
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized silver nanoparticles for SERS amplification with enhanced reproducibility and for ultrasensitive optical fiber sensing in environmental and biochemical assays†
Nguyen Tran Truc Phuongab,
Vinh Quang Dang ab,
Le Van Hieuab,
Ta Ngoc Bachc,
Bui Xuan Khuyenc,
Hanh Kieu Thi Taab,
Heongkyu Jud,
Bach Thang Phanbe and
Nhu Hoa Thi Tran*ab
ab,
Le Van Hieuab,
Ta Ngoc Bachc,
Bui Xuan Khuyenc,
Hanh Kieu Thi Taab,
Heongkyu Jud,
Bach Thang Phanbe and
Nhu Hoa Thi Tran*ab
aFaculty of Materials Science and Technology, University of Science, Ho Chi Minh City, Vietnam. E-mail: ttnhoa@hcmus.edu.vn
bVietnam National University, Ho Chi Minh City, Vietnam
cInstitute of Materials Science, Vietnam Academy of Science and Technology, Hanoi, Vietnam
dDepartment of Physics, Gachon University, Seongnam, Gyeonggi-do 13120, Republic of Korea
eCenter for Innovative Materials and Architectures (INOMAR), HoChiMinh City, Viet Nam
First published on 1st November 2022
Abstract
Plasmonic sensors have broad application potential in many fields and are promising to replace most bulky sensors in the future. There are various method-based chemical reduction processes for silver nanoparticle production with flexible structural shapes due to their simplicity and rapidity in nanoparticle fabrication. In this study, self-assembled silver nanoparticles (Ag NPs) with a plasmon peak at 424 nm were successfully coated onto –NH2-functionalized glass and optical fiber sensors. These coatings were rapidly produced via two denaturation reactions in plasma oxygen, respectively, and an APTES ((3-aminopropyl)triethoxysilane) solution was shown to have high strength and uniformity. With the use of Ag NPs for surface-enhanced Raman scattering (SERS), excellent results and good stability with the detection limit up to 10−10 M for rhodamine B and 10−8 M for methylene blue, and a signal degradation of only ∼20% after storing for 30 days were achieved. In addition, the optical fiber sensor with Ag NP coatings exhibited a higher sensitivity value of 250 times than without coatings to the glycerol solution. Therefore, significant enhancement of these ultrasensitive sensors demonstrates promising alternatives to cumbersome tests of dye chemicals and biomolecules without any complicated process.
1. Introduction
Nowadays, silver is predominately used in engineered nanomaterials because of its outstanding properties such as antibacterial, low toxicity, and high sensitivity.1 The plasmonic property makes it one of the best materials to enhance the sensitivity of optical sensors. The plasmonic property is the main feature of localized surface plasmon resonance (LSPR) effects, which occurs only in precious noble metal nanoparticles. The most popular are gold and silver.2 The ability of enhancing the Raman signal in the electromagnetic field (EM) of silver is 2–3 times higher than that of gold.3 At the same time, silver produces 10 to 100 times higher SERS signals than those of similar gold nanostructured signals. This is because its band gap d–s is in the UV region, causing less damping of the plasmon mode.4 However, the sensitivity of LSPR properties to the change in refractive index of the medium also makes Ag nanoparticles receive much attention; this is used in increasing the sensitivity of sensing system optics.5 Consequently, it is applied in fabricating biosensors to detect biomolecules at extremely low concentrations.However, Surface-Enhanced Raman Scattering (SERS) is one of the most rapid and sensitive techniques to detect probe molecules at ultralow concentrations as the fingerprint.6 SERS and fiber-optic optical sensors used in conjunction with metal nanoparticles are increasingly showing strengths in the rapid detection of molecules in real time with extreme accuracy and responsiveness that meet the actual requirements of social development.7,8 Compared with other analysis methods such as fluorescence sensing, high-performance liquid chromatography (HPLC), spectrophotometry, and enzyme-linked immunosorbent assays (ELISA), SERS and fiber optic sensing found some advantages in facile sample preparation, low chemical usage, and excellent stability. Therefore, the LSPR-applied fiber optic sensor of metal nanoparticles (NPs) is a promising candidate in the detection and treatment of diseases such as cancer, diabetes,9 and biomedical diagnosis,10 and in other regions such as environmental monitoring11 and food industry.12
Many studies aimed at optimizing sensor sensitivity and stability by creating multilayer films,6 and fabricating nanoparticles with hollow structures13 and alloy forms,14 have achieved many remarkable results. However, these methods make the sensors more complicated, increase the cost, and are affected by many factors, and hence, ensuring stability requires high technology.15,16 To overcome these outcomes, in this study, we focused on optimizing the strengthening ability of the self-assembled Ag NP layer by thoroughly investigating both the synthesis and functional processes of Ag NPs onto the glass to build a simple sensor system with high stability and performance.
Rhodamine B (RhB) and D-glucose are extensively chosen as substances for evaluating the sensitivity of SERS and optical fiber sensors, respectively. They are easily found in the laboratory and are represented in the food and health-monitoring sectors, in which RhB with the molecular formula C28H31ClN2O3 is used as a synthetic dye that has been identified as an illegal additive in food by the European Food Safety Authority (EFSA), because of its toxicity to the nervous system of humans and animals.17 Due to the cheap price of RhB, it is widely used for dyeing, especially in counterfeit chili powder.18 Therefore, the sensitive detection of this substance in food testing is very important. However, glucose is an important organic substance present in most organisms and its concentration greatly affects the functions of parts of the organism.19 Particularly, in biomedicine, blood glucose levels reflect many aspects of health conditions. The rapid and accurate detection of changes in glucose levels allows for diagnosing diabetes disease, which is a typical example that brings many complications and serious consequences to health.20,21 Currently, the detection of blood glucose levels is handled quickly and simply by enzymatic methods22 such as the copper reduction method (modified from Rotblatt et al.),23 the o-toluidine method,24 and the glucose oxidase and peroxidase (GOD-POD) method.25 In contrast, an ultra-sensitive low-concentration sensor system was discovered for non-invasive glucose monitoring via saliva or respiratory fluids to control glucose levels, which received much attention. Small-sized, simple, low-cost, and highly sensitive sensor chips are an urgent need to develop for the quick detection of these substances in healthcare applications.
In this study, Ag NPs were easily synthesized by reducing Ag+ in AgNO3 to form silver nuclei in the presence of ethylene glycol. The size and shape of particles were controlled based on the concentration of polyvinylpyrrolidone (PVP), which acted as both a stabilizer and a capping agent. The self-assembled layer of Ag NPs was also optimized with excellent enhancement in SERS with a detection area of 10−4 M to 10−10 M corresponding to a 1011-fold enhancement factor. The results reported in this study compared with a previously published by Huanyu Chi et al.26 showed a significant improvement in detection limit when the same Ag NPs were used as substrates. This shows the importance of optimizing the enhancement condition of the SERS substrate. Even the LOD reported in this study was equal to the LOD value on the superhydrophobic silver nanoparticles decorated with aligned silver nanowire films reported by Jianchao Wang et al.27 It can be seen that this structure is relatively much more complex, which means that it is difficult to control stability. The immobilization of silver nanoparticles on the substrate in this study also showed a detection limit 10 times higher than that previously reported in a study by Debanjan Das et al. on coating silver nanoparticles onto a cellulose paper by a mirror coating reaction.28 Meanwhile, the stability, high uniformity, and reproducibility of SERS substrates were surveyed by investigating the Raman signals after storing for two months under our laboratory conditions. The activity of the active substrate was not significantly decreased in comparison with the freshly prepared chips. It also affirmed a strong development in the creation of simple, fast, and low-cost sensors to prepare stable, highly sensitive, and reproducible SERS substrates by modulating this property of Ag NPs.
Ag NPs coatings are also used for optical fiber sensors that obtained a high sensitivity of 6 × 10−8 RIU when tested with glycerol. The –COOH group is modified on the surface of the Ag NP coating to increase the sensitivity to glucose. The calculated limit of detection (LOD) for glucose is 4 μM corresponding to a sensitivity of 3 × 10−7 RIU. In this study, it can be seen that the superior fiber optic LSPR sensor has a LOD 350 times better than that of the fluorescent sensor using conA as a recognition.29 In addition, LOD is 116 times higher than that of the SPR optical fiber sensor with a layer of glucose-sensitive membrane reported by Yuan et al., where GOx was embedded in PAM gel modified with SiO2 nanoparticles.30 In comparison to the previous studies for glucose detection, LSPR-based fibers provide label-free sensors for finding the concentration in real time, fast detection, fabrication clarity, and ultra-low quantitative analysis. This plasmonic sensor can deliver promising sensing capability, revealing the capability of non-invasive glucose testing through the respiratory fluid.
