
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignosulfonate in situ-modified reduced graphene oxide biosensors for the electrochemical detection of dopamine†
Ying Yuan,
Shuangxin Wang,
Ping Wu,
Tongqi Yuan and
Xiluan Wang*
and
Xiluan Wang*
Beijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing, 100083, P. R. China. E-mail: wangxiluan@bjfu.edu.cn
First published on 31st October 2022
Abstract
Lignosulfonate (LS), a biomass by-product from sulfite pulping and the paper-making industry, which has many excellent characteristics, such as renewable, environmentally friendly, amphiphilic nature, and especially the abundant content of hydrophilic functional groups in its architecture, making it highly reactive and can be used as a sensitive material in sensors to show changes in electrical signals. Herein, we report a one-step in situ method to fabricate lignosulfonate-modified reduced graphene oxide (LS–rGO) green biosensors, which can be used for the sensitive electrochemical detection of dopamine without interference from uric acid and ascorbic acid. The modified LS molecular layers act as chemical-sensing layers, while the rGO planar sheets function as electric-transmitting layers in the as-assembled dopamine biosensors. After the in situ-decoration of the LS modifier, the sensing performance of LS–rGO for the detection of dopamine was much higher than that of the pure rGO electrode, and the highest current response of the biosensor toward dopamine greatly improved from 11.2 μA to 52.07 μA. The electrochemical sensitivity of the modified biosensor was optimized to be 0.43 μA μM−1, and the detection limit was as low as 0.035 μM with a wide linear range (0.12–100 μM), which is better than that of most previously reported metal- and organic-based modified graphene electrodes. The newly designed biosensor has unique advantages including rapid, stable, sensitive and selective detection of dopamine without interference, providing a facile pathway for the synthesis of green resource-derived sensing materials instead of the traditional toxic and expensive modifiers.
Introduction
Lignin is considered the most abundant, eco-friendly and renewable aromatic compound resource on Earth,1–3 but its complex structure and poor dispersion make its direct utilization difficult. Lignosulfonate (LS), as one of the derivatives of lignin, is a type of by-product from the pulp and paper making industry,4–6 which not only can maintain the primary aromatic framework and multiple active functional groups of lignin, but simultaneously introduce multi-sulfonic functional groups during sulfite pulping.7,8 Based on the hydrophobic aromatic skeleton and hydrophilic sulfonated functionalized chains, LS presents typical amphiphilic behavior, further broadening the application scope of lignin. Even though, the green resources based on LS are mainly used in fields, such as construction, industry and agriculture, making the high-valued utilization of LS limited. The multi-functionalities of LS make it easy to interact with a variety of biological small molecules, which means that LS is a potential candidate for application in the biomedical engineering field, especially the electrochemical detection of biological small molecules.As an emerging carbon-based material, graphene-based electrode surface materials have been extensively studied owing to the unique properties, such as excellent electrochemical surface area,9,10 high charge carrier mobility,11–13 wide electrochemical window and good biocompatibility,14 making it an ideal candidate for electrochemical analysis. However, the absence of chemically active sites on the basal plane of rGO reduces the heterogeneous electron transfer between biomolecules and electrode surface during electrochemical detection.15 Thus, to address this, a range of modifiers has been used to improve its electrochemical sensing performance. The common modifiers include metal oxide nanoparticles (such as MnO2 (ref. 16) and Co3O4 (ref. 17)), metal nanoparticles (AuNPs18 and AgNPs19), conductive polymers (PPy20,21 and PANI22) and ionic liquids.23 Although these organic and metal-based modifiers can be employed to achieve sensitive detection, their significant threat to the environment and high consumption of chemical resources limit their wide application. Hence, it is essential to urgently develop greener modifiers instead of the expensive, nondegradable and toxic modifiers. The introduction of LS on the surface of graphene via supramolecular interaction not only can improve the dispersion of graphene, but also the multifunctional groups of LS increase the number of active sites present in graphene, which further promotes the sensitivity during electrochemical analysis. The application of LS–rGO composite materials in the field of biological small molecule detection, integrating the advantages of both, not only alleviates the energy and environment crisis caused by the use of heavy metals and petroleum-based materials, but simultaneously promotes the effective high-value utilization of lignin.
Dopamine (DA), as a type of catechin neurotransmitter,24,25 is one of the most important biological small molecules, playing a role in the central nervous26 and cardiovascular and hormone system.27,28 Furthermore, it is of great significance in the study of physiological function, diagnosis and treatment of mental illness, drug detection and food safety. Thus, the accurate detection of dopamine in biological fluids is essential. Currently, the mainstream analytical methods for the detection of dopamine include fluorescence spectrometry, high-performance liquid chromatography, capillary electrophoresis and colorimetry, which are usually accompanied by several problems during practical applications, and it is difficult to detect DA in vivo. Given that the DA molecule contains easily oxidizable phenolic hydroxyl groups, its quantification can also be achieved by electrochemical methods, which are fast, simple and enables in vivo testing. A comparison between electrochemical biosensors and other analytical methods are shown in Table S1.† Also, for the electrochemical detection of DA with high selectivity and sensitivity but without interference, numerous electrode surface materials have been developed.29–31
According to the above perspective, herein, an electrochemical sensing platform based on an LS–rGO composite material is reported for the detection of DA on a glassy carbon electrode (GCE) using a one-step in situ modification method. The conjugated large π bonds between LS and rGO in the LS–rGO composite are beneficial for electron transfer to DA through π–π stacking interactions. Meanwhile, the similar structure to catechol and large amounts of anionic groups of LS can raise the affinity for dopamine, enhancing the selectivity of the biosensor. Hence, the maximum response (Ipa) of the LS–rGO electrode reached 52.07 μA for the detection of 100 μM DA, which was nearly 12.8 times higher than that of the bare electrode and 4.65 times higher than that of the pure rGO electrode. Moreover, the LS–rGO electrode enabled the highly selective detection of dopamine in the presence of uric acid (UA) and ascorbic acid (AA) with a low detection limit of 0.035 μM, which is better than that of most previously reported metal- and organic-based modified graphene electrodes. Electrochemical biosensors with high sensitivity, selectivity and stability fit well with the green approach in replacing conventional metal- and organic-based electrode modifiers, which is of great strategic significance for sustainable development and extending the valorization of lignocellulosic biomass.
