
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light induced RAFT for asymmetric functionalization of silica mesopores†
Claire Förstera,
Lothar Veithb and
Annette Andrieu-Brunsen *a
*a
aMacromolecular Chemistry–Smart Membranes, Technische Universität Darmstadt, 64287 Darmstadt, German. E-mail: annette.andrieu-brunsen@.tu-darmstadt.de
bMax Planck Institute for Polymer Research, 55128, Mainz, Germany
First published on 23rd September 2022
Abstract
One key feature for bioinspired transport design through nanoscale pores is nanolocal, asymmetric as well as multifunctional nanopore functionalization. Here, we use a visible-light induced, controlled photo electron/energy transfer-reversible addition–fragmentation chain-transfer (PET-RAFT) polymerization for asymmetric polymer placement into mesoporous silica thin films including asymmetric polymer sequence design.
Despite the numerous examples of gated nanopore transport, transport direction as well as selectivity is still a challenge in technological pores.1,2 To date, directional transport has been experimentally demonstrated in Janus materials.3–6 With respect to individual nanopores transport direction and ion rectification has been shown for asymmetrically shaped nanoscale pores and channels.7 For ion track etched nanochannels, Siwy demonstrated that ionic current rectification takes place even in the presence of identical pH value and electrolyte concentration on both pore openings.8 The potential of asymmetric nanochannel polymer functionalization for selective transport direction has been demonstrated by modeling studies from Huang and Szleifer.9 They demonstrate the importance of the local placement and monomer sequence control of a pH-responsive copolymer precisely located at the pore entrances. By using a polyelectrolyte block located at the wall and a hydrophobic block located toward the pore center a multistimuli responsive nanogate was created.9 Furthermore, high ionic rectification factors above 1000 for asymmetric designed nanopores have been theoretically demonstrated10 by implementing two orthogonal gates at both entrances of a nanopore.10 Although controlled polymer functionalization of nanopores significantly advanced within the last years,1,2,11 experimental examples on local polymer placement and sequence control remain a challenge and need local polymer placement and controlled polymerization. Polymerization control in nanopores is most often achieved by surface-initiated atom transfer radical polymerization (SI-ATRP), SI-RAFT, or iniferter-initiated polymerizations. Using controlled SI-ATRP the low dispersity12,13 as well as influence of curvature on polymerization14 was demonstrated. By using SI-RAFT or iniferter-initiated polymerizations re-initiation was achieved.15–19 In addition, a few examples using SI-ring opening metathesis polymerization (SI-ROMP) have been demonstrated.20–22 Nanolocal nanopore functionalization has been e.g. achieved using contactless electro functionalization,23 which requires a (semi-) conducting membrane and a sandwich like composition.24 Using localized gold coatings and thiol chemistry locally limited functionalization was achieved.25 For inverse opal monolayers with a gold coating at the outer surface as well as on the pore bottom local placement of three functional units including two polymers has been demonstrated.26 Using visible light induced polymerization in combination with nanoscopically limited visible light emitted from surface plasmons of metallic nanoparticles integrated into mesoporous films allows to nanoscopically limit polymer functionalization in such mesoporous materials.27 To achieve polymer sequence design including local control along the mesoporous film cross section we perform a two-step visible light induced PET-RAFT. In a first step mesopores were filled with poly-2-(dimethylamino)ethyl methacrylate (PDMAEMA). In a subsequent re-initiation of the visible light induced PET-RAFT in the presence of (2-(methacryloyloxy)ethyl phosphate) MEP mainly the outer mesoporous film surface was functionalized with a block-co-oligomer of PDMAEMA-b-PMEP. Mesoporous silica films have been prepared using evaporation-induced self-assembly (EISA)28 and dip coating. Inspired by Dunphy et al.29 pore sizes in the range of 13 nm, a porosity of 52–66 vol% at 15% relative humidity, and film thicknesses between 500-600 nm, determined by TEM, ellipsometry and REM measurements, were obtained using Pluronic® F127 as template (Fig. 1a). A major advantage of the applied visible light induced PET-RAFT polymerization for locally resolved re-initiation is its robustness towards the presence of oxygen. By functionalization with PDMAEMA for two hours pore filling degrees of almost 90 vol% were achieved (Fig. 2 and S1†). The film thickness does not significantly increase upon PDMAEMA functionalization which seems consistent when considering the pore radius and the error bar of the ellipsometry measurement (Fig. 2a and b). In the presence of a second monomer MEP, re-initiation of the polymerization resulted into block-copolymer functionalization at the outer surface. The slight increase of the refractive index (up to 0.08, Table S3†) indicates only minor re-initiation inside the mesopores, while the film thickness increased significantly upon PMEP polymerization (Fig. 2a, blue to red). The RAFT agent at the outer mesoporous film surface was destroyed using a CO2-plasma treatment as described by Krohm et al.22 and Babu et al.30 (Fig. 2b). The comparison of CO2-plasma treated and non–CO2–plasma treated films reveals a larger increase of the film thickness (up to 112 nm, average 69 nm, Table S3†) upon polymerization without CO2-plasma treatment, while the pore filling degrees are comparable for polymer functionalized mesoporous films with and without CO2-plasma treatment. This observation supports local polymer re-initiation mainly at the outer mesoporous film surface which is in addition consistent with ATR-IR measurements (Fig. S2†).
