
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous LaFeO3 perovskite catalysts synthesized by different methods and their high activities for CO oxidation
Xuehui Huang *,
Xuefang Wang,
Xinke Yang,
Penghui Deng,
Wenzhen Chen and
Xiangao Hu
*,
Xuefang Wang,
Xinke Yang,
Penghui Deng,
Wenzhen Chen and
Xiangao Hu
School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China. E-mail: huangxh@whut.edu.cn; Tel: +86-027-87651779 Tel: +86-13071258702
First published on 23rd November 2022
Abstract
In this study, spherical α-Fe2O3 prepared by the hydrothermal method was used as a template for the first time; LaFeO3 perovskite catalysts were successfully synthesized by the molten salt method (M-LF-T), sol–gel method (S-LF-T), and co-precipitation method (C-LF-T), respectively. To determine the optimal synthesis method, X-ray diffraction patterns were obtained and showed that single phase LaFeO3 with good crystallinity was prepared by the molten salt method after calcination at 600 °C for 4 h. SEM and TEM images showed that the M-LF-600 catalyst preserved the spherical structure of α-Fe2O3 template. Compared with the catalysts synthesized by the sol–gel method and co-precipitation method, the M-LF-600 catalyst had the highest BET surface area of 16.73 m2 g−1. X-ray photoelectron spectroscopy analysis showed that the M-LF-600 catalyst had the highest surface Fe3+/Fe2+ molar ratio and the best surface oxygen adsorption capacity. The CO oxidation of the LaFeO3 catalyst demonstrated that the M-LF-600 catalyst had the best catalytic performance.
1. Introduction
With the rapid development of the automobile industry, the problem of environmental pollution has become more and more serious. Among automobile exhaust pollutants, CO has attracted considerable attention due to the fact that it causes much harm to human health and the environment. CO is the most abundant and widely distributed pollutant gas produced by insufficient combustion of carbon or carbon-containing substances.1 CO mainly comes from industrial and traffic emissions, of which traffic emissions account for two-thirds of CO emissions.2,3 CO catalytic oxidation has become one of the environmental problems that need to be solved urgently due to the serious impact of CO emissions on human beings and even the ecological environment. Among the catalysts suitable for catalytic oxidation of CO, precious metals were initially thought to be a good choice. However, precious metals are expensive and have few reserves. Previous studies have found that perovskite catalysts with an ABO3 structure show great potential due to their high oxygen mobility properties, chemical and thermal stability at high temperatures.4Conversion of CO into CO2 in air requires catalyst, the temperature range of CO burning in the air to form CO2 varies with the catalyst. For perovskite catalysts, the A-site is usually an alkaline earth metal ion or a lanthanide ion,5,6 and the B-site is usually a cation with high oxidation reaction efficiency, such as CO, Mn and Fe.7 In numerous previous studies, La was selected as the A-site element, which can generate more oxygen vacancies with the cations of the B-site elements.8 The catalytic activity of the catalyst is related to many factors, such as pore volume, micromorphology, specific surface area, etc., which are usually related to the method of preparation.9 For ABO3-type perovskite oxides with La occupying the A-site, LaFeO3 catalysts have higher structural and thermal stability compared to LaMnO3 and LaCoO3 catalysts, and it is more suitable to play its role in poor working conditions. In addition, the preparation raw materials for the preparation of LaFeO3 catalysts have less environmental pollution and are cheaper and easier to obtain. LaFeO3 is widely used in various fields, including the production of solid oxide fuel cells,10 sensors,11 oxygen permeable membranes,12 and environmental catalysts.13,14 Therefore, the study of LaFeO3 perovskite catalysts is of great significance in terms of economy and environmental protection.
So far, there have been a lot of studies on improving the physicochemical properties and catalytic performance of catalysts. Gosavi et al.15 prepared pure phase LaFeO3 perovskite oxides by co-precipitation method and compared them with the oxides synthesized by low-temperature combustion method and sol–gel method. Improved surface area was observed for all three methods as compared to the previously reported values. Zhao et al.16 used pomelo peel as the biological template to prepare hierarchically porous LaFeO3 perovskites for the catalytic oxidation of NO to NO2, the maximum NO conversions for LaFeO3 prepared with and without template were 90% at 305 °C and 76% at 313 °C, respectively. Wang et al.9 prepared porous LaMnO3 spheres and cubes by molten salt method using Mn2O3 template at 550 °C, the two catalysts all exhibited high catalytic performance for the combustion of toluene. Vradman et al.17 synthesized LaMnO3 samples using LiCl–KCl or NaCl–KCl as the molten salt at 500–800 °C for 3 h, the synthesized samples had different structures. Mihai et al.18 successfully prepared cubic LaFeO3 perovskite nanoparticles with high surface area using carbon nanotubes (CNTs) as the hard template, they found that CNTs can stabilize not only the perovskite cubic structure, but also the size. Huang et al.19 obtained porous La0.8Sr0.2Mn0.8Cu0.2O3 microspheres in eutectic NaNO3–KNO3 at 550 °C using MnCO3 with different morphologies as the template. The La0.8Sr0.2Mn0.8Cu0.2O3 catalyst showed good catalytic activity (T50 = 83.63 °C, T90 = 97.40 °C) and stability. According to relevant studies, there were few reports on the preparation of LaFeO3 perovskite catalysts with specific morphologies by self-template method.