Results and discussion
The characteristics of LS–rGO composite
 | ||
| Fig. 1 (a) Schematic illustration of the synthetic route for LS–rGO/GCE. (b) 20 mL rGO aqueous solution (0.5 mg mL−1) before and after one week. (c) 20 mL LS–rGO (0.5 mg mL−1) aqueous solution before and after two months. SEM images of (d) rGO and (e) LS–rGO surface. | ||
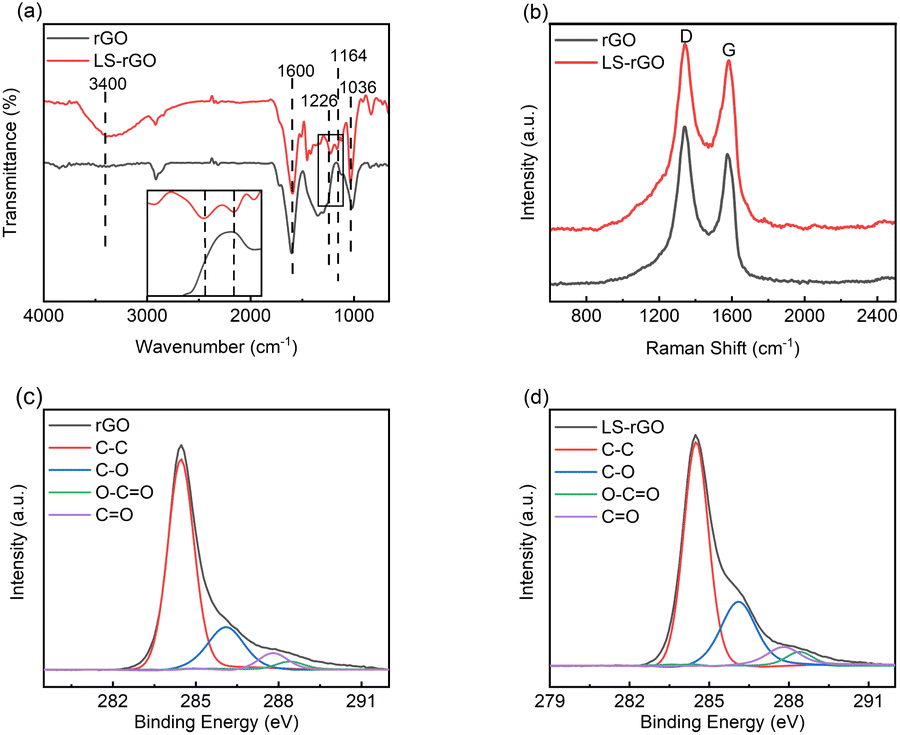 | ||
| Fig. 2 (a) FT-IR spectra of rGO and LS–rGO samples. The inset image shows a partial enlargement of the FT-IR spectra. (b) Raman spectra of rGO and LS–rGO samples. High-resolution XPS spectra for (c) rGO sample and (d) LS–rGO samples. | ||
The intensity ratio of the D and G bands (ID/IG) can be used to investigate the degree of structural disorder in carbon-based materials.35 The ID/IG ratio decreased from 1.22 to 1.07 after rGO was decorated with LS. This indicates that the noncovalent interaction formed between the LS chains and rGO sheets not only ameliorated the uniform dispersion, but also enhanced the structural integrity of graphene after reduction.36–38 The great sp2 character can result in a better performance towards the detection of DA.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.8 eV) and O–CO (288.4 eV) for LS–rGO increased compared to that of rGO.39–41 The main reason for this is that LS can be considered as a phenolic macromolecule, which is rich in oxygen-containing groups such as hydroxyl, carbonyl and carboxyl groups. The introduction of LS increased the content of related multifunctional groups in the composite. Table S2† exhibits that the elemental content ratio of O and C increased from 14.66% for rGO to 25.12% for LS–rGO, which is owing to the incorporation of LS, whose branched chains are rich in oxygen-containing functional groups. Thus, it offered the abundant active sites to interact with DA molecules.
O (287.8 eV) and O–CO (288.4 eV) for LS–rGO increased compared to that of rGO.39–41 The main reason for this is that LS can be considered as a phenolic macromolecule, which is rich in oxygen-containing groups such as hydroxyl, carbonyl and carboxyl groups. The introduction of LS increased the content of related multifunctional groups in the composite. Table S2† exhibits that the elemental content ratio of O and C increased from 14.66% for rGO to 25.12% for LS–rGO, which is owing to the incorporation of LS, whose branched chains are rich in oxygen-containing functional groups. Thus, it offered the abundant active sites to interact with DA molecules.Electrochemical activity of LS–rGO/GCE towards the oxidation of DA