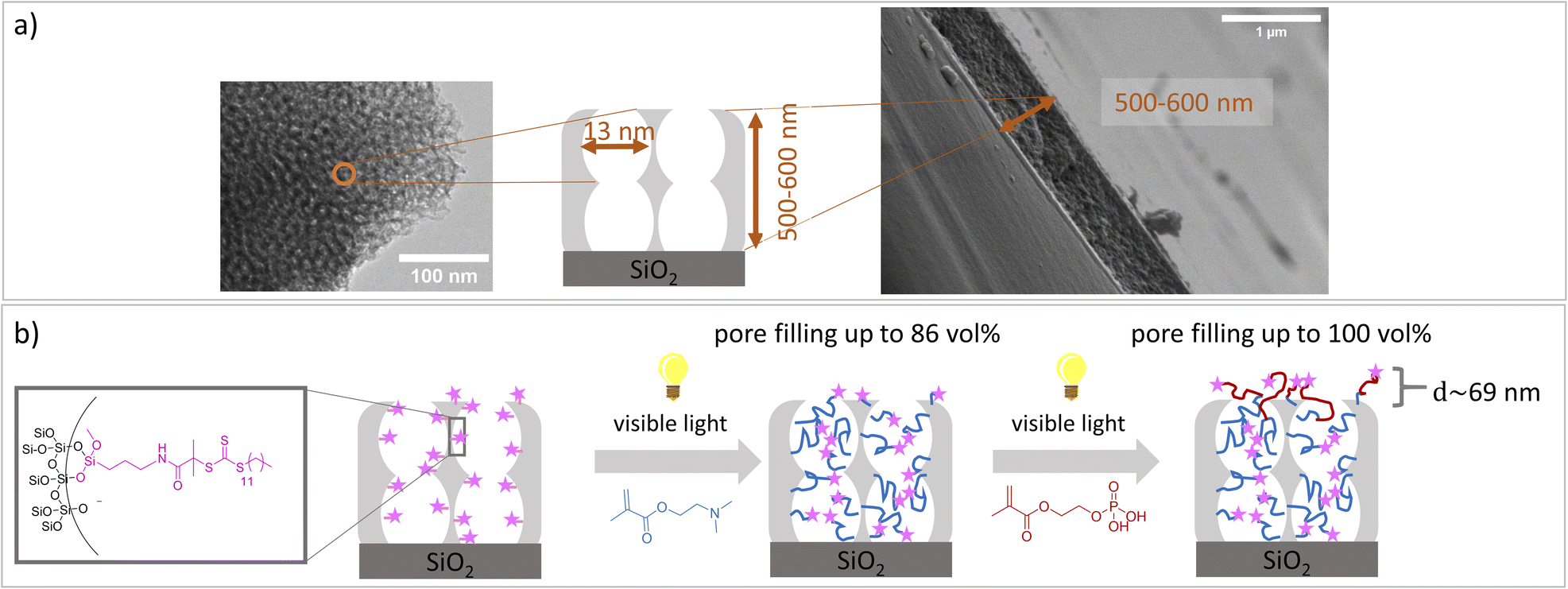 | ||
| Fig. 1 (a) Mesoporous silicafilm with film thicknesses between 500–600 nm (SEM image) and pore sizes in the range of 13 nm (TEM image). (b) Visible light induced PET-RAFT to functionalize mesoporous silica films with PDMAEMA-b-PMEP. Pore fillings and film thicknesses were determined by ellipsometry. | ||
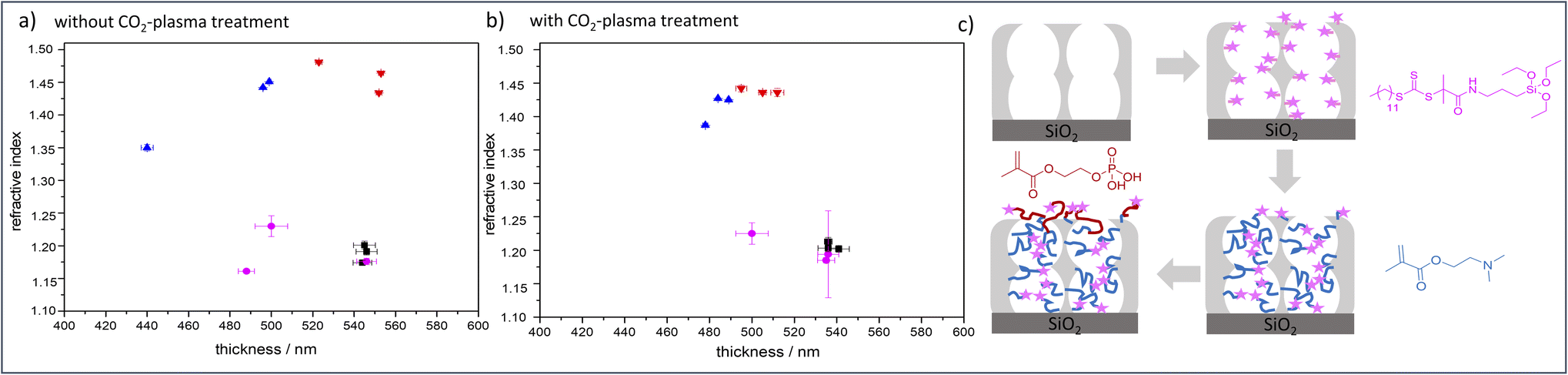 | ||
| Fig. 2 Results of the ellipsometry measurements at 15% relative humidity (a) without CO2-plasma treatment and (b) with CO2-plasma treatment. (c) Schematic figure, showing the different functionalization steps black = mesopores silicafilm, magenta = DDMAT functionalized film, red = PDMAEMA functionalized film, blue = PDMAEMA-b-PMEP functionalized film. Shown are each one samples, which were measured on three different spots. | ||
ToF-SIMS depth profiling analysis (Fig. 3a and b) shows a significantly higher signal intensity for PO3− (diagnostic ion for PMEP) on the outer film surface than inside the mesoporous silicafilm. On the topmost layer a ratio of CN−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PO3− of approximately 1:23.5 was detected, while the ratio inside of the mesoporous silicafilm is approximately 1:0.05. The intensity of S− resulting from the RAFT agent DDMAT, also is most intense at the outer surface. After removing the first layers, corresponding to the polymer at the outer planar surface of the mesoporous film, the S− intensity reduces but stays almost constant (Si: S ratio approximately 1:0.03) until the SiO2 substrate below the mesoporous film is reached. This proofs the presence of DDMAT along the entire