
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new integrated method for tissue extracellular vesicle enrichment and proteome profiling†
Miaomiao Zhang ab,
Tong Liub,
Zhuokun Dub,
Hang Li‡
*b and
Weijie Qin*ab
ab,
Tong Liub,
Zhuokun Dub,
Hang Li‡
*b and
Weijie Qin*ab
aDepartment of Immunology, Medical College of Qingdao University, Qingdao, Shandong 266071, PR China. E-mail: aunp_dna@126.com
bNational Center for Protein Sciences Beijing, State Key Laboratory of Proteomics, Beijing Proteome Research Center, Beijing Institute of Lifeomics, Beijing 102206, PR China. E-mail: hang.li@bit.edu.cn
First published on 22nd November 2022
Abstract
Extracellular vesicles (EVs) are membranous vesicles released by cells that carry a number of biologically important components such as lipids, proteins, and mRNAs. EVs can mediate cancer cell migration, invasion, angiogenesis, and cell survival, greatly contributing to cell-to-cell communication in the tumor microenvironment. Additionally, EVs have been found to have diagnostic and prognostic significance in various cancers. However, the direct isolation of pure EVs remains challenging, especially from tissue samples. Currently available EV isolation approaches, e.g., ultracentrifugation, are time-consuming, instrumental dependent, and have a low recovery rate with limited purity. It is urgent to develop rapid and efficient methods for enriching tissue EVs for biological and clinical studies. Here, we developed a novel isolation approach for tissue EVs using an extraction kit combined with TiO2 microspheres (kit-TiO2). The EVs were first precipitated from the tissue fluid using a precipitation agent and then further enriched using microspheres based on the specific interaction between TiO2 and the phosphate groups on the lipid bilayer of the EVs. Kit-TiO2 approach led to improved purity and enrichment efficiency of the isolated EVs, as demonstrated by western blot and proteomic analysis, compared with previously reported methods. A total of 1966 protein groups were identified from the tissue EVs. We compared the proteomic profiles of the liver tissue EVs from healthy and hepatocellular carcinoma (HCC) bearing-mice. Twenty-five significantly upregulated and 75 downregulated protein groups were found in the HCC EVs. Among the differentially expressed proteins, Atic, Copa, Cont3, Me1, Anxa3, Fth1, Anxa5, Phb1, Acaa2, ATPD, and Glud1 were reported to be highly relevant to HCC. This novel isolation strategy has provided a powerful tool for enriching EVs directly from tissues, and may be applied in biomarker discovery and drug screening of HCC.
1. Introduction
EVs are cell-derived membranous vesicles with phospholipid bilayers, ranging from 30 to 1000 nm in diameter.1,2 These cell-derived vesicles exist in a variety of body fluids, e.g., serum, urine, saliva, milk, bile, and other fluids.3–6 EVs share the same topology as cells and contain various biologically important molecules, including proteins, RNA, and DNA.7 EVs have play major roles in intercommunication during biological processes, particularly in cancer initiation and progression.8–10 It is worth noting that the characterization of EVs has evolved from in vitro investigation of cell culture supernatants to more recently in vivo studies on directly isolated EVs from animal or human body fluids, e.g., plasma, urine, and saliva.11–13While EVs are found in large quantities in the circulatory system, EVs also exist in the interstitial tissues and play a key role in cell-to-cell communication. In tumors, interstitial EVs are essential in modulating the tumor microenvironment for cell seeding and metastatic growth.14 Compared to EVs obtained from cell cultures, EVs directly isolated from tissues show great advantages in tissue specificity and close relevance to the microenvironment.12 Therefore, optimized techniques for EV extraction from solid tissue specimens are needed to enable in vivo study of tumor EVs and in-depth exploration of their underlying biological mechanisms.
Hepatocellular carcinoma (HCC) is the third most common cancer in the world, accounting for 85–90% of primary hepatic tumors.15–17 HCC cells can interfere with the biological behaviors of different types of cells by releasing EVs. For example, secreted EVs regulate the epithelial-mesenchymal transition (EMT) in the microenvironment to transform the surroundings into an inflammatory state, coordinate with neighboring tumor cells to increase invasiveness, and induce the conversion of adjacent fibroblasts and macrophages to CAFs and TAMs.18 Moreover, HCC-released EVs might interfere with immune cells and endothelial cells to induce immune escape and angiogenesis.19,20 HCC-secreted EVs also mediate signaling pathways and regulatory factors involved in intercellular interactions.21
Several studies have revealed that differentially expressed EV miRNAs might be used as biomarkers for diagnosing HCC. Additionally, the delivery of EV miRNAs and lncRNAs may significantly inhibit cancer development.22,23 Therefore, EV components are potential diagnostic markers and therapeutic targets for HCC.