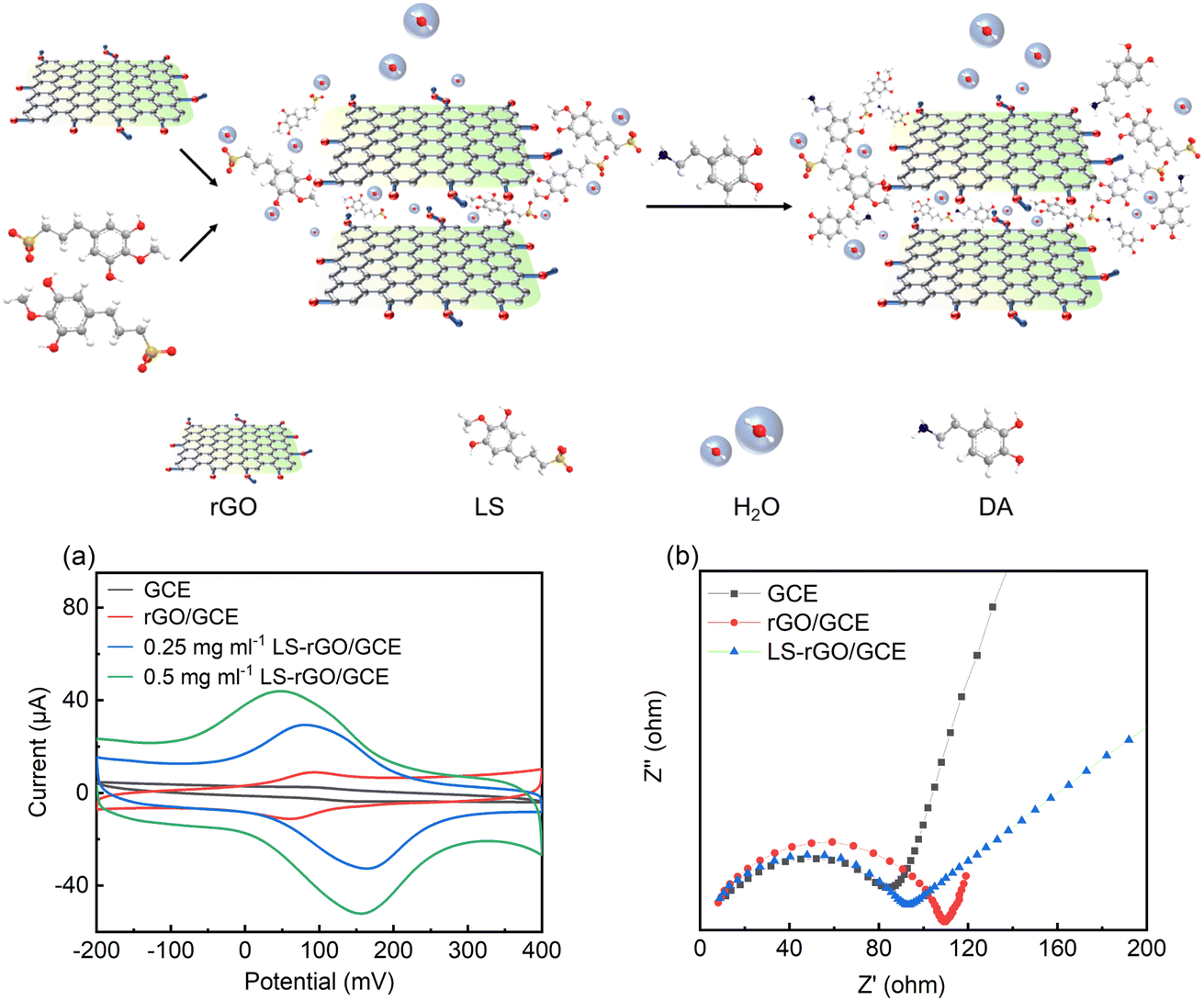 | ||
| Fig. 3 Top: schematic explaining the sensing mechanism of LS–rGO/GCE sensors. (a) CV curves of 100 μM DA in 0.1 M PBS (pH = 7.0) at a scan rate of 50 mV s−1 on GCE, rGO/GCE (0.5 mg mL−1), LS–rGO/GCE (0.25 mg mL−1), and LS–rGO/GCE (0.5 mg mL−1). (b) Nyquist plots of 5 mM [Fe (CN)6]4−/3− and 0.1 M KCL in 0.1 M PBS on GCE, rGO/GCE, and LS–rGO. Frequency range of 0.1 Hz to 1000 kHz. The amplitude of 5 mV was applied. | ||
Electrochemical impedance spectroscopy
Electrochemical impedance spectroscopy (EIS) was used to further evaluate the electrochemical impedance of the bare GCE, rGO/GCE, and LS–rGO/GCE. The electronic transfer capacity at the interface between the solution and electrode varies inversely to charge transfer resistance (Rct), which is represented as the diameter of the high-frequency semicircles in the Nyquist plots.42 As depicted in Fig. 3b, semicircles are observed in high-frequency region, confirming that the electron transfer between the interfaces is impeded and reflecting the existence of Rct. Compared to rGO/GCE, the Rct value of LS–rGO/GCE decreased from 110 Ω to 92 Ω, which indicates that the modified electrode had accelerated electron transfer owing to the restoration of the sp2 conjugation network. This is ascribed to the less stacking of the rGO sheets in the presence of LS. Furthermore, the formation of three-dimensional channels in the LS macromolecule is conducive for electron transfer.Electrocatalytic mechanism of DA oxidation
To reveal the kinetic mechanism, the responsive currents at different scan rates (v) were investigated in the range of 50 to 500 mV s−1. Fig. 4a shows that both Ipa and Ipc increased dramatically with an increase in scan rate, while the oxidation potential moved toward the positive direction to some extent, which indicates the occurrence of a quasi-reversible reaction.43 Fig. 4b shows that the Epa and Epc are proportional to log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) v at high scan rates, where v > 100 mV, and the linear regression equations are as follows:
v at high scan rates, where v > 100 mV, and the linear regression equations are as follows:|
Epa = −7.86 + 74.86logv(R2 = 0.991)