film thickness and indicates the absence of re-initiation within the mesopores being due to confinement and pore filling but not due to removal of initiator for example. XPS measurements show a similar ratio of Si: S (Fig. 3e and Table S5†). Furthermore, the intensity decrease in the PO3−, CN− and S− intensities at the beginning of the depth profiling (Fig. 3b) and the increase in SiO2 intensity (Fig. S4†) indicates a polymer overlayer on the mesoporous silica, which is removed in the first 50 seconds of sputtering. The SiO2− intensity stays at a constant level after these initial sputtering cycles until the SiO2 substrate below the mesoporous film is reached (Fig. S3†). Finally, TGA (Fig. 3) and XPS (Fig. S5†) results confirm the successful functionalization of the mesoporous silicafilms with PDMAEMA-b-PMEP. After 2 h polymerization of DMAEMA a mass loss of ∼36% was obtained by TGA analysis. After subsequent re-initiation and functionalization with MEP a mass loss of ∼39% was achieved by TGA measurements (Fig. 3c, blue and red). The low mass loss increase obtained after re-initiation in comparison to the mass loss after DMAEMA polymerization in the first step is related to the re-initiation with MEP taking place mainly on the outer surface. In analogy to Pasetto et al.31 a grafting density of DDMAT of 0.2–0.6 molecules per nm and an estimation of the PDMAEMA chain length of ∼9.4 monomers per chain was calculated using the mass loss of DDMAT functionalized films and PDMAEMA functionalized films in TGA measurements (Fig. 3c, magenta, blue). A specific surface area of 2.93 × 1020–7.84 × 1020 nm2 g−1 was used (electronic ESI†).
