
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalisation of vitamin B12 derivatives with a cobalt β-phenyl ligand boosters antimetabolite activity in bacteria†
Christopher Brenig,
Paula Daniela Mestizo and
Felix Zelder *
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH 8057 Zurich, Switzerland. E-mail: felix.zelder@chem.uzh.ch; Web: https://www.felix-zelder.net Fax: +41 44 635 6803
First published on 7th October 2022
Abstract
This study describes the syntheses of four singly- and two doubly-modified vitamin B12 derivatives for generating antimetabolites of Lactobacillus delbrueckii (L. delbrueckii). The two most potent antagonists, a Coβ-phenyl-cobalamin-c,8-lactam and a 10-bromo-Coβ-phenylcobalamin combine a c-lactam or 10-bromo modification at the “eastern” site of the corrin ring with an artificial organometallic phenyl group instead of a cyano ligand at the β-site of the cobalt center. These two doubly-modified B12 antagonists (10 nM) inhibit fully B12-dependent (0.1 nM) growth of L. delbrueckii. In contrast to potent 10-bromo-Coβ-phenylcobalamin, single modified 10-bromo-Coβ-cyanocobalamin lacking the artificial organometallic phenyl ligand does not show any inhibitory effect. These results suggest, that the organometallic β-phenyl ligand at the Co center ultimately steers the metabolic effect of the 10-bromo-analogue.
1. Introduction
Non-functional analogues of vitamins and vitamin building blocks represent an important class of drugs and drug candidates for treating different classes of diseases ranging from bacterial and fungal infections to human cancer.1–3 In the first half of the 20th century, prontosil was introduced as the first commercially available antimicrobial agent and saved millions of lives.4 The sulfonamide-based drug targets effectively bacterial, but not human biosynthesis of vitamin B9 (folic acid) explaining its selective therapeutic effect. Modified folic acid derivatives such as methotrexate, trimetrexate or pemetrexed represent other examples of important antibacterial and anticancer agents by inhibiting folic acid-dependent enzymatic transformations.1,5,6 In contrast to these folate-based drugs, modified cobalamin (vitamin B12) derivatives have not been developed so far to approved antibacterial or anticancer drugs.7 Nevertheless, some singly-modified vitamin B12 derivatives showed promising inhibitory effects on B12-dependent pathways.8 For example, B-ring-modified hydroxycobalamin-c,8-lactam (Fig. 1B left) and β-ligand-modified 4-ethylphenylcobalamin (Fig. 1B right) decreased significantly the activities of liver L-methylmalonyl coenzyme A mutase and methionine synthase as indicated by elevated plasma methylmalonic acid concentration and total homocysteine concentration in rodents.9,10 In contrast to these important results, bi-functionalised B12 derivatives combining two instead of a single modification were not explored so far in a systematic fashion.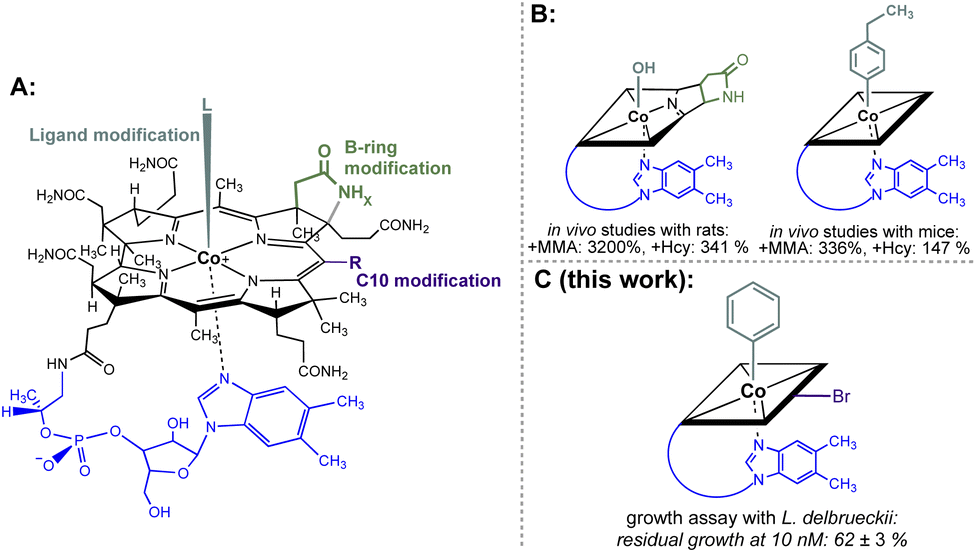 | ||
| Fig. 1 (A) General structural formula of cobalamins (Cbls; L = CN, R = H, x = 2 for vitamin B12, 1; the natural f-side chain of Cbls is depicted in blue). The upper (β-) axial ligand (turquoise), B-ring subunit (green) and the C10 position (violet) are highlighted as target sites for chemical modification. (B) Schematic depictions of B12 antimetabolites with a single modification at either the B-ring (left) or upper-ligand (right) modification. The effect on the increase of important biomarkers MMA (methylmalonic acid) and Hcy (homocysteine) of B12-dependent metabolism in mammals is indicated.9,10 (C) Schematic depiction of the most potent B12 antimetabolite of L. delbrueckii combining two chemical modifications at the upper ligand and the C10 position developed in this work. | ||
Herein we report on the antibacterial activity of doubly-modified B12 derivatives. It is demonstrated in a proof-of-concept study with L. delbrueckii, that a specific second structural modification (i.e. with an artificial organometallic phenyl instead of a cyano ligand at the β-side of the CoIII centre) steers the metabolic effect toward the desired activity as an antimetabolite.
