and
η indicates the charge-collection efficiency.
It is vital to note that the LHE of DSSCs is a significant element that affects their performance. In general, a large LHE generates the most significant amount of photocurrent. To compute the LHE, the following equation is used:77
where
f denotes the strength of the oscillator proportional to the
λmax of the dye. Indeed, this strength has a direct impact on the LHE.
Eqn (5) ref.
78 may be used to compute the injection driving force into the CB of the TiO
2.
| | |
ΔGinject = Edye* − ECB
| (5) |
where
Edye* signifies the oxidation potential energy of the molecule in the excited state and
ECB symbolizes the reduction potential of the conduction band of TiO
2. The value of
Edye* may be computed in the following manner:
79where
Edye defines the oxidation potential of the molecule in the neutral state (
Edye = −
EHOMO), and
E00 denotes the vertical transition energy corresponding to the
λmax.
80
The driving force for dye regeneration denoted by the symbol ΔGreg can be expressed as follows:81
| | |
ΔGreg = Edye − EElectrolytRedox
| (7) |
where
EElectrolytRedox is represented as the redox potential of the electrolyte I
−/I
3− (−4.80 eV).
75
The DFT approach can be used to optimize dyes' positive and negative states to calculate their total energy of reorganization.82 It is known that a significant value of Jsc requires a low total energy of reorganization; this can be confirmed by combining the hole energy of reorganization with the electron one,83 as represented by the following formula (8):84
| | |
λi = (E±0 − E±±) + (E0± − E0)
| (8) |
wherhe energy of the catie
E±0 represents ton (anion) calculated with the optimized structure of the neutral molecule,
E±± is the energy of the cation (anion) calculated with the optimized cation (anion) structure,
E0± denotes the energy of the neutral molecule calculated at the cationic (anionic) state, and
E0 is the energy of the neutral molecule at the ground state.
In addition, excellent nonlinear optical (NLO) characteristics may affect the short-circuit current density.85 According to eqn (9) and (10), the polarizability (α) and hyperpolarizability (βtot) are determined as:
| |
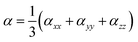 | (9) |
| |
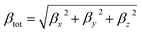 | (10) |
where
xx,
yy, and
zz represent the tensor coefficients of polarizability, and
βi = (
i =
x,
y,
z) combines the different quantities:
Indeed, different reactivity parameters have been studied to explain and predict various physicochemical phenomena that provide details about the electrical structure of the chemistry of target compounds,86 e.g. employing EHOMO and ELUMO can give the chemical potential (μ),87 chemical hardness (η),88,89 softness (S), and electronegativity (χ)90 by using the following formulas:
| |
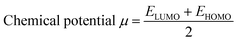 | (11) |
| |
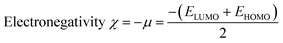 | (12) |
| |
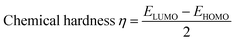 | (13) |
All the computations in this research were performed using the Gaussian 09 software package,91 and GaussView 6.0.16 for the simulated spectra and orbital density.
Selecting the test method
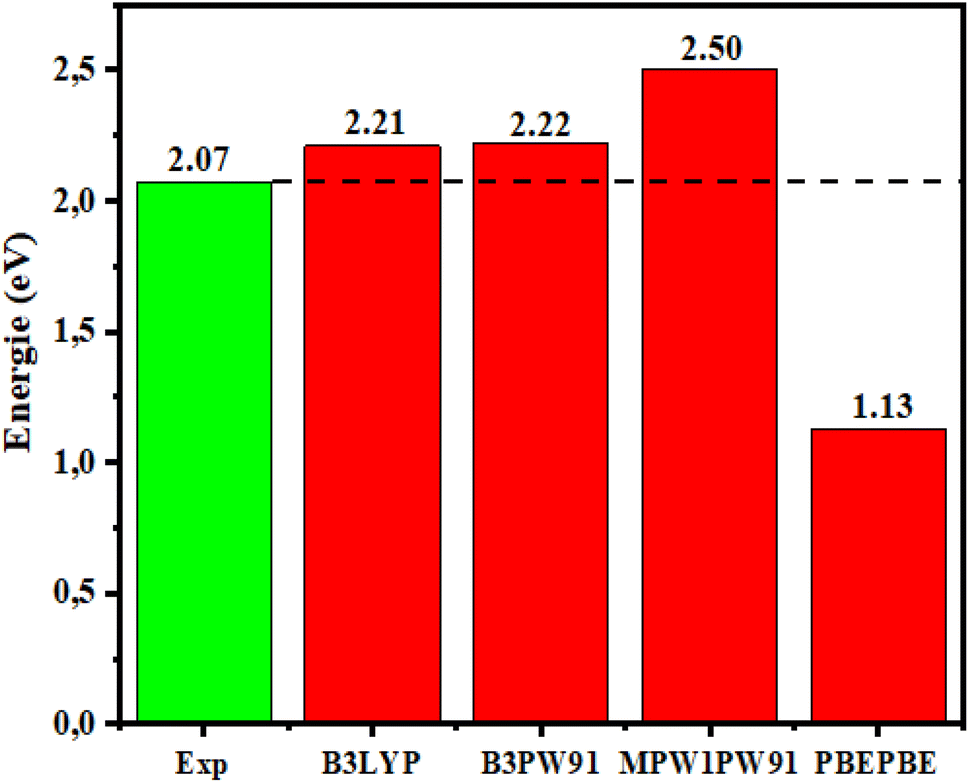
To examine the impact of different functionals on the electronic characteristics: the Eg of the dye D1 was studied theoretically using the PBEPBE,92 MPW1PW91,93 B3PW91,94 and B3LYP95 functionals combined with the 6-31G** basis set, and the data obtained are presented in Fig. 2, which shows the bandgap energies determined from B3LYP/6-31G**. The results were close to the experimental results with a reliable and acceptable error variance, while the error margin given by other functionals was higher. Therefore, the B3LYP/6-31G** functional was considered to be much more suitable for determining our developed molecules' structural and electronic characteristics.
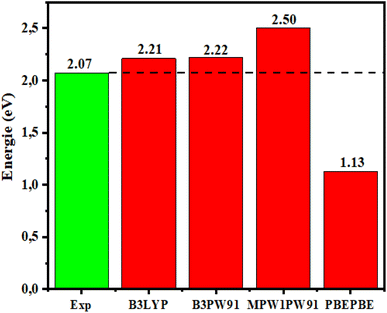 |
| | Fig. 2 Egap of D1 determined using DFT with different functionals and comparison with the experimental values. | |
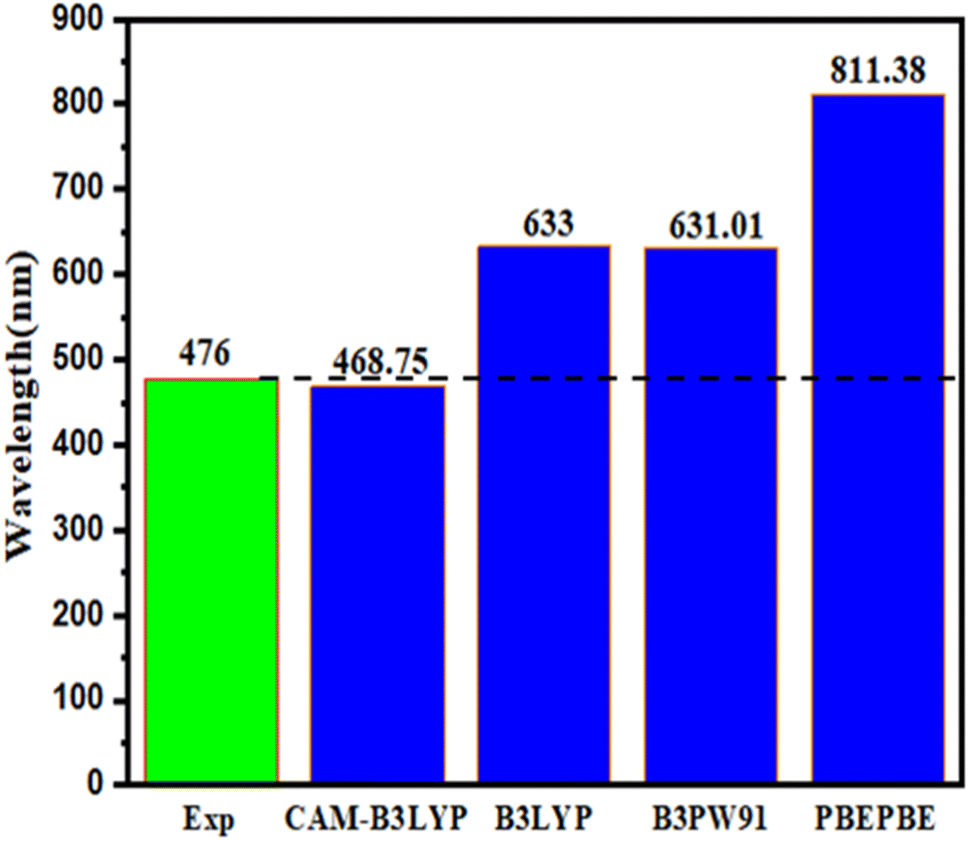
The choice of the TD-DFT theoretical function to describe the vertical transition energy of a potential sensitizer depends on the reference molecule's absorption wavelengths (λmax). Fig. 3 shows the absorption wavelengths (λmax) of the reference molecule examined using TD-DFT with various functionals, such as PBEPBE,92 CAM-B3LYP,96 B3LYP,95 and B3PW91 (ref. 97) combined with the 6-31G** basis set. The results indicated that the CAM-B3LYP with 6-31G** offered a maximum value of λ equal to 468.75 nm, similar to the experimental value (476 nm).58 Based on the findings, CAM-B3LYP with the 6-31G** level was chosen to compute the optical characteristics of the five proposed molecules (D1, D2, D3, D4, and D5) (Fig. 4).