:PO3− of approximately 1:23.5 was detected, while the ratio inside of the mesoporous silicafilm is approximately 1:0.05. The intensity of S− resulting from the RAFT agent DDMAT, also is most intense at the outer surface. After removing the first layers, corresponding to the polymer at the outer planar surface of the mesoporous film, the S− intensity reduces but stays almost constant (Si: S ratio approximately 1:0.03) until the SiO2 substrate below the mesoporous film is reached. This proofs the presence of DDMAT along the entire film thickness and indicates the absence of re-initiation within the mesopores being due to confinement and pore filling but not due to removal of initiator for example. XPS measurements show a similar ratio of Si: S (Fig. 3e and Table S5†). Furthermore, the intensity decrease in the PO3−, CN− and S− intensities at the beginning of the depth profiling (Fig. 3b) and the increase in SiO2 intensity (Fig. S4†) indicates a polymer overlayer on the mesoporous silica, which is removed in the first 50 seconds of sputtering. The SiO2− intensity stays at a constant level after these initial sputtering cycles until the SiO2 substrate below the mesoporous film is reached (Fig. S3†). Finally, TGA (Fig. 3) and XPS (Fig. S5†) results confirm the successful functionalization of the mesoporous silicafilms with PDMAEMA-b-PMEP. After 2 h polymerization of DMAEMA a mass loss of ∼36% was obtained by TGA analysis. After subsequent re-initiation and functionalization with MEP a mass loss of ∼39% was achieved by TGA measurements (Fig. 3c, blue and red). The low mass loss increase obtained after re-initiation in comparison to the mass loss after DMAEMA polymerization in the first step is related to the re-initiation with MEP taking place mainly on the outer surface. In analogy to Pasetto et al.31 a grafting density of DDMAT of 0.2–0.6 molecules per nm and an estimation of the PDMAEMA chain length of ∼9.4 monomers per chain was calculated using the mass loss of DDMAT functionalized films and PDMAEMA functionalized films in TGA measurements (Fig. 3c, magenta, blue). A specific surface area of 2.93 × 1020–7.84 × 1020 nm2 g−1 was used (electronic ESI†).