In recent years, the template method has been widely used because of its ability to synthesize perovskite-type oxides with large specific surface area and pore structures of different sizes.20 Ideally, the retained sample can inherit the morphology of the hard template after removing the template. However, this step is usually treated with acid or alkali solution, which may lead to the destruction or collapse of the morphology and structure, making it difficult for the sample to maintain the morphology of the hard template. At the same time, impurities may be left to affect its catalytic performance. Therefore, the template method is not conducive to large-scale preparation.
Li et al.21 prepared pure polycrystalline Fe3O4 phase by co-precipitation method. The strain and lattice distortion of magnetite decreases significantly with increasing average grain size. In this study, Fe2O3 provides the necessary conditions for catalysts due to its abundant surface active centers, large specific surface area, and high tunability of composition or structure. Therefore, we used spherical α-Fe2O3 prepared by hydrothermal method as the template for the first time, and LaFeO3 perovskite catalysts were synthesized by three different methods: molten salt method, sol–gel method, and co-precipitation method. The microstructure, CO catalytic performance, and high-temperature stability of the samples synthesized by three different methods were compared and explored.
2. Experimental
2.1 Chemicals
All chemical reagents in the experiment were analytical purity and used without any further purification. Ferric chloride hexahydrate (FeCl3·6H2O), citric acid monohydrate (C6H8O7·H2O), ammonia (NH3·H2O), polyvinylpyrrolidone (PVP), sodium nitrite (NaNO2), and absolute ethanol were supplied by Sinopharm Chemical Reagent Co. Ltd. Lanthanum nitrate hexahydrate (La(NO3)3·6H2O) was purchased from Shanghai Macklin Biochemical Co. Ltd.2.2 α-Fe2O3 template preparation
Spherical α-Fe2O3 was synthesized by hydrothermal method. At first, 1.2 g PVP and 1.08 g FeCl3·6H2O were added to 60 mL deionized water at room temperature. Then the mixture was stirred constantly for 10 min, the stirred solution was transferred to a 100 mL Teflon-lined autoclave, and the autoclave was placed in an electric thermostatic drying oven at 120 °C for 24 h. After the reaction, the samples were cooled to room temperature naturally. And the products were isolated by centrifugation, washed four times with absolute ethanol and deionized water. Finally, the samples were dried in an electric thermostatic drying oven at 60 °C for 24 h. The samples were denoted as SP-α-Fe2O3.2.3 Catalyst preparation
Using the synthesized SP-α-Fe2O3 microspheres as the template, the LaFeO3 samples were synthesized by three methods, namely molten salt method (M-LF-T), sol–gel method (S-LF-T), and co-precipitation method (C-LF-T), respectively. The detailed processes of preparing catalysts by three methods are as follows.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The above mixture was put into a 50 mL ceramic crucible and then heated to 600 °C, 700 °C and 800 °C respectively in a muffle furnace. To determine the optimal calcination temperature, the calcination time was 4 h, and the heating rate was 2 °C min−1. After the temperature in the oven reached room temperature, the mixture was dissolved in 50 mL deionized water and stirred at 50 °C for 2 h for subsequent isolation and purification. The residual molten salts in the products were removed by washing three times with deionized water and absolute ethanol. In the end, the samples were dried in an electric thermostatic drying oven at 60 °C for 8 h, thus obtaining the porous spherical LaFeO3 catalysts. The obtained samples were denoted as M-LF-T (T: calcination temperature).
:5. The above mixture was put into a 50 mL ceramic crucible and then heated to 600 °C, 700 °C and 800 °C respectively in a muffle furnace. To determine the optimal calcination temperature, the calcination time was 4 h, and the heating rate was 2 °C min−1. After the temperature in the oven reached room temperature, the mixture was dissolved in 50 mL deionized water and stirred at 50 °C for 2 h for subsequent isolation and purification. The residual molten salts in the products were removed by washing three times with deionized water and absolute ethanol. In the end, the samples were dried in an electric thermostatic drying oven at 60 °C for 8 h, thus obtaining the porous spherical LaFeO3 catalysts. The obtained samples were denoted as M-LF-T (T: calcination temperature).2.4 Material characterization
In this study, powder X-ray diffraction (XRD) was used for phase analysis of precursor and catalysts, XRD patterns were obtained by a D8 Advance X-ray diffractometer. The 2θ rotation range was 10° to 80°, and the scanning step was 0.02°. The microscopic morphologies of the samples were observed by field emission scanning electron microscope (FE-SEM) with a magnification of 30 to 800000 times. Since the samples in this experiment contained magnetism and had low conductivity, the samples were sprayed with metal before the test to enhance the conductivity. The sputtered metal was Pt SEM images were obtained by a HITACHI S4800 electron microscope (Japan). Transmission electron microscope (TEM) further confirmed the morphologies of the samples, TEM images were shown using a Tecnai G2 F20 S-TWIN (USA). The specific surface areas and pore size distributions of the samples were calculated by a Micromeritics ASAP 2460 M automatic specific surface area and porosity analyzer (USA) at −196 °C. The specific surface areas of the samples were determined by the Brumauer–Emmett–Teller (BET) model, and the porosity was characterized by the adsorption branches of the isotherms using the Barrett–Joyner–Holanda (BJH) model. Sample analyses were performed after outgassing in vacuo (residual pressure ∼10−2 mbar) for 5 h at 120 °C. ESCALAB 250 Xi X-ray photoelectron was used to detect the different valence states and relative contents of elements contained on the surface of the samples. XPS survey spectra were obtained by an ESCALAB 250 Xi system (Thermo, China) at room temperature under a 10−7 Pa (10−9 torr) vacuum. The excitation source was Al Kα ray (hν = 1486.6 eV), and the vacuum degree was 10−7 Pa. Calibration with Eb = 284.8 eV of C 1s was used as standard.