In terms of separating EVs from liver tissues or any other tissue, it is critical to properly disrupt the tissue while minimizing cellular disruption. One or more EV separation steps might be imposed to increase the EV purity with minimal cellular contamination.24 Previously published methods rely on tissue homogenization and filtration, which is likely to disrupt the cellular structure and mix intracellular vesicles with EVs.25,26 Current methods for EV isolation include ultracentrifugation, precipitation kits, immunoaffinity approaches, size exclusion chromatography (SEC), and ultrafiltration.27,28 However, ultracentrifugation is time-consuming and repeated centrifugation might lead to EV loss, resulting in limited throughput and recovery rates.29 The kit precipitation method is mainly based on polymer precipitation, but the purity of the extracted EV is relatively low. The introduced polymers are also difficult to remove and may lead to severe interference in downstream analysis.30,31 SEC shows a good recovery rate and reproducibility. However, the size of the purification column limits the quantity of the sample and the throughput of this method. Additionally, the elution step dilutes the sample, resulting in a low concentration of the final product. The immunoaffinity approach isolates EVs through the specific interactions between antibody and EV membrane proteins. The recovery of EVs is unpredictable, depending on the performance of the antibody, and the cost is relatively high.
In this report, we established a novel method to enrich EVs from liver tissue based on a two-step method via a combination of an extraction kit and TiO2 microspheres. In this method, the aqueous contents and complex components in the tissue are first removed by a kit method, and then the tissue EVs are extracted using TiO2 microspheres via the specific interaction between TiO2 and the phosphate groups on the EV lipid bilayers. The isolated EVs were characterized by their size, morphology, and proteomic profiles. The proposed enrichment approach has been demonstrated to have high efficiency and good reproducibility for enriching tissue EVs. Obviously improved purity and quantity of the enriched EVs was achieved as demonstrated by western blot and proteomic analysis compared with previously reported methods. Furthermore, we successfully applied this approach to analyze HCC samples. In total, 25 significantly upregulated proteins were found, 11 of which were associated with liver cancer, demonstrating the potential of the proposed new method for tissue EVs for cancer studies.
2. Materials and methods
2.1 Animal models
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Beijing Proteome Research Center, and approved by the Animal Ethics Committee of Beijing Proteome Research Center (license: IACUC-20211228-42). C57BL/6 mice were purchased from Beijing Weitong Lihua Inc. The HCC model mice (Available from Shanghai Model Organisms Center, Inc) were established as previously described.32 Briefly, 2 week-old C57 male mice (reared at SPF level 5) were injected intraperitoneally with DEN (Sigma N0258, 40 mg kg−1, configured with saline to 4 mg mL−1, 10 μL g−1) once, and 6 weeks later (8 week-old mice) they were injected with CCL4 (National Drug 10006464, configured with corn oil (ABCone, C67366) to 10%, 10 μL g−1) twice a week for 20 weeks. At the 16th week of administration, the liver was dissected and observed to check for primary liver cancer. During the experiment, the mice were observed daily for coat sheen, activity, feces, water, and food administration, and weighed once a week to monitor their health status.2.2 Liver tissue samples from the mice