| (1) |
|
Epc = −261.76 − 87.09logv(R2 = 0.986)
| (2) |
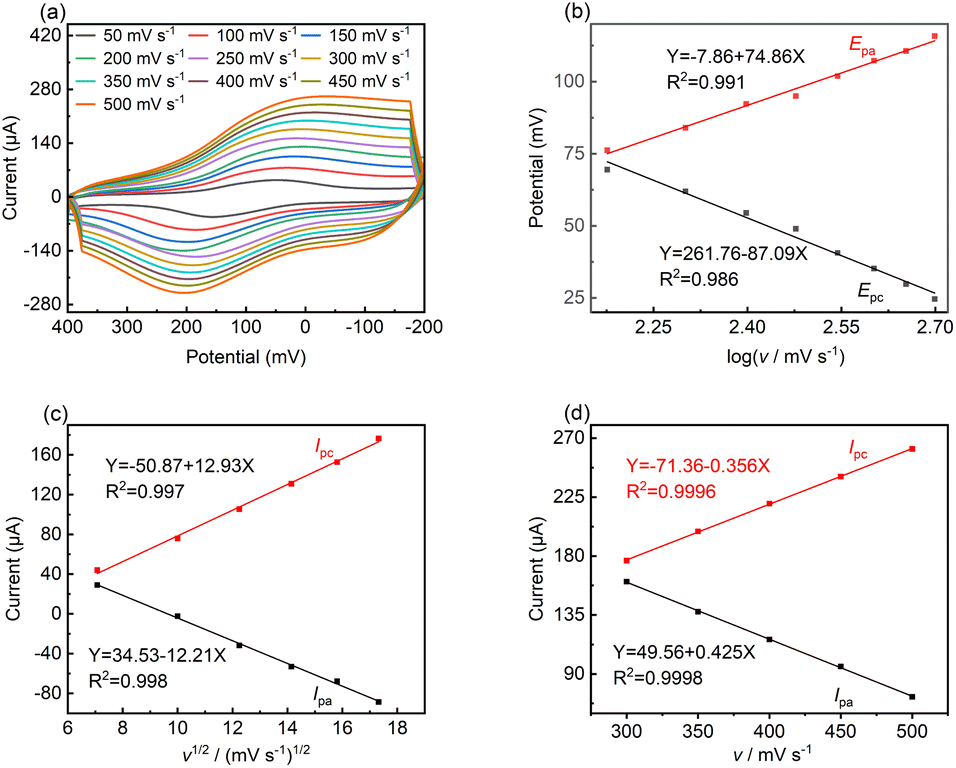 | ||
| Fig. 4 (a) CV curves of LS–rGO/GCE in 0.1 M PBS (pH = 7.0) containing 100 μM DA at various scan rates (50–500 mV s−1). (b) Plots of peak current (Epa and Epc) versus scan rate. (c) Plots of peak current (Ipa and Ipc) versus square root of scan rate in the range of 50–300 mV s−1. (d) Plots of peak current (Ipa and Ipc) versus scan rate in the range of 300–500 mV s−1. | ||
In the case of a quasi-reversible electrochemical process, the Laviron equation is widely used for the calculation of the electron transfer rate constant (KS) and other dynamic parameters.44 According to the linear regression equations and the Laviron equation, the dynamic parameters of the surface electrochemical processes were evaluated with the values of α, n, and KS to be 0.46, 1.47, and 2.69, respectively. Fig. 4c presents that Ipa and Ipc are proportional to the square root of the scan rate in the range of 50–300 mV s−1. The linear regression equations are as follows:
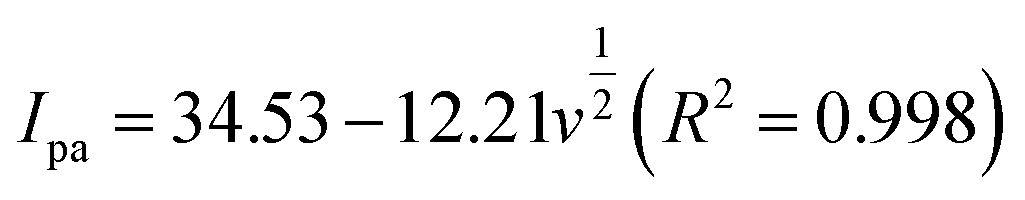 | (3) |
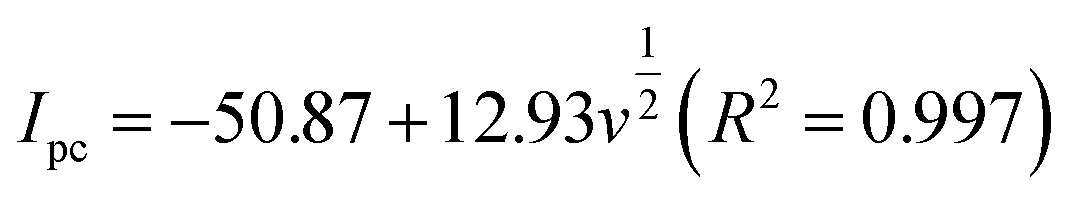 | (4) |
| Ipa = −71.36 − 0.356v(R2 = 0.9996) | (5) |
| Ipc = 49.56 + 0.425v(R2 = 0.9998) | (6) |
The pH condition of the working environment is crucial in the oxidation of DA on modified electrodes. Thus, the influence of pH value on the detection performance of the LS–rGO/GCE biosensor at 50 mV s−1 (Fig. 5) was investigated.
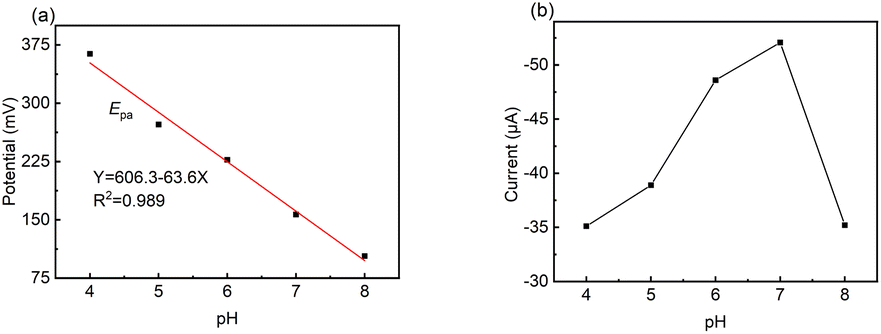 | ||
| Fig. 5 Relationship of (a) potential (Epa) and (b) CV current (Ipa) versus pH value toward 100 μM DA in 0.1 M PBS at a scan rate of 50 mV s−1 on LS–rGO/GCE. | ||
The linear regression equation of Ep versus pH value is presented as follows:
| Epa = 606.3 − 63.6pH (R2 = 0.989) | (7) |
The slope value of the calibration curve was calculated to be 63.6 mV pH−1, approaching 59 mV pH−1, which is the theoretical value of a proton-coupled electron transfer reaction at 25 °C. This shows that the electron transfer is often accompanied by the synchronous migration of the same quantities of protons in the oxidation process of DA on LS–rGO/GCE.49,50 Fig. 5b shows that in the pH range of 4.0–8.0, Ipa increased initially, and then decreased with a gradual increase in pH, reaching the maximum value when pH is 7.0. Hence, pH 7.0 was found to be optimal for the other studies.