2.5 CO catalytic oxidation evaluation
100 mg catalyst was evenly distributed on 500 mg quartz wool and wetted with ethanol to make the sample adhere. And then the catalyst-loaded quartz wool was placed in a drying oven at 60 °C and taken out after the evaporation of absolute ethanol. Quartz wool was used to carry the catalyst, and the quartz wool was put into the quartz tube (i.d. = 12 mm) with tweezers, so that the sample was in the center of the quartz tube. In order to prevent the flow of the catalyst from flushing the catalyst into the gas chromatograph and blocking the flow valve, clean quartz wool can be used to fix the catalyst in the center of the quartz tube. Finally, the quartz tube containing the catalyst was placed in the electric furnace. The electric furnace was connected with a thermocouple, and its temperature was adjusted by a temperature controller. In this study, the temperature range of the catalytic experiments was from room temperature to 350 °C, and the experimental data were collected at intervals of 25 °C. The concentration of CO at the outlet of the reactor as a function of temperature was measured by a gas chromatograph. The molar composition of the feed was CO/O2/Ar (balance) = 1/1.25/97.75 and the total flow rate of the gas mixture was 438 mL min−1, giving a space velocity (GHSV) of 12 000 h−1. The system outlet was connected to a gas chromatograph (GC-7890 II, China) equipped with FID and TCD double detectors to separate reactants and samples.3. Results and discussions
For the purpose of determining the optimal synthesis method and calcination temperature, and analyzing their influence on the crystallinity and phase transition of the catalysts. X-ray diffraction patterns of the spherical α-Fe2O3 template prepared by hydrothermal method and LaFeO3 catalysts prepared by three different methods are depicted in Fig. 1. It can be seen from Fig. 1a that eight diffraction peaks of the as-synthesized α-Fe2O3 were consistent with rhombohedral Fe2O3 (JCPDS no. 33-0664, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c). In addition, the half peak width of the XRD diffraction peak was narrow and the peak type was sharp, showing that hydrothermal method can obtain high-phase-purity α-Fe2O3. Furthermore, Fig. 1b–d clearly show that the samples prepared by three different methods using spherical α-Fe2O3 as the template corresponding to orthorhombic LaFeO3 (JCPDS no. 75-0541, Pmm). S-LF-T (T = 700 °C, 800 °C) and C-LF-T (T = 700 °C, 800 °C) perovskite catalysts prepared by sol–gel method (Fig. 1c) and co-precipitation method (Fig. 1d) contained trace monoclinic La2O3 phase (JCPDS no. 74-2430, P63/mmc). The oxide phase in the samples possibly owing to the formation of hydrated La2O3 during stirring, because hydrated La2O3 may be formed even at room temperature.22 In contrast, there were no additional diffraction peaks in the M-LF-T (T = 600 °C, 700 °C, 800 °C) catalyst synthesized by molten salt method (Fig. 1b). From the above results, it can be seen that molten salt method can obtain high-phase-purity LaFeO3. However, it was difficult to synthesize pure and single-phase LaFeO3 catalyst by sol–gel method and co-precipitation method under the same conditions. It can also be seen that the calcination temperature can be effectively reduced by using molten salt method, which was related to the fact that a eutectic reaction environment can be provided by the molten salt, so as to improve ion mobility and promote the reaction process. Due to the fact that no peaks consistent with the LaFeO3 phase were shown on the XRD patterns of the S-LF-600 and C-LF-600 catalysts, the above two samples were not considered in the subsequent experiments.
c). In addition, the half peak width of the XRD diffraction peak was narrow and the peak type was sharp, showing that hydrothermal method can obtain high-phase-purity α-Fe2O3. Furthermore, Fig. 1b–d clearly show that the samples prepared by three different methods using spherical α-Fe2O3 as the template corresponding to orthorhombic LaFeO3 (JCPDS no. 75-0541, Pmm). S-LF-T (T = 700 °C, 800 °C) and C-LF-T (T = 700 °C, 800 °C) perovskite catalysts prepared by sol–gel method (Fig. 1c) and co-precipitation method (Fig. 1d) contained trace monoclinic La2O3 phase (JCPDS no. 74-2430, P63/mmc). The oxide phase in the samples possibly owing to the formation of hydrated La2O3 during stirring, because hydrated La2O3 may be formed even at room temperature.22 In contrast, there were no additional diffraction peaks in the M-LF-T (T = 600 °C, 700 °C, 800 °C) catalyst synthesized by molten salt method (Fig. 1b). From the above results, it can be seen that molten salt method can obtain high-phase-purity LaFeO3. However, it was difficult to synthesize pure and single-phase LaFeO3 catalyst by sol–gel method and co-precipitation method under the same conditions. It can also be seen that the calcination temperature can be effectively reduced by using molten salt method, which was related to the fact that a eutectic reaction environment can be provided by the molten salt, so as to improve ion mobility and promote the reaction process. Due to the fact that no peaks consistent with the LaFeO3 phase were shown on the XRD patterns of the S-LF-600 and C-LF-600 catalysts, the above two samples were not considered in the subsequent experiments.
 | ||
| Fig. 1 XRD patterns of samples: (a) α-Fe2O3 template; (b) LaFeO3 samples synthesized by molten salt method at 600–800 °C; (c) LaFeO3 samples synthesized by sol–gel method at 600–800 °C; (d) LaFeO3 samples synthesized by co-precipitation method at 600–800 °C. | ||
Furthermore, it can be found that the intensity of the XRD diffraction peak became higher as the calcination temperature increased. It indicated that the higher the synthesis temperature, the better the crystallinity of the catalyst, but the higher the synthesis temperature, the less conducive to the improvement of the specific surface area and the maintenance of the original microstructure of the catalyst, which can be further demonstrated in subsequent tests. Therefore, various factors should be integrated to determine the optimal temperature parameters of the sample.
