
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of fully functionalised spiropyran pyrazolone skeletons via a formal [4 + 2] cascade process using β-nitro-styrene-derived MBH-alcohols†
Yeruva Pavankumar Reddy and
Shaik Anwar *
*
Department of Chemistry, School of Applied Sciences and Humanities, Vignan's Foundation for Science Technology and Research-VFSTR (Deemed to be University), Vadlamudi-522213, Guntur, Andhra Pradesh, India. E-mail: shaikanwarcu@gmail.com; drsa_sh@vignan.ac.in; Web: http://www.vignan.ac.in/bshanwar.php Tel: +91-8632344700
First published on 2nd December 2022
Abstract
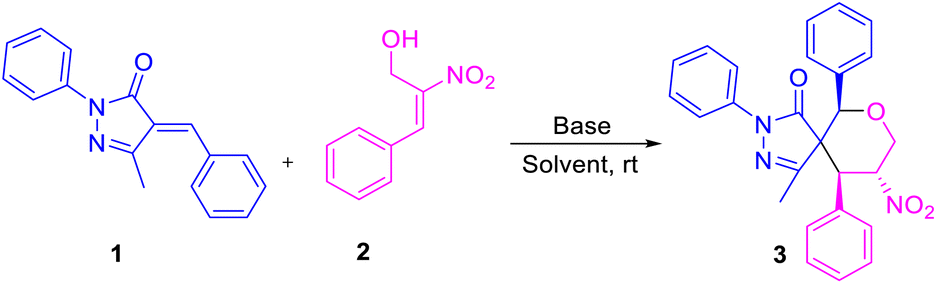
An efficient protocol was established to construct spiro pyrazolone tetrahydropyran scaffolds at ambient temperature under metal-free conditions. The reaction proceeded via formal [4 + 2] cyclisation of trans-β-nitro-styrene-derived Morita–Baylis–Hillman (MBH) alcohol with α-arylidene pyrazolone. The reaction followed an oxa-Michael/Michael cascade pathway, resulting in the formation of new C–C and C–O bonds. Organocatalytic synthesis of spiropyrazolones using quinine-derived catalyst resulted in 94% enantiomeric excess (ee) and excellent (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) diastereoselectivity.
:1) diastereoselectivity.
The nitrogen-containing pyrazolone compounds are highly efficient and amenable to their activity as antimicrobials, antitumor agents, and type 4 inhibitors of phosphodiesterase, and thereby play a crucial role in pharmaceutical and medicinal chemistry (Fig. 1).1 The synthesis of skeletons possessing spirocyclohexane pyrazolones,2 spiropyrazolone tetrahydroquinolines,3 spirobenzofuran pyrazoloedione,4 spiropyroloidinepyrazolones,5 spiro tetrahydrofuran pyrazolones,6 spiropyrazolone epoxide,7a spiro oxindole-fused spiropyrazolones,7b spirooxindole pyrrolidine pyrazolone,8 spiropyrazolonecyclohexene carbaldehydes,9 spiropyarazolonecyclohexanone,10 and fused pyrazolones such as dihydropyranopyrazoles11 and tetrahydropyranopyrazoles12 has received considerable interest in recent years. Arylidene pyrazolone has attracted considerable attention due to its unique 1,2-ambiphilic nature for the construction of elegant building blocks such as spirocycles,13 dispirocycles14 and fused heterocycles.15a–c Spiropyrazolones have been synthesized from the reaction of α-arylidene pyrazolone with various substrates, but the reaction using MBH adducts are scarce.15d To the best of our knowledge, there has been to date only one report on the synthesis of spiropyrazolone tetrahydropyran derivatives using alkylidene trimethylene carbonate,16 and no report on the synthesis of functionalized spiropyrazolones using MBH-alcohol with 5 contiguous stereocenters. Of the MBH adducts, β-nitro-styrene-derived MBH adducts were previously used by various research groups for the construction of spiropyrazolone skeletons (Scheme 1).