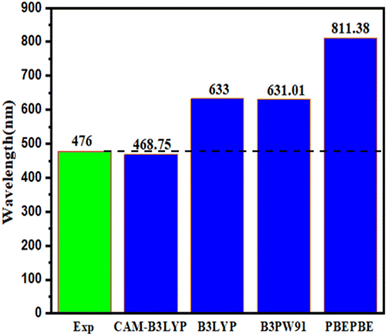 |
| | Fig. 3 Examining the functional used in the TD-DFT calculation of the maximum absorption wavelengths (λmax) for D1. | |
 |
| | Fig. 4 Structural optimization of the studied molecules (D1–D5). | |
Results and discussion
Molecular geometry
Table 1 gives the geometric characteristics of the potential sensitizer structures, where d explains the bond distance and θ represents the dihedral angle (Scheme 1). As illustrated in Table 1, all the parameters were determined at the DFT/B3LYP/6-31G** level. It is important to note that the short bond lengths facilitate the ICT in the D–π–A architecture.98 The optimized structures of all the studied dyes as sensitizers showed identical conformations, as confirmed by the bond length values all lying in the range of 1.43–1.46 Å. These bond lengths revealed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C character in all the dyes, which facilitated ICT between the donor fragment and the -spacer and the -bridge and the acceptor fragment. According to Table 1, the values of θ1, θ2, and θ3 for all the potential proposed dyes ranged between −175° and −179°, which revealed that all the compounds possessed a planar geometry with the dihedral angle θ2 of the D1 dye. This was slightly twisted due to the steric interactions between the π-spacer and acceptor fragments.99 Thus, altering the π-bridge influenced the planarity of these designed molecules, hence increasing their light-collecting effectiveness.
C character in all the dyes, which facilitated ICT between the donor fragment and the -spacer and the -bridge and the acceptor fragment. According to Table 1, the values of θ1, θ2, and θ3 for all the potential proposed dyes ranged between −175° and −179°, which revealed that all the compounds possessed a planar geometry with the dihedral angle θ2 of the D1 dye. This was slightly twisted due to the steric interactions between the π-spacer and acceptor fragments.99 Thus, altering the π-bridge influenced the planarity of these designed molecules, hence increasing their light-collecting effectiveness.
Table 1 Distances of di (Å) and the dihedral angles θi (°) of the studied dyes
| Dye |
d1 (Å) |
d2 (Å) |
d3 (Å) |
θ1 (°) |
θ2 (°) |
θ3 (°) |
| D1 |
1.46 |
1.46 |
1.45 |
−176.28 |
−150.61 |
−179.06 |
| D2 |
1.46 |
1.45 |
1.43 |
−175.45 |
−170.76 |
−179.09 |
| D3 |
1.46 |
1.45 |
1.43 |
−176.69 |
−175.60 |
−179.58 |
| D4 |
1.46 |
1.45 |
1.43 |
−178.05 |
−179.08 |
−179.99 |
| D5 |
1.46 |
1.45 |
1.43 |
−177.28 |
−179.64 |
−179.36 |
 |
| | Scheme 1 Studied geometric parameters. | |
Frontier molecular orbitals
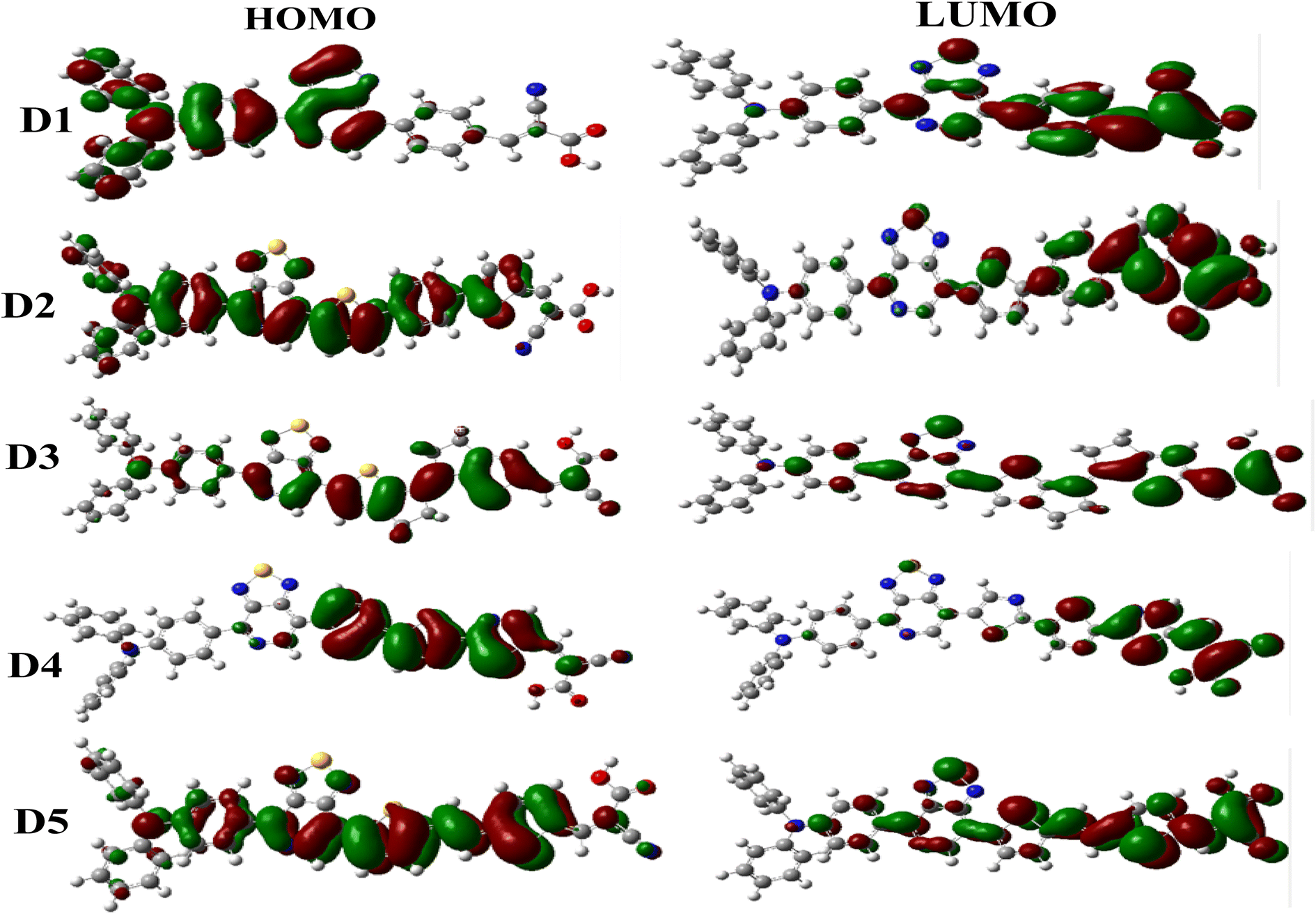
Frontier molecular orbitals (HOMO and LUMO) are quantum mechanical descriptors that give essential information about the optoelectronic properties of dyes.100 These orbitals assist in examining the distribution and stabilization of the charge density throughout the dyes. According to the literature, electron transfer (S0 → S1) must occur in each noticed dye from the ground state (HOMO) to the excited state (LUMO).101,102 The HOMO–LUMO band gap (Eg) indicates the decrease and increase in reactivity due to various π-spacers. Charge transfer is facilitated when the band gap is small and vice versa.103 Theoretical determination of the energy levels for the HOMO and LUMO for any dye is vital for understanding the construction of the electronic features in dye systems.104 This helps to determine the suitability of suggested dyes to fit the fundamental conditions for DSSC photosensitizer materials.105 The EHOMO, ELUMO, and the various Egap values of some currently investigated research molecules are illustrated in Fig. 6, and the FMO schematic is shown in Fig. 5.
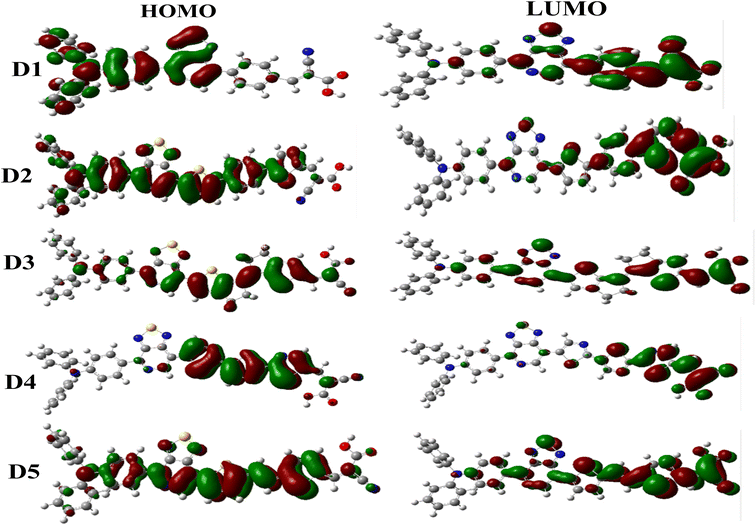 |
| | Fig. 5 Contour plots of the FMOs of all dyes examined. | |
In this work, the proposed dyes (D1–D5) comprised triphenylamine as electron-donor unit [1,2,5]-thiadiazolo[3,4-c]-pyridine as the auxiliary acceptor group, different π-linkers, and acceptor parts (cyanoacrylic acid). The analysis of the frontier orbitals demonstrated that the HOMO of dye D1 was concentrated on the donor triphenylamine and [1,2,5]-thiadiazolo[3,4-c]-pyridine. At the same time, the LUMO was distributed on the π-spacers and the acceptor regions (cyanoacrylic acid). For the dyes D2, D3, and D5, the HOMO electrons were widely distributed on these dyes with a maximum density on different π-spacers. In contrast, the LUMOs were principally situated on cyanoacrylic acid, with a minimum density on the π-spacers groups. Finally, the D4 dye showed a high HOMO on the π-spacers group and a strong LUMO on the cyanoacrylic acid group.