Fig. 2 shows SEM images of the α-Fe2O3 template, and LaFeO3 perovskite catalysts synthesized by molten salt method, sol–gel method, and co-precipitation method. As shown in Fig. 2a, the α-Fe2O3 precursor prepared by hydrothermal method presented a complete and rough spherical structure. The spheres were evenly distributed with a diameter of about 800 nm, and there were many closely packed nanoparticles on the surface of α-Fe2O3 spheres. Besides, there was no obvious agglomeration between the spheres. In Fig. 2b, a large number of complete and compact spherical structures can be observed in the M-LF-600 catalyst. There were lots of irregular pores between the spheres. It indicated that many pore structures were formed in the catalyst after the in situ reaction, thereby increasing the specific surface area of the catalyst. And the formation of pore structures also provided conditions for the retention of active sites and the increase of gas adsorption, thereby directly enhancing the catalytic performance of the catalyst.
 | ||
| Fig. 2 SEM images of (a) α-Fe2O3, (b) M-LF-600, (c) S-LF-700, and (d) C-LF-700. | ||
As shown in Fig. 2c, there was almost no spherical structure observed in the S-LF-700 sample synthesized by sol–gel method, probably because the preparation process of the sol–gel method required the mixture containing the spherical template to be continuously stirred at 80 °C for a long time, which may lead to the collapse of the original spherical structure. Similarly, it can be seen from Fig. 2d that the C-LF-700 catalyst synthesized by co-precipitation method was composed of many irregular particles stacked on each other. It may be due to the high synthesis temperature of the catalyst, leading to the decomposition of a good deal of particles at high temperature.
To sum up, LaFeO3 catalysts can be synthesized by molten salt method, sol–gel method, and co-precipitation method using spherical α-Fe2O3 as the template. Compared to the other two samples, the catalyst synthesized by molten salt method at 600 °C had good thermal stability, thus retaining the spherical morphology of the template.
As shown in Fig. 3, the TEM images further proved that the α-Fe2O3 template and the M-LF-600 perovskite catalyst exhibited porous spherical morphology. The size of α-Fe2O3 template and M-LF-600 catalyst were both in the micrometer scale. It can be seen from Fig. 3a-1 that α-Fe2O3 microspheres had a uniform morphology, while the spherical structure of the catalyst in Fig. 3b-1 was surrounded by abundant irregular particles, which may be related to the disintegration of the structure in the high-temperature calcination environment. In addition, it can be seen from Fig. 3a-2 that the lattice spacing of α-Fe2O3 sample was calculated to be 0.270 nm, which was in complete consistency with the (104) plane of rhodochrosite Fe2O3 (0.270 nm; JCPDS no. 33-0664, Rc). As shown in Fig. 3b-2, the lattice spacing of M-LF-600 sample was measured as 0.230 nm, which was almost identical to the (111) plane of orthorhombic LaFeO3 (0.226 nm; JCPDS no. 75-0541, Pmm). The above results corresponded to the XRD results.
 | ||
| Fig. 3 TEM images of (a-1 and a-2) α-Fe2O3, and (b-1 and b-2) M-LF-600. | ||
The nitrogen adsorption–desorption isotherms and related pore size distribution curves of the samples prepared by three different methods are shown in Fig. 4. By measuring these curves, the BET specific surface area and pore structure can be determined. As can be seen from Fig. 4a, according to IUPAC classification, all three catalysts displayed a type IV isotherm with H3 hysteresis loops in the range of relative pressure P/P0 of 0.6–1.0,23 indicating the existence of the mesoporous structures. It can also be seen that the M-LF-600 catalyst had the highest nitrogen adsorption quantity, followed by the S-LF-700 and C-LF-700 catalysts. It suggested the M-LF-600 catalyst had the most pore structure and thus had the largest specific surface area and pore volume, which can also be demonstrated with the data in Table 1. In addition, as shown in Fig. 4b, the hysteresis loops of the three catalysts had different relative pressure ranges, indicating that there existed differences in the pore-size distributions. C-LF-700 sample showed a spike at about 2.6 nm while M-LF-600 and S-LF-700 samples showed a spike at about 3.9 nm, which may be formed by aggregated nanocrystals. And there was a wide weak peak in the range of 5–60 nm, which may be due to the disintegration of the spherical structure and the reduction of the pore structure. It showed that the three catalysts were composed of micropores, mesopores, and a small quantity of macropores, which can be consistent with the results of SEM and TEM tests. In summary, the three catalysts had different average pore diameters and pore volumes, resulting in different specific surface areas. Compared with the S-LF-700 and C-LF-700 catalysts, the M-LF-600 catalyst possessed the largest specific surface area and pore volume. These factors were favorable for the attachment of the active sites and possibly enhance the catalytic performance of the catalyst.