2. Experimental section
2.1 Materials and reagents
De-ionized (DI) water (Thermo Scientific Easypure II, Göteborg, Sweden) was used for all processes with resistivity more sizeable than 16 MΩ cm−1. Silver nitrate (AgNO3, 99%), polyvinylpyrrolidone (PVP, Mw ∼ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 99%), mercaptosuccinic acid (MSA, HOOCCH(SH)CH2COOH, 97%), and (3-aminopropyl)triethoxysilane (APTES, 99%) were obtained from Sigma Aldrich. Ethylene glycol (EG, C2H6O2, >99.7%), sodium sulfide nonahydrate (Na2S·9H2O, >98%), and sodium hydroxide (NaOH, 96%) were supplied by Guangdong Guanghua Sci-Tech Co., Ltd (China). Ethanol (EtOH, C2H5OH, 99.8%) and D-glucose (C6H12O6, 99%) were provided by Fisher Ltd (UK), and glycerol (C3H8O3, 99%) was obtained from Duksan Pure Chemicals Co. (Ltd, Korea). Polydimethylsiloxane (PDMS, Sylgard 184) was obtained from Dow Corning Co., USA. All chemicals were of analytical grade and used without further purification. The sensor was assembled by aligning a multimode optical fiber (numerical aperture of 0.37, JFTLH-Polymicro Technologies) with a core diameter of 200 μm.
000, 99%), mercaptosuccinic acid (MSA, HOOCCH(SH)CH2COOH, 97%), and (3-aminopropyl)triethoxysilane (APTES, 99%) were obtained from Sigma Aldrich. Ethylene glycol (EG, C2H6O2, >99.7%), sodium sulfide nonahydrate (Na2S·9H2O, >98%), and sodium hydroxide (NaOH, 96%) were supplied by Guangdong Guanghua Sci-Tech Co., Ltd (China). Ethanol (EtOH, C2H5OH, 99.8%) and D-glucose (C6H12O6, 99%) were provided by Fisher Ltd (UK), and glycerol (C3H8O3, 99%) was obtained from Duksan Pure Chemicals Co. (Ltd, Korea). Polydimethylsiloxane (PDMS, Sylgard 184) was obtained from Dow Corning Co., USA. All chemicals were of analytical grade and used without further purification. The sensor was assembled by aligning a multimode optical fiber (numerical aperture of 0.37, JFTLH-Polymicro Technologies) with a core diameter of 200 μm.
2.2 Preparing SERS substrate
Ag NPs were synthesized by chemical reduction according to the following procedure. First, a three-necked flask containing 15 ml of EG and different amounts of PVP (100, 90, and 80 mg) was heated to 170 °C. Then, 1 ml of AgNO3 96 mg ml−1 and 70 μl Na2S 15 mg ml−1 were added through two necks of the flask, alternatively. As the Ag nuclei were formed, the color of the solution changed from caramel to dark green due to the growth of larger Ag NPs. After 100 min, the Ag NP solution was cooled down in an ultrasonic bath for 15 min, and then the Ag NPs were washed several times with acetone and collected by centrifugation at 4000 rpm for 10 min. The final product was dispersed in 30 ml of DI and stored in the darkness at 4 °C.APTES was used to anchor the functional group (–NH2) onto the glass surface after treatment in an oxygen plasma atmosphere for 2 min. This functional group has (–NH2) been proved to exhibit excellent binding ability with Ag NPs. The process of denaturing functional groups (–NH2) was investigated and proven by V. Thi Huong et al.8 The Ag NP coating was investigated according to the soaking time of the Ag NP solution on the modified glass for 1, 2, and 3 h. Finally, a drop of 50 μl of analyte solution, specifically RhB or methylene blue (MB), was dropped onto a glass surface coated with Ag NPs. The Raman spectra were recorded for further analyses. The fabrication process of the SERS substrate is depicted in Scheme 1.
 | ||
| Scheme 1 Schematic depiction of the synthesis of silver nanoparticles and the fabrication of SERS substrates on a glass surface. | ||
2.3 Characterizations
The properties of these different Ag NP solutions were investigated by UV-vis spectroscopy, X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), and dynamic light scattering (DLS). Then, the suitable coating was selected based on XRD and FESEM results. The Raman signal of the analyte was recorded immediately after the solution was drained using a HORIBA Europe GmbH instrument (HORIBA Scientific, HORIBA Ltd) at an excitation wavelength of 532 nm.2.4 Manufacturing process of fiber optic sensor chip LSPR
:1 ratio was used to remove the cladding (the surface of the fiber core after removing the jacket and cladding layer is observed in Fig. S4†). Ag NPs coatings were functionalized onto the core in a process similar to the coatings on SERS substrates via the (–NH2) group present in APTES. The fiber surface continues to activate the carboxyl group with an MSA solution of 0.1 mM in ethanol at room temperature (RT) for 16 h. The MSA denatured fraction was washed several times with ethanol and distilled water before storage or use.The surface modification for D-glucose detection included the generation of the carboxyl groups on the fiber core surface. The D-glucose concentrations were from 0.25 mM to 30 mM in DI.
:1 (v/v) to make a PDMS mold that has a blank space in the middle to load the analyte solution, which is pumped into and out of the chamber through the inlet and outlet. Then, the fiber with the denatured sensing area is placed into the blank space of the PDMS chamber. The open surface of the PDMS is tightly bonded to a glass (SiO2) through the –O– bridge to create a closed chamber. This bonding process was performed after the two bonding surfaces of the PDMS mold, and the functionalized glass substrate with free –OH groups by placing them in an oxygen plasma for 2 min. Finally, to improve the bonding quality, the complete microfluidic channel was heated at 70 °C for 30 min.3. Results and discussion
3.1 Preparation of Ag NP plasmonic coating
Fig. 1a shows the UV-vis spectra of Agb1, Agb2, and Agb3 samples, which were synthesized by different amounts of PVP 100, 90, and 80 mg, respectively. As a result, the effect of PVP concentrations on the particle size and morphology of AgNPs was clearly observed. The plasmon peaks of the solutions Agb1, Agb2, and Agb3 were represented by the absorption spectra of these solutions in the UV-vis region. The maximum absorption peaks at 416, 424, and 427 nm correspond to Agb1, Agb2, and Agb3 samples, respectively. Although there was no significant change in the maximum absorption wavelength, the disproportionation of the plasmon peak has demonstrated a large variation of the particles in different Ag NP solutions. This indicated the heterogeneity in the size and shape of particles in the solution. The particle size and their distribution in solutions are also important factors affecting the plasmon peak of precious metal nanoparticles. In the Mie model,31 the resolution of the Maxwell equations shows the redshift of the plasmon peaks of spherical metal nanoparticles with the increase in particle size. Increasing the size of the nanoparticles leads to a redshift of the peak absorption wavelength and broadens the plasmon-enhanced peak.32 This phenomenon occurred by the aggregation of small NPs. Therefore, for the Agb3 sample, we can see that the plasmon peak is relatively wide and disproportionate, which means that the particle size distribution in the Agb3 solution is likely to be highly identical. Especially for the Agb1 solution, the strong absorption region spanned almost the visible region, reflecting the interspersed existence of silver nanoparticles of different shapes in the solution.33,34 Depending on the metal nature, shape, size, and dielectric medium containing metal nanoparticles, the plasmon resonances peaked at different wavelengths. In the case of different shapes and sizes of a mixture of nanoparticles, the expanded UV-vis extinction spectra were received due to the superposition of spectra in individual shape sizes with each other.35 Therefore, the Agb2 solution was estimated to contain a more uniform size and shape of particles as compared to the Agb1 and Agb3 solutions, which is elucidated by the width and balance of the absorption spectrum.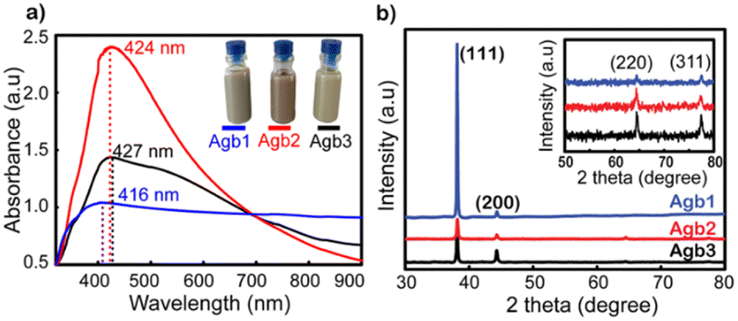 | ||
| Fig. 1 (a) UV-vis spectra and (b) XRD patterns of Agb1, Agb2, and Agb3 solutions. | ||
As shown in Fig. S1,† the DLS results presented the correlation between the size distributions in the solutions and the plasmon absorption peaks. The purity of the phase structure of Ag NPs solutions was demonstrated by X-ray diffraction results shown in Fig. 1b. The characteristic diffraction peaks of the face-centered cubic (FCC) structure of silver36 corresponding to 2θ angles at 38°, 44°, 65°, and 77° belonged to the crystal orientations of (111), (200), (220), and (311), respectively.37
As shown in Fig. 1b, the (111) orientation was drastically developed for all 3 types of solutions of Ag NPs. The intensity of the (111) orientation of Agb1 is 5 and 6 times higher than that of Agb2 and Agb3, respectively, soaked on glass substrates under the same conditions. As the ratio of PVP in the solution becomes larger, the XRD diffraction peaks tend to be wider and lower. From this result, the reducing and stabilizing role of PVP in the process of forming crystals in different orientations of Ag NPs can be confirmed.38 This is consistent with the result of the displacement of the maximum absorption wavelength observed in the UV-vis spectrum (Fig. 1a). The (111) orientation of Agb1 has high intensity, which can be explained by a large amount of PVP, which is not only limiting the growth of Ag in other directions but also helps to prevent the corrosion of Na2S to the (111) pole. Therefore, the Agb1 sample was strongly oriented in the (111) plane leading to the formation of long Ag bars in the solution.39,40 The sizes of the crystals growing in different directions were calculated according to Scherrer's formula,33 and are shown in Fig. S2.† The strong evolution of the (111) orientation demonstrates the formation of rods in the Agb1 solution. The FESEM results exhibited in Fig. S3† show that the visual shapes of the Agb1, Agb2, and Agb3 particles are in perfect agreement with the development of the crystal orientation. Agb1 is proven to contain a mixture of Ag NPs with many different shapes and sizes such as rod, triangular, and cube, in contrast with Agb2 and Agb3 consisting of only spherical silver nanoparticles. The size particle diameters of Agb2 and Agb3 are 80 and 120 nm, respectively. This result is completely consistent with the conclusion from the UV-vis spectrum and XRD data (Fig. 1).