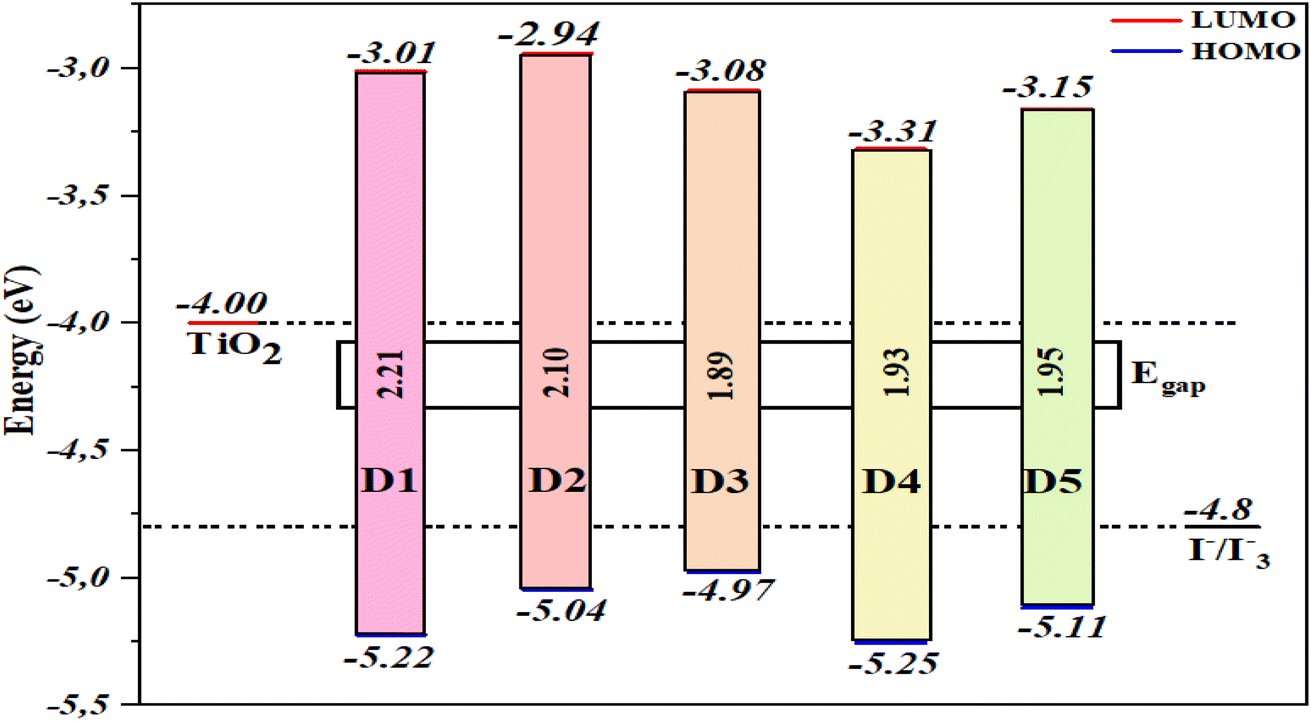
Based on DSSC devices' operating principle, photosensitizer materials should have appropriate energy levels to allow for electron injection and material regeneration. The energy levels of the D1 to D5 molecules were estimated at the B3LYP/6-31G** level, and their energy diagrams are shown in Fig. 6, showing that the HOMO levels of all the molecules were below the redox potential of I−/I3−. Therefore, the oxidized molecules (photosensitizers) could be reduced by electrons from the electrolyte (I−/I3−). Moreover, the molecules had LUMO values higher than the CB of TiO2 (−4.00 eV), which implied that the electrons could be injected quickly into the TiO2 semiconductor during the excitation of the molecules. As previously stated, the proposed dyes (D1–D5) are good photosensitizer selections and are suitable for DSSC applications. The HOMO energy values of the proposed dyes (D1–D5) followed the order: D3 (−4.979 eV) > D2 (−5.045 eV) > D5 (−5.118 eV) > D1 (−5.221 eV) > D4 (−5.255 eV), and the values implied a regeneration capacity and good performance. Hence, the proposed molecules D2, D3, and D5 had greater regeneration capacities than D1. Meanwhile, the LUMO levels of the molecules D2–D5 and D1 were −2.942, −3.086, −3.316, −3.159, and −3.011 eV, respectively. The results showed no significant difference except for D2.
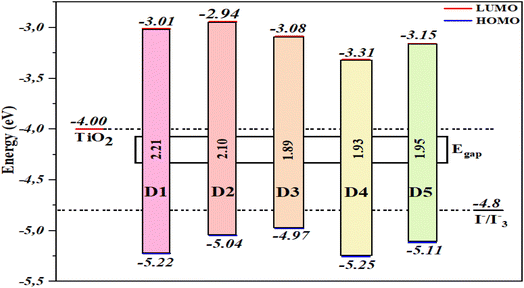 |
| | Fig. 6 Illustration of the energy levels of all molecules studied, TiO2, and the electrolyte (I−/I3−). | |
The energy gap has a considerable effect on the activity of molecules; whereby a sensitizer with a small bandgap energy is more favourable for electron excitation, and has enhanced absorption properties. For that reason, sensitizers may absorb more photons in the visible region, increasing the Jsc and PCE.106,107 The findings in Fig. 6 show that the bandgap energy of the proposed molecules could be modulated by inserting various aromatic spacers. D3 offered the lowest Eg of 1.89 eV, followed by D4 (1.93 eV), D5 (1.95 eV), D2 (2.10 eV), and D1 (2.21 eV). The proposed molecules (D2–D5) had smaller Eg values than D1, implying that D2–D5 would have a better short-circuit current density (Jsc) than D1. Due to a smaller energy gap, the UV-vis spectra of D2–D5 may be red-shifted relative to D1.
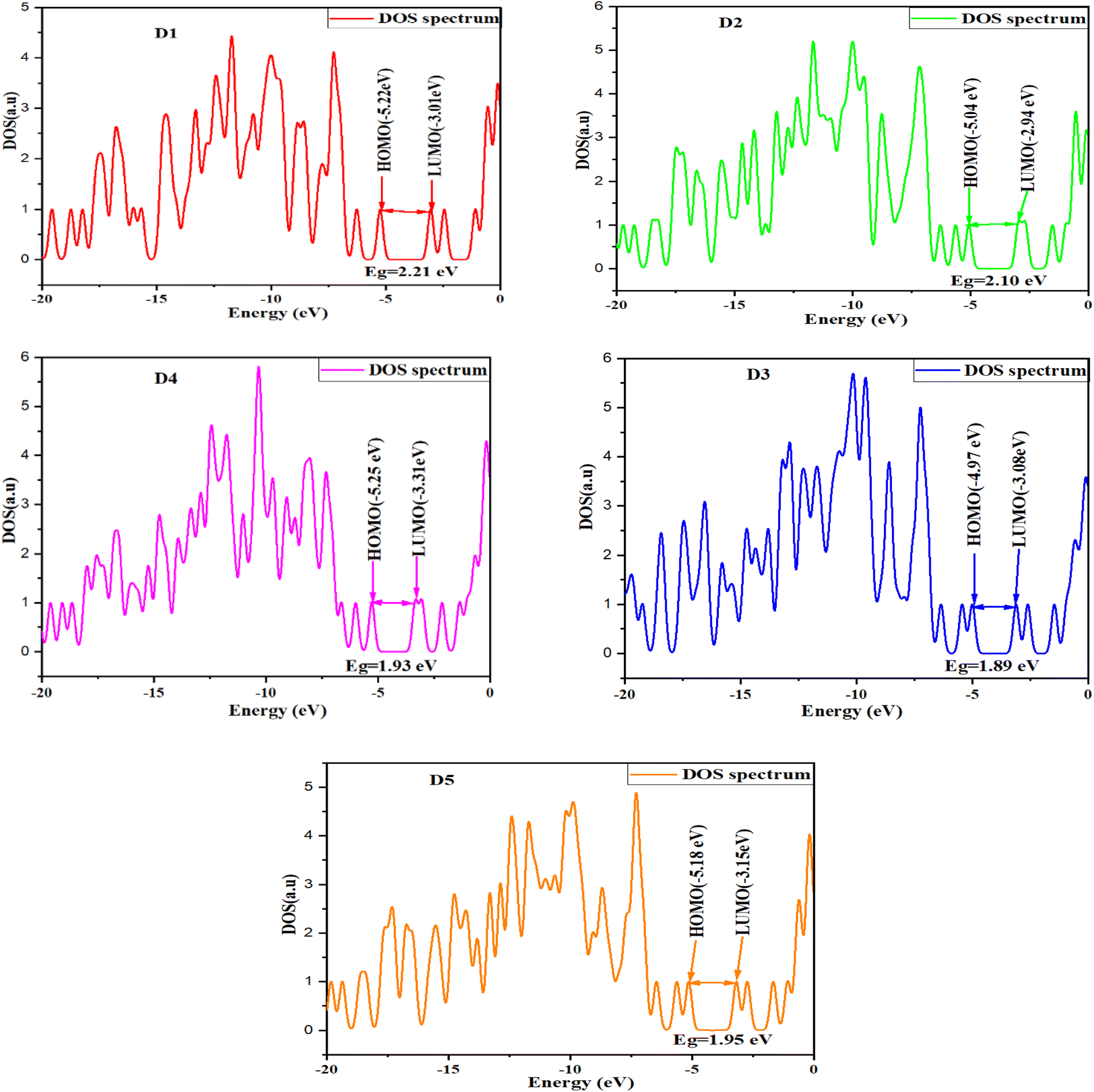
Density of states (DOS)
Numerous studies have shown that examination of the density of states can explain the charge-transfer characteristics of organic semiconductors.108 Indeed, it also provides the contribution of the MOs of each molecule's different components to the overall system. The GaussSum can be used to determine the density of states (DOS) function.109 Fig. 7 shows the DOS results for predicting the alpha and beta free electrons for the valence and conduction bands, respectively.110 The energy of the band gap increased in parallel with the DOS of the proposed molecules. In DSSCs, the voltage is associated with the energy gap between the Fermi level (next to the conduction-band level) of the TiO2 semiconductor electrode and the redox potential in the electrolyte. Evidently, when EF (Fermi energy) is near the midgap, the charge is transported from/to the metal electrode and organic material. Hence, the potential energy profile stays flat whenever EF approaches either the HOMO or LUMO level, while charge transfer is significant.33
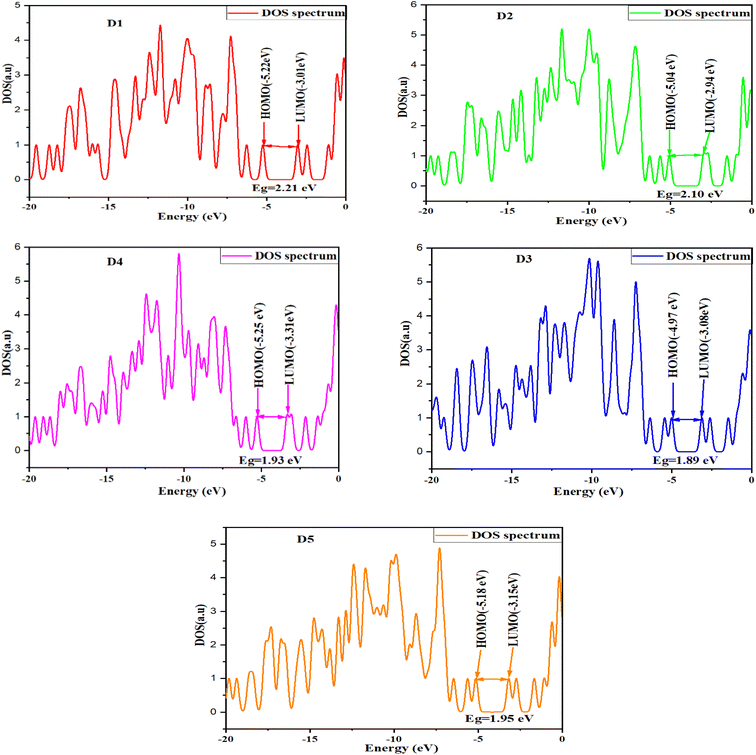 |
| | Fig. 7 Density of states for the examined molecules D1–D5. | |
Quantum chemical reactivity indices