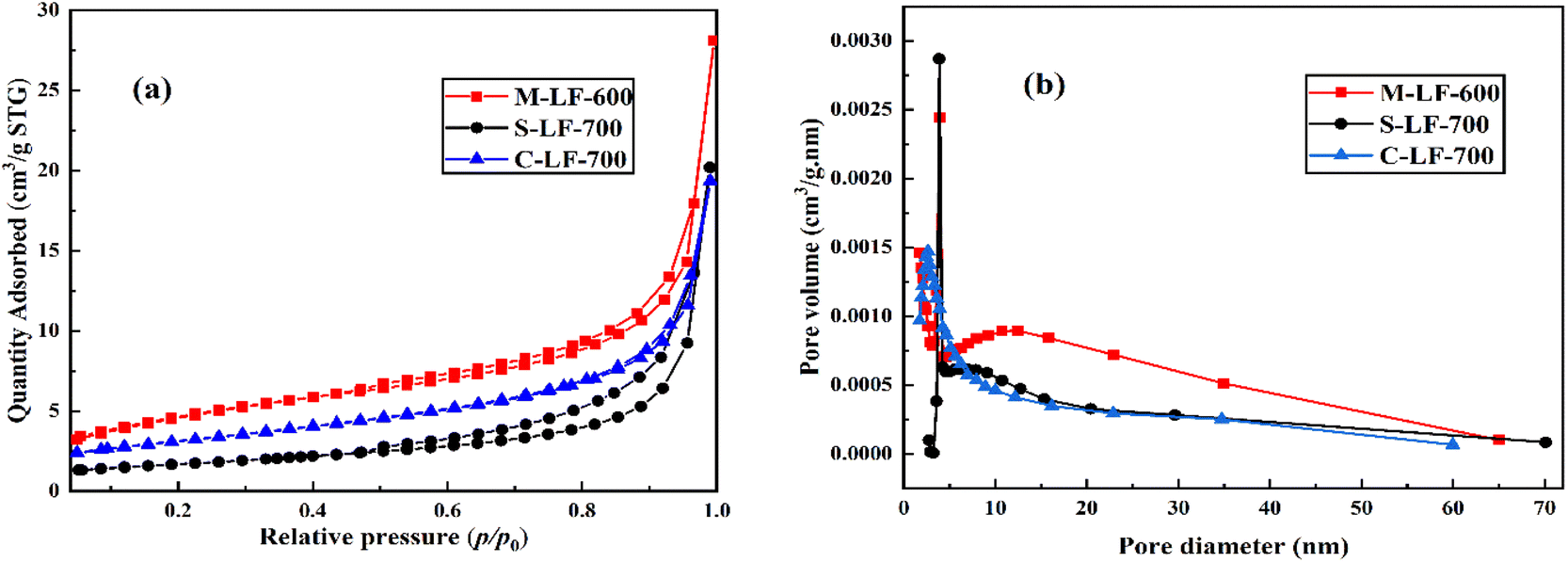 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution curves of samples. | ||
| Sample | Specific surface area (m2 g−1) | Average pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| M-LF-600 | 16.726 | 13.367 | 0.054 |
| S-LF-700 | 5.968 | 20.932 | 0.031 |
| C-LF-700 | 11.011 | 10.874 | 0.030 |
The XPS spectrum of the three samples (M-LF-600, S-LF-700, C-LF-700) are shown in Fig. 5 to analyze the chemical valence states and surface compositions. The binding energies of Fe 2p and O 1s of the three samples were listed in Table 2. In Fig. 5a, all three catalysts contained peaks corresponding to La, Fe, and O elements. In addition, Fe 2p possessed two binding energies in Fig. 5b, which were about 722.6 eV (2p1/2) and 708.9 eV (2p3/2).24 The curve has been further peak-fitted, and the Fe 2p3/2 XPS peak can be divided into two parts at around 709.934 and 708.461 eV. The former corresponded to the surface Fe3+ species, while the latter corresponded to the surface Fe2+ species,25 indicating that Fe2+ and Fe3+ ions were contained in all three samples. Similarly, as shown in Fig. 5c, the O 1s XPS peak can be divided into 3 parts at about 527.473 eV, 529.318 eV, and 530.694 eV, which corresponded to surface lattice oxygen (Olat), adsorbed oxygen (Oads), and hydration of oxygen (Oβ).26–29 The relative contents of Fe2+ and Fe3+ as well as the relative contents of adsorbed oxygen (Oads) and lattice oxygen (Olat) are important factors affecting the catalytic performance of CO. It is generally believed that the higher the molar ratio of Fe3+/Fe2+, the better the redox cycle capacity of Fe ions and the catalytic activity of the catalyst. In addition, for perovskite oxides, the oxidation reaction is mainly related to the adsorbed oxygen species, and the concentration of oxygen vacancies is related to the amount of oxygen adsorbed on the surface. The catalytic activity of the catalyst increases with the concentration of oxygen vacancies.30,31 As shown in Table 2, since the M-LF-600 catalyst contained the highest surface Fe3+/Fe2+ molar ratio and Oads/Olat molar ratio, it may have the highest CO catalytic activity.