The Agb2 solution was selected to create a coating to enhance the Raman signal in SERS sensors and increase the sensitivity in optical fiber sensors. Ag NPs dispersed in DI exhibited low stability for enhancing the signal in SERS,18 and hence, the self-assembled Ag NPs coating is considered a solution to increase signal stability in SERS. Ag nanoparticles immobilized on the SiO2 surface via the –NH2 groups with different immersion times (1, 2, and 3 h). In all investigated samples, the purity of the crystal phase and the increase in density were maintained, as evidenced by the XRD results shown in Fig. 2a. The characteristic diffraction peaks of the Ag NPs coatings have no shift as well as the appearance of other diffraction peaks, indicating that the crystal structure of the Ag nanoparticles remains stable at different immersion times. However, an obvious increase in the intensity of the diffraction peaks shows that the self-assembly process continues and there is no sign of saturation at the survey times.
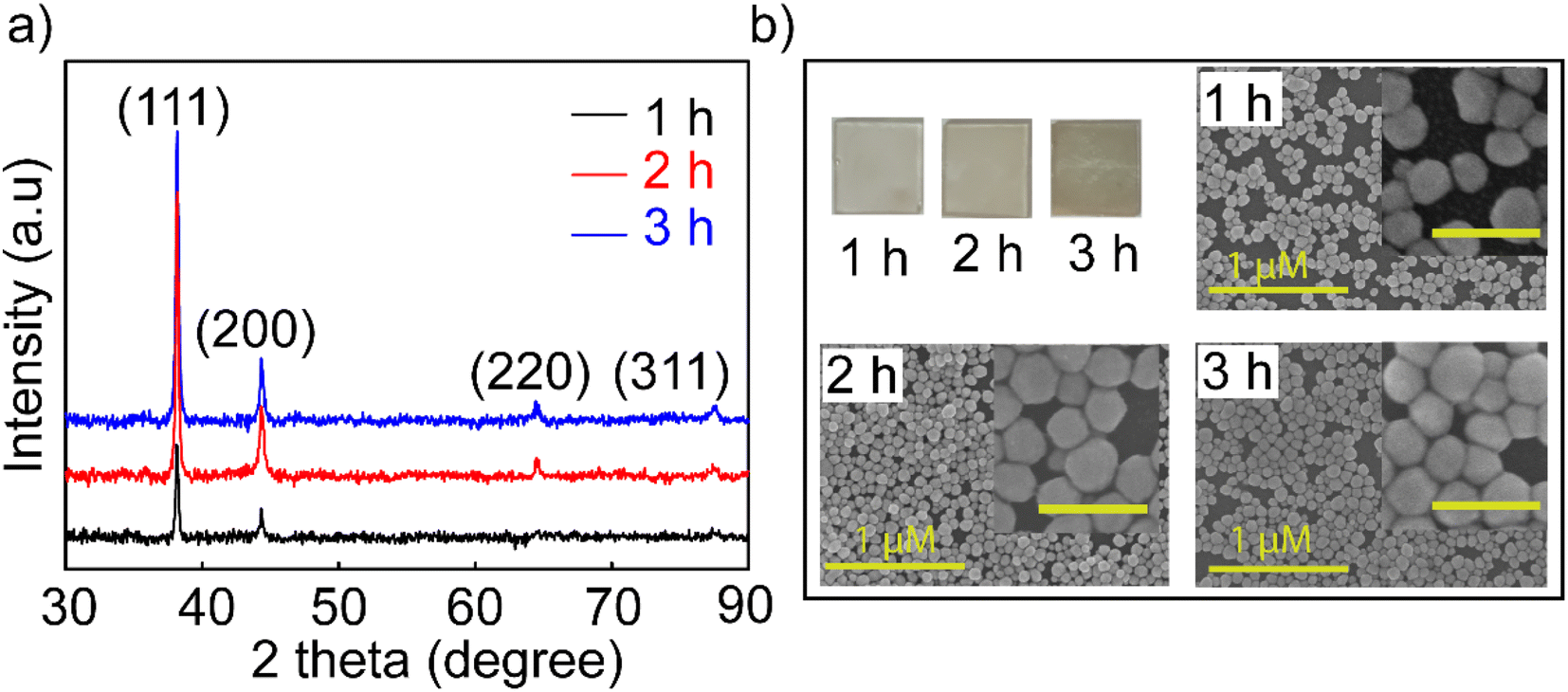 | ||
| Fig. 2 (a) XRD patterns and (b) FESEM images of investigated Ag NP coatings over time of 1, 2, and 3 h with the inset image scale of 200 nm. | ||
The FESEM images (Fig. 2b) of the coatings with the corresponding time also showed an increase in the particle density of Ag NPs on the SiO2 substrate. Many theories supported the creation of nanoparticle dimers, trimers, or clusters to create hot spots with larger enhanced electromagnetic fields,41,42 but it is difficult to manipulate these clusters evenly all over the substrate. In this experiment, the 3 h coating was observed for the agglomeration and overlapping of Ag NPs, but the surface of SiO2 substrates was exposed to voids not covered by Ag NPs. The fact that the Ag NPs become dense and overlapping leads to a decrease in the surface effect of the nanoparticles. Because signal enhancement occurs at the interface of Ag NPs and the analysis molecule when the Ag NPs are overlapped, there is no space left for the analyte molecules to enter the empty slot between Ag NPs due to the limited surface area, and hence, the Raman signal is not enhanced at its best and this is elucidated in Fig S5.† Many studies indicated that the distance between nanoparticles is 2 nm,43 and the analytical molecules can absorb and fully contact the surface of the nanoparticles. A model using FDTD simulation for coating Ag NPs with a particle diameter of 80 nm and a distance between particles of 60 nm was reported by Vahid Eskandari et al.44 showing the electric field distribution on the surface, and their intensity for application in SERS sensors. Therefore, the 2 hours coating sample has a high uniformity on the surface and less overlap between particles, and it was selected for the highest performance in the sensor. For greater certainty, a survey measuring the SERS signal enhancement of the coatings in 1, 2, and 3 h was performed. The results shown in Fig. S5† proved to harmonize with the theory stated about the density of nanoparticles in SERS enhancement and the FESEM results shown in Fig. 2b. Ag NPs 2 h coating shows stronger enhancement and lower signal attenuation even at a low concentration of 10−10 M, and characteristic RhB oscillations are still observed. This is another reason that supports the decision to choose symmetrical polygonal Ag NPs although many theories have demonstrated that Ag NPs have a particular morphology containing spikes such as stars and leaves needles for higher electromagnetic field focus.45 Because the interaction and support of local plasmons with each other is also an essential factor. They form nanogaps between the nanoparticles and the nanogap site has been shown to provide superior signal enhancement compared to that on the top surface of the nanocoating.46 The selection of Ag NPs with a polygonal shape will help to create a good coating with high particle density but still leave enough gaps for the analyte to absorb for a good SERS enhancement effect. For these reasons, the Ag NPs-2 h coating has been improved for suitable use as a substrate in SERS and as a coating in fiber optic sensors.