Phosphate-buffered saline (PBS) (VIVICUM, VCM3008) was preheated in a 37 °C water bath, and all surgical instruments were sprayed with 70% ethanol and wiped dry. The pump tubing was flushed by running 70% ethanol through and rinsed twice with running water to remove any residue. A sterile 5 cm Petri dish with RPMI-1640 medium (VIVICUM, VCM5140) was prepared to hold the digested liver after perfusion. The mice were injected intraperitoneally with 400 μL of anesthetic (3% sodium pentobarbital) and were checked to ensure complete anesthesia through a skin pinch clamp response or toe stimulation response. All four limbs of the mouse were held down with pins. The abdominal skin was cleaned by spraying with 70% ethanol and wiping with gauze and an alcohol pad, avoiding mouse fur contamination. The skin was cut from the anterior to the pelvic bone using sterile scissors. The portal vein (PV) and inferior vena cava (IVC) were exposed by gently applying a cotton-tipped applicator, and the intestine was moved to the left side of the animal. Then, a lagging needle was inserted carefully from the portal vein. Once the blood returned, the inferior portal cava was perfused with preheated irrigation solution at a flow rate of 3.5–4 mL min−1 for 4 min. During the perfusion, the inferior vena cava was severed and clamped with forceps for 7–10 s until the liver was enlarged. To ensure that the liver was swollen and relaxed, the procedure was repeated at 30 s intervals. After 5–7 perfusions, the liver blood was completely removed through the inferior vena cava as the liver turned white. Then, the liver was cut out with the gallbladder carefully removed and placed in a prechilled Petri dish containing PBS.2.3 Preparation of mouse liver tissue fluid
The liver tissue was gently sliced into small sections (2–4 mm3) and incubated for 20 min at 37 °C in the RPMI-1640 medium supplemented with collagenase D (Sigma, c5138-1G, 2 mg mL−1) and DNase I (Solarbio, no. D8071, 40 U mL−1). The incubated tissue blocks were gently pressed through a cell filter on top of a conical tube to remove any undigested tissue. The dissociated tissues were then centrifuged at 300 × g for 10 min at 4 °C. The generated pellet (“liver homogenate with collagenase, LHC”) was stored at −80 °C prior to protein extraction. The supernatant was transferred to a fresh tube and centrifuged at 2000 × g for 20 min at 4 °C to remove cell debris and aggregates. The separated supernatant was further centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min at 4 °C to remove any large or apoptotic vesicles. The obtained tissue fluid was stored at −80 °C prior to EV isolation.
000 × g for 30 min at 4 °C to remove any large or apoptotic vesicles. The obtained tissue fluid was stored at −80 °C prior to EV isolation.
2.4 Isolation of tissue EVs by kit combined with TiO2 strategy
The tissue fluid sample (1 mL) was added to 250 μL of EV precipitant (Umibio EV extraction and purification kit) and incubated at 4 °C for 2 h. The solution was centrifuged at 10000 × g for 1 h and the supernatant was removed. The EV precipitate was resuspended and mixed with phenol red-free DMEM (VIVICUM, VCM50520). The supernatant was then mixed with 20 mg TiO2 microspheres (I.D. = 5 μm, GL Sciences) and incubated for 10 min at 4 °C on a shaker (THZ-C, Thermo Fisher Scientific) for sufficient enrichment. The sample was then centrifuged at 1000 × g for 5 min at 4 °C. The precipitate was washed with 200 μL 150 mM sodium chloride and centrifuged at 1000 × g for 5 min at 4 °C. After washing three times, the obtained precipitate was TiO2 microspheres enriched with tissue EVs.
2.5 Isolation of tissue EVs by TiO2
The above tissue fluid sample (1 mL) was directly mixed with 20 mg TiO2 microspheres and incubated for 10 min at 4 °C on a constant temperature shaker for adequate enrichment. After incubation, the samples were centrifuged at 1000 × g for 5 min at 4 °C. Next, the separated precipitate was washed three times with 200 μL 150 mM sodium chloride and centrifuged at 1000 × g for 5 min at 4 °C. Finally, the precipitated TiO2 microspheres with tissue EVs attached were obtained.2.6 Isolation of tissue EVs by precipitation reagent
Using the Umibio EV extraction and purification kit, the tissue fluid sample (1 mL) was added to 250 μL EV precipitant and incubated at 4 °C for 2 h. The sample was centrifuged at 10000 × g for 1 h and the supernatant was removed. The enriched EV pellet was suspended in PBS for downstream experiments.
2.7 Isolation of tissue EVs by ultracentrifugation
According to a previously described method,33,34 the tissue fluid sample was ultracentrifuged at 110000 × g for 70 min at 4 °C. The precipitated pellet was washed twice with PBS by ultracentrifuging the mixture at 110000 × g for 70 min at 4 °C. The obtained EV pellet was stored at −80 °C until further analysis.