 | ||
| Fig. 5 (a) XPS survey spectra, (b) Fe 2p, and (c) O 1s XPS spectra of LaFeO3 samples. | ||
| Sample | Fe 2p | O 1s | Oads/Olat | Fe3+/Fe2+ | |||
|---|---|---|---|---|---|---|---|
| Fe | Eb/eV | Area | Eba/eV | Area | |||
| a Oβ: hydration of oxygen; Oads: adsorbed oxygen; Olat: surface lattice oxygen. | |||||||
| M-LF-600 | 2p1/2 | 722.531 | 9415.290 | 530.694(Oβ) | 13165.730 | 2.78 | 0.99 |
| 2p3/2 | 709.934 | 9036.730 | 529.318(Oads) | 32786.670 | |||
| 2p3/2 | 708.461 | 9107.620 | 527.473(Olat) | 15010.930 | |||
| 2p1/2 | 722.683 | 4180.640 | 531.072(Oβ) | 8198.380 | |||
| S-LF-700 | 2p3/2 | 710.145 | 5510.360 | 529.935(Oads) | 34825.180 | 2.56 | 0.96 |
| 2p3/2 | 708.702 | 5737.900 | 527.860(Olat) | 12918.330 | |||
| 2p1/2 | 722.723 | 8894.070 | 531.262(Oβ) | 2862.070 | |||
| C-LF-700 | 2p3/2 | 710.477 | 7702.350 | 529.988(Oads) | 45175.380 | 2.45 | 0.75 |
| 2p3/2 | 708.736 | 10211.490 | 528.014(Olat) | 16441.550 | |||
The catalytic performance and high-temperature stability of the LaFeO3 samples prepared by three different methods in CO oxidation are shown in Fig. 6. The temperature range of the test was 50 °C–250 °C, and the temperature range for collecting data was 25 °C. In Fig. 6a, the CO catalytic activity of the three samples all improved significantly with the increase of experimental temperature. And the sequence of CO catalytic performance of the fresh LaFeO3 samples was M-LF-600 > S-LF-700 > C-LF-700 > S-LF-800 > M-LF-700 > C-LF-800 > M-LF-800. The results showed that the fresh LaFeO3 sample synthesized by molten salt method at 600 °C possessed the best catalytic performance, followed by the samples synthesized by sol–gel method and co-precipitation method at 700 °C. And it also indicated that under the same preparation method, the catalytic activity of the LaFeO3 catalyst decreased with the increase of the synthesis temperature. On the one hand, increasing the calcination temperature can increase the activation temperature of the sample, thereby increasing the activation energy required for the catalytic reaction and reducing the catalytic efficiency. On the other hand, the increase of the temperature accelerated the ion migration rate in the reaction, resulting in excessive crystal growth. It inhibited the formation of pore structure and even led to the structure collapse, thereby reducing the specific surface area of the sample. At the same time, the increase of the temperature also led to the formation of impurity phase without catalytic properties, which directly reduced the catalytic performance of the sample. From the previous specific surface area test results, it can be seen that the M-LF-600 catalyst had the largest specific surface area and pore volume, followed by S-LF-700 and C-LF-700 catalysts. In general, increasing the specific surface area is conducive to promoting the attachment of active sites, so that the space for oxidation reactions can be enlarged. These conditions are conducive to improving the catalytic performance of the catalysts. However, the specific surface area is only one of the factors affecting the catalytic activity rather than a decisive factor. From the previous XPS analysis, it can be seen that the Fe3+/Fe2+ surface molar ratio (1.06) and Oads/Olat molar ratio (0.75) of the M-LF-600 catalyst were significantly larger than those of the S-LF-700 and C-LF-700 catalysts. The catalytic activity of perovskite catalyst is affected by the active center and specific surface area. If the active center is blocked and the specific surface area decreases, the perovskite catalyst may be deactivated.32 In conclusion, the M-LF-600 catalyst had the largest specific surface area and the most active sites, which was directly related to improving the catalytic activity of the M-LF-600 catalyst. Furthermore, the high-temperature catalytic stability of the catalyst is also an important index for practical application and evaluation. Therefore, on-stream reaction experiments were performed on LaFeO3 catalysts at 600 °C for 14 h to study the catalytic stability. The results shown in Fig. 6b indicated that the catalytic performance of M-LF-600, S-LF-700, and C-LF-700 catalysts was all higher than 99.95%, and CO was almost completely converted into CO2. Among them, CO oxidation reaction of the M-LF-600 catalyst was the most stable at 600 °C, which was hardly affected by time and temperature changes in the experimental process. M-LF-600 sample showed a weak fluctuation, with changes of about 0.0083% detected over 14 h at 600 °C. This indicated that compared with the S-LF-700 and C-LF-700 catalysts, the M-LF-600 catalyst synthesized by molten salt method possessed the best high-temperature catalytic stability in CO oxidation. From the previous scanning results, more spherical structures could be observed in the catalyst prepared by molten salt method, and the spherical structure was a relatively stable structure. The three catalysts contained different spherical structures, which may lead to different catalytic stabilities. In real life, catalysts are reused. The catalytic activity results of CO oxidation after three-time cycling tests on M-LF-600, S-LF-700, and C-LF-700 catalysts are shown in Fig. 6c and d. Compared with the first activity test, the M-LF-600, S-LF-700, and C-LF-700 samples all showed higher catalytic activities in subsequent catalytic cycles. This was similar to the results observed in the work of Huang et al.33 In addition, the M-LF-600 catalyst still maintained superior stability regardless of cycling times. In contrast, although the catalytic effect of S-LF-700 and C-LF-700 catalysts was better than the first time, their catalytic activities were still lower than that of fresh M-LF-600 catalyst, which may be due to the disintegration of the non-spherical structures synthesized by sol–gel method and co-precipitation method, resulting in the reduction of specific surface area and the number of active sites. These behaviors were consistent with the results of SEM, TEM, BET, and XPS analysis.