3.2 SERS sensor
Fig. 3 shows the SERS spectrum of RhB on Ag-SERS substrates of Ag NPs with RhB concentrations from 10−4 to 10−10 M. The Raman intensity was decreased proportionally with the decrease in RhB concentrations. Obviously, the peaks at 621, 1192, 1280, 1357, 1508, and 1648 cm−1 were assigned to ring deformation. More specifically, the Raman peak at a wave number of 638 cm−1 was assigned to the C–H in-plane bending vibration, and peaks at 1192 and 1355 cm−1 were ascribed to the aromatic C–C skeleton stretching vibration. The peaks at wavenumbers of 1280 and 1507 cm−1 were assigned to the C–C stretching of the ring. Finally, the vibration of the strong C–C stretching band in the xanthene ring was recorded at 1648 cm−1.47,48 At a concentration of 10−10 M, characteristic RhB oscillations can still be observed, this demonstrates the ability to detect the existence of RhB at this concentration. Among the characteristic oscillations, the C–C tensile oscillation at 1648 cm−1 shows the most obvious signal intensity. Therefore, the standard curve calibrated of peak intensities at 1648 cm−1 was chosen to build from the logarithmic signal attenuation of the RhB concentrations with the function y = −959x + 11250 corresponding to the reliability R2 = 0.9924 (Fig. 3). The analysis enhancement factor (AEF) calculated49 from the above spectrum is 1011-fold. The evidence about the specific SERS signal for RhB, and R2, the proportion of the variance above, is the irrefutable proof for the enhanced capacity of Ag NPs located on our simply modified glass surfaces at an optimal incubation time of 2 h. The resulting substrates could be used to strengthen the credible quantification not only for RhB but also for other organic hybrids.
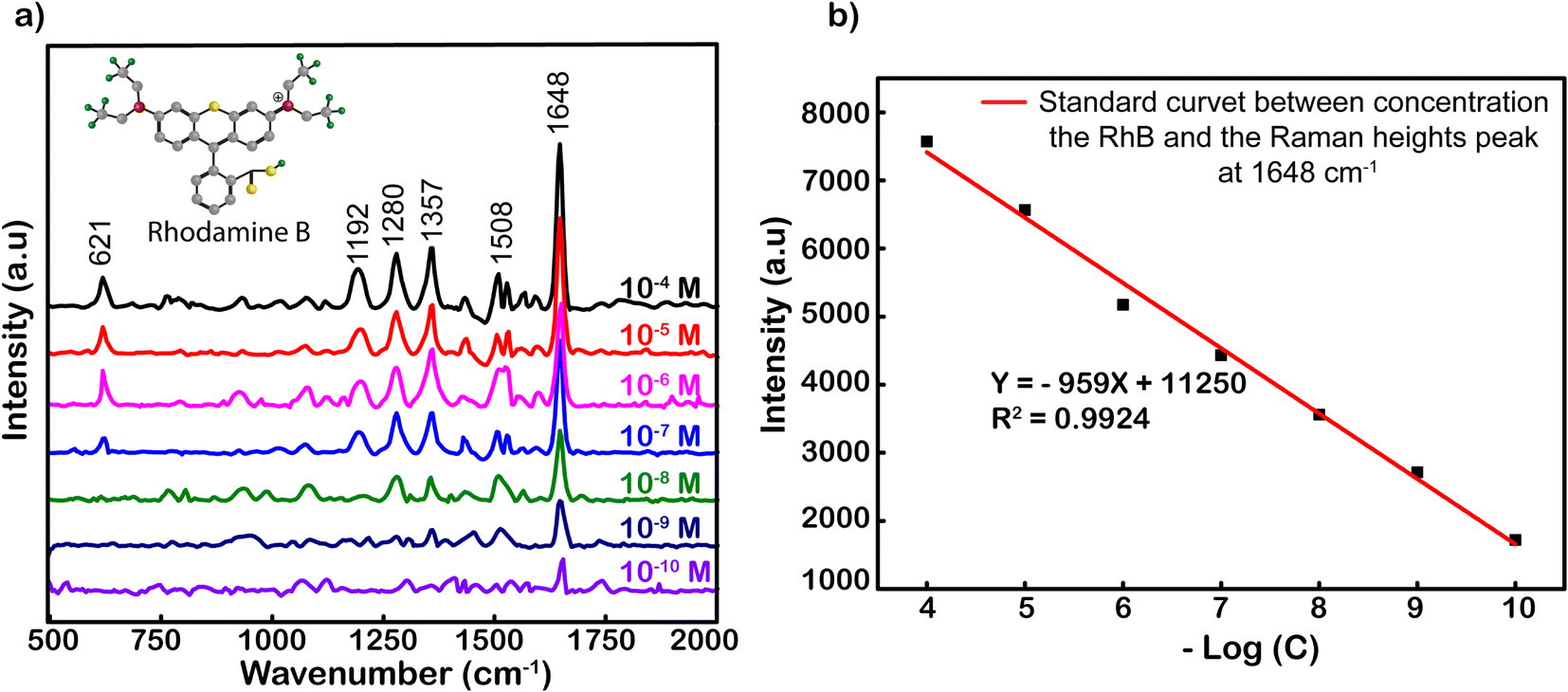 | ||
| Fig. 3 (a) Raman spectra of RhB adsorbed on Ag-SERS substrate thin films with different concentrations. (b) Standard curve calibrated by the linear relationship between the logarithm of RhB concentration (logC) and heights peaks at 1648 cm−1. | ||
Another popular dye was also used to demonstrate the excellent performance enhancement of Ag NP substrates up to the concentration of 10−8 M (Fig. 4a). Typical movements include weak skeletal deformation of C–N–C vibration, weak vibration of in-plane bending of C–H, in-plane ring deformation of C–H, medium vibration of symmetrical stretching of C–N, and strong vibration of ring stretching of C–C. These fluctuations peaked at wavenumbers 448, 501, 770, 1154, 1390, and 1624 cm−1, respectively.50 The tensile oscillation of C–C at 1624 cm−1 has the strongest intensity, and hence, it was selected to plot out the standard curve correlation between the log-scale of the RhB concentration and the Raman intensity (Fig. 4b). The decrease in peak intensity by logarithmic concentration is linearly represented by the function y = −159.5x + 1591. The reliability value (R2) was remarkably high at 0.9891, showing the accuracy and reliability of the measurement. The analysis enhancement factor of Ag NPs for MB was also calculated from the under spectrum as 1.3 × 108 fold. This is also a promising result in the research and development of substrates to enhance SERS. The analytical enhancement factor calculated based on the powder Raman spectra of RhB and MB is presented in Fig. S6.†
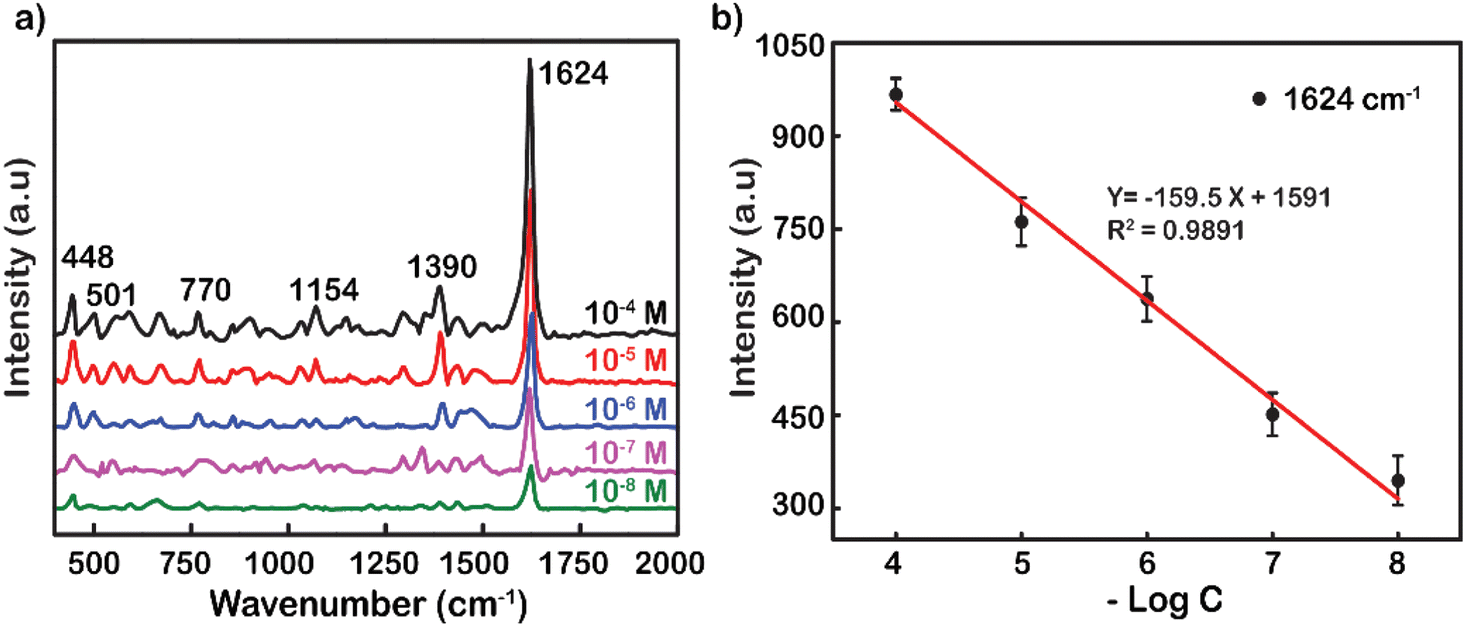 | ||
| Fig. 4 (a) Raman spectra of MB adsorbed on Ag-SERS substrate thin films with different concentrations. (b) Standard curve calibrated by the linear relationship between logC and peak intensity at 1624 cm−1. | ||
Except for the sensitivity, the homogeneity and reproducibility of the Ag NPs substrate are important factors in practical applications as well, which is interesting in this study. Fig. 5a shows the SERS spectrum of RhB (concentration is 10−8 M), with randomly chosen separated 11 spots on the Ag NP substrate, where the intensity of characteristic peaks is the same from different locations. To further strengthen the reliability, SERS mapping at 1648 cm−1 of RhB on glass/Ag NP substrates was performed (Fig. 5b). From the SERS mapping, it can be seen that the signal intensity is relatively uniform when scanning the spectrum on the membrane surface. This means that the surface coverage of the Ag NP coating on the film is high. To directly compare the fluctuation of the peaks, the intensity distributions of 1192, 1280, 1355, 1508, and 1648 cm−1 peaks of the RhB molecule from these Raman spectra are shown in Fig. 5c. The graph shows a clearer comparison of the intensities of 1192, 1280, 1355, 1508, and 1648 cm−1 peaks of RhB at 11 randomly selected sites on the Ag-SERS substrates. This result displays the intensity of the peaks almost on the same horizontal line. The enhanced Raman peaks at 1192, 1280, 1355, 1508, and 1648 cm−1 have relative deviations (RSDs) of 11.18, 12.50, 5.70, 12.60, and 12.07%, respectively, which is much lower than Natan's report on the scientific standard (20%) of bound nanostructures.51 Fig. 5d shows the magnitude of the maximum resonance peak at 1648 cm−1 at 11 different locations randomly selected on the membrane with error bars showing that the deviation of these intensities from the mean value calculated from the above graph is 2900 a.u., corresponding to the mean standard deviation (RSD) of 12% from that. This result proves that the Ag NP coating has uniformity and reproducibility, increasing the reliability of the sensing devices.