2.8 Liver homogenate preparation
For proteomic analysis, liver tissue was ground in cold PBS for 60 s using a cryogenic tissue grinder (Thermo Fisher Scientific) to obtain collagenase-treated (LHC) liver homogenates. The ground tissue was added to 4% SDS lysis buffer and sonicated on ice for 20 min. The homogenate was then centrifuged at 14000 × g for 10 min at 4 °C. The resulting protein supernatant was stored at −80 °C until analysis.
2.9 Characterization of the isolated tissue EVs
2.10 Proteomic analysis
The isolated EVs attached to TiO2 microspheres were dissolved in 40 μL of 4% SDS buffer containing 1% protease inhibitor. The resulting suspension was sonicated on ice for 20 min and then centrifuged at 10000 g at 4 °C for 5 min. The separated supernatant was processed immediately for the filter-aided sample preparation (FASP) digestion. The supernatant was diluted with 8 M urea to a final concentration of 0.1% SDS, and the diluted solution was transferred into a 30 kDa ultrafiltration tube to remove the SDS. Then, the proteins were reduced using 10 mM dithiothreitol for 4 h at 37 °C and alkylated with 20 mM iodoacetamide in the dark at RT for 40 min. The protein concentrations were measured with a Nanodrop 2000 (Thermo Scientific). Trypsin was added at a 1:50 (w/w) ratio and incubated overnight at 37 °C. The final obtained peptide solution was thermally dried using a vacuum rotary drier. Subsequent stable isotope dimethylation labeling the of peptides was conducted. Liver tissue samples from healthy and HCC mice were used for EV protein identification and relative quantification. The peptides were first redissolved in 200 μL of 100 mM TEAB buffer solution. Then, 4 μL of 4% formaldehyde-D2 solution was added to the HCC group and 4 μL of 4% formaldehyde solution was added to the healthy group. Additionally, 4 μL of 0.6 M sodium cyanoborohydride was added to each sample, vortexed and mixed. The reaction was carried out at room temperature for 1 h. Next, 16 μL of 1% ammonia solution acidified with formic acid was added to terminate the reaction. Finally, the light and heavy isotope-labeled groups were mixed and desalted using StageTip C18. The desalted peptides were concentrated and dried using a vacuum spin dryer, and then stored at −80 °C until analysis. The obtained peptides (500 ng) were used for LC-MS analysis.
LC-MS analysis was performed using a Q-Exactive HF-X mass spectrometer (Thermo Fisher Scientific, USA) equipped with an UltiMate 3000 LC system. A 15 cm-long LC column packed with 1.9 μm C18 packing particles was used for the peptide separation. The mobile-phase solvents were HPLC water acidified by 0.1% FA for phase A and 80% acetonitrile acidified by 0.1% FA for phase B. An 88 min gradient of 6–45% buffer B was used for peptide elution at a flow rate of 600 nL min−1. A high voltage of 2.3 kV was applied to the electrospray. The MS system was operated in positive ion mode at a resolution of 120 000 (at m/z 200) using data-dependent acquisition (DDA) and full scans were acquired with a scan range of m/z 350–1550 with a target of 3 × 106 ions for the automated gain control (AGC) and 20 ms maximum injection time. For the MS/MS scans, the normalized collision energy was set to 27%, the resolution was 15000 at m/z 200, accumulated for a maximum of 30 ms or until the AGC target of 2 × 104 ions was reached.
The raw LC-MS/MS data files were analyzed using MaxQuant (version 1.6.17.0) with the spectra searched against the UniProt mouse database (updated on February, 2022). For the identification of peptides, the mass tolerances were set to 20 ppm for the initial precursor ions and 0.5 Da for the fragmented ions. Two missed cleavages in tryptic digests were allowed. Cysteine carbamidomethylation was set a static modification. Oxidation of methionine and N-terminal acylation was accepted as the variable modification. Filtering for the protein and peptide identification was set at a 1% false discovery rate (FDR).