The chemical potential (μ), electronegativity (χ), and chemical hardness (η) of the proposed molecules were obtained from eqn (11)–(13) using the B3LYP method with the 631G** basis set, and the findings are tabulated in Table 2. According to the data in Table 2, the TiO2 compound had the lowest chemical potential (−4.66 eV)111 compared with the potential chemical values calculated for the proposed molecules D1–D5. This suggests the electron transfer from the dyes as electron donors to the TiO2 semiconductors as electron acceptors would be relatively straightforward.112 Furthermore, the chemical hardness (η) of all the studied molecules was lower than that of TiO2 (η = 3.78 eV), indicating that all the dyes could release one or more electrons when compared to the TiO2 semiconductor. Additionally, the electronegativity of TiO2 (−4.66 eV) was higher than for the dyes, implying that TiO2 may readily accept electrons from these dyes.
Table 2 Calculated values for the electronic and chemical parameters of the five dyes studied
| Dye |
EHOMO (eV) |
ELUMO (eV) |
Egap (eV) |
μ (eV) |
η (eV) |
χ (eV) |
| D1 |
−5.223 |
−3.012 |
2.21 |
−4.12 |
1.10 |
4.12 |
| D2 |
−5.045 |
−2.942 |
2.10 |
−3.99 |
1.05 |
3.99 |
| D3 |
−4.979 |
−3.086 |
1.89 |
−4.06 |
0.94 |
4.06 |
| D4 |
−5.255 |
−3.316 |
1.93 |
−4.28 |
0.97 |
4.28 |
| D5 |
−5.118 |
−3.159 |
1.95 |
−4.14 |
0.98 |
4.14 |
| TiO2 |
— |
− 4.00 |
— |
−4.66 |
3.78 |
4.66 |
Natural bond orbital (NBO) analysis
NBO analysis is a key factor for studying the charge transfer or conjugative interactions of molecular systems. It offers a good platform for examining intramolecular and intermolecular bonds and bond interactions.113 Therefore, NBO analysis was carried out here at the DFT/B3LYP/6-31G** level to determine charge populations for the proposed molecules, and the data are reported in Table 3. The donor and π-linker groups of all the molecules exhibited a positive NBO charge, indicating that they were efficient electron-donating groups. On the other hand, the negative NBO value of the supplementary acceptor and main acceptor groups suggested they were good electron-withdrawing groups. During photoexcitation, the NBO charges on the donor groups of each molecule were arranged as follows: D4 (0.136e) > D5 (00.134e) > D1 (0.124e) > D3 (0.117e) > D2 (0.111e), suggesting that D4 and D5 had a better electron-donating capacity than the reference molecule D1. For the acceptor, the NBO charges for D1–D5 were −0.102, −0.145, −0.198, −0.110, and −0.173e, respectively, indicating that the insertion of thiophene derivatives as π-spacers could augment the electron density in the acceptor. In addition, Δq(D–A) indicates the difference between the electron donor and acceptor. According to Table 3, D3 had a maximum q(D–A) (0.315e), followed by D5 (0.307e), D2 (0.256e), D4 (0.246e), and D1 (0.226e), signifying that all the dyes had a greater charge separation than D1.
Table 3 NBO analysis for all the proposed molecules (D1–D5)
| Dyes |
D |
A |
π |
A |
Δq(D–A) |
| D1 |
0.124 |
−0.107 |
0.085 |
−0.102 |
0.226 |
| D2 |
0.111 |
−0.133 |
0.167 |
−0.145 |
0.256 |
| D3 |
0.117 |
−0.141 |
0.222 |
−0.198 |
0.315 |
| D4 |
0.136 |
−0.128 |
0.102 |
−0.110 |
0.246 |
| D5 |
0.134 |
−0.139 |
0.178 |
−0.173 |
0.307 |
Electrostatic potential (ESP)
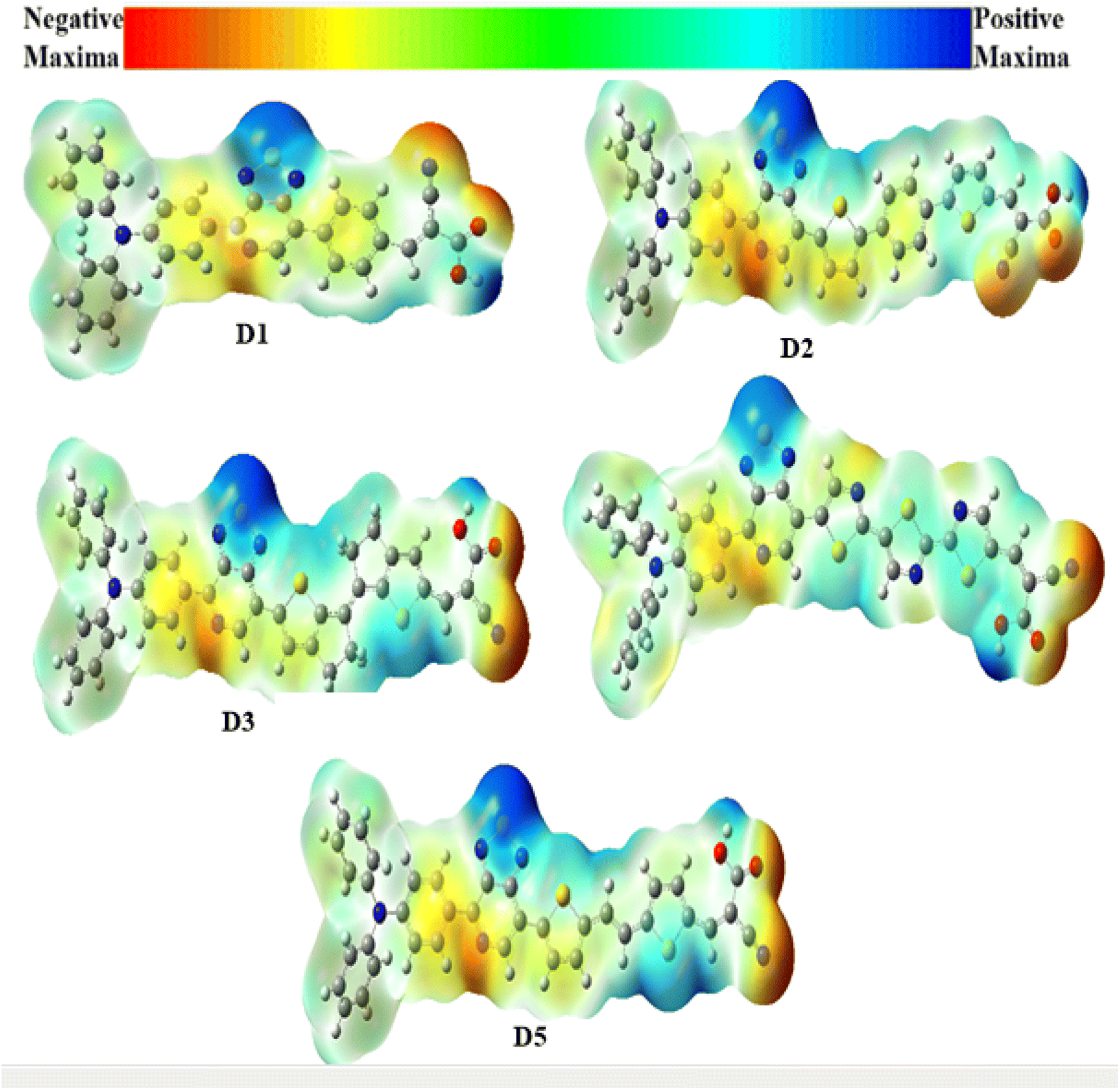
The electron density is an essential parameter for predicting both the reactivity of electrophilic and nucleophilic sites (i.e. an explanation of chemical reactions) and the interactions through hydrogen bonding.114 The molecular electrostatic potentials (MEPs) for the D1–D5 dyes are shown in Fig. 8, where the red area corresponds to the negative potentials (the electron-rich field), which characterizes the reactive zone for an electrophilic attack. In contrast, the blue area relates to the positive potentials (electron-poor fields), which determine the reactive zone for nucleophilic attack. Thus, a specific colour defined each MEP value in the following order: red < orange < yellow < blue. As illustrated in Fig. 8, the MEP revealed that all the dyes' maximum positive area (blue colour) was centred on the carboxyl hydrogen atom and the sulfur atoms of the auxiliary acceptor, mainly for D1, D4, and D5. In contrast, the negative area (red colour) was primarily centred around the nitrogen atoms of the acceptor groups (–CN) in the D1–D5 dyes, which were the most favourable sites for the electrolyte.