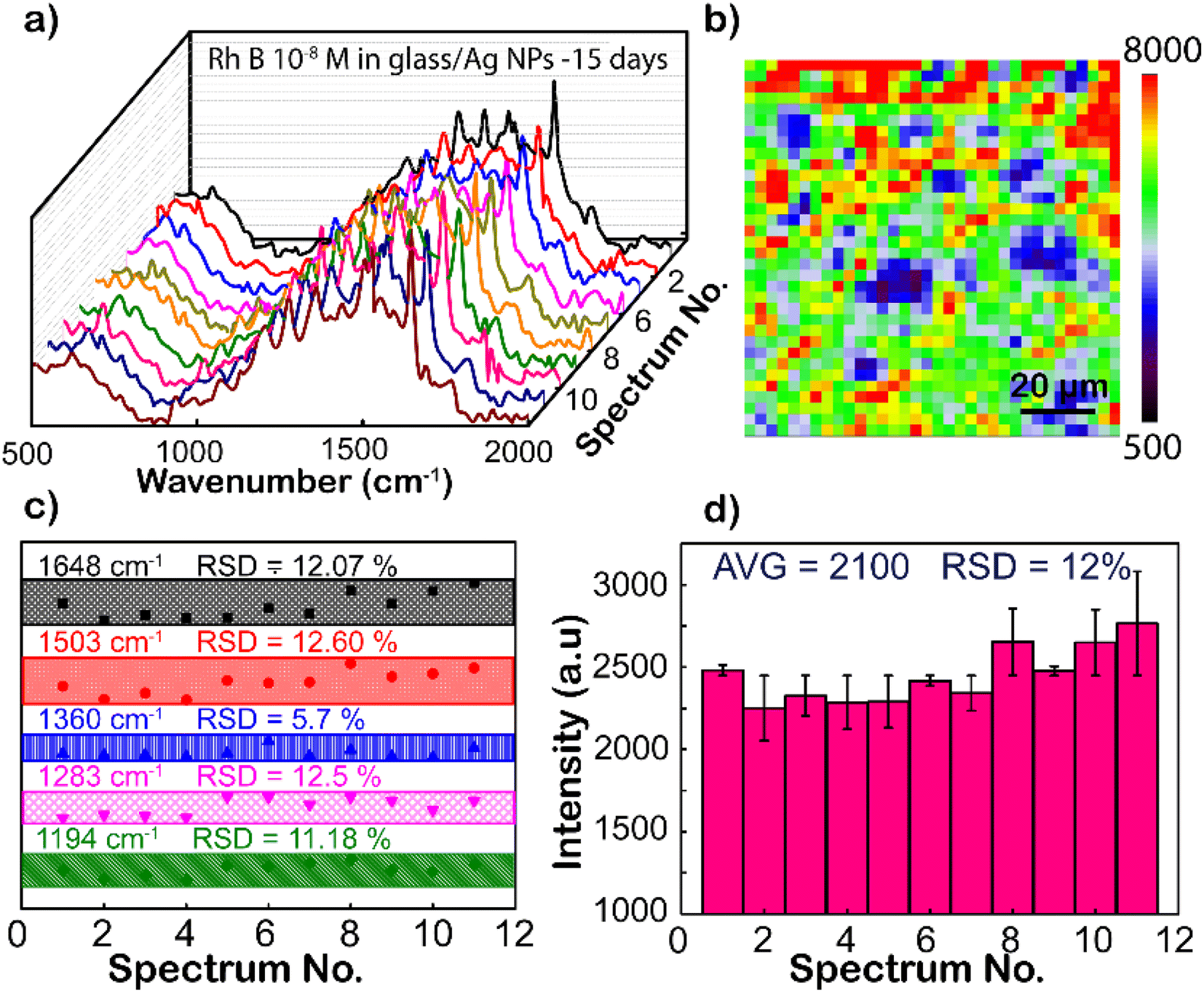 | ||
| Fig. 5 Investigation of the uniformity and repeatability of Ag-SERS substrates. (a) SERS spectra at 11 different positions. (b) SERS mapping at a wave number of 1648 cm−1 of RhB at concentrations of 10−4 M. (c) Intensities allocation of 1194, 1283, 1360, 1503, and 1648 cm−1 peaks from SERS results. (d) Intensity dispensation of 1648 cm−1 peaks. | ||
To investigate the stability and the reproducibility of the sample, the Ag NP thin film was used to adsorb RhB at 10−8 M after storing in the desiccator under the avoid-light condition within 0, 15, 30, and 60 days after the fabrication. The same preparation protocol was completed with those four surveyed products, and their Raman spectra of 10−8 M RhB solution are plotted in Fig. 6a. For the long-term storage, the Raman intensity of RhB gradually decreased. Fig. 6b illustrates the reduced amount of Raman intensity of 10−8 M RhB on Ag-SERS substrates by different storage times at a wavenumber of 1648 cm−1. After storing for 15 days, the created film was almost unchanged which was proved by 89% remaining peak intensity compared with the fresh one. The remaining peak intensity percentages underwent a cutting down to about 80 and 48% by keeping in 30 and 60 days, respectively. As shown in Fig. 6b, the intensity of RhB was left at approximately 50% after storing for 60 days. The silver nanoparticles were reported to react easily with the surrounding environment to affect the SERS signal; however, the obtained results showed that the Ag NPs coated on the glass surface showed high stability still maintaining 80% signal strength after 30 days of storage. This may be due to the protection of PVP on the surface of silver nanoparticles and the Ag NP coatings stored in an environment without the influence of temperature or other oxidizing agents other than a small amount of oxygen in the air. Based on this result, the lifetime is long enough for SERS-active substrates and recommended for a month of expiration for the best performance in this detection technique.