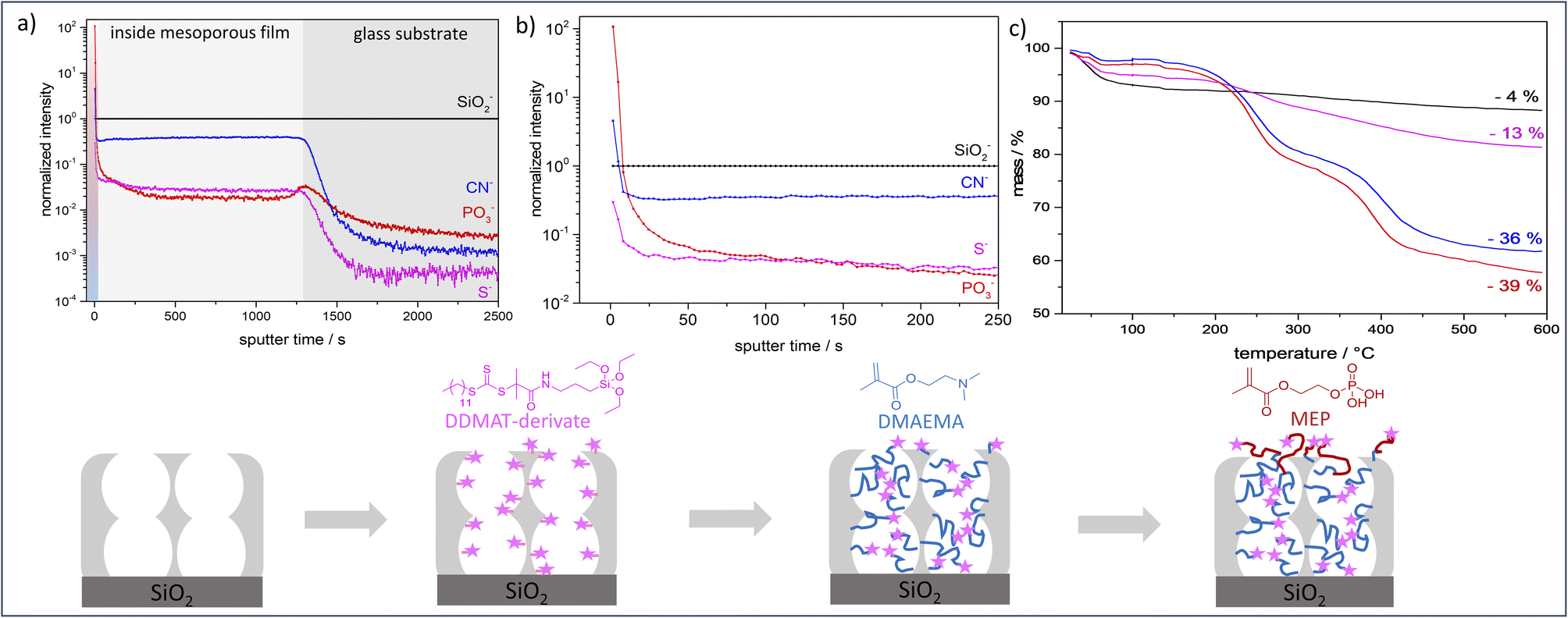 | ||
| Fig. 3 (a and b) Results of the ToF-SIMS depth profiling of the PDMAEMA-b-PMEP-functionalized film reveals the intensity ratios of the diagnostic ions for PDAMEMA (CN, blue), PMEP (PO3, red) and DDMAT (S, magenta). The shading of the profile reveals the film on the mesoporous silica film (red-blue), the silica film (light grey) and the silica substrate (dark grey). Signal intensities are point-to-point normalized to the signal intensity of SiO2−. (c) TGA measurements of mesoporous silicafilm (black), DDMAT-functionalized film (magenta), PDMAEMA-functionalized film (blue) and PDMAEMA-b-PMEP-functionalized film (red). Schematic figure, showing the different functionalization steps, represents the legend for the colour code. | ||
The PDMAEMA-b-PMEP copolymer functionalized mesoporous films show a static contact angle (CA) of approximately 72° and an advancing CA up to 67–95° (Fig. S6b and d†). The hydrophobic regime of mesopores, in which water is excluded from entering the pores is usually reported for CA values higher than 90°32,33 static or advancing CA although reports on 65° exist.34–36 As we observed that water was not able to imbibe into the mesopores under the applied experimental conditions (even not if the polymer functionalized mesoporous film was incubated in aqueous KCl solution prior to the experiment) reliable molecular transport characterization from aqueous solution was measured after addition of ethanol (0.06vol%, Fig. 4, S6 and Fig. S7†). For PDMAEMA functionalization (Fig. 4a) the cyclic voltammograms show the expected ionic pore accessibility governed by the PDMAEMA pH-dependent charge: cation exclusion at acidic pH due to the positive charge of PDMAEMA (pKa value in solution around pH 7.537), and a minor cation accessibility at basic pH is observed. The most prominent change induced by PDMAEMA functionalization is the anion pre-concentration at acidic pH. A reduction in peak current density for anion pore accessibility at basic as compared to acidic pH is observed (Fig. 4a red dashed line and red line). As expected PMEP functionalization (Fig. 4b) results into an exclusion of anions at basic pH and thus negatively charged PMEP with only minor anionic pore accessibility at acidic pH 