Nonlinear optical properties (NLO)
NLO characteristics were measured via the intra-molecular charge delocalization in conjugated molecular systems. Hence, these properties might be utilized to assess the efficiency of the hole-electron transfer of the sensitizer, which can also affect the Voc and the solar cell performance. It has been shown that conjugated molecular systems possess excellent NLO properties. Therefore, it often offers excellent photoelectric conversion characteristics.114–117 The α and βtot of the molecules are computed employing the B3LYP approach and 6-31G** basis set, and the calculation results are listed in Table 4. Based on the values in Table 4, the polarizability (α) of all dyes is arranged as follows: D3 (148.77 × 10−24 esu) > D5 (140.42 × 10−24 esu) > D4 (132.88 × 10−24 esu) > D2 (130.06 × 10−24 esu) >D1 (87.42 × 10−24 esu). These results indicate that D2–D3 performs better in electron charge transfer than D1. For the hyperpolarizability (βtot), D2–D5 have greater first hyperpolarizability than D1, and values are in the following order: D4 > D5 > D2 > D3 > D1. It indicates that the proposed D2–D5 dyes will present a more efficient photocurrent response and perform better than D1 due to their excellent NLO characteristics.
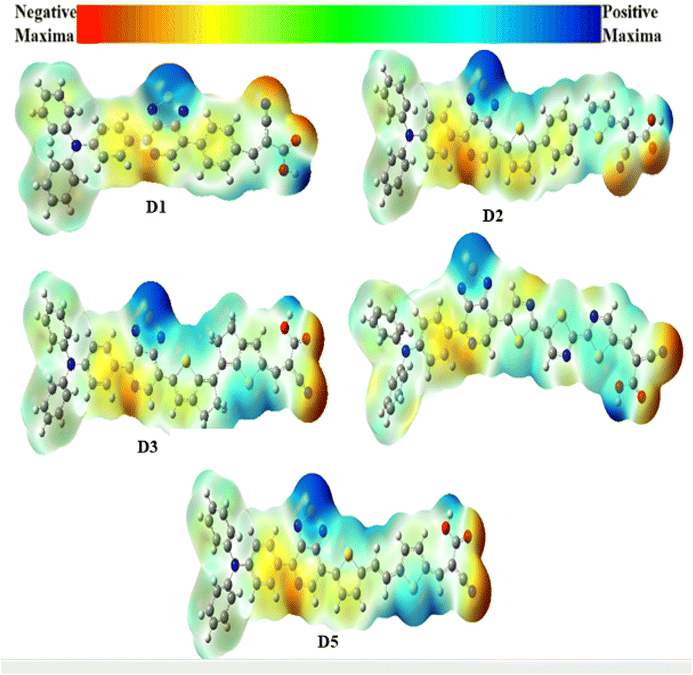 |
| | Fig. 8 Calculated electrostatic potentials on the molecular surfaces of the studied dyes. | |
Table 4 Calculated average isotropic polarizability 〈α〉 and the first hyperpolarizability (βtot) for D1–D5 and their constituents applying the B3LYP/6-31G** set
| Parameter |
D1 |
D2 |
D3 |
D4 |
D5 |
| αxx |
1127.90 |
1826.55 |
2200.52 |
1883.56 |
2119.14 |
| αxy |
24.07 |
6.94 |
24.00 |
−11.03 |
−6.22 |
| αyy |
437.46 |
521.44 |
550.56 |
555.79 |
487.52 |
| αxz |
−0.16 |
10.55 |
−4.12 |
−11.68 |
0.92 |
| αyz |
−12.34 |
−30.75 |
−9.01 |
2.33 |
−16.86 |
| αzz |
204.27 |
284.85 |
260.63 |
250.67 |
235.99 |
| 〈α〉 (a.u.) |
589.88 |
877.61 |
1003.90 |
896.67 |
947.55 |
| 〈α〉 (10−24 esu) |
87.42 |
130.06 |
148.77 |
132.88 |
140.42 |
| βxxx |
83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 624.40 624.40 |
−127466.00 |
−108879.00 |
−177392.00 |
−155029.00 |
| βxxy |
4543.67 |
−9817.01 |
−9986.1 |
15466.20 |
−10344.30 |
| βxyy |
−113.06 |
1108.60 |
1254.72 |
−5037.69 |
994.63 |
| βyyy |
−63.76 |
−69.38 |
−170.44 |
810.49 |
−142.33 |
| βxxz |
1854.24 |
1092.00 |
2854.64 |
1835.06 |
2672.25 |
| βxyz |
417.07 |
407.57 |
127.05 |
−1096.19 |
153.20 |
| βyyz |
35.34 |
22.73 |
8.679 |
268.18 |
35.59 |
| βxzz |
48.42 |
−275.22 |
137.68 |
25.50 |
149.13 |
| βyzz |
22.90 |
−98.71 |
−10.98 |
18.76 |
−19.46 |
| βzzz |
17.75 |
16.73 |
6.76 |
−5.30 |
10.85 |
| βtot (a.u.) |
83702.73 |
127030.72 |
108004.56 |
183142.65 |
154267.40 |
| βtot (10−30 esu) |
723.133 |
1097.457 |
933.084 |
1582.225 |
1332.624 |
Optical properties
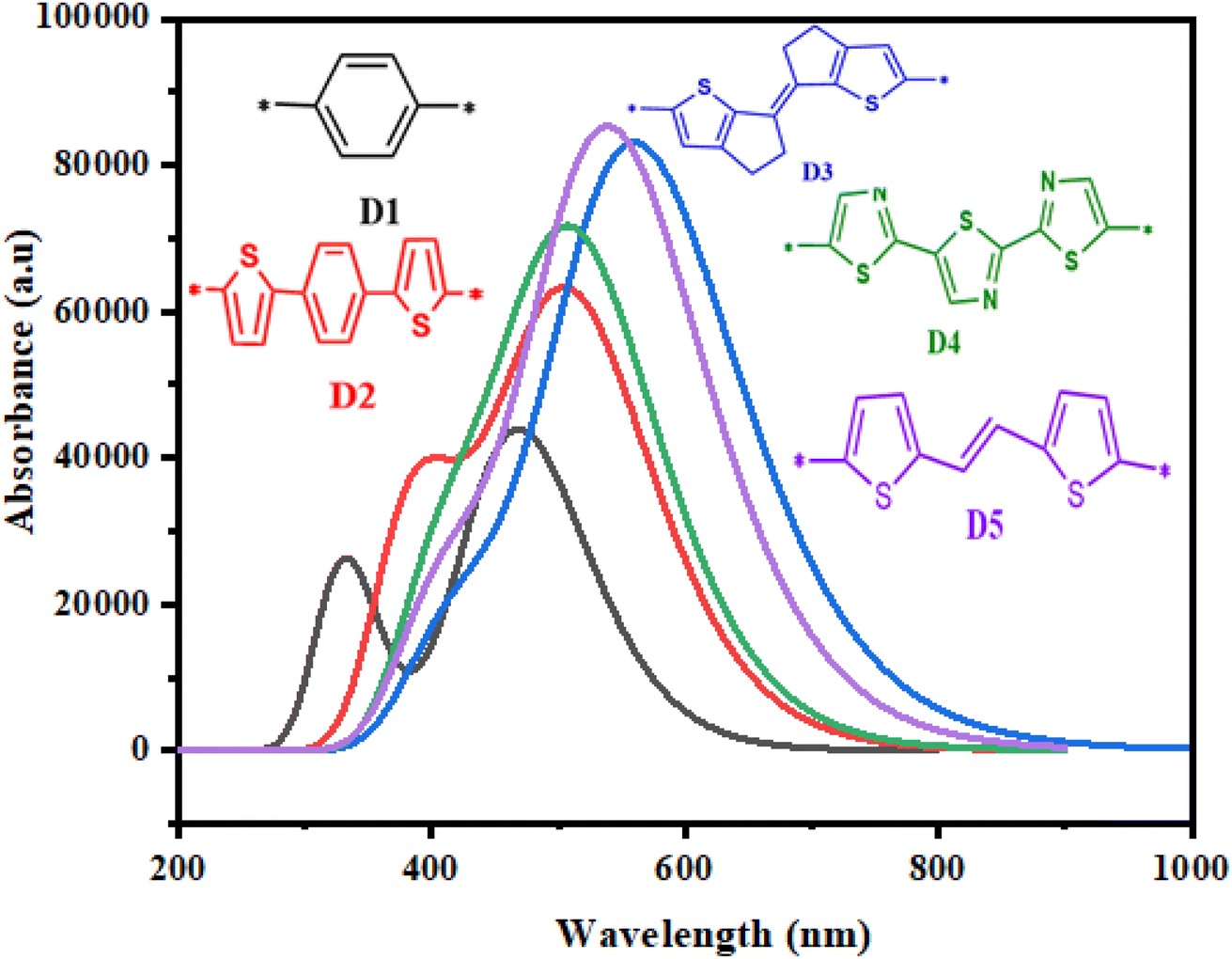
Photovoltaic characteristics are strongly related to the absorption of photons by dyes. An excellent organic solar cell must possess a broad and strong visible absorption spectrum.104 To examine the effects of various π-spacers of the proposed dyes (D2–D5) based on the molecule (D1) and to get a clearer idea of the possible electronic transitions of the five studied dyes, the proposed dyes' electronic transitions and absorption characteristics were calculated using TD-DFT/CAM-B3LYP/6-31G**,118 in the gas phase. The absorption spectra of the reference molecule (D1) and the investigated molecules (D2–D5) are shown in Fig. 9, and the results are tabulated in Table 5. From the results in Fig. 9 and Table 5, the maximum absorption values obtained for the molecule (D1) and its derivatives D2, D3, D4, and D5 were 468.749, 508.840, 563.103, 514.051, and 542.007 nm, respectively. The proposed dyes showed high absorption between 400 and 800 nm in the visible zone. These values correlated with the lowest energy and were ascribed to an electronic transition from the ground state S0 to the excited state S1. In addition, the maximum absorption wavelengths of these dyes were 542.007 nm and 563.103 nm for D5 and D3, respectively. Indeed, a dominant transition with low energy (2.20–2.64 eV) and a higher value of oscillation force (1.08–2.06) accompanied the absorption spectra.