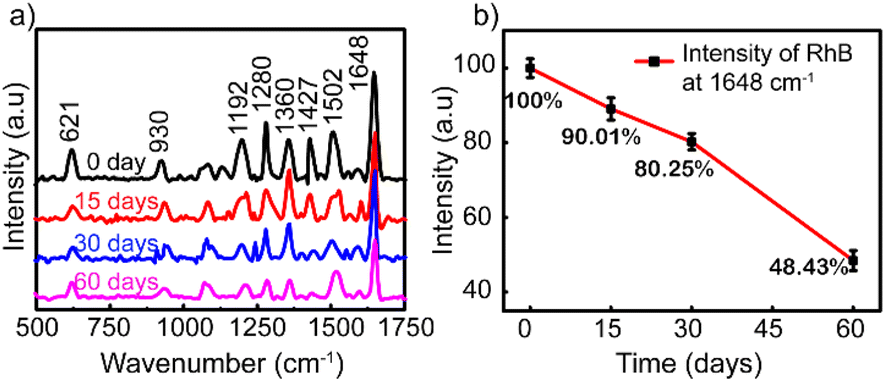 | ||
| Fig. 6 (a) Raman spectra of RhB (10–8 M) adsorbed on the Ag NP thin film. (b) Graph showing the Raman intensities after storing for 0, 15, 30, and 60 days. | ||
An RhB detection test was also performed on chili powder at a concentration of 2.4 × 10−7 g g−1 (10 ml of RhB 10−8 M in 0.2 g of chili powder) to check the method's feasibility. The RhB sample was mixed into chili powder using the extraction procedure referenced from the report of Yingying Sun et al.52 The test results on chili powder are compared with the SERS spectrum of RhB in DI presented in Fig. S7.† It can be observed that characteristic spectral peaks of RhB are kept similar to those of RhB in DI, but the intensity of the recorded spectrum is weaker and has less separation. Table 1 summarizes the results obtained from previous studies providing an overview of the development of SERS substrates for the detection of rhodamine B. As can be seen in this study, using a simple substrate with optimized survey conditions to achieve the results can be seen as an improvement compared to previous studies.
| Substrate | Synthesis/fabrication | EF or AEF (fold) | LOD | Reproducibility | Ref. |
|---|---|---|---|---|---|
| Ag NPs solutions | Chemical reduction method | 1.6 × 107 | 0.08 μg L−1 | — | 53 |
| Composite CNF-AgNPs | Hydrothermal | — | 5 × 10−7 M | — | 54 |
| Silver/gold alloy | Chemical reduction method | 1010 | 10−11 M | 3.1% (RSD of 8 points at 1650 cm−1) | 55 |
| AgNPs aggregates | Spray-drying method | 3.7 × 108 | >10−10 M | — | 56 |
| B-Bi2O3/Bi2O2CO3 substrate | Hydrothermal | — | 10−4 | — | 57 |
| Ag NPs/cellulose paper | Silver-mirror | 6.59 × 107 | 10−9 | 1.59% (RSD of 50 points at 1650 cm−1) | 28 |
| Ag NPs/glass | Chemical reduction method | 1011 | 10−10 M | 12.07% (RSD of 11 points at 1648 cm−1) | This study |
3.3 Optical fiber sensor
Glycerol diluted in water from 0 to 30% (v/v) was used to test the sensitivity of the fiber modified with Ag NPs because glycerol is a substance that is easily calibrated into a refractive index. Total internal reflection (TIR) is the main principle of light transmission in fibers. For conventional fibers, the cladding layer has a larger extraction than the core of the fiber, so total reflection occurs internally between the cladding layer and the core layer. For modified fiber, the cladding layer has been removed, because the air has a lower index of refraction than that of the core (nair = 1 < ncore = 1.457). The number of attenuated total reflections (ATRs) was calculated using eqn (1):58
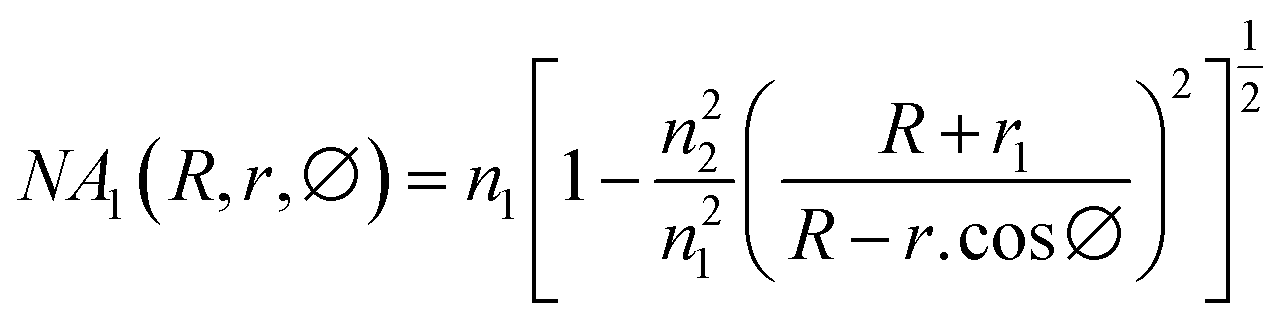 | (1) |
As described in the experimental section, removing both the protective and cladding layers of the modal fiber and then replacing them with a plasmonic layer of material help to increase the sensitivity of the sensor. At that time, the optical intensity when transmitted through the sensing area will be changed due to the interaction of the analytes and the plasmonic material layer, which changes the dielectric conditions of the medium.
In turn, low to high concentrations of glycerol are injected into the microfluidic channel through a mechanical pump at a constant rate. When the analyte solution fills the channel and the sensing area is completely covered with the glycerol solution, signal acquisition begins. Factors such as the light intensity of the fiber input, the flow rate through the channel, and the amount of the solution passing through the channel are kept constant throughout the measurement. The average value of 60 data points recorded continuously for 60 s of each concentration of glycerol varied from 0 to 30% (v/v). The results show a non-linear decline in energy output corresponding to the increase in glycerol concentration (Fig. 7b). This difference in light output power is caused by the difference in the refractive index of the analyte solutions at different concentrations. The refractive index is one of the strong factors that lead to a significant change in the output signal. The first is to cause a numerical aperture mismatch at the interface between the liquid cladding waveguide and the solid cladding waveguide. The connecting two waveguides of numerical aperture (NA) are leading to an increase or partial loss of energy. However, the change in solution refractive index also causes a shift in the plasmon peak of the metal nanoparticles. The sensitivity of the LSPR peaks to this refractive index enhances the sensitivity of fiber-optic optical sensors. As the concentration of glycerol injected through the flow cell increases, the number of glycerol molecules absorbed into the surface of the fiber sensor-guided light also increases.59 This makes them easily accessible, causing changes in the plasmon resonance conditions. The higher the glycerol concentration, the more molecules are adsorbed onto the sensor surface. This causes a strong variation in the LSPR resonance condition. At the same time, the power output energy loss is caused by the absorption of glycerol molecules. Because the number of absorption oscillators per unit volume increases, it causes a loss of light energy as it travels through the fiber. Besides that, the number of absorption vibrations is proportional to the concentration of molecules attached to the sensor surface of the optical fiber. Therefore, the power output is attenuated with the increase in the concentration of glycerol. In particular, when the glycerol concentration increased from 0 to 30%, the output signal decreased by 88 μW and the strongest signal reduction was 6 μW corresponding to the glycerol concentration increasing only 0.001%. It is the plasmonic effect of energy dissipation, enhanced by multiple reflections of light guided along the core of the Ag NP-coated fiber sensor.60 Another important factor that could be responsible for the decrease in the sensor's optical output for the increase in glycerol concentration is the change in LSPR as the glycerol concentration increases. As mentioned earlier, the shape of the nanoparticles has a great influence on their LSPR effect. In this case, increasing the concentration of glycerol induced the formation of anisotropic silver nanoparticles, leading to the formation of absorption peaks at the long wavelength.61,62 This is the main cause of the deep decrease in glycerol concentration. The limit of detection (LOD) of glycerol calculated from the results shown in Fig. 7b is 1.34 × 10−4%, corresponding to 6.1 × 10−8 RIU as a minimum resolvable change in the bulk refractive index.8 This result proves that the sensitivity of the fiber optic sensor, when coated with Ag NPs, has been shown to be 250 times higher than that reported previously.63
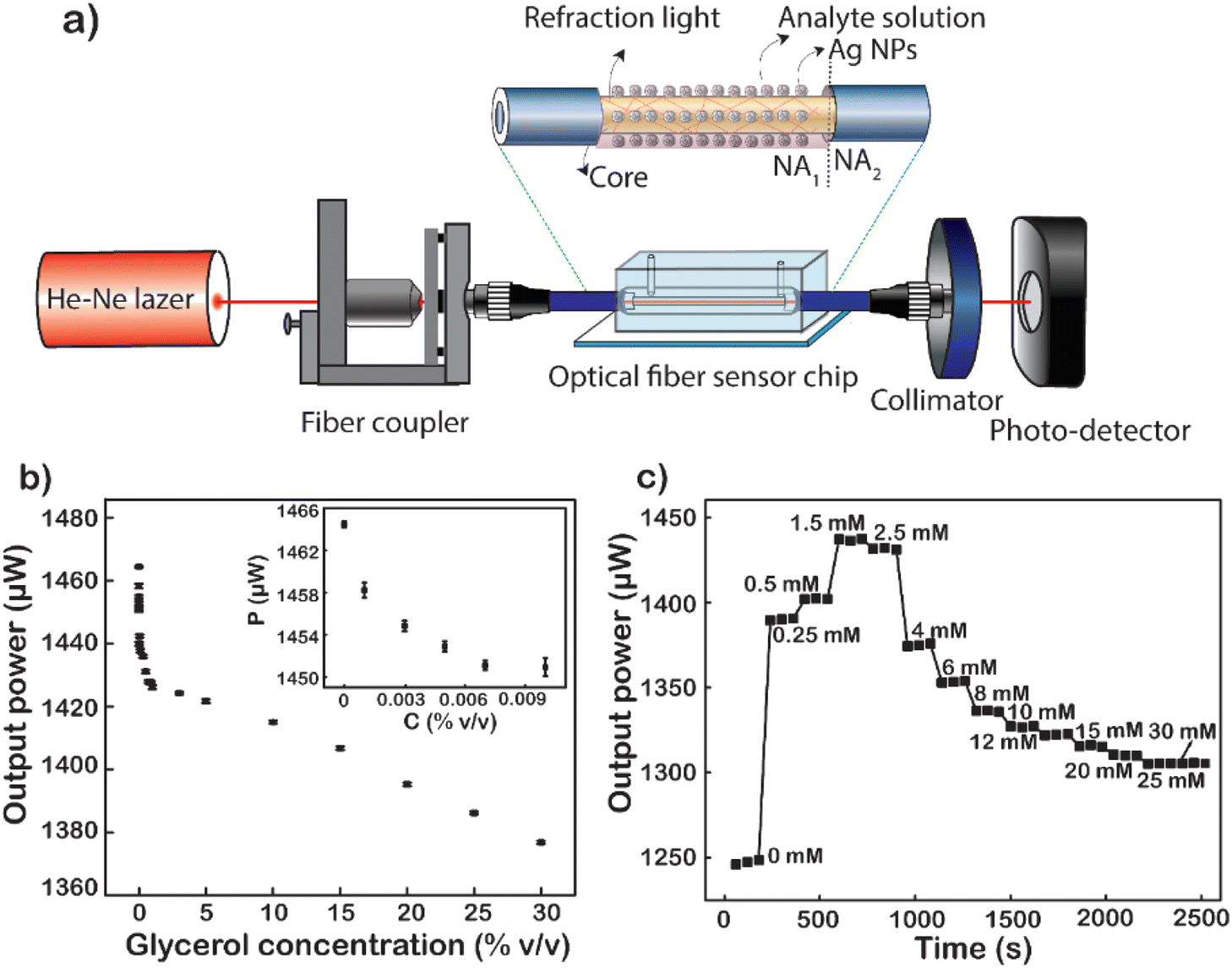 | ||
| Fig. 7 (a) Illustration of the system for measuring signals using a fiber optic sensor. (b) Sensitivity of the sensor using a glycerol solution. (c) Time-dependent optical power transmitted through the fiber sensor upon injection of serial D-glucose concentrations. | ||