2. Results and discussion
2.1 Syntheses and characterisation of modified vitamin B12 derivatives
Starting from vitamin B12 (1; Fig. 1A) we prepared two novel bi-functionalized Cbls (4 and 7; Scheme 1) using established synthetic protocols.11 In particular, we combined in the analogues either a c,8-lactam11 (Scheme 1 left) or C10–Br12 (Scheme 1 right) modification at the corrin ring with a β-phenyl ligand at the Co-center (Scheme 1, bottom line). These combinations were purposefully chosen because the selected transformations (i) induced biological effects in previous studies when incorporated into the Cbl scaffold (Fig. 1B) and (ii) provide analogs in high purity under mild conditions.9,10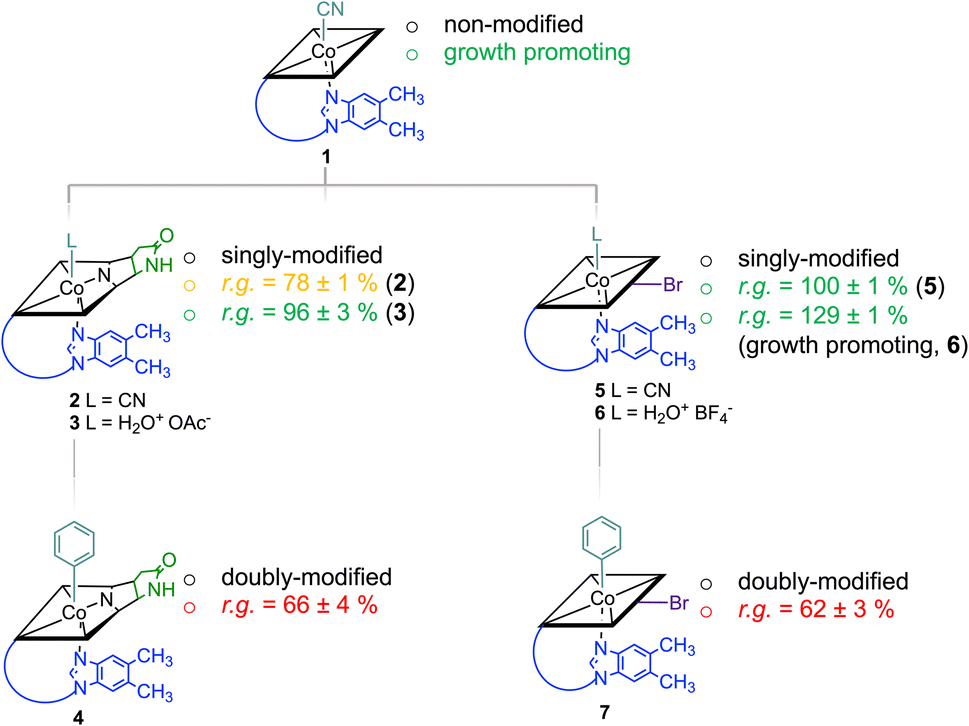 | ||
| Scheme 1 Cascade of multiple chemical modifications of Cbls and their physiologic effect (promotion of growth or activity as antimetabolite; r.g. = residual bacterial growth compared to a B12-only control group) on a L. delbrueckii bacterial culture (the strength of the effect is indicated with colors; green: growth promoting; orange: medium inhibition of growth; red: strong inhibition of growth). The scheme does not indicate the course of chemical reactions. Charges omitted for clarity. | ||
Syntheses, isolation, and characterization of the two novel doubly-modified derivatives 4 and 7 and four singly-modified Cbls (2, 3, 5 and 6) are described in more detail below and in the Experimental section. In this section, we outline only the preparation and characterization of unprecedented compounds Coβ-phenyl-Cbl-c,8-lactam (4; Scheme 2 top) and 10-bromo-Coβ-phenylcobalamin (7; Scheme 2 bottom).
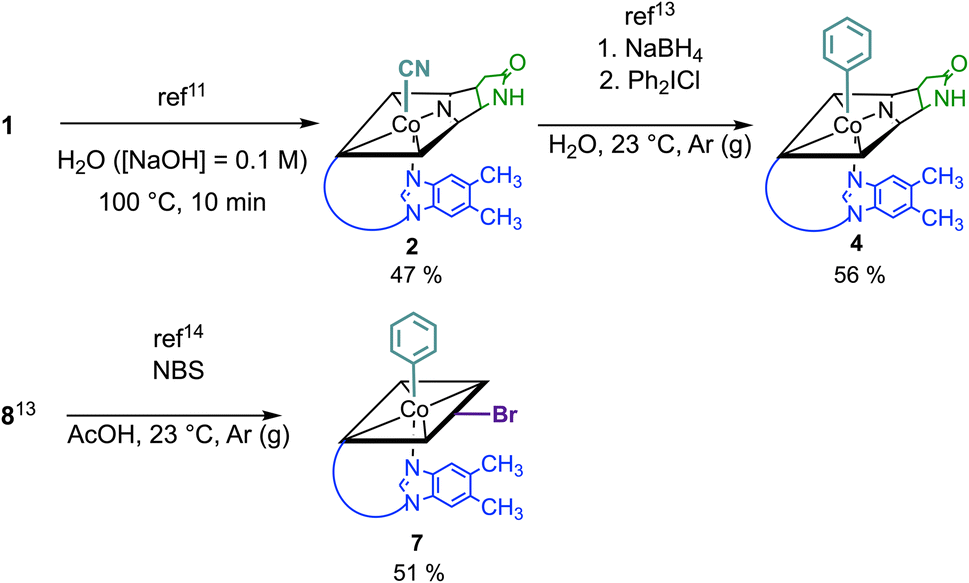 | ||
| Scheme 2 Top: synthesis of Coβ-phenyl-Cbl-c,8-lactam (4) from vitamin B12 (1) in a two-step procedure via Coβ-cyano-Cbl-c,8-lactam (2) applying selective and high-yielding chemical transformations in aqueous media. Bottom: synthesis of 10-bromo-Coβ-phenylcobalamin (7) starting from Coβ-phenylcobalamin (8). | ||
Vitamin B12 analogue 4 was synthesized in two steps starting from commercially available B12 in a total isolated yield of 27%. First, c,8-lactam 2 was prepared under basic conditions at 100 °C following a procedure of Todd et al.11 After reduction of its CoIII center with NaBH4 (10 equiv.) and subsequent treatment with diphenyl iodonium chloride (2 equiv.) in H2O,13 the organometallic target 4 was obtained. The occurrence of a pseudo molecular ion at m/z = 1404.56 ([M + H]+, m/zcalc: 1404.60 for C68H92CoN13O14P+) in the ESI-MS spectrum of 4 indicated successful arylation at the Co center of 2, supported by the observation of a hypsochromically shifted γ-band (Δλ = 18 nm) with diminished intensity (Δlog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε = 0.22) in the UV/vis spectrum. This spectral behavior is typical for organometallic Cbls featuring a CoIII–C bond (Fig. S13†).13 For the synthesis of 10-bromo-Coβ-phenylcobalamin (7; Scheme 2 bottom), we considered that Cbls bearing good leaving groups are prone towards reducing agents. Therefore, 10-bromo-Coβ-cyanocobalamin (5) is not compatible with arylation conditions using NaBH4.14 Having this in mind, we brominated the previously described Coβ-phenylcobalamin (8)13 with N-bromosuccinimide (NBS) at its C10 position according to a method of Wagner.14 ESI-MS analysis confirmed successful formation of 7 by the presence of its adduct ion peak at m/z = 1485.53 ([M + H]+) and the characteristic bromine isotopic pattern. Bromination at position C10 of 7 was further proven by the absence of the signal of the proton at C10 in the 1H-NMR spectrum and the absorption spectrum of 7 exhibited a characteristic redshift of the αβ-band to 537 nm.12