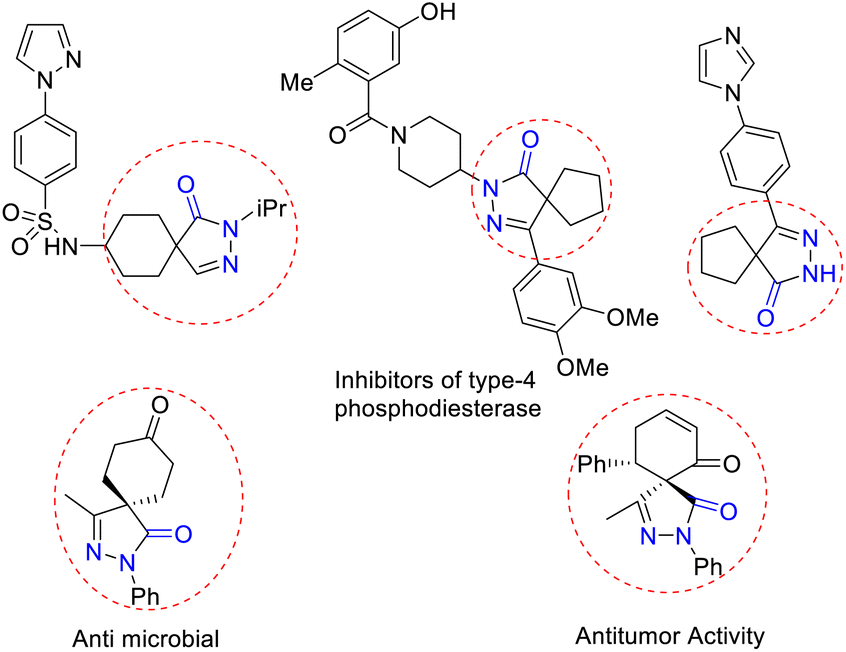 | ||
| Fig. 1 Biologically active spiropyrazolone skeletons. | ||
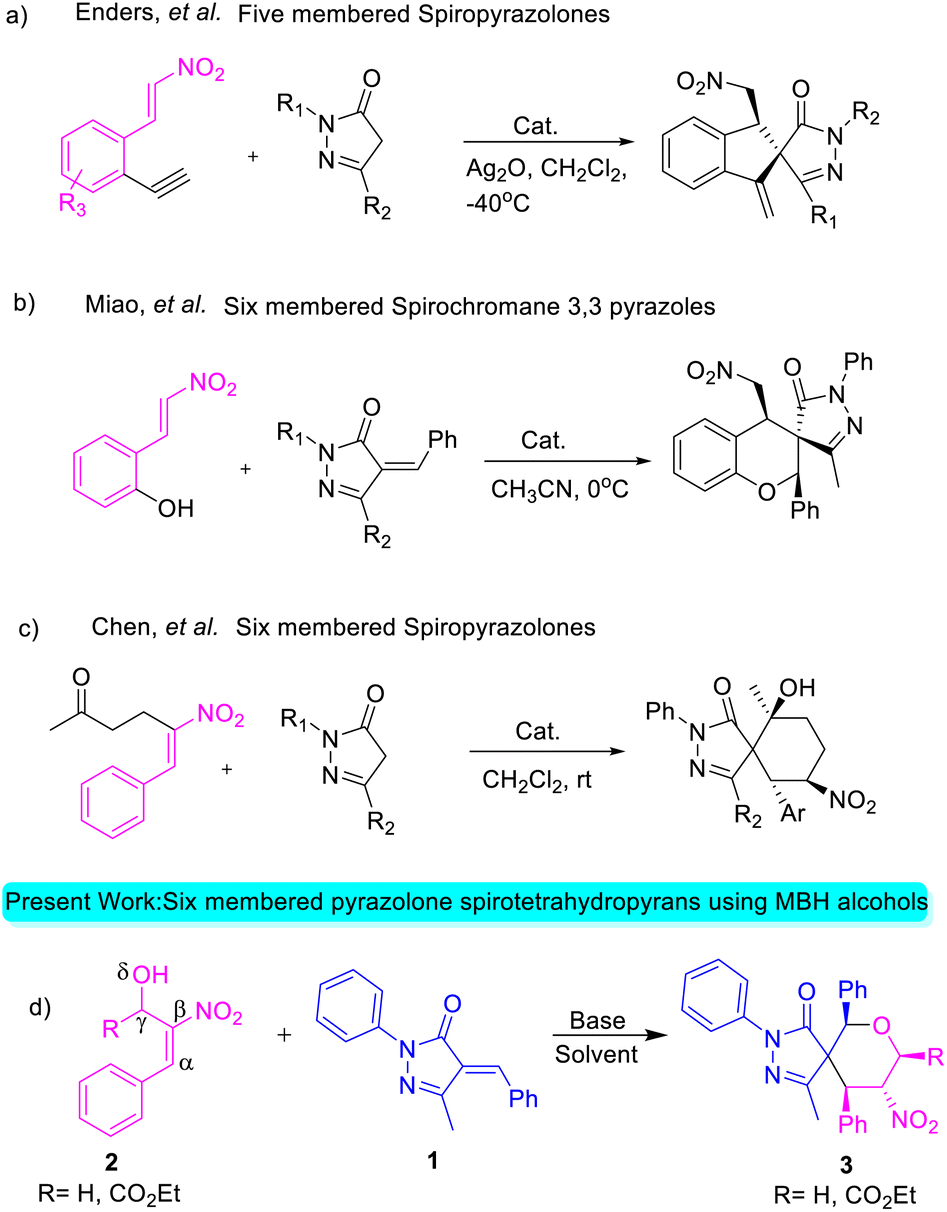 | ||
| Scheme 1 Annulation reactions using β-nitro-styrene-derived adducts. | ||
Enders and his team carried out a sequential organo- and silver catalysis for the synthesis of spiropyrazolones using alkyne-tethered nitroalkenes17 (eqn (a), Scheme 1). Miao et al. reported the synthesis of spirochromane 3,3 pyrazoles using 2-nitro vinyl phenols18 (eqn (b), Scheme 1). Chen and co-workers constructed spiranopyrazoles19 using 5-nito-6-phenyl-hex-5-en-2-one (eqn (c), Scheme 1). On the other hand, the utility of nitro-styrene-derived MBH alcohols as 1,4-bis-ambiphiles (α-C, δ-O) has been less extensively investigated.20 In continuation of our efforts towards the synthesis of various spirocyclic systems,21 herein we report the synthesis of spiropyrazolone tetrahydropyran scaffolds using β-nitro-styrene-derived MBH alcohols, resulting in the formation of the desired products with 4 to 5 contiguous chiral centers through [4 + 2] annulation (eqn (d), Scheme 1).