Relationship between dopamine concentration and response current
Fig. 6a shows the cyclic voltammograms for LS–rGO/GCE at different concentrations of DA in 0.1 M PBS (pH = 7.0). With an increase in the concentration of DA in the range of 10 μM to 100 μM, the peak current increased linearly, and the equation is as follows:| I/μA = 0.219C/μM + 20.94 (R2 = 0.993) | (8) |
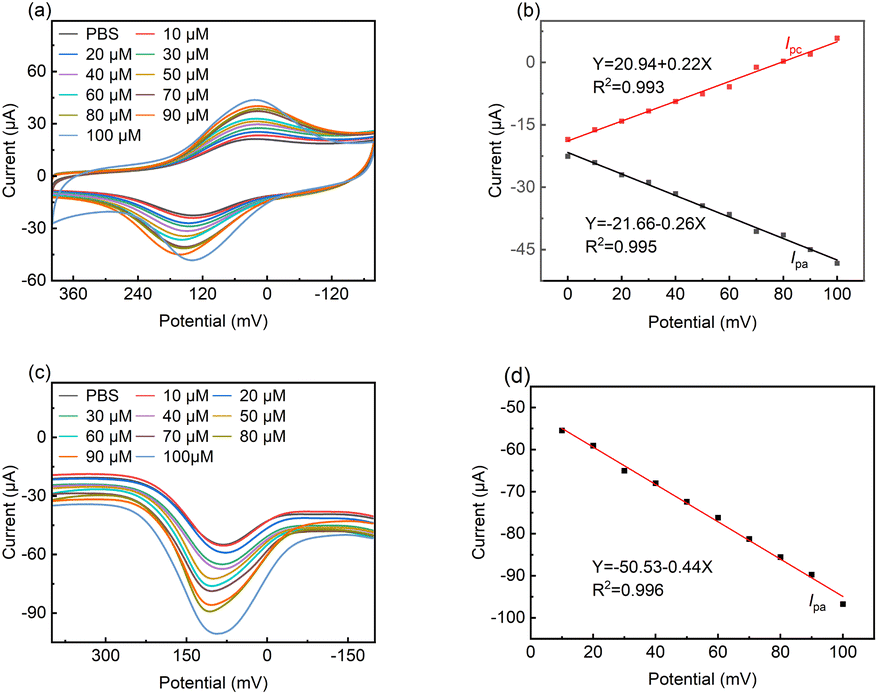 | ||
| Fig. 6 (a) CV curves of LS–rGO/GCE in 0.1 M PBS (pH = 7.0) containing various concentrations of DA (0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 μM) at a scan rate of 50 mV s−1. (b) Linear relationship of peak potential with concentration. (c) DPV curves of LS–rGO/GCE in 0.1 M PBS (pH = 7.0) containing various concentrations of DA (0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 μM). DPV settings: step potential: 4 mV; amplitude: 50 mV; pulse period: 0.2 s; and amplitude: 50 mV. (d) Linear relationship between peak potential (Ipa) with concentration. | ||
The linear correlation (Fig. 6b) confirms that the electrochemical oxidation of DA follows first-order kinetics with respect to DA concentration.
Differential pulse voltammetry (DPV) is a type of electrochemical measurement method widely used for analysis, which has the advantages of high sensitivity and high selectivity. Thus, DPV was also used to detect DA with a concentration in the range of 10 μM to 100 μM at the LS–rGO/GCE surface (Fig. 6c).
It can be observed in Fig. 6d that the oxidation peak currents are also proportional to its concentrations, with the linear relationship equation as follows:
| I/μA = −0.44C/μM − 50.53 (R2 = 0.996) | (9) |
The detection limit was estimated to be 0.035 μM, which is better than that of most previously reported metal- and organic-based modified graphene electrodes (Table S3†), suggesting the high sensitivity of the LS–rGO/GCE biosensor.
Repeatability and stability of the sensor
To test the reversibility of the modified electrode, the repeated current response of the LS–rGO/GCE was measured for 100 μM DA in 0.1 M PBS (pH = 7) at a scan rate of 50 mV s−1. Fig. 7a shows the current response (Ipa) under multiple cycles, and the relative standard deviation (RSD) of the current response (Ipa) was calculated to be 1.10% for 7 measurement times, indicating that the modified electrode has excellent repeatability. Then, to evaluate the storage stability of LS–rGO/GEC, the Ipa for the detection of 100 μM DA at 50 mV s−1 was measured for 6 weeks and the histogram shows that the Ipa could still maintain 97.3% compared with the initial response current after storage for 6 weeks.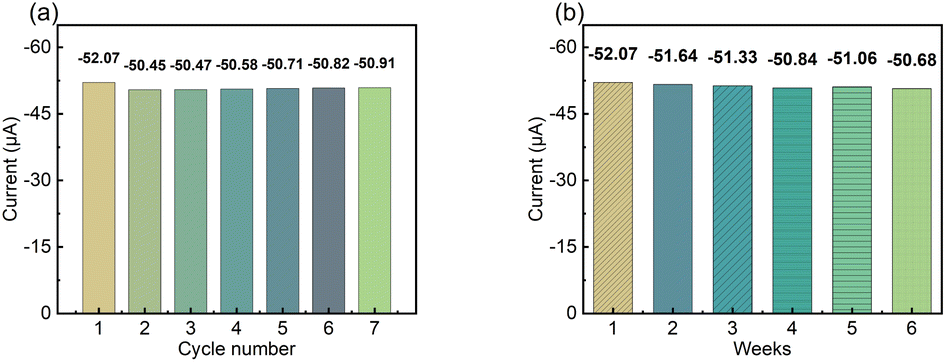 | ||
| Fig. 7 (a) Histogram of peak current (Ipa) for LS–rGO/GCE tested repeatedly seven times. (b) Histogram of peak current (Ipa) on the LS–rGO/GCE under continuous monitoring for 6 weeks. | ||
Selectivity of the sensor