ε = 0.22) in the UV/vis spectrum. This spectral behavior is typical for organometallic Cbls featuring a CoIII–C bond (Fig. S13†).13 For the synthesis of 10-bromo-Coβ-phenylcobalamin (7; Scheme 2 bottom), we considered that Cbls bearing good leaving groups are prone towards reducing agents. Therefore, 10-bromo-Coβ-cyanocobalamin (5) is not compatible with arylation conditions using NaBH4.14 Having this in mind, we brominated the previously described Coβ-phenylcobalamin (8)13 with N-bromosuccinimide (NBS) at its C10 position according to a method of Wagner.14 ESI-MS analysis confirmed successful formation of 7 by the presence of its adduct ion peak at m/z = 1485.53 ([M + H]+) and the characteristic bromine isotopic pattern. Bromination at position C10 of 7 was further proven by the absence of the signal of the proton at C10 in the 1H-NMR spectrum and the absorption spectrum of 7 exhibited a characteristic redshift of the αβ-band to 537 nm.12
2.2 Bacterial growth assays
The inhibitory potential of the small library of four singly-, and two doubly modified B12 derivatives (2–7) was assessed with bacterial growth assays using L. delbrueckii.These Gram-positive bacteria are ideal for such proof-of-concept studies since they possess ribonucleotide reductase (RNR) as the only Cbl-dependent enzyme (see Fig. 2 for details).15–17 Small concentrations of B12 (0.1 nM) in the medium are sufficient to support growth of L. delbrueckii (positive control; Fig. 3).
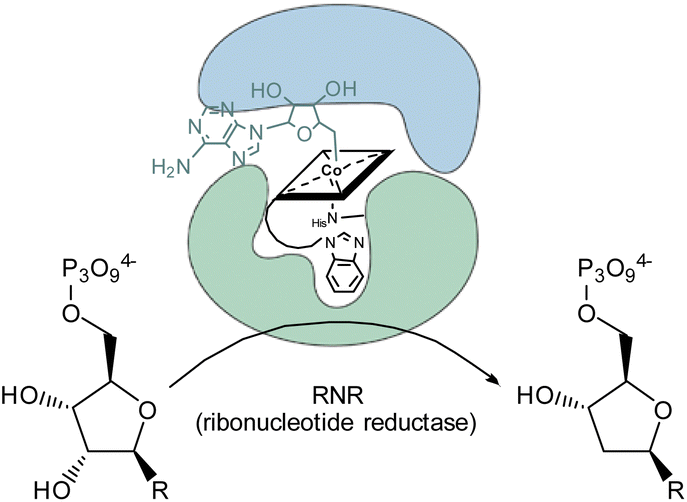 | ||
| Fig. 2 Schematic representation of the conversion of ribonucleotides to deoxyribonucleotides catalyzed by 5′-adenosylcobalamin-dependent ribonucleotide reductase in the metabolism of L. delbrueckii.15 | ||
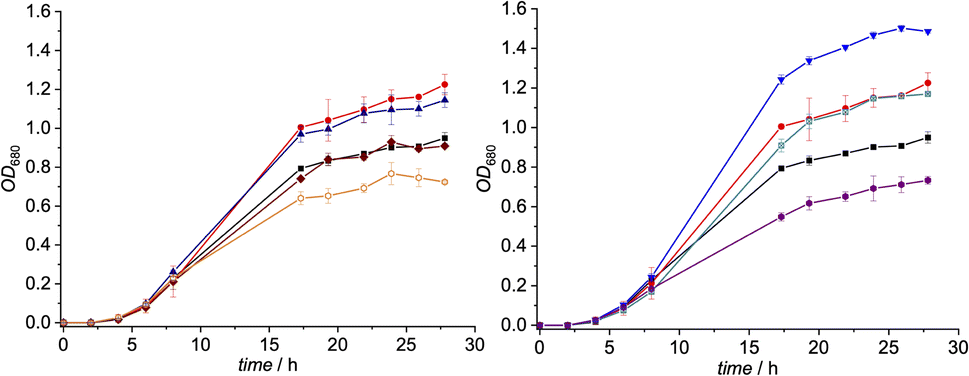 | ||
| Fig. 3 Left: growth of a suspension culture of L. delbrueckii in presence of derivatives 2 (brown rhombs), 3 (blue triangles) and 4 (orange hollow hexagons) (c = 10 nM) and B12 (1, c = 0.1 nM) vs. two control groups (positive control: red dots; containing only 1 (0.1 nM), negative control: black squares; containing only medium) at 37 °C (n = 3). Right: growth of a suspension culture of L. delbrueckii in presence of derivatives 5 (turquoise marked squares), 6 (inverse blue triangles) and 7 (violet hexagons) (c = 10 nM) and B12 (1, c = 0.1 nM) vs. two control groups (positive control: red dots; containing only 1 (0.1 nM), negative control: black squares; containing only medium) at 37 °C (n = 3). | ||
Notably, when exogenous B12 was absent in the assay medium (negative control), substantial residual growth (r.g.; 80 ± 1%) was still observed suggesting contamination of the B12-free medium with little amounts of Cbl.17–19 In competition assays with a 100-fold excess of analogs 2–7 (10 nM) over B12, c,8-lactam modified CNCbl 2 showed 22% inhibition (r.g. = 78 ± 1%; Fig. 3 left) of B12-triggered growth. Bi-functionalization of analog 2 with an additional organometallic β-phenyl ligand (i.e., analog 4) strengthened further the antimicrobial activity (r.g. = 66 ± 4%; Fig. 3 left). Inhibition in this competition assay was stronger than residual growth in the absence of exogeneous B12 (i.e., negative control) suggesting that total B12 in the medium was effectively outcompeted by the presence of analogue 4 (Fig. 3 left). In contrast to single modified c,8-lactam 2, single-modified 10-BrCNCbl (5) had no evident effects on B12-dependent growth (r.g. = 100 ± 1%; Fig. 3 right). This biological lethargy changed drastically upon further modification with a β-phenyl group at the CoIII center. Bi-functionalized 7 with a 10-Br modification was a comparably strong inhibitor (r.g. = 62 ± 3%) as bi-functionalized 4 with a c,8-lactam group (Fig. 3). The importance of the β-phenyl functionality at the CoIII center of inhibitors 4 and 7 for triggering the antimetabolic effect was further supported by studying the corresponding Coβ-aqua derivatives 3 and 6. These analogues lacking the organometallic phenyl ligand had either negligible (r.g. (3) = 96 ± 3%, Fig. 3-left) or even growth-promoting effects (r.g. (6) = 129 ± 1%, Fig. 3 right).