3. In agreement with this observation cations show pore accessibility at acidic pH and pre-concentration at basic pH under the applied conditions. Interestingly, cyclic voltammograms for asymmetric PDMAEMA-b-PMEP functionalized mesopores with mainly PDMAEMA inside the mesopores and PDMAEMA-b-PMEP mainly on the outer surface (Fig. 4c) show ionic pore accessibility being controlled by both polymers. Anionic [Fe(CN)6]3−/4− can access the pores at basic pH while anions are strongly pre-concentrated at acidic pH at which the PDMAEMA is expected to be positively charged. Quantitatively, this pre-concentration is comparable to the one of mesopores only functionalized with PDMAEMA. At the same time, cationic [Ru(NH3)6]2+/3+ probe molecules (Fig. 4a blue) are almost fully excluded at acidic pH which is consistent with the PDMAEMA positive charge and the observed anion pre-concentration. Interestingly, [Ru(NH3)6]2+/3+ cations are pre-concentrated at basic pH. As this pre-concentration was not observed in only PDMAEMA functionalized pores but for PMEP functionalized pores, this cation pre-concentration at basic pH is not exclusively due to remaining silanol groups at the pore wall, but has to be due to the PMEP at the outer surface.
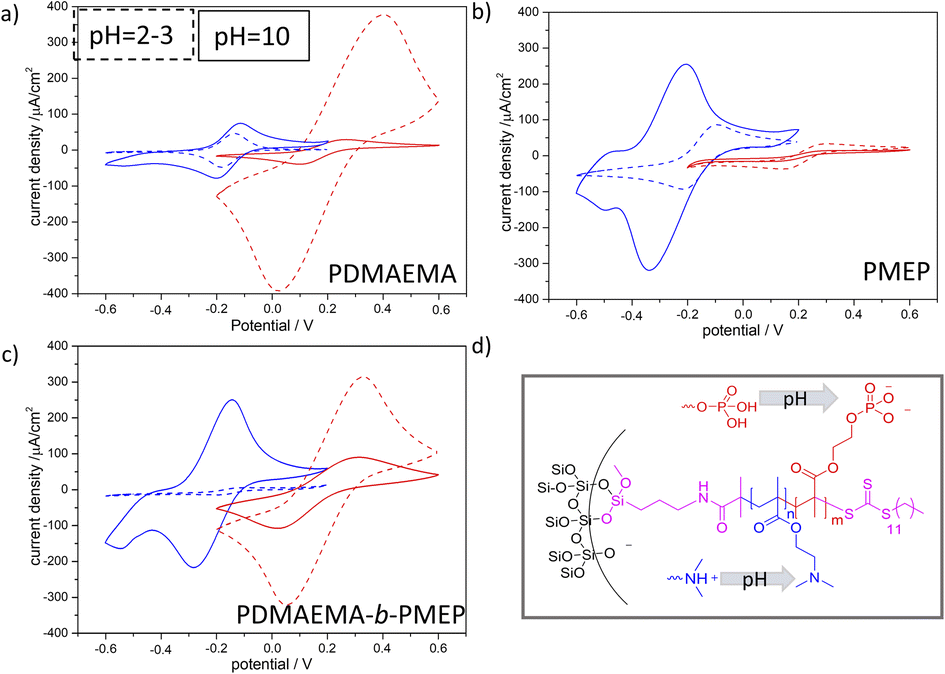 | ||
| Fig. 4 CV measurements (100 mVs−1) of (a) PDMAEMA functionalized (2 h irradiation), (b) PMEP functionalized (2 h irradiation), (c) PDMAEMA-b-PMEP functionalized mesoporous silicafilm using 0.06 vol% of ethanol. blue) [Ru(NH3)6]2+/3+ as probe molecule; red) [Fe(CN)6]3−/4− as probe molecule at concentration of 1 mM in 100 mM KCl electrolyte solution. (d) Schematic illustration of the porous silicafilm in dependence of the pH-value. Cyclic voltammetry measurements of PDMAEMA-b-PMEP functionalized mesoporous films were performed on four sets of independently synthesized mesoporous silicafilms. | ||
Angelomé and coworkers38 used CV measurements to analyze diffusion in mesoporous TiO2 films, where they predicted the diffusion layer for [Fe(CN)6]3-/4- to be in a range of 20–200 µm using scan rates of 5–500 mVs−1. This agrees with our observation that PMEP functionalization on the outer surface of a 500–600 nm mesoporous silicafilm significantly influences the detected pore accessibility.