The selectivity of LS–rGO/GCE for DA was in the presence of interference from other coexisting biological macromolecules in the body. Obviously, there was no peak-overlap with fluid interferences, such as UA and AA. As shown in Fig. 8b and c, the peak interval between DA for UA is about 138 mV and 195 mV for AA, respectively. Thus, coexisting species and the interference caused by the presence of UA and AA in the detection of DA can be disregarded.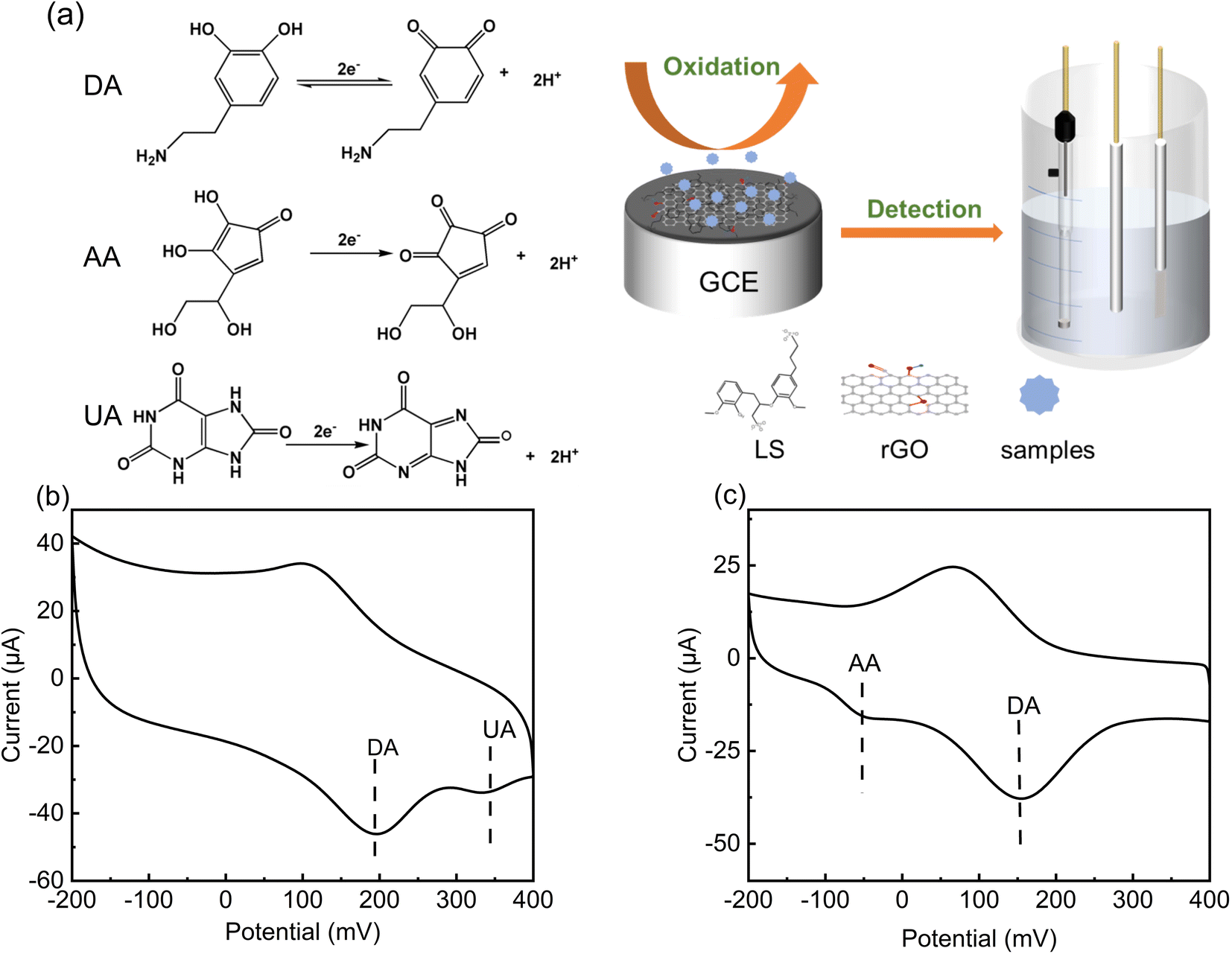 | ||
| Fig. 8 (a) CV curve of LS–rGO/GCE in 0.1 M PBS (pH = 7.0) containing 0.2 mM UA, 0.1 mM DA at a scan rate of 100 mV s−1. (b) CV curve of LS–rGO/GCE in 0.1 M PBS (pH = 7.0) containing 0.2 mM AA, 0.1 mM DA at a scan rate of 100 mV s−1. (c) Schematic representation of electrocatalysis of DA, AA, and UA at LS–rGO/GCE. | ||
UA, AA and DA are all electrically active substances, and thus during electrochemical detection, when the potential changes to the critical potential of their redox potential, the corresponding reactions will occur. In human serum, these three biomolecules have similar electrochemical oxidation potentials, which often hinders the accurate detection of DA. However, the acidity coefficients (pKa) of UA and AA are 5.4 and 4.1 respectively, while that of DA is 8.61. Thus, in different pH solutions, they exist in different ionic forms.
When pH = 7, DA mainly exists in the form of a cation, while UA and AA mainly exist in the form of anions. As shown in Table S4,† due to the ionization of the phenolic hydroxyl and amide groups in DA molecule, it has multiple ionic states in different pH aqueous solutions, and 99% of DA exists in the form of cation in neutral solution. As an anionic surfactant, lignosulfonate has strong cation selective permeability due to the abundant sulfonic groups on its side chain. In the aqueous solution of pH = 7.0 PBS, the positively charged –NH3+ group is easily absorbed on LS, which contains extensive negatively charged –SO32− through electrostatic interaction. In addition, the aromatic structure of LS–rGO and DA can generate a large conjugation area, and the interface electronic transmission of DA can be boosted by π–π interactions, which can restrain the attachment of UA and AA onto the electrode surface. Thus, the modifier is beneficial for the selective detection of DA and these results are reasonable to explain the anti-interference of LS–rGO/GCE in coexisting species.
To further illustrate the selectivity of the electrodes, the interference from substances significantly co-existed with DA in biological fluids, such as NaCl, KCl and Glu, was excluded by performing a chronoamperometry test. As shown in Fig. S2,† a slight change in current was observed with the addition of 1 μM of NaCl, KCl and Glu, and when the concentration of interfering substances was increased to 5 μM, no significant change in current was observed.