The latter result suggests, that aqua derivative 7 is still recognized, internalized, and metabolized by the microorganism to a growth promoting, enzymatically active organometallic AdoCbl cofactor (Fig. 3). In contrast, this reductive biological transformation is apparently not possible for 2, 4 and 7 containing σ-donating cyano or phenyl ligands in addition to a strucurally altered Cbl scaffold.20 Although we propose herein inhibition of RNR with inhibitors 2, 4 and 7 (Fig. 3), the actual biological target(s) and modes of action of these B12 analogues have still to be unraveled in future biological studies. So far, our proof-of-concept studies clearly demonstrate that the second, ligand-centered modification of the analogues with a β-phenyl ligand significantly steers antimetabolite activity. Replacement of this ligand with a weakly coordinating aqua ligand effected complete reversal of the antimetabolite activity of both bi-functionalized derivatives.
3. Conclusions
We have synthesized and tested antibacterial activity of four single- and two novel bi-functionalised B12 analogues. B12-dependent growth studies with L. delbrueckii showed strikingly that doubly modified Coβ-phenyl-cobalamin-c,8-lactam and 10-bromo-Coβ-phenylcobalamin were the most potent antagonists. Of note, inhibition was even stronger than residual growth in the absence of exogenous B12. Moreover, these studies demonstrated strikingly, that the second, β-axial modification significantly steers the metabolic effect. In particular, the incorporation of an organometallic β-phenyl ligand at the cobalt center either empowered the inhibitory potential of CNCbl-c,8-lactam or more interestingly, induced antimetabolic activity in an erstwhile innocent 10-brominated Cbl analogue.4. Experimental section
4.1 General
Chemicals were of reagent grade quality or better and obtained from Sigma-Aldrich, ACROS Organics, Merck or Fluka and used without further purification unless otherwise indicated. Vitamin B12 was obtained from Sigma-Aldrich or received as a generous gift from DSM Nutritional Products AG (Basel/Switzerland) and Prof. Em. Bernhard Jaun (ETH Zurich, Switzerland). All solvents were of reagent, analytical, HPLC or LC-MS grade, respectively, and obtained from commercial suppliers. Bi-distilled H2O was used in all reactions. H2O from a Milli-Q (Merck-Millipore) water purification system was used for UV/vis spectroscopy, mass spectrometry and when indicated. Reactions were carried out under N2 (g) or Ar (g) in oven-dried (100 °C) glass equipment and monitored for completion by analysing a small sample (after suitable workup) by LC-MS. Evaporation of the solvents in vacuo was done with the rotary evaporator (Büchi) at the given bath temperature and pressure. SepPak® RP-18 cartridges (Waters) were applied for solid phase extraction. The compounds were dissolved in H2O, transferred to the adsorbent, washed with H2O or the indicated aq. soln, followed by H2O, and eluted with CH3OH.4.2 Chromatography
000 resolution (full width half-maximum); automatic gain control (AGC) target of 3.00 × 106; maximum allowed ion transfer time (IT) 30 ms; mass calibration to <2 ppm accuracy with Pierce® ESI calibration solns (Thermo Fisher Scientific, Rockford, USA); lock masses: ubiquitous erucamide (m/z 338.34174, (+)-ESI) and palmitic acid (m/z 255.23295, (−)-ESI).4.3 Spectroscopy
ε) in nm. Both 1H- and 13C-NMR spectra were carried out at 298 K in D2O or CD3OD and at 500 MHz or 126 MHz, respectively. The 1H-NMR spectra were performed in an AVANCE NEO 500 MHz spectrometer (Bruker) using a 5 mm-z-gradient RT-BBI probehead; δ in ppm relative to HDO (δ 4.79; corresponds to TMS (δ 0.00)) or CHD2OD (δ 3.31; corresponds to TMS (δ 0.00)), J in Hz. Spectra in D2O were presaturated. The 13C-NMR spectra were performed in an AVANCE NEO 500 MHz spectrometer (Bruker) using a 5 mm z-gradient CP-BBO probehead; δ in ppm relative to CD3OD (δ 49.0; corresponds to TMS (δ 0.0)), J in Hz.4.4 Bacterial growth assays