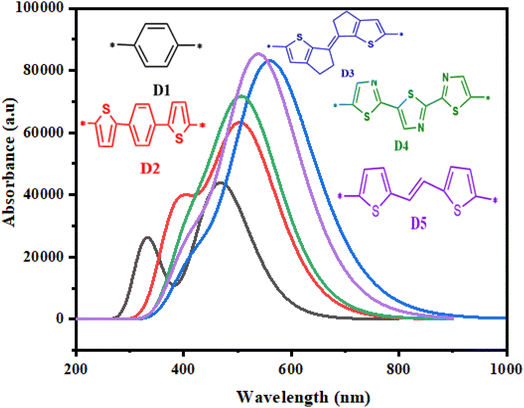 |
| | Fig. 9 Absorption spectra of the studied dyes. | |
Table 5 Absorption spectral data calculated at the TD-DFT/CAM-B3LYP/6-31G** level
| Dyes |
λmax (nm) |
Eex (eV) |
f |
MO/character |
| D1 |
468.749 |
2.64 |
1.08 |
HOMO → LUMO (83%) |
| 338.338 |
3.66 |
0.20 |
HOMO−1 → LUMO (77%) |
| 329.920 |
3.75 |
0.44 |
HOMO → LUMO+1 (53%) |
| D2 |
508.840 |
2.43 |
1.52 |
HOMO → LUMO (81%) |
| 395.307 |
3.13 |
0.78 |
HOMO−1 → LUMO (31%) |
| 368.605 |
3.36 |
0.16 |
HOMO−1 → LUMO (49%) |
| D3 |
563.103 |
2.20 |
2.01 |
HOMO → LUMO (89%) |
| 438.804 |
2.82 |
0.37 |
HOMO → LUMO+1 (65%) |
| 402.245 |
3.08 |
0.17 |
HOMO−1 → LUMO (65%) |
| D4 |
514.051 |
2.41 |
1.67 |
HOMO → LUMO (49%) |
| 413.363 |
2.99 |
0.69 |
HOMO−1 → LUMO (38%) |
| 367.851 |
3.37 |
0.01 |
HOMO−1 → LUMO (27%) |
| D5 |
542.007 |
2.28 |
2.06 |
HOMO → LUMO (83%) |
| 421.542 |
2.94 |
0.55 |
HOMO → LUMO+1 (49%) |
| 384.554 |
3.22 |
0.10 |
HOMO−1 → LUMO (52%) |
Major parameters affecting the photovoltaic performance of DSSCs
The influence of multiple π-linkers on the effectiveness of DSSCs was determined by investigating some essential parameters, such as ΔGinject, ΔGreg, LHE, and the excited-state lifetime (τ), which were used to examine and assess the Jsc and Voc. ΔGinject and LHE are essential parameters influencing the Jsc obtained from eqn (6). Suitable dyes must have a significant LHE that reflects the absorption ability of most photons in the UV-visible region and subsequently injects photoexcited electrons into the CB of TiO2. Table 6 summarizes the LHE, ΔGinject, ΔGreg, and Voc values, where the LHE value for D3 and D5 exhibited the largest value (0.99), followed by D2 (0.96), D4 (0.98), and D1 (0.9), suggesting that the developed dyes D2–D5 showed higher light-absorption potentials compared to D1. Hence, D2–D5 will present higher Jsc values because of their superior LHE.
Table 6 Calculated electronic properties of the studied dyes (in eV)
| Dyes |
EHOMO |
ELUMO |
E00 |
Edyes |
Edyes* |
ΔGinject |
ΔGreg |
LHE |
Voc |
| D1 |
−5.223 |
−3.012 |
2.64 |
5.22 |
2.58 |
−1.42 |
0.42 |
0.91 |
0.99 |
| D2 |
−5.045 |
−2.942 |
2.43 |
5.04 |
2.61 |
−1.39 |
0.24 |
0.96 |
1.06 |
| D3 |
−4.979 |
−3.086 |
2.20 |
4.97 |
2.78 |
−1.22 |
0.17 |
0.99 |
0.92 |
| D4 |
−5.255 |
−3.316 |
2.41 |
5.25 |
2.84 |
−1.16 |
0.45 |
0.98 |
0.69 |
| D5 |
−5.118 |
−3.159 |
2.28 |
5.11 |
2.83 |
−1.17 |
0.31 |
0.99 |
0.85 |
The negative ΔGinject values demonstrated that electrons could be injected easily into TiO2. The ΔGinject values in Table 6 show that D1 and D2 had greater values than D3, D4, and D5, indicating that electron injection would be easier into TiO2 for them. To get fast electron transfer, ΔGreg must be as low as possible; as shown in Table 6, except for D4, the ΔGreg values of all the developed compounds were lower than that of the reference molecule D1, indicating that those dyes had a greater power conversion efficiency.
The charge-transfer rate (k) also influences a DSSC device's efficiency, which may be defined as follows:26
| |
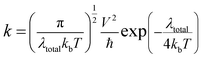 | (14) |
where
V is the charge-transfer coupling constant,
T is the temperature,
kb is the Boltzmann constant, and the
λtotal is the reorganization energy. Except for the
λtotal, all the parameters in
eqn (14) are believed to be constants; while a lower
λtotal will expedite electron transport and boost the
Jsc.
λtotal is the sum of the electron-reorganization energies (
λe) and the hole-reorganization energies (
λh); these energies were determined using
eqn (8), and obtained values are listed in
Table 7, where the computed
λtotal for the proposed dyes (D1–D5) increased in the following order: D4 < D5 < D2 < D1 < D3. So, due to the low values of
λtotal, the molecules D2, D4, and D5 would have good electron-transfer efficiency compared to molecule D1 (the reference molecule).
Table 7 Reorganization energies, λtotal values (eV), and excited-state lifetime τ (ns) of the molecules (D1–D5)
| Dyes |
λe (eV) |
λh (eV) |
λtotal (eV) |
τ (ns) |
| D1 |
0.1 |
0.20 |
0.30 |
0.19 |
| D2 |
0.09 |
0.17 |
0.26 |
0.16 |
| D3 |
0.20 |
0.32 |
0.52 |
0.15 |
| D4 |
0.06 |
0.13 |
0.19 |
0.15 |
| D5 |
0.08 |
0.15 |
0.23 |
0.14 |
Also, (τ) has an essential effect on the charge-transport characteristics of a material. Indeed, a longer lifetime of the excited state enables charge transport and reduces the loss of energy119 eqn (15) was used to determine the (τ) of the dyes:120
| |
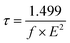 | (15) |
where
E denotes the transition energy between various excited states, and
f indicates the oscillator strength.
Table 7 shows the data for (
τ) of all the molecules. From
Table 7, the values of the excited-state lifetimes were in the following order: D1 (0.19 ns) > D2 (0.16 ns) > D3 (0.15 ns) = 4 (0.15 ns) > D5 (0.14 ns).
Conclusion
In this research, four novel molecules (D2–D5) with different π-bridges were developed based on previous research-based experiment results of the dye D1. DFT and TD-DFT were applied as theoretical methods to test the critical parameters associated with the Jsc and Voc values of the dyes. The results demonstrated that all the developed compounds (D2–D5) possessed lower energy gaps, thus resulting in their absorption spectra being red-shifted by a different degree than D1, particularly for D3 and D5. It is important to note that D2–D5 presented larger absorption bands, indicating a higher light-absorption possibility. The more outstanding Voc and Jsc of the proposed dyes D2–D5 may be partly attributable to their lower λtotal, Greg, η, Ginject, and larger LHEs.
On the other hand, D2–D5 exhibited higher charge-transfer efficiencies and a reduced charge recombination protocol compared to D1. According to this investigation, the modifications of the π-spacer units of the dye reference affected the overall performance of the studied dyes. This study should give theoretical guidance for experiments to develop and test highly effective D–A–π–A type sensitizer materials.
Author contributions
A. Azaid: paper writing; T. Abram, R. Kacimi, M. Raftani, Y. Khaddam, and D. Nebbach: graph preparation, analyzed and interpreted the data; M. Bouachrine and M. Alaqarbeh: analyzed the DFT calculations and supervision of the work; A. Sbai and T. Lakhlifi: computational part of the study and DFT calculations. All authors commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to express sincere thanks to the crews in the Molecular Chemistry and Natural Substances Laboratory (University Moulay Ismail, Meknes) for the support.