Experimental
Preparation of LS–rGO
GO was prepared via the oxidation of natural graphite powder (325 meshes) according to a modified Hummers' method51 (Hummers, 1958). A GO dispersion was diluted to 0.5 mg mL−1. Then a certain amount of LS powder was added to 100 mL GO solution to obtain an LS–GO aqueous, followed by ultrasonication for 10 min to obtain a homogeneous dispersion, in which the mass ratios of LS and GO were 25:1, 10:1, 5:1, 1:1, 1:5, 1:10, and 1:25, respectively. In a typical procedure for the chemical conversion of GO to rGO according to Li52 (Li et al., 2008), 48.4 μL hydrazine hydrate (98 wt%) was added to the obtained brown dispersion with the weight ratio of hydrazine to GO of 7:10. Then, ammonia (28 wt%) was added to the solution to adjust the pH to about 10, and after being vigorously shaken or stirred, the solution was reacted at 95 °C in a water bath for 1 h.
Fabrication of LS–rGO/GCE
The bare GCE was polished using an α-Al2O3 slurries with different sizes (1.0, 0.3 and 0.05 μm). Then, the GCE was washed thoroughly with ethyl alcohol and DI water under ultrasonication and blown with ultrapure N2 gas for the next step. The LS–rGO-modified electrode was prepared via the drop-coating method. 5 μL of the reduced LS–rGO dispersion was deposited onto the surface of the polished GCE via drop casting with a pipette gun. After the solvent evaporated and solidified, a coating was formed, which bonded to the electrode surface to achieve the purpose of loading. A pure rGO electrode was prepared for comparison in the same way and air-dried for subsequent testing.Characterization of the LS–rGO
AFM (Bruker multimode 8) was used to measure the thickness and transverse diameter of GO. The AFM analysis was conducted using a drop of dialyzed GO solution, which had been diluted and ultrasonicated for 1 h on a mica substrate. The prepared LS–rGO and rGO solutions were dropped on a copper sheet and observed through SEM (Zeiss Sigma 300, Germany) after the solvent was completely dried to contrast the surface morphology and the folding degree. The Raman spectra of the freeze-dried LS–rGO and rGO samples were recorded on a Senterra R200-L apparatus (Bruker Optics) with an excitation wavelength of 532 nm. The FT-IR measurements (Bruker tensor II) were performed in the wavenumber range of 680–4000 cm−1 to explore the chemical structure of the freeze-dried LS–rGO and rGO samples. The XPS analysis was performed on a Thermo Scientific K-Alpha photoelectron spectrometer using Al Kα (1486.6 eV) radiation.All electrochemical measurements were performed on an electrochemical workstation (CHI-760E, Austin, Texas) with a standard three-electrode system, as shown in Fig. 1a. Details can be found in the ESI.†
Conclusions
Herein, an LS–rGO-based biosensor that can be used for the detection of DA was developed through a simple one-step in situ-modification method. The maximum current response (Ipa) of the as-prepared LS–rGO/GCE was 52.07 μA, which is almost 12.8 times and 4.65 times higher than that of the bare GCE and rGO/GCE, respectively. The electrochemical sensitivity of the modified biosensor was optimized to be 0.43 μA μM−1 with a detection limit as low as 0.035 μM in a wide linear range (0.12–100 μM), which is better than that of most metal- and organic-based modified graphene electrodes. Moreover, LS–rGO/GCE exhibited high selectivity in the presence of coexisting species, such as UA and AA. In addition, the resulting biosensors show good repeatability and stability for the determination of DA. The novel designed biosensor derived from lignin modifier has potential to be used for the trace detection of DA, and simultaneously it provides a new idea for the detection of other trace elements using lignin as a sensing material, helping the resource utilization of lignin-based materials.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51603012).References
- A. E. Kazzaz and P. Fatehi, Ind. Crops Prod., 2020, 154, 112732 CrossRef.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- C. Zhao, Z. Hu, L. Shi, C. Wang, F. Yue, S. Li, H. Zhang and F. Lu, Green Chem., 2020, 22, 7366–7375 RSC.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. C. Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- Z. H. Feizi, A. E. Kazzaz, F. Kong and P. Fatehi, Sep. Purif. Technol., 2019, 222, 254–263 CrossRef CAS.
- M. Parit and Z. Jiang, Int. J. Biol. Macromol., 2020, 165, 3180–3197 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- C. Wang, J. Zhang, C. Liu, X. Song and C. Zhang, Chem. Eng. J., 2021, 412, 128609 CrossRef CAS.
- Z. Liu, L. Wang, G. Ma, Y. Yuan, H. Jia and W. Fei, J. Mater. Chem. A, 2020, 8, 18933–18944 RSC.
- H. Zhang, D. Yang, A. Lau, T. Ma, H. Lin and B. Jia, Small, 2021, 17, 2007311 CrossRef CAS.
- A. A. Balandin, ACS Nano, 2020, 14, 5170–5178 CrossRef CAS PubMed.
- P. Su, F. Wang, Z. Li, C. Y. Tang and W. Li, J. Mater. Chem. A, 2020, 8, 15319–15340 RSC.
- Y. Sun, X. Liu, X. Lv, T. Wang and B. Xue, J. Cleaner Prod., 2021, 295, 126406 CrossRef CAS.
- Z. Jiang, B. Feng, J. Xu, T. Qing, P. Zhang and Z. Qing, Biosens. Bioelectron., 2020, 166, 112471 CrossRef CAS.
- S. Tiwari, R. Patil, S. K. Dubey and P. Bahadur, Adv. Colloid Interface Sci., 2020, 281, 102167 CrossRef CAS PubMed.
- Q. He, J. Liu, X. Liu, G. Li, D. Chen, P. Deng and J. Liang, Electrochim. Acta, 2019, 296, 683–692 CrossRef CAS.
- L. Bao, T. Li, S. Chen, C. Peng, L. Li, Q. Xu, Y. Chen, E. Ou and W. Xu, Small, 2017, 13, 16020775 CrossRef.
- Y. Zhou, M. Ma, H. He, Z. Cai, N. Gao, C. He, G. Chang, X. Wang and Y. He, Biosens. Bioelectron., 2019, 146, 111751 CrossRef CAS.