Culture medium was prepared by dissolving MRS broth (Difco) for Lactobacilli (5.5 g) in Milli-Q H2O (100 mL) and subsequently filtered through a sterile 2.0 μm filter. The culture medium (14 mL) was inoculated with Lactobacillus delbrueckii subsp. Lactis, DSM 20355 from a micro-ring culture (previously stored at −70 °C). The closed tubes were incubated at 30 °C for 24 h. Afterwards a small sample (0.5 mL) was taken out and OD680 nm was determined and typically yielded values around 1.0 after 24 h. A second culture was inoculated by addition of the inoculate (200 μL) to fresh MRS broth (14 mL), followed by incubation at 30 °C for 24 h, resulting in OD680 nm = 1.5 prior to the conduction of the assay. The bacterial culture was centrifuged (5000 rpm/5 min), and the remaining pellet was suspended in H2O ([NaCl] = 0.9%, 14.0 mL) and incubated at 37 °C for 30 min. The resulting suspension was centrifuged again (5000 rpm/5 min), followed by two washing steps in H2O ([NaCl] = 0.9%, 14.0 mL) to remove remaining traces of the growth medium. Afterwards, H2O ([NaCl] = 0.9%, 5.0 mL) was added to the pellet and the bacterial suspension was stored at 37 °C. Vitamin B12 assay medium (Sigma-Aldrich, 41.5 g) was dissolved in Milli-Q H2O (500 mL) and the mixt. was heated to 40 °C under stirring until everything was dissolved, before Tween® 80 (1.0 mL) was added and everything was thoroughly homogenized. The pH was adjusted to 6.0 by addition of H2O ([NaOH] = 0.5 M) and the medium was filtered through a sterile 2.0 μm filter. Subsequently, sterilized bacterial assay glass tubes (Fisher Scientific, total volume: 7.0 mL) were filled with of B12 assay medium (6.5 mL), a sterile stock solution of vitamin B12 in Milli-Q H2O (6.5 μL, 1.0 nM), except for the negative control, and a sterile soln of the respective test compound in H2O (6.5 μL, 100 nM to 0.1 μM), except for the positive control. All tubes were finally inoculated with 40 μL of the bacterial suspension, tightly closed, and incubated at 37 °C for 28–60 h. OD680 nm was monitored photometrically every 2–8 h (after through mixing of the tubes), until saturation of growth was detected. All assays were performed in triplicates and average values of OD680 nm (±2σ) were obtained and plotted vs. time (t) in h (hours) to obtain growth curves. Residual growth values (r.g.) were estimated after 26 h and are given in% relative to the growth of the positive control group.4.5 Experimental procedures
UV-vis (H2O, c = 4.1 × 10−5 M): 279 (3.92), 308 (3.69), 321 (3.63), 360 (4.20), 518 (3.64), 549 (3.66). UPLC: tret = 1.70 min (method 2). ESI-MS (H2O/MeCN): m/z = 677.58 (100, [M + 2H]2+), 1353.55 (10, [M + H]+, m/zcalc: 1353.56 for C63H87CoN14O14P+).1H-NMR (D2O, c = 4.1 × 10−5 M): 7.27 (s, HC7N), 7.09 (s, HC2N), 6.45 (s, HC4N), 6.31 (d, J = 3.0, HC1R), 5.93 (s, HC10), 4.67 (d, J = 3.8, ribose-CHOH), 4.30–4.20 (m, 2 corrin-CH), 4.11 (d, J = 8.3, corrin-CH), 4.07–3.99 (m, 2 corrin-CH), 3.91–3.85 (d-like m, Ha of H2C5R), 3.70 (dd, J = 12.8, 3.8, Hb of H2C5R), 3.55 (d, J = 14.3, Ha of H2C175), 3.36–3.27 (m, HC13), 2.95–2.85 (m, Hb of H2C175, corrin-CH), 2.79–2.41 (m, 5 corrin-CH2) superimposed by 2.54 (s, H3C151) and 2.52 (s, H3C51), 3.40–2.31 (m, 5 corrin-CH2), 2.10–1.73 (m, 4 corrin-CH2) superimposed by 2.24 (s, dmbi-CH3), 2.22 (s, dmbi-CH3) and 1.84 (s, H3C7A), 1.42 (s, H3C12A), 1.37 (s, H3C2A), 1.34–1.23 (m, corrin-CH2) superimposed by 1.32 (s, H3C17B), 1.21 (d, J = 6.0, H3C177), 1.12 (s, H3C12B), 0.45 (s, H3C1A). Assignments were made in comparison with data from lit.21,22