The fiber after being modified with Ag NPs was been modified with MSA to form a –COOH group on the surface. This way, the D-glucose molecules will be easily trapped on the surface of the sensing area. Fig. 7c shows the graph of real-time normalized output power when different D-glucose concentrations are pumped over the microfluidic channel. At a given concentration is the mean of 60 data points corresponding to a retention time of 60 s. The results are normalized to the power value when measured with the DI corresponding to a solution with zero glucose concentration. It can be observed that when starting to pump a very low concentration of D-glucose solution, there is a sudden increase in the output signal compared to the signal when only DI and this signal continue to increase to 1.5 mM. When at too low a concentration less than 1.5 mM, glucose molecules stick little or not on the surface of the fiber sensor area. The changes in both the bulk refractive index and the surface index are almost negligible,64 leading to no change in LSPR resonance conditions. Nevertheless, an increase in the refractive index of the liquid reduces the NA difference between the liquid-coated waveguide and the silica-coated waveguide. The decrease in the NA dissimilarity between these two parts leads to an increase in the output power of the fiber when the solution containing glucose is really low. However, when the glucose concentration is large enough, the fixation of glucose on the core surface of the fiber takes place, gradually increasing the amount of glucose on the surface (nearly) and increasing the surface enhancement index. When the concentration of D-glucose injected into the sensing area increases, the greater number of molecules attracted and absorbed down the fiber surface creates more chemical interactions on the surface that creates the LSPR condition change.65 This proves that the more molecules fill the sensor surface or the higher the index of each liquid field, the closer these molecules will be to the metal surface. This causes the effect of plasmonic resonance to become stronger leading to significant attenuation of the output power of fibers, which explains the strong attenuation of intensity at 4 mM in the data. To increase the reliability of the results, 4 recycles at a glucose concentration from 0 to 1.5 mM were performed, and the results are presented in Fig. S8.† This result also proves the reusability of the fiber optic sensor and the stability of the measurements, since no significant deviation was observed over 4 recycles.
The baseline of the transmitted power was obtained via the normalized transmitted power with D-glucose concentrations presented in Fig. 7c. The measured standard deviation (SD) of signals at each concentration was gained very small compared to the change in the transmitted power, which led us to reach a LOD of 4 μM corresponding to a sensitivity of 2.95 × 10−7 RIU. The fiber optic LSPR sensor has a LOD 350 times better than that of the fluorescent sensor when using conA as a recognition.29 In addition, it is 116 times higher than that of the SPR optical fiber sensor with a layer of glucose-sensitive membrane reported by Yuan et al., where GOx was embedded in PAM gel modified with SiO2 nanoparticles.30 This is a fast glucose detection sensor without the use of GOx or other labels making the system simple and less toxic. In comparison to the previous studies for glucose detection, LSPR-based fiber provides label-free sensors for point-of-care testing in real time, rapid detection, fabrication simplicity, and ultra-low quantitative analysis. This plasmonic sensor can deliver promising sensing capability, revealing the capability of quantitating D-glucose.
4. Conclusion
In summary, Ag NPs with a plasmon peak at 424 nm have been successfully synthesized with high stability by a simple reduction method with ethylene glycol. Ag NP coatings with high storage and uniformity have been investigated and demonstrated in increasing the sensitivity and detection limit of SERS sensors and optical fiber sensors. SERS substrates using self-assembled Ag NPs optimized for the spacing of the particles have achieved excellent results and good selectivity, as evidenced by successfully detecting rhodamine B with detection limits up to 10−10 M corresponding to an enhancement factor of 1011-fold and for methylene blue 10−8 M corresponds to a 1.6 × 108-fold enhancement. The storage time for the coating to maintain good SERS enhancement performance is about 30 days corresponding to the average signal intensity value reaching 80%. Besides, surface homogeneity with a standard deviation not exceeding 15% has been demonstrated. In the optical fiber sensor, the sensitivity value with the addition of Ag NP coating is 250 times higher than that without the coating, as investigated with the glycerol solution. The limit of detection measured with glucose is 4 μM, and this result is compared to give a better signal than some other methods performed previously.These results show that the desirable ability to fabricate a facile sensing system utilizing single-component nanoparticles with simple structures can still achieve such high performance by optimizing the factors of size, shape, and density of the coating to developing maximizes the enhanced capacity of the material. Moreover, this study opens up the direction for developing sensor devices with compact size, fast response, and cost-effective application in health monitoring or clinical testing. Optical sensors are proving to be outstanding alternatives to cumbersome test systems with complex sample preparation processes currently in use.
Conflicts of interest
There are no conflicts to declare.Author contributions
N. T. T. P., L. V. H: conceptualization, methodology, writing – original draft preparation, data curation, formal analysis; V. Q. D., T. N. B., B. X. K.: data curation, formal analysis; H. J., T. K. T. L., B. T. P.: data curation, methodology, N. H. T. T.: supervision, funding acquisition; investigation; methodology, writing – reviewing and editing, funding acquisition.Acknowledgements
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under grant number DS2022-18-01. We would like to gratefully acknowledge the Vietnam National University in Ho Chi Minh City, Center for Innovative Materials and Architectures (Laboratory for Optics and Sensing).References
- G. A. Sotiriou and S. E. Pratsinis, Curr. Opin. Chem. Eng., 2011, 1, 3 CrossRef CAS PubMed.
- P. Tan, H. S. Li, J. Wang and S. C. B. Gopinath, Biotechnol. Appl. Biochem., 2021, 68, 1236–1242 CAS.
- F. J. G. De Abajo, Rev. Mod. Phys., 2007, 79, 1267–1290 CrossRef.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- A. Jouyban and E. Rahimpour, Talanta, 2020, 217, 121071 CrossRef CAS PubMed.
- V. D. Phung, W. S. Jung, T. A. Nguyen, J. H. Kim and S. W. Lee, Nanoscale, 2018, 10, 22493–22503 RSC.
- P. Q. T. Do, V. T. Huong, N. T. T. Phuong, T. H. Nguyen, H. K. T. Ta, H. Ju, T. B. Phan, V. D. Phung, K. T. L. Trinh and N. H. T. Tran, RSC Adv., 2020, 10, 30858–30869 RSC.
- V. Thi Huong, H. K. Thi Ta, N. X. D. Mai, T. T. Van Tran, B. X. Khuyen, K. T. L. Trinh, N. Y. Lee, B. T. Phan and N. H. T. Tran, Nanotechnology, 2021, 32, 335505 CrossRef PubMed.
- S. Kumar, R. Singh, B. K. Kaushik, N. K. Chen, Q. S. Yang and X. Zhang, IEEE Sens. J., 2019, 19, 7399–7406 CAS.