Both healthy mouse and HCC mouse samples were used in this study. Three biological replicates were performed for each group. Protein expression was quantified for each group using a dimethyl labeling quantitative approach. The intensities of the EV proteins from healthy and HCC mice were extracted from the MaxQuant result files to represent the final expression of specific proteins in the samples. Any missing values were filled according to the minimum value filling principle. The protein expression matrix was processed with the egdreR package for normalization and differential expression analysis. An adjusted P value of <0.01 and log2|fold change|> 1.5 were used to indicate proteins that were significantly differentially expressed.35 The obtained data were screened for differential proteins by the following principles: (1) proteins identified in at least two of the three experiments. (2) The protein contained at least 1 unique peptide. All statistical analyses were performed in the R statistical programming language (version 4.1.1). GO analysis using online DAVID Bioinformatics Resources 6.8 software.36
3. Results and discussions
Our approach for purifying EVs is to combine an EV extraction kit with TiO2 microspheres. First, we used the purification kit for the crude separation of tissue EVs to remove aqueous contents and reduce the complexity of the tissue fluid. Then, the EVs were further enriched by exploiting the binding between TiO2 and phosphate groups on the phospholipid bilayer of EVs. Due to the flexibility of the lipid bilayer, strong multisite interactions between the TiO2 microspheres and the EVs were achieved.It is difficult for TiO2 microspheres to seize all EVs in crude tissue fluid due to the relatively low concentration of EVs. Additionally, any components with phospholipid bilayer structures in the tissue fluid tend to bind to TiO2 microspheres, resulting in interference with EV isolation. Therefore, a combination of an extraction kit first followed by TiO2-based isolation was developed in this work to enrich EVs with high purity from tissues (Fig. 1).
 | ||
| Fig. 1 The workflow of EVs isolation by kit-TiO2 microspheres method. | ||
3.1 Characterization of the isolated EVs
To characterize the enriched EVs, we compared kit-TiO2 enrichment strategy with conventional enrichment approaches using SEM. As shown in Fig. 2A, the TiO2 microspheres show a clean and smooth surface, with a uniform diameter of 5 μm before incubation with the tissue fluid samples. After being incubated with the kit extracted tissue fluid samples, the surface of the TiO2 microspheres was covered with a large number of round vesicles (Fig. 2B). The attached vesicles on the microsphere surface were further characterized by TEM. As shown in Fig. 2C, numerous vesicles were present on the surface of the TiO2 microspheres. Under alkaline conditions (pH 10–12), the coordinated binding between the phosphate group and TiO2 microspheres is destroyed. We introduced 10% ammonia to elute the enriched EVs for TEM and NTA analysis. After elution, intact, round vesicle structures were observed (Fig. 2D). This result suggested that the lipid bilayer was preserved during separation and elution. | ||
| Fig. 2 Characterization of the isolated EV (A) SEM image of TiO2 microsphere; (B) SEM image of TiO2 microsphere after incubation with kit extracted tissue EVs; (C) TEM image of the tissue EVs adsorbed on the TiO2 microsphere; (D) TEM images of EVs isolated by kit-TiO2 method was pointed by red arrows; (E) the size distribution of the isolated EVs separated by different methods and measured by NTA; (F) concentration of the EVs were measured by NTA for the different extraction methods, n = 3; (G) quantification of total protein by BCA protein assay for the EVs isolated by different methods, n = 3; (H) western blot of marker protein TSG101, CD63, and syntenin-1 for EVs extracted using the three different enrichment methods, including extraction kit only, TiO2 only, and kit combined TiO2; (I) western blot analysis of cellular interfering protein VDAC, calnexin, and GM130 in the extracted liver tissue EV and liver tissue homogenate; (H + I) 20 μg protein was loaded per lane. | ||
We performed NTA particle size, number analysis and protein quantification (BCA) on the eluted EV. As shown in Fig. 2E, the size distribution of EVs isolated by the three methods were analyzed by NTA. The average particle size was 175.9 nm, 192.6 nm, and 239.2 nm for EVs extracted using kit-TiO2, kit only, and TiO2 only approaches, respectively. EVs extracted using the kit only and TiO2 only method displayed a broad distribution of vesicles with several peaks. However, the EVs extracted using the kit-TiO2 approach only had one major peak, indicating its narrower size distribution. As shown in Fig. 2F and G. The number of particles and protein abundance of EVs were measured by NTA and BCA protein assay kit. The results indicated that the concentration of EVs particles extracted using kit-TiO2 was significantly higher than that of the other two methods. Interestingly, we found the protein of EVs extracted using the kit method was higher than that using kit-TiO2. A possible explanation is that the EVs enriched by the kit method might contain more high abundant contaminate proteins.37–39
We also conducted western blotting experiments to confirm the above results. EVs were isolated by the kit-TiO2 approach, TiO2 alone and the kit alone to simultaneously validate the EV markers TSG101, CD63 and syntenin-1. As shown in Fig. 2H, the intensity of the maker protein syntenin-1 and CD63 were significantly higher in EVs isolated by the kit-TiO2 approach than that obtained by the other two methods. Next, western blot analysis was performed to assess the purity of the extracted EVs. Typical cellular proteins, e.g., Golgi (GM130), endoplasmic reticulum (Calnexin), and mitochondrial proteins (VDAC), were detected in high abundance in liver tissue homogenates, but were absent in the EV samples extracted by the combined method (Fig. 2I), suggesting that cell debris was completely removed and that the isolated EVs were of high purity.