UV/vis (H2O, c = 2.2 × 10−5 M): 276 (sh., 4.38), 290 (sh., 4.29), 349 (4.39), 404 (sh., 3.76), 495 (3.96), 523 (3.94). UPLC: tret = 1.30–1.60 min (method 2). ESI-MS (H2O/MeCN): m/z = 664.02 (100, [M–H2O + H]2+, m/zcalc: 663.78 for C62H87CoN13O14P2+).1H-NMR (CD3OD, c = 7.3 × 10−3 M): 7.87 (d, J = 3.7, OH), 7.22 (s, CH7N), 6.96 (s, HC2N), 6.59 (s, HC4N), 6.21 (d, J = 3.1, HC1R), 6.19 (s, HC10), 4.71–4.63 (m, ribose-CHOH), 4.36 (d, J = 8.2 Hz, corrin-CH), 4.19–4.15 (m, corrin-CH, ribose-CH), 4.13–4.09 (m, corrin-CH), 3.95 (dd, J = 12.7, 2.9, Ha of H2C5R), 3.85 (d, J = 9.5, Ha of corrin-CH2), 3.78 (dd, J = 12.7, 4.1, Hb of H2C5R), 3.75–3.69 (m, corrin-CH), 3.51 (d, J = 10.7, Hb of corrin-CH2), 3.36 (s, superimposed by CHD2OD signal), 3.11–3.07 (m, corrin-CH), 3.04 (d, J = 17.7, corrin-CH), 2.88 (d, J = 17.7, corrin-CH), 2.85–2.73 (m, corrin-CH2), 2.72 (s, corrin-CH3), 2.69–2.48 (m, 5 corrin-CH2) superimposed by 2.63 (2s, corrin-CH3, CH3COO), 2.45–2.37 (m, Ha/b of corrin-CH2), 2.33 (s, dmbi-CH3), 2.29 (s, dmbi-CH3), 2.23–2.10 (m, 3 corrin-CH2), 1.97 (s, corrin-CH3), 1.94–1.82 (m, corrin-CH2, Ha/b of corrin-CH2), 1.66 (d, J = 7.6, Ha/b of corrin-CH2), 1.58 (s, corrin-CH3), 1.53 (s, corrin-CH3), 1.51–1.41 (m, corrin-CH2), 1.38 (s, corrin-CH3), 1.34 (s, corrin-CH3), 1.29 (d, J = 6.4, H3C177), 0.53 (s, H3C1A). Assignments were made in comparison with data from lit.22
:1 ratio, corresponding to the isomeric forms of 4. The raw products were isolated using SPE. Subsequent purification via prep. HPLC (method A), followed by crystallization from aqueous acetone, delivered Coβ-phenylcobalamin-c,8-lactam (4, 14.5 mg, 10.3 μmol, 56%) as pale red crystals and, after precipitation from MeOH/ethyl acetate, its side product 4a (Scheme S3, ESI†) (1.85 mg, 1.30 μmol, 7%) as an orange powder. 4a (Scheme S3, ESI†) was tentatively assigned according to ref. 13 and not further characterized.UV-vis (H2O, c = 3.9 × 10−5 M): 283 (4.09), 342 (3.98), 370 (3.80), 470 (sh., 3.55), 517 (3.73). UPLC: tret = 2.45 min (method 1). HRMS (ESI+): m/z = 702.79905 (100, [C68H91O14N13CoP + 2H]2+, m/zcalc: 702.80150), m/z = 1404.59518 (50, [M + H]+, m/zcalc = 1404.59564). 1H-NMR (D2O, c = 1.2 × 10−2 M) δ 7.34 (s, HC2N), 7.26 (s, HC7N), 6.81 (d, J = 5.7 Hz, HC4L), 6.76 (t, J = 6.9 Hz, HC3L, HC5L), 6.66 (s, HC4N), 6.29 (d, J = 3.1 Hz, HCR1), 5.89 (s, HC10), 5.79 (d, J = 7.8 Hz, HC2L, HC6L), 4.70 (td, J = 8.4, 4.3 Hz, HC3R), 4.42–4.33 (m, HC176), 4.30 (t, J = 3.7 Hz, HC2R), 4.20 (d, J = 8.4 Hz, HC3), 4.18–4.13 (m, HC4R), 3.97 (dd, J = 12.9, 2.5 Hz, Ha of H2C5R), 3.78 (dd, J = 13.0, 4.2 Hz, Hb of H2C5R), 3.57 (dt, J = 14.0, 2.3 Hz, Ha of H2C175), 3.43 (d, J = 10.3 Hz, HC13), 3.36 (d, 10.0 Hz, HC19), 3.05–2.90 (m, Ha and Hb of H2C71, Hb of H2C175), 2.79 (s, H3C51), 2.76–2.74 (t-like, J = 5.9 Hz, HC18), 2.70 (s, H3C151), 2.68–2.61 (m, Ha and Hb of H2C132, Ha of H2C171), 2.58–2.47 (m, H2C32, Ha of H2C172), 2.44 (d, J = 5.9 Hz, H2C181), 2.34 (s, H3C10N), 2.31–2.23 (t-like, H3C11N, Hb of H2172), 2.19–1.99 (m, H2C82, Ha of H2C131, H2C31), 1.97–1.73 (m, Hb of H2C131, H2C21, Hb of H2C171) superimposed by 1.92 (s, H3C7A), 1.60–1.46 (m, Ha of H2C81), 1.42 (s, H3C12A), 1.29 (s, H3C2A), 1.24 (d, J = 6.3 Hz, H3C177), 1.18 (s, H3C17B), 1.16–1.09 (m, Hb of H2C81), 0.93 (s, H3C12B), 0.55 (s, H3C1A). 13C-NMR (D2O, c = 1.2 × 10−2 M) δ 178.2 (C133), 178.0 (C33), 177.6 (C11), 176.5 (C72), 176.3 (C16), 176.3 (C83), 176.1 (C22), 176.0 (C182), 175.7 (C4), 174.9 (C173), 168.1 (C9), 163.8 (C14), 162.3 (C6), 144.3 (C1L), 142.4 (C2N), 137.7 (C8N), 133.9 (C5N), 133.0 (C2L, C6L), 131.8 (C6N), 130.1 (C9N), 127.3 (C3L, C5L), 124.8 (C4L), 118.8 (C4N), 110.9 (C7N), 106.5 (C5), 105.9 (C15), 90.4 (C10), 86.8 (C1R), 85.6 (C1), 81.7 (C4R), 75.6 (C19), 74.8 (C8), 72.9 (C3R), 72.5 (C176), 68.7 (C2R), 60.3 (C5R), 59.0 (C17), 56.0 (C3), 53.2 (C13), 50.9 (C7), 47.6 (C12), 46.1 (C2), 45.2 (C175), 43.7 (C71), 41.5 (C21), 38.4 (C18), 35.2 (C32), 34.4 (C132), 32.8 (C171), 32.2 (C172), 31.7 (C181), 29.7 (C12B), 29.4 (C81), 29.0 (C82), 27.9 (C131), 25.2 (C31), 21.0 (C1A), 20.0 (C7A), 19.9 (C12A), 19.8 (C10N), 19.1 (C11N), 18.8 (C177), 17.1 (C17B), 16.8 (C51), 16.2 (C2A), 15.2 (C151). Assignments were made based on 2D NMR studies (DQF-COSY, HSQC, HMBC, NOESY) and comparison with data from lit.13