Initially, we carried out an optimization of conditions for the construction of spiro pyrazolone tetrahydropyran scaffolds using various types of solvents and bases at room temperature. Treatment of unsaturated arylidene pyrazolone and nitro-styrene-derived primary MBH alcohol using DABCO in the presence of acetonitrile (CH3CN) furnished the desired product in 16% yield (entry 1, Table 1). Performing the reaction instead in a polar solvent, e.g., THF, did not improve the yield (entry 2, Table 1). And performing the reaction instead in a chlorinated solvent, e.g., CHCl3 or DCM, also did not considerably enhance the yield of product 3a (entries 3 and 4, Table 1). An increase in yield was observed by shifting to an inorganic base, i.e., Cs2CO3, which together with using CH2Cl2 as the solvent gave the product 3a in 60% yield (entry 5, Table 1); and here, use of the polar aprotic solvent CH3CN instead of CH2Cl2 increased the yield to 66% (entry 6, Table 1). The best reaction conditions were obtained when using K2CO3 as an inorganic base in CH3CN to obtain product 3a in 93% yield (entry 7, Table 1). A decrease in the yield for product formation was observed when heating the reaction mixture at 60 °C (entry 8, Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) | drb |
| a Unless otherwise noted, reactions were carried out with (0.19 mmol of) 1 with (0.28 mmol of) 2 using 0.47 mmol of base in 1.5 ml of CH3CN solvent.b Determined from a 1H-NMR analysis of a crude reaction mixture.c Reaction carried out at 60 °C. | |||||
| 1 | DABCO | CH3CN | 8 | 16 | n.d |
| 2 | DABCO | THF | 7 | 22 | n.d |
| 3 | DABCO | CHCl3 | 7 | 27 | >20:1 |
| 4 | DABCO | DCM | 7 | 25 | n.d |
| 5 | Cs2CO3 | CH2Cl2 | 1 | 60 | > 20:1 |
| 6 | Cs2CO3 | CH3CN | 1 | 66 | >20:1 |
| 7 | K2CO3 | CH3CN | 1 | 93 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1 |
| 8c | K2CO3 | CH3CN | 1 | 76 | >20:1 |
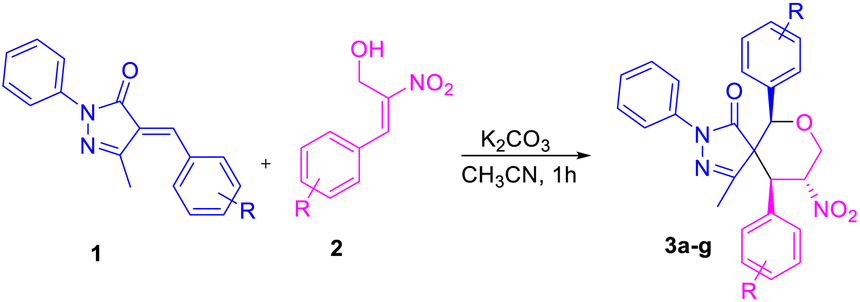