In conclusion, using a visible light induced and oxygen-tolerant PET-RAFT, and polymerization re-initiation with two oppositely charged monomers an asymmetric polymer placement and an asymmetric polymer chain sequence design with a PDMAEMA-b-PMEP functionalization on top of the mesoporous silica layers and a PDMAEMA pore filling was fabricated. The key to this asymmetric block-copolymer functionalization was a relatively high pore filling of almost 90 vol% after the first polymerization step with an estimated chain length of 9 monomers per chain. Transport characterization of these asymmetric and block-co-oligomer functionalized mesoporous silicafilms clearly shows the influence of both polymer blocks on ionic pores accessibility. Altogether, the defined design of polymer functionalization in mesoporous silica layers combined with orthogonal characterization techniques and transport characterization clearly demonstrates the influence of outer surface functionalization on the transport performance of mesoporous layers and represents a first step towards experimentally designing and understanding the potential of asymmetric porous layer functionalization with polymer sequence control. Managing the local control of polymer composition/chain architecture along the transport direction of mesoporous films is a first important experimental step to improve transport performances, which have been theoretically proposed.9,10
Author contributions
Claire Förster: data collection, analysis and manuscript writing. Lothar Veith: measurement and interpretation of ToF-SIMS. Annette Andrieu-Brunsen: acquisition of funding, supervision of project, and assistance with manuscript writing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 803758) and acknowledge support by the Deutsche Forschungsgemeinschaft (DFG – German Research Foundation) and the Open Access Publishing Fund of Technical University of Darmstadt. The authors further thank Ulrike Kunz and Prof. Kleebe (TU-Darmstadt) for TEM measurements, Karl Kopp and Prof. Hess (TU-Darmstadt) for XPS measurements and Joanna Mikolei for argon and krypton adsorption experiments. The authors further thank Samet Varol for fruitful discussions and support with interpretation of the ToF-SIMS data.Notes and references
- R. Brilmayer, C. Förster, L. Zhao and A. Andrieu-Brunsen, Curr. Opin. Biotechnol., 2020, 63, 200 CrossRef CAS PubMed.
- R. Pardehkhorram and A. Andrieu-Brunsen, Chem. Commun. (Camb), 2022, 58, 5188 RSC.
- Z. Wang, X. Yang, Z. Cheng, Y. Liu, L. Shao and L. Jiang, Mater. Horiz., 2017, 4, 701 RSC.
- Ç. S. Koşak, S. Trosien and M. Biesalski, ACS Appl. Mater. Interfaces, 2018, 10, 37478 CrossRef PubMed.
- G. Huang, Y. Liang, J. Wang, X. Zeng, Z. Li and X. Zhang, Mater. Lett., 2019, 246, 76 CrossRef CAS.
- H. Zhou, H. Wang, H. Niu and T. Lin, Sci. Rep., 2013, 3, 2964 CrossRef PubMed.
- Z. Zhang, L. Wen and L. Jiang, Chem. Soc. Rev., 2018, 47, 322 RSC.
- Z. S. Siwy, Adv. Funct. Mater., 2006, 16, 735 CrossRef CAS.
- K. Huang and I. Szleifer, J. Am. Chem. Soc., 2017, 139, 6422 CrossRef CAS PubMed.
- S. Qin, K. Huang and I. Szleifer, ACS Nano, 2021, 15(11), 17678–17688 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105 CrossRef CAS PubMed.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, Macromolecules, 2008, 41, 8584 CrossRef CAS.
- M. Kruk, Isr. J. Chem., 2012, 52, 246 CrossRef CAS.