UV-vis (H2O, c = 4.9 × 10−5 M): 283 (3.69), 290 (3.71), 367 (4.13), 416 (sh., 3.15), 553 (3.51), 577 (3.55). UPLC: tret = 2.05 min (method 1). ESI-MS (H2O/MeOH): m/z = 718.21 (100, [M + 2H]2+), 1435.45 (11, [M + H]+, m/zcalc: 1435.48 for C63H88BrCoN14O14P+). 1H-NMR (D2O, c = 4.2 × 10−3 M): 7.27 (s, HC7N), 7.09 (s, HC2N), 6.48 (s, HC4N), 6.34 (d, J = 3.0, HC1R), 4.74–4.70 (m, ribose-CHOH), 3.37 (dd, J = 8.3, 6.8, ribose-CHOH), 4.31–4.24 (m, corrin CH, ribose-CHOH), 4.20 (d, J = 9.0, corrin-CH), 4.03 (t, J = 9.0, 2H), 3.93–3.87 (m, 1H), 3.73 (dd, J = 14.2, 3.4), 3.59 (d, J = 14.3 Hz, corrin-CH), 3.35 (d, J = 9.0, corrin-CH), 2.94 (dd, J = 14.3, 9.8, corrin-CH), 2.76–2.59 (m, 5 corrin-CH2), 2.58–2.52 (m, 2 corrin-CH2) superimposed by 2.57 (s, corrin-CH3) and 2.54 (s, corrin-CH3), 2.40–2.15 (m, 2 corrin-CH2), superimposed by 2.25 (s, corrin-CH3) and 2.23 (s, corrin-CH3), 2.11–1.75 (m, 3 corrin-CH2) superimposed by 1.89 (s, corrin-CH3) and 1.79 (s, corrin-CH3), 1.36 (s, corrin-CH3), 1.35 (s, corrin-CH3), 1.29 (s, corrin-CH3), 1.23 (d, J = 6.0, H3C177), 1.21–1.06 (m, corrin-CH2), 0.35 (s, corrin-CH3). Data is in agreement with lit.12
UV/vis (H2O, c = 1.1 × 10−5 M): 280 (4.2), 289 (4.2), 357 (4.4), 421 (3.6), 532 (3.9), 555 (3.9). UPLC: tret = 1.75 min (method 1). ESI-MS (H2O/MeCN): m/z = 704.81 (100, [M–H2O + 2H]2+, m/zcalc: 704.74 for C62H88BrCoN13O14P2+). 1H-NMR (D2O, c = 1.1 × 10−3 M): 7.08 (s, HC7N), 6.44 (s, HC2N), 6.35 (s, HC4N), 6.16–6.14 (d-like m, HC1R), 4.23–4.10 (m, HC19, HC176, HC2R, HC8), 3.91 (d, J = 8.3, HC4R), 3.79 (d, J = 12.0, Ha of H2C5R), 3.62 (d, J = 12.0, Hb of H2C5R), 3.56–3.46 (m, Ha of C175, HC13), 2.90–2.77 (m, Hb of H2C175, HC18), 2.71–2.38 (m, H2C181, H2C132 Ha of H2C171, H2C172) superimposed by 2.60 (s, H3C151) and 2.54 (s, H3C51), 2.36–2.27 (m, H2C32, Ha of H2C71), 2.22–1.69 (m, H2C21, H2C31, Ha of H2C81, Hb of H2C171, H2C172, H2C71) superimposed by 2.17 (s, H3C10N), 2.13 (s, H3C11N), 1.87 (s, H3C7A) and 1.75 (s, H3C12A), 1.42–1.10 (m, Hb of H2C81, H2C82) superimposed by 1.38 (s, H3C2A), 1.33 (s, H3C17B), 1.29 (s, H3C12B) and 1.15 (d, J = 6.0, H3C177), 0.31 (s, H3C1A). Chemical shifts were identical with those published earlier.20
UV-vis (H2O, c = 3.5 × 10−5 M): 284 (4.04), 348 (4.00), 380 (sh., 3.79), 471 (sh., 3.47), 537 (br., 3.67). UPLC: tret = 2.65 min (method 1). HRMS (ESI+): m/z = 742.76424 (100, [C68H92O14N13BrCoP + 2H]2+, m/zcalc: 742.76481), m/z = 1484.52168 (30, [M + H]+, m/zcalc = 1484.52125). 1H-NMR (CD3OD, c = 8.7 × 10−3 M): δ 8.30 (bs, HC2N), 7.41 (s, HC7N), 7.17 (s, HC4N), 6.74 (t, J = 7.0 Hz, HC4L), 6.60 (t, J = 7.6 Hz, HC3L–HC5L), 6.34 (d, J = 4.2 Hz, HC1R), 5.32 (d, J = 7.3 Hz, HC2L–HC6L), 4.62 (dd, J = 2.8, 7.5 Hz, HC3R), 4.53 (t-like, HC2R), 4.45–4.38 (m, HC4R), 4.38–4.30 (m, HC176), 3.88 (dd, J = 12.4, 3.1 Hz, Ha of H2C5R), 3.76 (dd, J = 12.4, 4.1 Hz, Hb of H2C5R), 3.57–3.46 (m, Ha of H2C175, HC13, HC19), 3.05 (dd, J = 13.9, 7.5 Hz, Hb of H2C175), 3.02–2.96 (m, HC18), 2.74 (s, H3C51), 2.70–2.66 (d, Ha of H2C71) superimposed by 2.67 (s, H3C151), 2.65–2.53 (m, Ha of H2C171, H2C132), 2.49–2.33 (m, H2C32, corrin-CH2, Ha of H2C172) superimposed by 2.40 (s, H3C10N) and 2.37 (s, H3C11N), 2.28–2.14 (m, Hb of H2C171, Ha of H2C82, Ha of corrin-CH2, Hb of H2C172), 2.10–2.00 (m, H2C31, Hb of H2C82), 1.95 (d, Ha of H2C21) superimposed by 1.96 (s, H3C7A), 1.90–1.87 (d, Hb of H2C21) superimposed by 1.87 (s, H3C12A), 1.79–1.67 (m, Hb of corrin), 1.64 (d, J = 14.1 Hz, Hb of H2C71), 1.35 (s, H3C2A), 1.23 (d, J = 6.2 Hz, H3C177), 1.18 (s, H3C17B), 1.14 (s, H3C12B), 0.90 (s, H3C1A). 13C-NMR (CD3OD, c = 8.7 × 10−3 M): δ 178.1, 177.9, 177.6, 177.5, 176.9, 175.9, 175.4, 174.9, 174.6, 173.0, 165.9, 164.0, 142.6 (C2N), 135.4, 134.1 (C2L, C6L), 131.8, 130.3, 128.2 (C3L, C5L), 126.3 (C4L), 118.0 (C4N), 112.9 (C7N), 111.2, 107.9, 94.2 (C10), 88.2, 87.6 (C1R), 85.7, 77.2 (C19), 75.8 (C3R), 74.5, 73.1, 72.3, 62.8, 60.9, 58.2, 57.8, 57.7, 57.5, 57.3, 57.1 (C3), 57.1 (C13), 52.2, 47.1, 46.5 (C175), 43.5 (C71), 42.4 (C21), 39.8 (C18), 36.4 (C32), 34.4, 34.3 (C132), 34.2 (C172), 33.4, 33.0 (C171), 28.9, 28.7 (C82), 27.8 (C12B), 26.9 (C31), 23.9 (C12A), 23.6 (C1A), 20.7 (C10N), 20.5 (C11N), 20.0 (C177), 19.8 (C7A), 19.1 (C17B), 17.3 (C51), 17.2 (C2A), 17.0 (C151). Assignments were made based on 2D NMR studies (HSQC and HMBC) and comparison with data of 4.