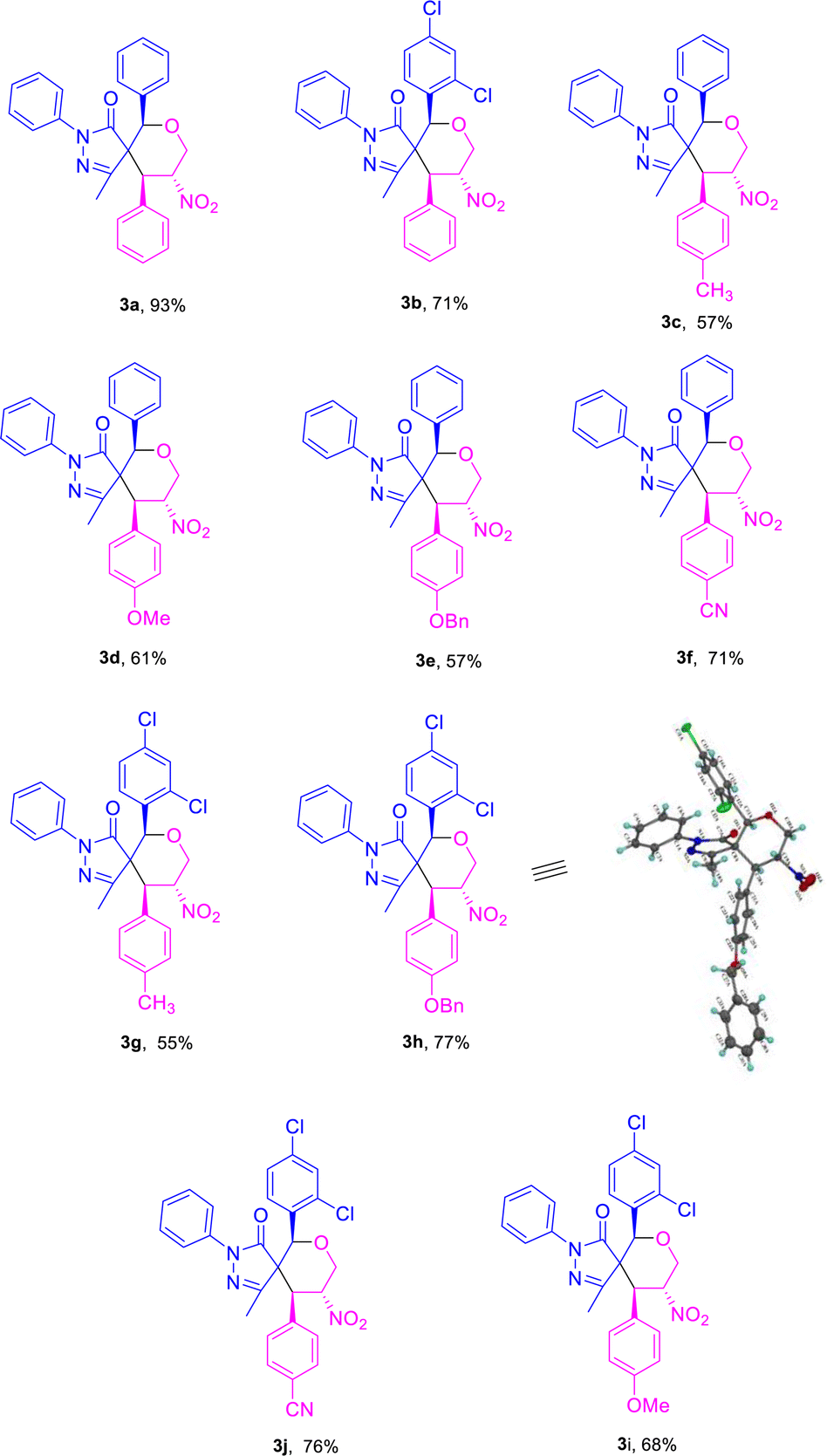
Based on the best optimized conditions, we studied the scope of different substituents at the aryl ring of pyrazolone 1 as well as primary MBH alcohol 2. The 2,4-dichloro-substituted arylidene pyrazolone 1b gave the desired product 3b in 71% yield (Table 2). Use of the electron-donating group –CH3 at the para position of the MBH alcohol furnished the corresponding product 3c in 57% yield. Electron-rich donating groups –OMe and –OBn gave 3d–e in 61 and 57% yields, respectively. An electron-withdrawing group at the para position also gave a good yield for product 3f. We examined the yield and functional group tolerance by changing the substituents at arylidene pyrazolones and MBH alcohols; here, products 3g–j were obtained in moderate to good yields.
 |
 |
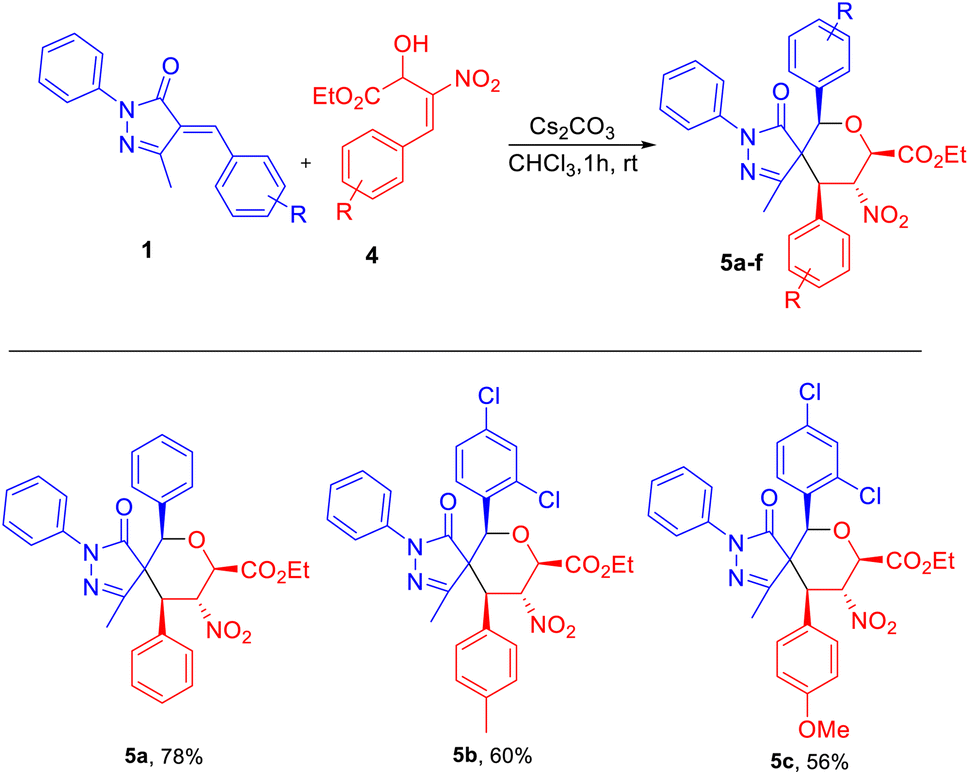
We next focused on building fully substituted spiro pyrazolone tetrahydropyrans scaffolds 5a–c using β-nitro-styrene-derived secondary (2°)-MBH alcohols with arylidene pyrazolones. With the best optimized set of conditions obtained previously, we carried out the construction of fully substituted spiropyran pyrazolone using K2CO3 in CH3CN to give 5a in 67% yield (entry 1, Table 3). The chlorinated solvents CH2Cl2, CHCl3, and CCl4 gave 5a in only 41–56% yields (entries 2–4, Table 3). And a further decline in yield was observed when using instead THF as solvent (entry 5, Table 3). We found that Cs2CO3 in the presence of CHCl3 was the best base–solvent combination for the formation of product 5a, with a 78% yield and good diastereoselectivity (entry 6, Table 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield | drb |
| a Unless otherwise noted, reactions were carried out with (0.19 mmol of) 1 with (0.28 mmol of) 4 using 0.47 mmol% of base in 1.5 ml of CHCl3 solvent.b Diastereomeric ratio was determined from 1H-NMR analysis of a crude reaction mixture. | |||||
| 1 | K2CO3 | CH3CN | 1 | 67 | >20:1 |