The EVs enriched by the combined method were examined by SDS-PAGE and Coomassie Blue staining. The supernatant, washing buffers (first wash, second wash, and third wash solution), and tissue EVs eluted from the TiO2 microspheres were all collected and examined. Protein signals were negligible in the washing buffer. Significant differences were detected between the tissue proteins (LHC) and EV proteins (Fig. 3A and B), indicating the heterogeneity between tissue cells and EVs. These results demonstrated that the EVs were effectively isolated through the binding to TiO2.
 | ||
| Fig. 3 (A) Separation of tissue EV proteins by SDS-PAGE (blue-stained), in which EVs were isolated by the kit-TiO2 approach; (B) distribution of tissue EVs proteins and liver tissue proteins (LHC: liver homogenates collagenase) by SDS-PAGE (20 μg protein was loaded per lane). | ||
To assess the reproducibility of this kit-TiO2 isolation strategy, three biological replicates were tested. EVs were extracted from three identical tissue fluid samples using the proposed strategy. The obtained EVs were lysed with 4% SDS, and EV proteins were collected. Then, the isolated EV proteins were characterized by SDS–PAGE. As shown in Fig. S1,† similar proteomic distribution patterns were observed for the three replicates in lanes 1, 2 and 3, suggesting that good reproducibility was achieved.
3.2 Optimized enrichment strategy
To further improve the enrichment of the EVs, we evaluated the amount of TiO2 microspheres used and the incubation time for EV isolation. Using 200 μL tissue fluid as the sample, the abundance of the EV protein marker TSG101 was traced before and after EV enrichment by western blotting. The protein intensity was relatively quantified based on the grayscale and normalized to the maximum value.40 As illustrated in Fig. 4A, the abundance of the protein marker was extremely low when using 1 mg TiO2. As the amount of TiO2 microspheres increased, the marker intensity was enhanced and then saturated at 20 mg. This might be due to insufficient dispersion of TiO2 in the solution for amounts higher than 20 mg. As a result, the optimal amount of TiO2 microspheres was 20 mg for enriching EVs from 200 μL tissue fluid.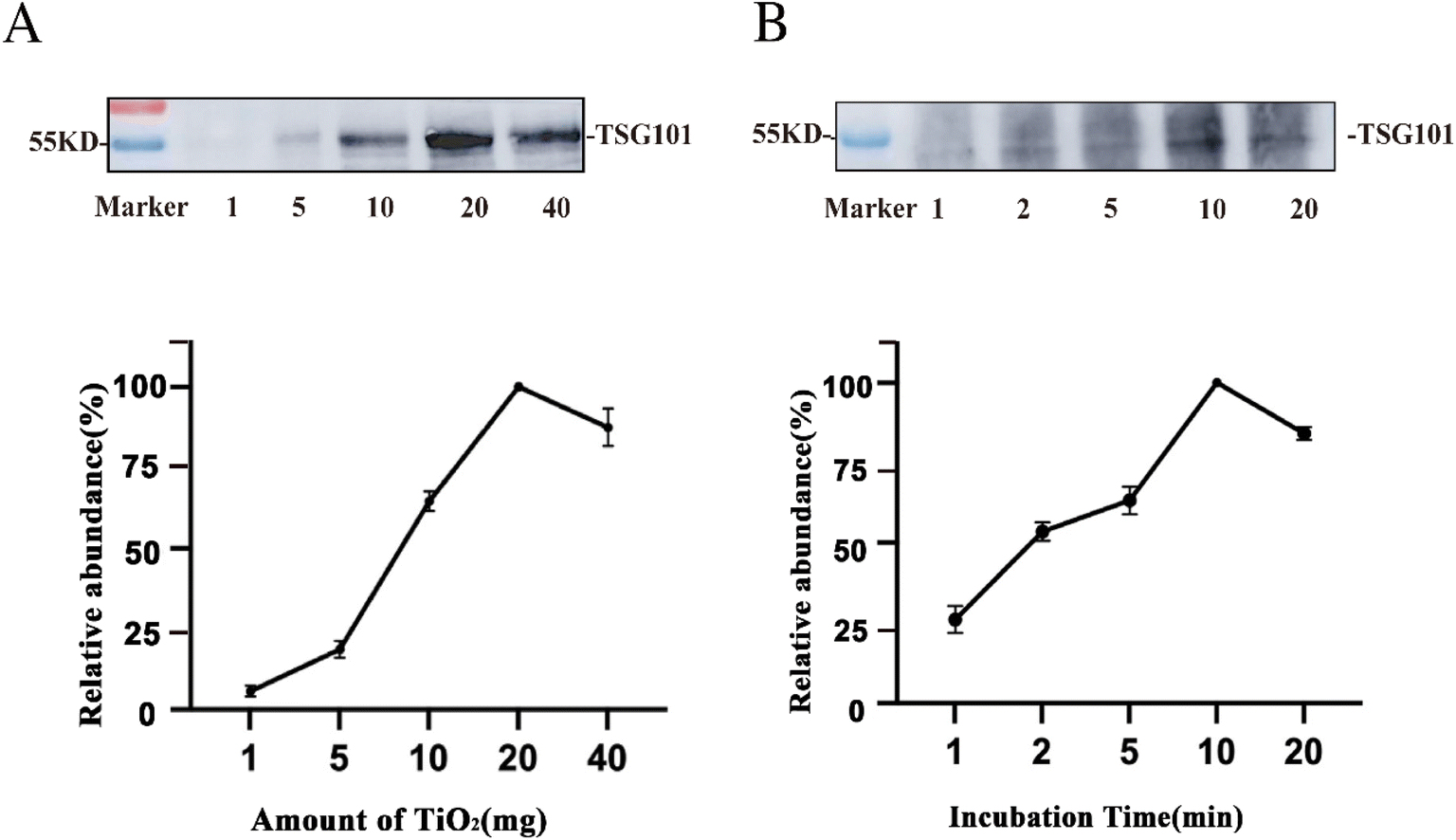 | ||