UV-vis (H2O, c = 6.4 × 10−5 M): 267 (3.95), 283 (3.93), 342 (3.83), 374 (3.69), 475 (sh., 3.42), 520 (br., 3.58). UPLC: tret = 2.52 min (method 1). ESI-MS (H2O/MeCN): m/z = 703.96 (100, [M + 2H]2+), 1406.63 (7, [M + H]+, m/zcalc: 1406.61 for C68H94CoN13O14P+). 1H-NMR (D2O, c = 3.2 × 10−5 M): 7.22 (s, HC2N), 7.16 (s, HC7N), 6.79 (t, J = 6.7, HC4L), 6.74 (t, J = 7.3, HC3L, HC5L), 6.59 (s, HC4N), 6.23 (d, J = 3.0, HC1R), 5.96 (s, HC10), 5.83 (d, J = 7.5, HC2L, HC6L), 4.69–4.64 (m, HC3R), 4.30 (d, J = 7.5, HC176), 4.24–4.21 (m, HC3, HC2R), 4.11–4.09 (m, HC4R), 3.92 (app. d, J = 11.3, Ha of H2C5R), 3.73 (dd, J = 12.8, 3.8, Hb of H2C5R), 3.52 (app. d, J = 14.3, Ha of H2C175), 3.41–3.32 (m, 3 corrin-CH), 2.96 (dd, J = 14.7, 8.7 Hz, Hb of H2C175), 2.73–2.56 (m, corrin-CH, 2 corrin-CH2) superimposed by 2.73 (s, H3C51) and 2.63 (s, H3C151), 2.49–2.40 (m, 2 corrin-CH2), 2.28 (s, H3C10N), 2.22 (s, H3C11N), 2.15–2.03 (m, 2 corrin-CH2), 1.96–1.93 (m, corrin-CH2), 1.89–1.63 (m, 4 corrin-CH2) superimposed by 1.85 (s, H3C7A), 1.40 (s, H3C12A), 1.24 (s, H3C2A), 1.21–1.09 (m, Hb of H2C82) superimposed by 1.19 (d, J = 6.8, H3C177) and 1.12 (s, H3C17B), 0.97 (d, J = 9.8, Hb of H2C81), 0.90 (s, H3C12B), 0.47 (s, H3C1A). Assignments were made in comparison with data from lit.13
Author contributions
F. Z. and C. B. designed the experiments and wrote the manuscript. C. B. and P. D. M. performed synthesis and characterization. C. B. executed the biological studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A generous gift of vitamin B12 from DSM Nutritional Products AG (Basel/Switzerland) is acknowledged. This work was, in part, financially supported by the UZH ‘Forschungskredit’ (grant-no. FK-17-088 to C. B. and FK-20-096 to P. D. M.).References
- F. Zelder, M. Sonnay and L. Prieto, ChemBioChem, 2015, 16, 1264–1278 CrossRef CAS PubMed.
- J. C. Somogyi and K. Trautner, in Vitamine, ed. R. Ammon, W. Dirscherl and G. Thieme, Stuttgart, 1974, 3, 10–138 Search PubMed.
- I. M. Kompis, K. Islam and R. L. Then, Chem. Rev., 2005, 105, 593–620 CrossRef CAS PubMed.
- G. Domagk, Angew. Chem., 1935, 48, 657–667 CrossRef CAS.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS PubMed.
- W. D. Zhou, E. W. Scocchera, D. L. Wright and A. C. Anderson, MedChemComm, 2013, 4, 908–915 RSC.
- F. Zelder and R. Alberto, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Elsevier Science, San Diego, 2012, vol. 25, 83–130 Search PubMed.
- F. Zelder, K. Zhou and M. Sonnay, Dalton Trans., 2013, 42, 854–862 RSC.
- S. P. Stabler, E. P. Brass, P. D. Marcell and R. H. Allen, J. Clin. Invest., 1991, 87, 1422–1430 CrossRef CAS PubMed.
- E. Mutti, M. Ruetz, H. Birn, B. Krautler and E. Nexo, PLoS One, 2013, 8, 1–9, DOI:10.1371/journal.pone.0075312.
- R. Bonnett, J. R. Cannon, V. M. Clark, A. W. Johnson, L. F. J. Parker, E. L. Smith and A. Todd, J. Chem. Soc., 1957, 1158–1168 RSC.
- L. Prieto, J. Rossier, K. Derszniak, J. Dybas, R. M. Oetterli, E. Kottelat, S. Chlopicki, F. Zelder and F. Zobi, Chem. Commun., 2017, 53, 6840–6843 RSC.
- C. Brenig, M. Ruetz, C. Kieninger, K. Wurst and B. Kräutler, Chem.–Eur. J., 2017, 23, 9726–9731 CrossRef CAS PubMed.
- F. Wagner, Proc. R. Soc. A, 1965, 288, 344–347 CAS.
- K. Zhou, R. Oetterli, H. Brandl, F. Lyatuu, W. Buckel and F. Zelder, ChemBioChem, 2012, 13, 2052–2055 CrossRef CAS PubMed.
- R. M. Oetterli, L. Prieto, B. Spingler and F. Zelder, Org. Lett., 2013, 15, 4630–4633 CrossRef CAS PubMed.
- J. A. Valu, Appl. Microbiol., 1965, 13, 486–490 CrossRef CAS PubMed.
- L. Prieto, M. Neuburger, B. Spingler and F. Zelder, Org. Lett., 2016, 18, 5292–5295 CrossRef CAS PubMed.
- K. Zelenka, H. Brandl, B. Spingler and F. Zelder, Dalton Trans., 2011, 40, 9665–9667 RSC.
- C. Brenig, L. Mosberger, K. Baumann, O. Blacque and F. Zelder, Helv. Chim. Acta, 2021, 104, 1–9, DOI:10.1002/hlca.202100067.
- K. Kurumaya and M. Kajiwara, Chem. Pharm. Bull., 1989, 37, 9–12 CrossRef CAS.
- Z. Rzepka, M. Respondek, J. Rok, A. Beberok, K. O. Proinsias, D. Gryko and D. Wrzesniok, Int. J. Mol. Sci., 2018, 19 DOI:10.3390/ijms19092845.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05748d |
| This journal is © The Royal Society of Chemistry 2022 |