| 2 | K2CO3 | CH2Cl2 | 1 | 56 | n.d |
| 3 | K2CO3 | CHCl3 | 1 | 47 | >20:1 |
| 4 | K2CO3 | CCl4 | 1 | 41 | >10:1 |
| 5 | K2CO3 | THF | 1 | 36 | n.d |
| 6 | Cs2CO3 | CHCl3 | 1 | 78 | >20:1 |
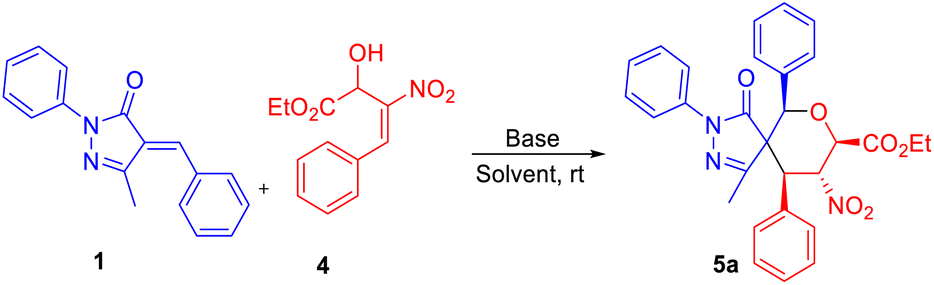
Use of the electron-donating groups methyl and methoxy at the para position of the MBH alcohol resulted in 56–60% yields of product, i.e., of 5c and 5b (Table 4). Furthermore, all the compounds 3a–j and 5a–c were confirmed from the results of IR, 1H, 13C NMR, HRMS, and NOESY analyses. The compound 3h was further confirmed using single-crystal XRD (Table 2).22
 |
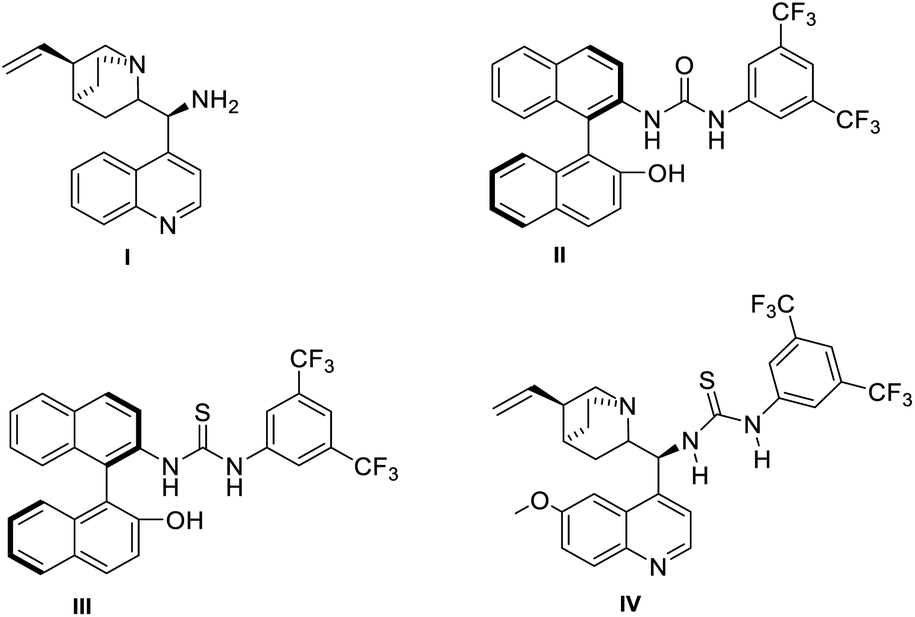
We further pursued our studies towards asymmetric synthesis of spiropyrazolones 3a using various chiral catalysts (I–IV). We observed a poor enantiomeric excess for the product formation in the presence of cinchona catalyst I (entry 1, Table 5). Using NOBIN-based catalysts II and III resulted each in a 10% enantiomeric excess (entries 3–4, Table 5). Interestingly, we obtained an excellent enantiomeric excess (94% ee) with high diastereoselectivity (>20:1) when using the thiourea-based hydrogen bonding catalyst IV (entry 4, Table 5; see ESI† for information on the transition state).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yield (%) | eeb (%) | drc |
| a All the reactions were carried out with (0.19 mmol of) 1, (0.11 mmol of) 2 and 10 mol% of catalyst in 1 ml of CH3CN solvent.b Enantiomeric excess determined from HPLC analysis.c Diastereomeric ratio was determined from 1H-NMR analysis of a crude reaction mixture. | |||||
| 1 | I | 1 | 35 | 50 | >20:1 |
| 2 | II | 1 | >10 | 17 | >20:1 |
| 3 | III | 1 | >15 | 11 | >20:1 |
| 4 | IV | 1 | 61 | 94 | >20:1 |
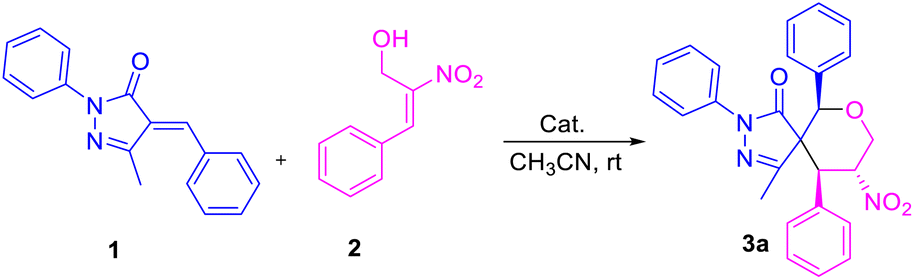
To further demonstrate the practical and scalable utility of our protocol, we carried out gram scale preparation of spiro pyrazolone tetrahydropyrans 3a and 5a, and achieved yields of 81% and 66% (Scheme 2).