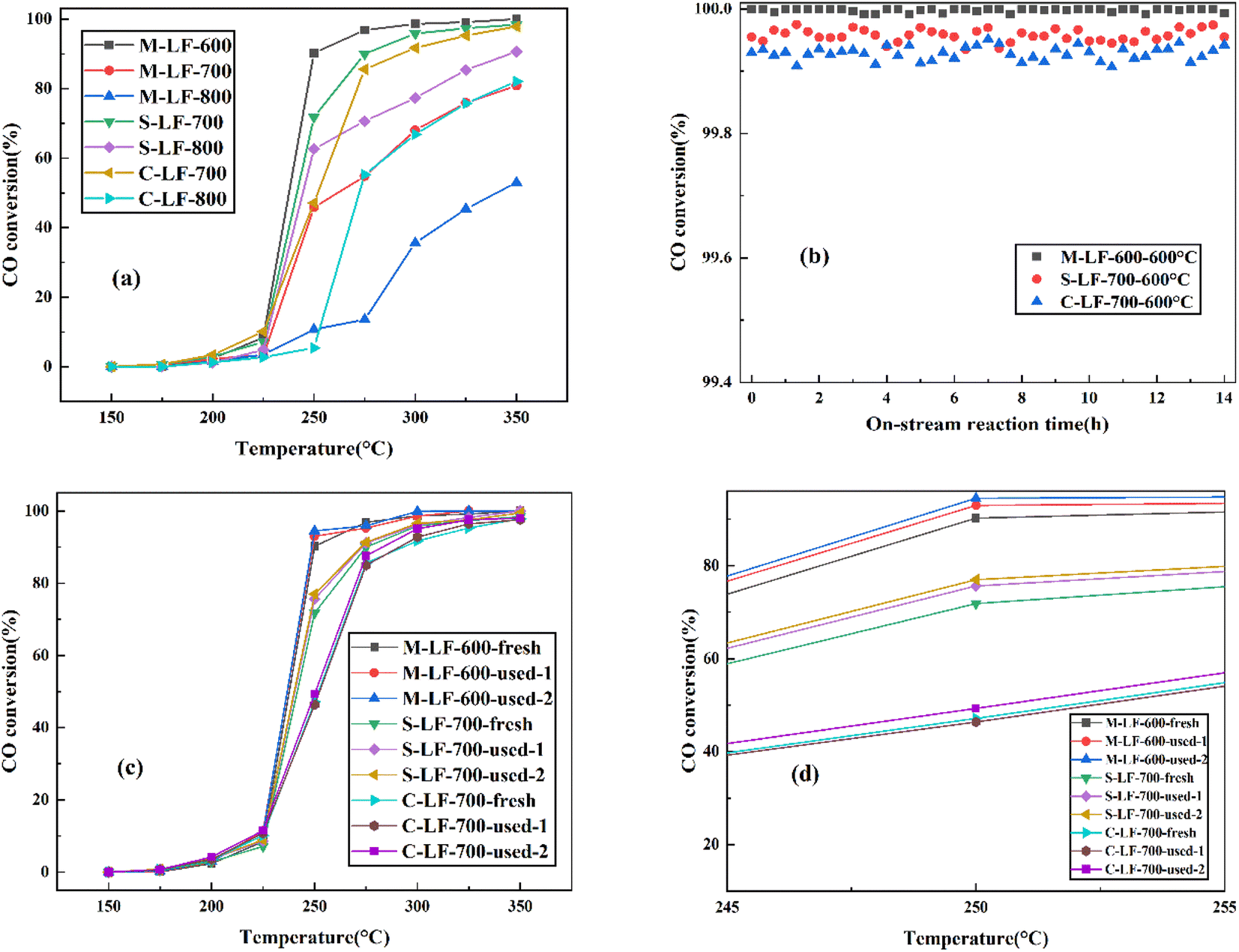 | ||
| Fig. 6 (a) Catalytic performance of all fresh catalysts in CO oxidation. Catalytic performance is given as a function of on-stream reaction time over LaFeO3 catalysts over 14 h at (b) 600 °C after 14 h on-stream reaction at the 100% conversion temperature. (c) Catalytic performance in CO oxidation of fresh and used LaFeO3 catalysts. (d) Magnified view of (c) between 245 and 255 °C. | ||
4. Conclusion
In conclusion, porous LaFeO3 catalysts with high crystallinity, large specific surface area, excellent catalytic performance in CO oxidation were successfully synthesized by molten salt method, sol–gel method, and co-precipitation method, using spherical α-Fe2O3 prepared by hydrothermal method as the template for the first time. Compared to the samples synthesized by sol–gel method and co-precipitation method, the porous LaFeO3 (M-LF-600) synthesized by molten salt method had the largest specific surface area and the highest Fe3+/Fe2+ surface molar ratio and Oads/Olat molar ratio, showed the best catalytic performance and high-temperature stability in CO oxidation. Based on these results, the molten salt method can be considered as a more promising method for the preparation of LaFeO3 catalysts.Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- L. Pahalagedara, D. A. Kriz, N. Wasalathanthri, C. Weerakkody, Y. Meng, S. Dissanayake, M. Pahalagedara, Z. Luo, S. L. Suib, P. Nandi and R. J. Meyer, Appl. Catal., B, 2017, 204, 411–420 CrossRef CAS.
- S. Dey, G. C. Dhal, D. Mohan and R. Prasad, Mater. Discovery, 2017, 8, 18–25 CrossRef.
- S. Dey, G. C. Dhal, D. Mohan and R. Prasad, Mater. Discovery, 2017, 8, 26–34 CrossRef.