References
- T. Zahra, K. S. Ahmad, A. G. Thomas, C. Zequine, M. A. Malik and R. K. Gupta, RSC Adv., 2020, 10, 9854–9867 RSC
 .
. - Y.-H. P. Zhang, Energy Environ. Sci., 2009, 2, 272 RSC .
- M. Marszalek, S. Nagane, A. Ichake, R. Humphry-Baker, V. Paul, S. M. Zakeeruddin and M. Grätzel, RSC Adv., 2013, 3, 7921 RSC .
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef .
- B. W. H. Saes, M. M. Wienk and R. A. J. Janssen, RSC Adv., 2020, 10, 30176–30185 RSC .
- E. Muchuweni, B. S. Martincigh and V. O. Nyamori, RSC Adv., 2020, 10, 44453–44469 RSC .
- L. Zhang, X. Yang, W. Wang, G. G. Gurzadyan, J. Li, X. Li, J. An, Z. Yu, H. Wang, B. Cai, A. Hagfeldt and L. Sun, ACS Energy Lett., 2019, 4, 943–951 CrossRef CAS .
- Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC .
- M. Peng, K. Yan, H. Hu, D. Shen, W. Song and D. Zou, J. Mater. Chem. C, 2015, 3, 2157–2165 RSC .
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS PubMed .
- Ö. Birel, S. Nadeem and H. Duman, J. Fluoresc., 2017, 27, 1075–1085 CrossRef PubMed .
- S. Ananthakumar, D. Balaji, J. Ram Kumar and S. Moorthy Babu, SN Appl. Sci., 2019, 1, 186 CrossRef CAS .
- M. Freitag and G. Boschloo, Curr. Opin. Electrochem., 2017, 2, 111–119 CrossRef CAS .
- S. Thomas, T. G. Deepak, G. S. Anjusree, T. A. Arun, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 4474–4490 RSC .
- A. Yella, R. Humphry-Baker, B. F. E. Curchod, N. Ashari Astani, J. Teuscher, L. E. Polander, S. Mathew, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, Md. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2733–2739 CrossRef CAS .
- X. Qian, H.-H. Gao, Y.-Z. Zhu, B. Pan and J.-Y. Zheng, Dyes Pigm., 2015, 121, 152–158 CrossRef CAS .
- J. Jia, Y. Chen, L. Duan, Z. Sun, M. Liang and S. Xue, RSC Adv., 2017, 7, 45807–45817 RSC .
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC .
- X. Yang, J. Zhao, L. Wang, J. Tian and L. Sun, RSC Adv., 2014, 4, 24377 RSC .
- H.-Q. Xia, C.-P. Kong, J. Wang, F.-Q. Bai and H.-X. Zhang, RSC Adv., 2014, 4, 50338–50350 RSC .
- B. Liu, B. Wang, R. Wang, L. Gao, S. Huo, Q. Liu, X. Li and W. Zhu, J. Mater. Chem. A, 2014, 2, 804–812 RSC .
- M.-D. Zhang, H.-X. Xie, X.-H. Ju, L. Qin, Q.-X. Yang, H.-G. Zheng and X.-F. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 634–641 RSC .
- H. A. Z. Sabek, A. M. M. Alazaly, D. Salah, H. S. Abdel-Samad, M. A. Ismail and A. A. Abdel-Shafi, RSC Adv., 2020, 10, 43459–43471 RSC .
- K. D. Seo, B. S. You, I. T. Choi, M. J. Ju, M. You, H. S. Kang and H. K. Kim, J. Mater. Chem. A, 2013, 1, 9947 RSC .
- L.-Y. Lin, C.-H. Tsai, F. Lin, T.-W. Huang, S.-H. Chou, C.-C. Wu and K.-T. Wong, Tetrahedron, 2012, 68, 7509–7516 CrossRef CAS .
- H. Roohi and N. Mohtamadifar, RSC Adv., 2022, 12, 11557–11573 RSC .
- S. J. Akram, N. M. A. Hadia, J. Iqbal, R. F. Mehmood, S. Iqbal, A. M. Shawky, A. Asif, H. H. Somaily, M. Raheel and R. A. Khera, RSC Adv., 2022, 12, 20792–20806 RSC .
- Y. Liu, L. Yin, B. Xie, C. Liu and Y. Li, Mater. Adv., 2022, 3, 6496–6505 RSC .
- H. Wu, Z. Huang, T. Hua, C. Liao, H. Meier, H. Tang, L. Wang and D. Cao, Dyes Pigm., 2019, 165, 103–111 CrossRef CAS .
- A. Azaid, T. Abram, R. Kacimi, A. Sbai, T. lakhlifi and M. Bouachrine, Mater. Today: Proc., 2021, 45, 7363–7369 CAS .
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed .
- G. C. dos Santos, E. F. Oliveira, F. C. Lavarda and L. C. da Silva-Filho, J. Mol. Model., 2019, 25, 75 CrossRef PubMed .
- H. Sadki, M. Bourass, M. N. Bennani and M. Bouachrine, Res. Chem. Intermed., 2018, 44, 6071–6085 CrossRef CAS .
- S. A. A. Shah, M. H. Sayyad, F. Wahab, K. A. Khan, M. A. Munawar, H. Elbohy and Q. Qiao, J. Mater. Sci.: Mater. Electron., 2016, 27, 4501–4507 CrossRef CAS .
- J. Preat, J. Phys. Chem. C, 2010, 114, 16716–16725 CrossRef CAS .
- M. R. S. A. Janjua, M. U. Khan, M. Khalid, N. Ullah, R. Kalgaonkar, K. Alnoaimi, N. Baqader and S. Jamil, J. Cluster Sci., 2021, 32, 243–253 CrossRef CAS .
- M. Yoosuf, S. C. Pradhan, M. M. Sruthi, S. Soman and K. R. Gopidas, Sol. Energy, 2021, 216, 151–163 CrossRef CAS .
- S. ElKhattabi, M. Hachi, A. Fitri, A. T. Benjelloun, M. Benzakour, M. Mcharfi and M. Bouachrine, J. Mol. Model., 2019, 25, 9 CrossRef CAS PubMed .
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC .
- E. Tanaka, M. S. Mikhailov, N. S. Gudim, E. A. Knyazeva, L. V. Mikhalchenko, N. Robertson and O. A. Rakitin, Mol. Syst. Des. Eng., 2021, 6, 730–738 RSC .
- M. Yoosuf, S. C. Pradhan, M. M. Sruthi, S. Soman and K. R. Gopidas, Sol. Energy, 2021, 216, 151–163 CrossRef CAS .
- Y.-S. Yen, J.-L. Hsu, J.-S. Ni and J. T. Lin, Dyes Pigm., 2021, 188, 109220 CrossRef CAS .
- K. Pei, Y. Wu, W. Wu, Q. Zhang, B. Chen, H. Tian and W. Zhu, Chem.–Eur. J., 2012, 18, 8190–8200 CrossRef CAS PubMed .
- X. Lu, Q. Feng, T. Lan, G. Zhou and Z.-S. Wang, Chem. Mater., 2012, 24, 3179–3187 CrossRef CAS .
- M. Velusamy, K. R. Justin Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Org. Lett., 2005, 7, 1899–1902 CrossRef CAS PubMed .
- W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS .
- J. Jie, Q. Xu, G. Yang, Y. Feng and B. Zhang, Dyes Pigm., 2020, 174, 107984 CrossRef CAS .
- X. Zhang, Z. Ma, Y. Yang, X. Zhang, X. Jia and Y. Wei, J. Mater. Chem. C, 2014, 2, 8932–8938 RSC .
- D. Zhang, T. Yang, H. Xu, Y. Miao, R. Chen, R. Shinar, J. Shinar, H. Wang, B. Xu and J. Yu, J. Mater. Chem. C, 2021, 9, 4921–4926 RSC .
- Q. Feng, X. Lu, G. Zhou and Z.-S. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7993 RSC .
- H. Sun, B. Liu, Z. Wang, S. Ling, Y. Zhang, G. Zhang, Y. Wang, M. Zhang, B. Li, W. Yang, J. Wang, H. Guo, F. Liu and X. Guo, J. Mater. Chem. C, 2020, 8, 4012–4020 RSC .
- X. Hu, S. Cai, G. Tian, X. Li, J. Su and J. Li, RSC Adv., 2013, 3, 22544 RSC .
- Y. Cui, Y. Wu, X. Lu, X. Zhang, G. Zhou, F. B. Miapeh, W. Zhu and Z.-S. Wang, Chem. Mater., 2011, 23, 4394–4401 CrossRef CAS .
- J. Mao, F. Guo, W. Ying, W. Wu, J. Li and J. Hua, Chem.–Asian J., 2012, 7, 982–991 CrossRef CAS PubMed .
- S. M. Wagalgave, M. Al Kobaisi, S. V. Bhosale and S. V. Bhosale, J. Electroanal. Chem., 2022, 915, 116341 CrossRef CAS .
- A. K. Palai, A. Kumar, K. Sim, J. Kwon, T. J. Shin, S. Jang, S. Cho, S.-U. Park and S. Pyo, New J. Chem., 2016, 40, 385–392 RSC .
- K. Chaitanya, B. M. Heron and X.-H. Ju, Dyes Pigm., 2017, 141, 501–511 CrossRef CAS .
- W. Ying, X. Zhang, X. Li, W. Wu, F. Guo, J. Li, H. Ågren and J. Hua, Tetrahedron, 2014, 70, 3901–3908 CrossRef CAS .
- B. S. Arslan, B. Arkan, M. Gezgin, Y. Derin, D. Avcı, A. Tutar, M. Nebioğlu and İ. Şişman, J. Photochem. Photobiol., A, 2021, 404, 112936 CrossRef CAS .