- M. Roushani, M. Ghanbarzadeh, F. Shandost-Fard, R. Sahraei and E. Soheyli, Mater. Sci. Eng., C, 2020, 108, 110507 CrossRef CAS PubMed.
- Y. Xie, X. Tu, X. Ma, M. Xiao, G. Liu, F. Qu, R. Dai, L. Lu and W. Wang, Electrochim. Acta, 2019, 311, 114–122 CrossRef CAS.
- X. Yang, L. Cao, J. Wang and L. Chen, ACS Sustainable Chem. Eng., 2020, 8, 10726–10739 CAS.
- N. R. Tanguy, M. Arjmand and N. Yan, Adv. Mater. Interfaces, 2019, 6, 1900552 CrossRef.
- J. Xie, C. Wang, J. Patra, P. C. Rath, Y. A. Gandomi, Q. Dong and J. Chang, ACS Sustainable Chem. Eng., 2019, 7, 16233–16240 CrossRef CAS.
- J. A. Behan, F. Grajkowski, D. R. Jayasundara, L. Vilella-Arribas, M. Garcia-Melchor and P. E. Colavita, Electrochim. Acta, 2019, 304, 221–230 CrossRef CAS.
- X. Wei, Z. Zhang and Z. Wang, Microchem. J., 2019, 145, 55–58 CrossRef CAS.
- Y. Huang, Y. Zhang, D. Liu, M. Li, Y. Yu, W. Yang and H. Li, Talanta, 2019, 201, 511–518 CrossRef CAS PubMed.
- S. Hannah, M. Al-Hatmi, L. Gray and D. K. Corrigan, Bioelectrochemistry, 2020, 133, 107480 CrossRef CAS PubMed.
- F. Xing, X. Hu, J. Jiang, Y. Ma and A. Tang, Int. J. Cardiol., 2016, 222, 1003–1011 CrossRef PubMed.
- Y. Cho, J. Park, K. Lee, T. Lee, Z. Luo and T. Kim, Nano Convergence, 2020, 7, 40 CrossRef CAS.
- Q. He, J. Liu, X. Liu, G. Li, P. Deng and J. Liang, Sensors, 2018, 18, 199 CrossRef PubMed.
- A. Numan, M. M. Shahid, F. S. Omar, S. Rafique, S. Bashir, K. Ramesh and S. Ramesh, Microchim. Acta, 2017, 184, 2739–2748 CrossRef CAS.
- C. Liu, Z. Li, P. Yu, H. Wong and Z. Gu, ACS Appl. Energy Mater., 2018, 1, 3973–3983 CrossRef CAS.
- Z. Ding, X. Mei and X. Wang, Nanoscale Adv., 2021, 3, 2529–2537 RSC.
- Y. Qian, Y. Zhou, M. Lu, X. Guo, D. Yang, H. Lou, X. Qiu and C. F. Guo, Small Methods, 2021, 5, 2001311 CrossRef CAS.
- A. Y. Lee, K. Yang, D. A. Nguyen, C. Park, S. M. Lee, T. G. Lee and M. S. Jeong, Appl. Surf. Sci., 2021, 536, 147990 CrossRef CAS.
- F. Li, X. Wang, T. Yuan and R. Sun, J. Mater. Chem. A, 2016, 4, 11888–11896 RSC.
- Y. Liu, L. Gao and J. Sun, J. Phys. Chem. C, 2007, 111, 1223–1229 CrossRef CAS.
- M. Nar, H. R. Rizvi, R. A. Dixon, F. Chen, A. Kovalcik and N. D'Souza, Carbon, 2016, 103, 372–383 CrossRef CAS.
- C. Chen, X. Wang, M. Li, Y. Fan and R. Sun, Sens. Actuators, B, 2018, 255, 1569–1576 CrossRef CAS.
- P. Li, G. Liao, S. R. Kumar, C. Shih, C. Yang, D. Wang and S. J. Lue, Electrochim. Acta, 2016, 187, 616–628 CrossRef CAS.
- Q. Zhu, B. Liang, Y. Cai, Q. Cao, T. Tu, B. Huang, L. Fang and X. Ye, Talanta, 2018, 190, 70–77 CrossRef CAS PubMed.
- W. He, R. Liu, P. Zhou, Q. Liu and T. Cui, Biosens. Bioelectron., 2020, 167, 112473 CrossRef CAS.
- T. Thomas, R. J. Mascarenhas, B. E. K. Swamy, P. Martis, Z. Mekhalif and B. S. Sherigara, Colloids Surf., B, 2013, 110, 458–465 CrossRef CAS PubMed.
- Q. Huang, X. Lin, L. Tong and Q. Tong, ACS Sustainable Chem. Eng., 2020, 8, 1644 CrossRef.
- X. Yan, Y. Gu, C. Li, B. Zheng, Y. Li, T. Zhang, Z. Zhang and M. Yang, Sens. Actuators, B, 2018, 257, 936–943 CrossRef CAS.
- Q. Yuan, Y. Liu, C. Ye, H. Sun, D. Dai, Q. Wei, G. Lai, T. Wu, A. Yu, L. Fu, K. W. A. Chee and C. Lin, Biosens. Bioelectron., 2018, 111, 117–123 CrossRef CAS.
- Q. Huang, H. Zhang, S. Hu, F. Li, W. Weng, J. Chen, Q. Wang, Y. He, W. Zhang and X. Bao, Biosens. Bioelectron., 2014, 52, 277–280 CrossRef CAS.
- A. Numan, M. M. Shahid, F. S. Omar, K. Ramesh and S. Ramesh, Sens. Actuators, B, 2017, 238, 1043–1051 CrossRef CAS.
- Q. Li, C. Huo, K. Yi, L. Zhou, L. Su and X. Hou, Sens. Actuators, B, 2018, 260, 346–356 CrossRef CAS.
- Q. Guo, T. Wu, L. Liu, H. Hou, S. Chen and L. Wang, J. Mater. Chem. B, 2018, 6, 4610–4617 RSC.
- W. S. A. O. Hummers, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05635f |
| This journal is © The Royal Society of Chemistry 2022 |