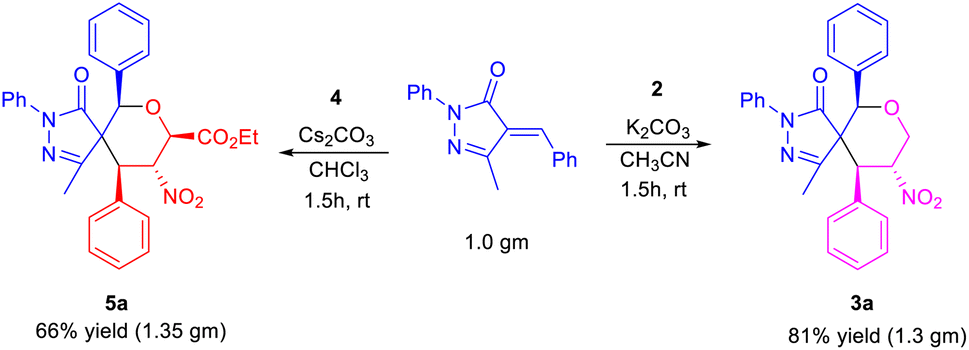 | ||
| Scheme 2 Gram scale synthesis of spiro pyrazolone tetrahydropyrans 3a and 5a. | ||
We investigated the feasibility of carrying out a triple-cascade reaction for the construction of spiro pyrazolone tetrahydropyrans 3a and 5a via the Knoevenagel/oxa-Michael/Michael process. To our delight, the reaction was amenable to a one-pot [1 + 1 + 4] formal cyclization to give the products 3a and 5a in 58% and 62% yields (Scheme 3).
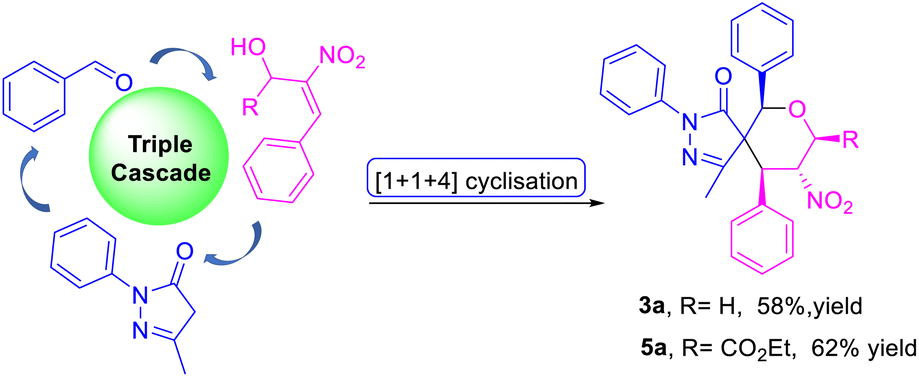 | ||
| Scheme 3 Approaching a triple cascade for the construction of spiro pyrazolone tetrahydropyran in a three-component manner. | ||
Most of the annulation reactions using MBH adducts involve the use of inorganic bases for proton abstraction, in line with a previous literature report,23 and in the current work a plausible mechanism for the construction of spiropyrazolone tetrahydropyran scaffolds was derived. According to this proposed mechanism, the initial reaction of the alkali carbonate serving as a base (i.e., K2CO3/Cs2CO3) with MBH alcohol generated the nucleophilic oxygen intermediate A. Attack by intermediate A from the “rear” position onto the benzylic carbon of α-arylidine pyrazolone via an oxa-Michael reaction generated a new O–C bond. And finally according to the proposed mechanism, further rearrangement of the resulting enol to a ketone and subsequent attack on the electrophilic olefinic site of MBH alcohol via formal [4 + 2] annulation resulted in the formation of a new C–C bond through a Michael reaction (Scheme 4). In situ Raman studies carried out for the reaction mixture shows the presence of keto group (i.e. 1495 cm−1) of α-arylidene pyrazolone at the beginning of the reaction. This corresponding peak of 1495 cm−1 gradually disappeared after the initial oxa-Michael addition to form intermediate B. The intermediate B on reaction with benzyl bromide resulted in the disappearance of corresponding keto group peak, before completion of the final cyclisation via Michael Addition (S5 page of ESI†).