- Y. K. Eom, J. Y. Hong, J. Kim and H. K. Kim, Dyes Pigm., 2017, 136, 496–504 CrossRef CAS .
- K. K. Chenab and M.-R. Zamani-Meymian, Mater. Sci. Semicond. Process., 2022, 151, 107018 CrossRef CAS .
- Y. S. Tingare, C. Su, M.-T. Shen, S.-H. Tsai, S.-Y. Ho and W.-R. Li, Molecules, 2020, 25, 2159 CrossRef CAS PubMed .
- C.-H. Yang, S.-H. Liao, Y.-K. Sun, Y.-Y. Chuang, T.-L. Wang, Y.-T. Shieh and W.-C. Lin, J. Phys. Chem. C, 2010, 114, 21786–21794 CrossRef CAS .
- D. Devadiga, M. Selvakumar, P. Shetty, M. Santosh, R. S. Chandrabose and S. Karazhanov, Int. J. Energy Res., 2021, 45, 6584–6643 CrossRef CAS .
- S. S. M. Fernandes, M. C. R. Castro, D. Ivanou, A. Mendes and M. M. M. Raposo, Coatings, 2021, 12, 34 CrossRef .
- M. S. Abusaif, M. A. Abu-Saied, M. Fathy, A. A. El-Sherif, A. B. Kashyout, M. R. Selim and Y. A. Ammar, J. Iran. Chem. Soc., 2021, 18, 949–960 CrossRef CAS .
- M. S. Abusaif, M. Fathy, M. A. Abu-Saied, A. A. Elhenawy, A. B. Kashyout, M. R. Selim and Y. A. Ammar, J. Mol. Struct., 2021, 1225, 129297 CrossRef CAS .
- B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed .
- X. Zhang, F. Gou, D. Zhao, J. Shi, H. Gao, Z. Zhu and H. Jing, J. Power Sources, 2016, 324, 484–491 CrossRef CAS .
- T.-F. Lu, W. Li and H.-X. Zhang, Org. Electron., 2018, 59, 131–139 CrossRef CAS .
- M. Y. Mehboob, R. Hussain, M. Adnan, Saira, U. Farwa, Z. Irshad and M. R. Saeed Ashraf Janjua, J. Phys. Chem. Solids, 2022, 162, 110508 CrossRef CAS .
- L.-W. Ma, Z.-S. Huang, S. Wang, H. Meier and D. Cao, Dyes Pigm., 2017, 145, 126–135 CrossRef CAS .
- A. Slimi, A. Fitri, A. Touimi Benjelloun, S. Elkhattabi, M. Benzakour, M. Mcharfi and M. Bouachrine, J. Electron. Mater., 2019, 48, 4452–4462 CrossRef CAS .
- T. Zhang, W. Guan, S. Wen, T. Ma, L. Yan and Z. Su, J. Phys. Chem. C, 2014, 118, 29623–29628 CrossRef CAS .
- P. Singh and N. M. Ravindra, Sol. Energy Mater. Sol. Cells, 2012, 101, 36–45 CrossRef CAS .
- X. Zuo, K. Chang, J. Zhao, Z. Xie, H. Tang, B. Li and Z. Chang, J. Mater. Chem. A, 2016, 4, 51–58 RSC .
- W. Sharmoukh, Z. M. Hassan, B. A. Ali, M. M. Elnagar, R. M. Abdo and N. K. Allam, J. Photochem. Photobiol.,
A, 2018, 367, 128–136 CrossRef CAS .
- R. Katoh, A. Furube, T. Yoshihara, K. Hara, G. Fujihashi, S. Takano, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 4818–4822 CrossRef CAS .
- K. O. Kzar Al-Masoodi, I. Rafiq, A. E. Assyry and A. Derouiche, J. Phys.: Conf. Ser., 2021, 1963, 012012 CrossRef .
- R. Kacimi, M. Bourass, T. Toupance, N. Wazzan, M. Chemek, A. El Alamy, L. Bejjit, K. Alimi and M. Bouachrine, Res. Chem. Intermed., 2020, 46, 3247–3262 CrossRef CAS .
- P. Pounraj, V. Mohankumar, M. S. Pandian and P. Ramasamy, J. Mol. Model., 2018, 24, 343 CrossRef CAS PubMed .
- J. K. Roy, S. Kar and J. Leszczynski, J. Phys. Chem. C, 2019, 123, 3309–3320 CrossRef CAS .
- M. Bourass, A. Touimi Benjelloun, M. Benzakour, M. Mcharfi, M. Hamidi, S. M. Bouzzine, F. Serein-Spirau, T. Jarrosson, J.-M. Sotiropoulos and M. Bouachrine, C. R. Chim., 2017, 20, 461–466 CrossRef CAS .
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339–2350 CrossRef CAS PubMed .
- M. P. Balanay and D. H. Kim, Curr. Appl. Phys., 2011, 11, 109–116 CrossRef .
- H. Wang, X. Wang, H. Wang, L. Wang and A. Liu, J. Mol. Model., 2006, 13, 147–153 CrossRef PubMed .
- M. Rocha, A. Di Santo, J. M. Arias, D. M. Gil and A. B. Altabef, Spectrochim. Acta, Part A, 2015, 136, 635–643 CrossRef CAS PubMed .
- N. Islam and D. C. Ghosh, Int. J. Mol. Sci., 2012, 13, 2160–2175 CrossRef CAS PubMed .
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS .
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS .
- M. Frisch, et al., Gaussian 09, Revision d. 01, Gaussian, Inc., Wallingford, CT, 2009, p. 201 Search PubMed .
- M. Khajehzadeh and M. Moghadam, Spectrochim. Acta, Part A, 2017, 180, 51–66 CrossRef CAS PubMed .
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS .
- M. Govindarajan, K. Ganasan, S. Periandy and S. Mohan, Spectrochim. Acta, Part A, 2010, 76, 12–21 CrossRef CAS PubMed .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed .
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS .
- F. London, J. Phys. Radium, 1937, 8, 397–409 CrossRef CAS .
- M. Javed, A. Farhat, S. Jabeen, R. A. Khera, M. Khalid and J. Iqbal, Comput. Theor. Chem., 2021, 1204, 113373 CrossRef CAS .
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC .
- M. Adnan, J. Iqbal, S. BiBi, R. Hussain, M. N. Akhtar, M. A. Rashid, B. Eliasson and K. Ayub, Zeitschrift für Physikalische Chemie, 2017, 231, 1127–1139 CrossRef CAS .
- A. Fitri, A. T. Benjelloun, M. Benzakour, M. Mcharfi, M. Hamidi and M. Bouachrine, Spectrochim. Acta, Part A, 2014, 132, 232–238 CrossRef CAS PubMed .
- N.-J. Zhao, Y.-W. Wang, Q. Liu, Z.-H. Lin, R. Liang, L.-M. Fu, X.-C. Ai, Z. Bo and J.-P. Zhang, Nanoscale, 2016, 8, 18390–18399 RSC .
- C. Chitpakdee, S. Namuangruk, K. Suttisintong, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk, K. Sirithip, V. Promarak and N. Kungwan, Dyes Pigm., 2015, 118, 64–75 CrossRef CAS .
- W.-L. Ding, D.-M. Wang, Z.-Y. Geng, X.-L. Zhao and W.-B. Xu, Dyes Pigm., 2013, 98, 125–135 CrossRef CAS .
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261 RSC .
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC .
- S. V. Kershaw, L. Jing, X. Huang, M. Gao and A. L. Rogach, Mater. Horiz., 2017, 4, 155–205 RSC .
- A. Stankevych, A. Vakhnin, D. Andrienko, L. Paterson, J. Genoe, I. Fishchuk, H. Bässler, A. Köhler and A. Kadashchuk, Phys. Rev. Appl., 2021, 15, 044050 CrossRef CAS .
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed .
- M. Raftani, T. Abram, N. Bennani and M. Bouachrine, Results Chem., 2020, 2, 100040 CrossRef CAS .
- M. Souilah, M. Hachi, A. Fitri, A. T. Benjelloun, S. El Khattabi, M. Benzakour, M. Mcharfi and H. Zgou, Res. Chem. Intermed., 2021, 47, 875–893 CrossRef CAS .
- A. O. Ukpene, J. C. Morka and O. C. Molua, J. Appl. Sci. Environ. Manage., 2022, 26, 1409–1413 Search PubMed .
- V. K. Rajan and K. Muraleedharan, Food Chem., 2017, 220, 93–99 CrossRef CAS PubMed .
- M. Hachi, A. Slimi, A. Fitri, S. ElKhattabi, A. T. Benjelloun, M. Benzakour and M. Mcharfi, Mol. Phys., 2020, 118, e1662956 CrossRef .
- M. Prakasam, R. Baskar, K. Gnanamoorthi and K. Annapoorani, Int. J. Adv. Sci. Eng., 2019, 5, 1118–1124 CAS .
- D. S. Patil, K. C. Avhad and N. Sekar, Comput. Theor. Chem., 2018, 1138, 75–83 CrossRef CAS .
- O. Britel, A. Fitri, A. T. Benjelloun, A. Slimi, M. Benzakour and M. Mcharfi, J. Photochem. Photobiol., A, 2022, 428, 113870 CrossRef CAS .
- A. Szemik-Hojniak, K. Oberda, I. Deperasińska, Y. P. Nizhnik and L. Jerzykiewicz, Polyhedron, 2015, 88, 190–198 CrossRef CAS .
- T.-F. Lu, W. Li, J. Chen, J. Tang, F.-Q. Bai and H.-X. Zhang, Electrochim. Acta, 2018, 283, 1798–1805 CrossRef CAS .
- R. M. Pallares, X. Su, S. H. Lim and N. T. K. Thanh, J. Mater. Chem. C, 2016, 4, 53–61 RSC .
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c,
Rchid Kacimia,
Tayeb Abram
*c,
Rchid Kacimia,
Tayeb Abram