- A. Tarjomannejad, A. Farzi, M. J. I. Gómez, A. Niaei, D. Salari and V. Albaladejo-Fuentes, Catal. Lett., 2016, 146, 2330–2340 CrossRef CAS.
- X. Wei, P. Hug, R. Figi, M. Trottmann, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2010, 94, 27–37 CrossRef CAS.
- C. Jin, X. Cao, L. Zhang, C. Zhang and R. Yang, J. Power Sources, 2013, 241, 225–230 CrossRef CAS.
- Y. Ding, S. Wang, L. Zhang, Z. Chen, M. Wang and S. Wang, Catal. Commun., 2017, 97, 88–92 CrossRef CAS.
- L. Wang, S. Fang, N. Feng, H. Wan and G. Guan, Chem. Eng. J., 2016, 293, 68–74 CrossRef CAS.
- Y. Wang, S. Xie, J. Deng, S. Deng, H. Wang, H. Yan and H. Dai, ACS Appl. Mater. Interfaces, 2014, 6, 17394–17401 CrossRef CAS PubMed.
- X. Chen, M. Zhang, H. Qin, J. Zhou, Q. Shen, K. Wang, W. Chen, M. Liu and N. Li, Sep. Purif. Technol., 2022, 280, 119751 CrossRef CAS.
- F. Li, S. Wang, Z. Wu, X. Xiong, J. Li, J. Zhou and X. Gao, J. Mater. Sci.: Mater. Electron., 2021, 32, 27587–27595 CrossRef CAS.
- X. Zhu, C. Shi, K. Li, K. Zhai, H. Wang, Y. Wei, D. Tian and C. Zeng, Int. J. Hydrogen Energy, 2017, 42, 19776–19787 CrossRef CAS.
- L. T. T. Nguyen, G. E. J. Poinern, H. T. Le, T. A. Nguyen, C. M. Tran and Z. Jiang, Asia-Pac. J. Chem. Eng., 2021, 16, 5 Search PubMed.
- H. Ren, Z. Wang, X. Chen, Z. Jing, Z. Qu and L. Huang, Chemosphere, 2021, 285, 131473 CrossRef CAS PubMed.
- P. V. Gosavi and R. B. Biniwale, Mater. Chem. Phys., 2010, 119, 324–329 CrossRef CAS.
- S. Zhao, L. Wang, Y. Wang and X. Li, J. Phys. Chem. Solids, 2018, 116, 43–49 CrossRef CAS.
- L. Vradman, J. Zana, A. Kirschner and M. Herskowitz, Phys. Chem. Chem. Phys., 2013, 15, 10914–10920 RSC.
- O. Mihai, S. Raaen, D. Chen and A. Holmen, J. Mater. Chem. A, 2013, 1, 7006–7011 RSC.
- X. Huang, H. Pan, K. Chen, Z. Yin and J. Hong, CrystEngComm, 2018, 20, 7020–7029 RSC.
- Q. Duan, J. Wang, C. Ding, S. Guo, Y. Jia, P. Liu and K. Zhang, Fuel, 2017, 193, 112–118 CrossRef CAS.
- J. Li, A. C. H. Huan, L. Wang, Y. Du and D. Feng, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 6876–6878 CrossRef CAS.
- M. Kumar, S. Srikanth, B. Ravikumar, T. C. Alex and S. K. Das, Mater. Chem. Phys., 2009, 113, 803–815 CrossRef CAS.
- F. Huang, X. Sun, Y. Zheng, Y. Xiao and Y. Zheng, Mater. Lett., 2018, 210, 287–290 CrossRef CAS.
- V. M. Gaikwad, J. R. Sheikh and S. A. Acharya, J. Sol-Gel Sci. Technol., 2015, 76, 27–35 CrossRef CAS.
- R. Rajagopalan, B. Chen, Z. Zhang, X. Wu, Y. Du, Y. Huang, B. Li, Y. Zong, J. Wang and G. H. Nam, Adv. Mater., 2017, 29, 1605694 CrossRef PubMed.
- S. Petrović, A. Terlecki-Baričević, L. Karanović, P. Kirilov-Stefanov, M. Zdujić, V. Dondur, D. Paneva, I. Mitov and V. Rakić, Appl. Catal. B, 2008, 79, 186–198 CrossRef.
- S. Phokha, S. Pinitsoontorn, S. Maensiri and S. Rujirawat, J. Sol-Gel Sci. Technol., 2014, 71, 333–341 CrossRef CAS.
- S. Phokha, S. Pinitsoontorn, S. Rujirawat and S. Maensiri, J. Nanosci. Nanotechnol., 2015, 15, 9171–9177 CrossRef CAS PubMed.
- G. Deng, Y. Chen, M. Tao, C. Wu, X. Shen, H. Yang and M. Liu, Electrochim. Acta, 2010, 55, 1120–1124 CrossRef CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei and Y. Sun, RSC Adv., 2014, 4, 48888–48896 RSC.
- X. Tang, F. Gao, Y. Xiang, H. Yi, S. Zhao, X. Liu and Y. Li, Ind. Eng. Chem. Res., 2015, 54, 9116–9123 CrossRef CAS.
- F. Martinovic, Q. Tran, F. Deorsola, S. Bensaid, R. Palkovits, W. Paulus, B. Bonelli, F. D. Renzo and R. Pirone, Catal. Sci. Technol., 2020, 10, 2193–2202 RSC.
- X. Huang, X. Shang and P. Niu, Acta Phys. Chim. Sin, 2017, 33, 1462–1473 CAS.
| This journal is © The Royal Society of Chemistry 2022 |