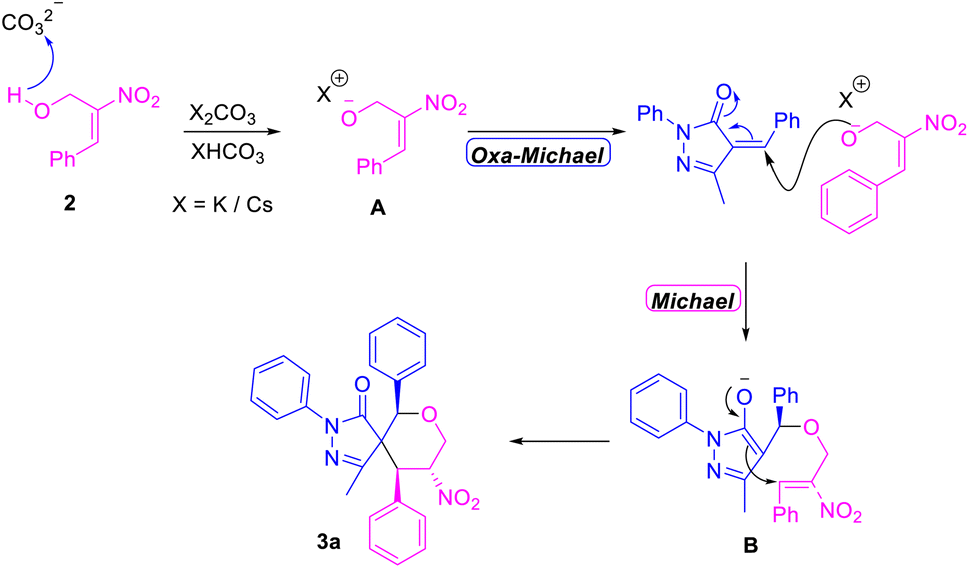 | ||
| Scheme 4 Plausible reaction mechanism for the synthesis of spiro pyrazolone tetrahydropyrans. | ||
Conclusions
In conclusion, the 1,4 ambiphilicity of β-nitro-styrene-derived MBH alcohols was investigated for achieving an efficient synthesis of tetrahydrospiropyrazolones via formal [4 + 2] cyclization at room temperature within 1 h. β-Nitro-styrene-derived 1° MBH alcohol gave tetrasubstituted spiropyrazolones when using K2CO3 whereas 2° MBH alcohol gave fully substituted spiropyrazolones when using Cs2CO3. The reaction tolerated various electron-withdrawing and electron-donating groups on the aryl ring of the arylidene pyrazolone as well as β-nitro-styrene-derived MBH alcohols to result in the desired products in high yields. Organocatalytic synthesis using quinine-derived thiourea catalyst resulted in desired spiropyrazolones with >94% enantiomeric excess and >20:1 dr. Interestingly, a triple-cascade three-component reaction produced the same spiropyrazolone tetrahydropyrans via the Knoevenagel/oxa-Michael/Michael process.
Author contribution
All authors contributed to the conception and design of the study. Material preparation, data collection, and analysis were performed by Yeruva Pavankumar Reddy. Shaik Anwar contributed additional analysis required to address the comments and issues from the reviewers. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SA thanks DST-SERB for providing the financial support under a Fast Track scheme (SB/FT/CS/079-2014). We are indebted to Vignan’s Foundation for Science, Technology and Research for providing research facilities at CoExAMMPC.Notes and references
- (a) M. S. Chande, P. A. Barve and V. Suryanarayan, J. Heterocycl. Chem., 2007, 44, 49–53 CrossRef CAS; (b) S. Wu, Y. Li, G. Xu, S. Chen, Y. Zhang, N. Liu, G. Dong, C. Miao, H. Su, W. Zhang and C. Sheng, Eur. J. Med. Chem., 2016, 115, 141 CrossRef CAS PubMed; (c) I. Schlemminger, B. Schmidt, D. Flockerzi, H. Tenor, C. Zitt, A. Hatzelmann, D. Marx, C. Braun, R. Kuelzer, A. Heuser, H.-P. Kley and G. J. Sterk, Germany Patent WO2010055083, 2008; (d) B. Schmidt, C. Scheufler, J. Volz, M. P. Feth, R.-P. Hummel, A. Hatzelmann, C. Zitt, A. Wohlsen, D. Marx, H.-P. Kley, D. Ockert, A. Heuser, J. A. M. Christiaans, G. J. Sterk and W. M. P. B. Menge, Germany Patent WO2008138939, 2010.
- J.-Y. Liu, J. Zhao, J.-L. Zhang and P.-F. Xu, Org. Lett., 2017, 19, 1846 CrossRef CAS PubMed.
- J.-H. Li and D.-M. Du, Chem. Asian J., 2004, 9, 3278 CrossRef PubMed.
- X.-L. Zhang, C.-K. Tang, A.-B. Xia, K.-X. Feng, X.-H. Du and D.-Q. Xu, Eur. J. Org. Chem., 2017, 2017, 3152 CrossRef CAS.
- J.-H. Li, H. Wen, L. Liu and D.-M. Du, Eur. J. Org. Chem., 2016, 2016, 2492 CrossRef CAS.