- H. Bayat, M. Raoufi, I. Zamrik and H. Schönherr, J. Colloid Interface Sci., 2020, 36, 2663 CAS.
- J. Chen, M. Liu, L. Huang, H. Huang, Q. Wan, J. Tian, Y. Wen, F. Deng, X. Zhang and Y. Wei, J. Taiwan Inst. Chem. Eng., 2018, 91, 570 CrossRef CAS.
- J. Chen, M. Liu, H. Huang, F. Deng, L. Mao, Y. Wen, L. Huang, J. Tian, X. Zhang and Y. Wei, J. Mol. Liq., 2018, 259, 179 CrossRef CAS.
- J. C. Tom, R. Brilmayer, J. Schmidt and A. Andrieu-Brunsen, Polymers, 2017, 9, 539 CrossRef PubMed.
- R. Brilmayer, C. Hess and A. Andrieu-Brunsen, Small, 2019, 15, e1902710 CrossRef.
- R. Brilmayer, S. Kübelbeck, A. Khalil, M. Brodrecht, U. Kunz, H. J. Kleebe, G. Buntkowsky, G. Baier and A. Andrieu Brunsen, Adv. Mater. Interfaces, 2020, 7, 1901914 CrossRef CAS.
- M. R. Buchmeiser, N. Atzl and G. K. Bonn, J. Am. Chem. Soc., 1997, 119, 9166 CrossRef CAS.
- S. Ma, J. Liu, Q. Ye, D. Wang, Y. Liang and F. Zhou, J. Mater. Chem. A, 2014, 2, 8804 RSC.
- F. Krohm, J. Kind, R. Savka, M. Alcaraz Janßen, D. Herold, H. Plenio, C. M. Thiele and A. Andrieu-Brunsen, J. Mater. Chem. C, 2016, 4, 4067 RSC.
- A. Bouchet, E. Descamps, P. Mailley, T. Livache, F. Chatelain and V. Haguet, Small, 2009, 5, 2297 CrossRef CAS PubMed.
- J. Liu, P. Pham, V. Haguet, F. Sauter-Starace, L. Leroy, A. Roget, E. Descamps, A. Bouchet, A. Buhot, P. Mailley and T. Livache, Anal. Chem., 2012, 84, 3254 CrossRef CAS.
- L. Velleman, D. Losic and J. G. Shapter, J. Membr. Sci., 2012, 411–412, 211 CrossRef CAS.
- M. Ochs, R. Mohammadi, N. Vogel and A. Andrieu-Brunsen, Small, 2020, 16, e1906463 CrossRef PubMed.
- D. John, M. Stanzel and A. Andrieu‐Brunsen, Adv. Funct. Mater., 2021, 31, 2009732 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- D. R. Dunphy, P. H. Sheth, F. L. Garcia and C. J. Brinker, Chem. Mater., 2015, 27, 75 CrossRef CAS.
- D. J. Babu, S. Yadav, T. Heinlein, G. Cherkashinin and J. J. Schneider, J. Mater. Chem. C, 2014, 118, 12028 CAS.
- P. Pasetto, H. Blas, F. Audouin, C. Boissière, C. Sanchez, M. Save and B. Charleux, Macromolecules, 2009, 42, 5983 CrossRef CAS.
- Y. Ma, X. Cao, X. Feng, Y. Ma and H. Zou, Polymer, 2007, 48, 7455 CrossRef CAS.
- G. O. Berim and E. Ruckenstein, J. Colloid Interface Sci., 2011, 359, 304 CrossRef CAS PubMed.
- E. A. Vogler, Adv. Colloid Interface Sci., 1998, 74, 69 CrossRef CAS PubMed.
- Y. Tian and L. Jiang, Nat. Mater., 2013, 12, 291 CrossRef CAS PubMed.
- X. Zhang, H. Liu and L. Jiang, Adv. Mater, 2019, 31, e1804508 CrossRef PubMed.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063 CrossRef CAS.
- P. Y. Steinberg, F. M. Zanotto, G. J. A. A. Soler-Illia, S. A. Dassie and P. C. Angelomé, J. Mater. Chem. C, 2021, 125, 23521 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05422a |
| This journal is © The Royal Society of Chemistry 2022 |