- S. Sharma and B. D. Gupta, J. Lightwave Technol., 2018, 36, 5956–5962 CAS.
- P. Dhara, R. Kumar, L. Binetti, H. T. Nguyen, L. S. Alwis, T. Sun and K. T. V. Grattan, IEEE Sens. J., 2019, 19, 8720–8726 CAS.
- N. Khansili, G. Rattu and P. M. Krishna, Sens. Actuators, B, 2018, 265, 35–49 CrossRef CAS.
- J. Li, J. Zhou, T. Jiang, B. Wang, M. Gu, L. Petti and P. Mormile, Phys. Chem. Chem. Phys., 2014, 16, 25601–25608 RSC.
- M. K. Singh, P. Chettri, J. Basu, A. Tripathi, B. Mukherjee, A. Tiwari and R. K. Mandal, Mater. Res. Express, 2020, 7, 015052 CrossRef CAS.
- D. J. Lee and D. Y. Kim, Sci. Rep., 2021, 11, 1–9 CrossRef PubMed.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- R. Jain, M. Mathur, S. Sikarwar and A. Mittal, J. Environ. Manage., 2007, 85, 956–964 CrossRef CAS PubMed.
- N. Tran, T. Phuong, T. Xoan, N. La, N. Tran and L. Gia, Spectrochim. Acta, Part A, 2021, 263, 120179 CrossRef PubMed.
- X. Liu and C. Lu, Indian J. Med. Res., 2021, 154, p760–p761 CrossRef PubMed.
- D. Bruen, C. Delaney, L. Florea and D. Diamond, Sensors, 2017, 17(8), 1866 CrossRef PubMed.
- C. Pimouguet, M. Le Goff, R. Thiébaut, J. F. Dartigues and C. Helmer, Can. Med. Assoc. J., 2011, 183, E115 CrossRef PubMed.
- E. R. Froesch and A. E. Renold, Diabetes, 1956, 5, 1–6 CrossRef CAS PubMed.
- M. D. Rotblatt and M. A. Koda-Kimble, Diabetes Care, 1987, 10, 103–110 CrossRef CAS PubMed.
- K. M. Dubowski, Clin. Chem., 2008, 54, 1919–1920 CrossRef CAS PubMed.
- S. P. Sharma, A. Prakash Anjankar and A. Kale, Int. J. Clin. Biochem. Res., 2017, 4, 6–10 Search PubMed.
- H. Chi, C. Wang, Z. Wang, H. Zhu, V. S. D. Mesias, X. Dai, Q. Chen, W. Liu and J. Huang, Analyst, 2020, 145, 5158–5165 RSC.
- J. Wang and G. Yi, Nanoscale Res. Lett., 2019, 14, 1–10 CrossRef PubMed.
- D. Das, S. Senapati and K. K. Nanda, ACS Sustainable Chem. Eng., 2019, 7(16), 14089–14101 CrossRef CAS.
- M. Aloraefy, T. Joshua Pfefer, J. C. Ramella-Roman and K. E. Sapsford, Sensors, 2014, 14, 12127–12148 CrossRef CAS PubMed.
- Y. Yuan, X. Yang, D. Gong, F. Liu, W. Hu, W. Cai, J. Huang and M. Yang, Opt. Express, 2017, 25, 3884 CrossRef CAS PubMed.
- W. Hergert and T. Wriedt, 35-The Mie Theory: Basics and Applications, ed.; Springer: Verlag Berlin and Heidelberg GmbH & Co. K., 2012 Search PubMed.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2013, 4, 3974–3983 RSC.
- W. H. Eisa, Y. K. Abdel-Moneam, A. A. Shabaka and A. E. M. Hosam, Spectrochim. Acta, Part A, 2012, 95, 341–346 CrossRef CAS PubMed.
- L. G. Bousiakou, H. Gebavi, L. Mikac, S. Karapetis and M. Ivanda, Croat. Chem. Acta, 2019, 92, 479–494 CrossRef CAS.
- Y. Yang, S. Matsubara, L. Xiong, T. Hayakawa and M. Nogami, J. Phys. Chem. C, 2007, 111, 9095–9104 CrossRef CAS.
- S. Khorrami, A. Zarrabi, M. Khaleghi, M. Danaei and M. R. Mozafari, Int. J. Nanomed., 2018, 13, 8013–8024 CrossRef CAS PubMed.
- Y. Cai, X. Piao, W. Gao, Z. Zhang, E. Nie and Z. Sun, RSC Adv., 2017, 7, 34041–34048 RSC.
- B. Khodashenas and H. R. Ghorbani, Arabian J. Chem., 2019, 12, 1823–1838 CrossRef CAS.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- H. Hofmeister, G. L. Tan and M. Dubiel, J. Mater. Res., 2005, 20, 1551–1562 CrossRef CAS.
- L. Xu, M. Sun, W. Ma, H. Kuang and C. Xu, Mater. Today, 2016, 19, 595–606 CrossRef CAS.
- L. Piantanida, D. Naumenko and M. Lazzarino, RSC Adv., 2014, 4, 15281–15287 RSC.
- S.-Y. Ding, E.-M. You, Z.-Q. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- V. Eskandari, H. Sahbafar, L. Zeinalizad, R. Mahmoudi, F. Karimpour, A. Hadi and H. Bardania, Arabian J. Chem., 2022, 15, 104005 CrossRef CAS.
- W. Kim, N. Kim, J. W. Park and Z. H. Kim, Nanoscale, 2016, 8, 987–994 RSC.
- E. E. Bedford, S. Boujday, C. M. Pradier and F. X. Gu, RSC Adv., 2015, 5, 16461–16475 RSC.
- S. N. Chen, X. Li, S. Han, J. H. Liu and Y. Y. Zhao, RSC Adv., 2015, 5, 99914–99919 RSC.
- J. Wang, H. Luo, M. Zhang, X. Zu, Z. Li and G. Yi, Nanoscale Res. Lett., 2017, 12, 587 CrossRef PubMed.
- C. H. Sun, M. L. Wang, Q. Feng, W. Liu and C. X. Xu, Russ. J. Phys. Chem. A, 2015, 89, 291–296 CrossRef CAS.
- C. Li, Y. Huang, K. Lai, B. A. Rasco and Y. Fan, Food Control, 2016, 65, 99–105 CrossRef CAS.
- M. J. Natan, Faraday Discuss., 2006, 132, 321–328 RSC.
- Y. Sun, W. Li, L. Zhao, F. Li, Y. Xie, W. Yao, W. Liu and Z. Lin, Food Chem., 2021, 357, 129741 CrossRef CAS PubMed.
- F. Liang, D. Jin, P. Ma, D. Wang, Q. Yang, D. Song and X. Wang, Anal. Lett., 2015, 48, 1918–1929 CrossRef CAS.
- S. W. Chook, C. H. Chia, C. H. Chan, S. X. Chin, S. Zakaria, M. S. Sajab and N. M. Huang, RSC Adv., 2015, 5, 88915–88920 RSC.
- T. T. Ha Pham, N. D. Dien and X. H. Vu, RSC Adv., 2021, 11, 21475–21488 RSC.
- C. Matsumoto, M. Gen, A. Matsuki and T. Seto, Sci. Rep., 2022, 12, 1–10 CrossRef PubMed.
- A. Saroj, U. Sharma, S. Das and V. Ramanathan, Spectrochim. Acta, Part A, 2022, 280, 121576 CrossRef CAS PubMed.
- A. T. Moraleda, C. V. García, J. Z. Zaballa and J. Arrue, Sensors, 2013, 13, 13076 CrossRef CAS PubMed.
- V. T. Tran, W. J. Yoon, J. H. Lee and H. Ju, J. Mater. Chem. A, 2018, 6, 23894–23902 RSC.
- A. K. Sharma and B. D. Gupta, Appl. Opt., 2006, 45, 151–161 CrossRef PubMed.
- T. Liu, D. R. Baek, J. S. Kim, S. W. Joo and J. K. Lim, ACS Omega, 2020, 5, 16246–16254 CrossRef CAS PubMed.
- P. Nalawade, T. Mukherjee and S. Kapoor, Adv. Nanopart., 2013, 2013, 78–86 CrossRef.
- V. T. Tran, N. H. T. Tran, T. T. Nguyen, W. J. Yoon and H. Ju, Micromachines, 2018, 9(9), 471 CrossRef PubMed.
- A. Belay and G. Assefa, J. Lasers Opt. Photonics, 2018, 5, 2 Search PubMed.
- T. T. Vu Nu, N. H. T. Tran, E. Nam, T. T. Nguyen, W. J. Yoon, S. Cho, J. Kim, K. A. Chang and H. Ju, RSC Adv., 2018, 8, 7855–7862 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The results provide information about the size and morphology of the investigated AgNPs including DLS analysis results, FESEM images, description of crystal size changes from the XRD pattern, and the ability to enhance in the SERS of the substrates over time of coating. See DOI: https://doi.org/10.1039/d2ra06074d |
| This journal is © The Royal Society of Chemistry 2022 |