- B. Mondal, R. Maity and S. Pan, J. Org. Chem., 2018, 83, 8645 CrossRef CAS PubMed.
- (a) S. Meninno, A. Roselli, A. Capobianco, J. Overgaard and A. Lattanzi, Org. Lett., 2017, 19, 5030 CrossRef CAS PubMed; (b) Y. Lin, B.-L. Zhao and D.-M. Du, J. Org. Chem., 2019, 84, 10209 CrossRef CAS PubMed.
- C. Wang, D. Wen, H. Chen, Y. Deng, X. Liu, X. Liu, L. Wang, F. Gao, Y. Guo, M. Sun, K. Wang and W. Yan, Org. Biomol. Chem., 2019, 17, 5514 RSC.
- A. Zea, R. Alba, A. Mazzanti, A. Moyanoa and R. Rios, Org. Biomol. Chem., 2011, 9, 6519 RSC.
- J.-X. Zhang, N.-K. Li, Z.-M. Liu, X.-F. Huang, Z.-C. Geng and X.-W. Wang, Adv. Synth. Catal., 2013, 355, 797 CrossRef CAS.
- (a) H. Zhang, H. Lv and S. Ye, Org. Biomol. Chem., 2013, 11, 6255 RSC; (b) J. Li and D. Du, Chin. J. Chem., 2015, 33, 418 CrossRef CAS.
- S. Wang, C. Rodriguez-Escrich and M. Pericàs, Org. Lett., 2016, 18, 556 CrossRef CAS PubMed.
- (a) C. Zhao, K. Shi, G. He, Q. Gu, Z. Ru, L. Yang and G. Zhong, Org. Lett., 2019, 21, 7943 CrossRef CAS PubMed; (b) W. Yang, Y. Zhang, S. Qiu, C. Zhao, L. Zhang, H. Liu, L. Zhou, Y. Xiao and H. Guo, RSC Adv., 2015, 5, 62343 RSC; (c) P. Sun, C.-Y. Meng, F. Zhou, X.-S. Li and J.-W. Xie, Tetrahedron, 2014, 70, 9330 CrossRef CAS.
- (a) B.-D. Cui, S.-W. Li, J. Zuo, Z.-J. Wub, X.-M. Zhanga and W.-C. Yuana, Tetrahedron, 2014, 70, 1895 CrossRef CAS; (b) N. Chen, L. Zhu, L. Gan, Z. Liu, R. Wang, X. Cai and X. Jiang, Eur. J. Org. Chem., 2018, 2018, 2939 CrossRef CAS.
- (a) L. Liu, Y. Zhong, P. Zhang, X. Jiang and R. Wang, J. Org. Chem., 2012, 77, 10228 CrossRef CAS PubMed; (b) A.-B. Xia, X.-L. Zhang, C.-K. Tang, K.-X. Feng, X.-H. Dua and D.-Q. Xu, Org. Biomol. Chem., 2017, 15, 5709 RSC; (c) s. wang, J. Izquierdo, R.-E. Carles and A. Pericàs, ACS Catal., 2017, 7, 2780 CrossRef CAS; (d) J.-Y. Liu, J. Zaho, J.-L. Zhang and P.-F. Xu, Org. Lett., 2017, 19, 1846 CrossRef CAS PubMed.
- B. Mao, H. Liu, Z. Yan, Y. Xu, J. Xu, W. Wang, Y. Wu and H. Guo, Angew. Chem., Int. Ed., 2020, 59, 11316 CrossRef CAS PubMed.
- D. Hack, Alexander B. Durr, K. Deckers, P. Chauhan, N. Seling, L. Rubenach, L. Martens, G. Raabe, F. Schoenebeck and D. Enders, Angew. Chem., Int. Ed., 2016, 55(5), 1797–1800 CrossRef CAS PubMed.
- W. Zheng, J. Zhang, S. Liu and Z. Miao, RSC Adv., 2015, 5, 91108 RSC.
- M. Amireddy and K. Chen, RSC Adv., 2016, 6, 77474 RSC.
- (a) Y. P. Reddy, V. Gudise, P. C. Settipalli and S. Anwar, ChemistrySelect, 2021, 6, 4456 CrossRef CAS; (b) V. Gudise, P. C. Settipalli, E. K. Reddy and S. Anwar, Eur. J. Org. Chem., 2019, 2019, 2234 CrossRef CAS.
- (a) V. Gudise, P. C. Settipalli, Y. P. Reddy and S. Anwar, ChemistrySelect, 2021, 6, 13589 CrossRef CAS; (b) S. Anwar, L.-T. Lin, V. Srinivasadesikan, V. B. Gudise and K. Chen, RSC Adv., 2021, 11, 38648 RSC; (c) P. C. Settipalli, Y. P. Reddy, V. Gudise and S. Anwar, ChemistrySelect, 2021, 6, 47 CrossRef CAS.
- CCDC 2215596 for 3h, possess the crystallographic data for this manuscript. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- (a) E. Gopi and I. N. N. Namboothiri, J. Org. Chem., 2014, 79, 7468 CrossRef CAS PubMed; (b) T. Kumar, S. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 4964–4972 CrossRef CAS; (c) W.-Y. Huang, Y.-C. Chen and K. Chen, Chem. Asian J., 2012, 7, 688–691 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2215596. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06076k |
| This journal is © The Royal Society of Chemistry 2022 |