| Fig. 4 Optimization of experimental conditions for EVs isolation. The protein marker TSG101 was relatively quantified based on the greyscale of western blot. Relative abundance of TSG101 was a function of the quantity of TiO2 (A) and incubation time (B). Protein intensity was normalized to the maximum intensity value. | ||
Furthermore, the incubation time of the tissue fluid with microspheres was assessed. As suggested above, the optimal amount of TiO2 (20 mg) was used for 200 μL of tissue fluid, and different incubation times of 1 min, 2 min, 5 min, 10 min, and 20 min were tested. Fig. 4B shows that the protein marker intensity increased with increasing incubation time and was saturated at 10 min of enrichment. Prolonged incubation might result in irreversible adsorption of EVs on the surface of TiO2 microspheres, and lead to a lower recovery rate of EVs. Therefore, 10 min was the optimized enrichment time and was used for subsequent experiments for isolating EVs from 200 μL tissue fluid.
3.3 Proteomic analysis of EVs from mouse liver tissue
To apply the proposed EV isolation strategy, mouse liver tissue was selected as the model system. Tissue EVs were enriched by the kit-TiO2 approach and characterized using mass spectrometry. As shown in Fig. 5A, we compared the proteins identified by different EV isolation methods with the mouse EV proteins in the existing EV database Exocarta.41 The kit-TiO2 method isolated 1317 EV proteins, while the kit alone, TiO2 alone, and the gold standard ultracentrifugation method isolated 1135, 1036, and 1166 EV proteins. The greater number of EV proteins isolated by kit-TiO2 methods indicated its advantages for tissue EV studies. Further comparison in Fig. 5B showed that a total of 1966 protein groups were present in EVs isolated from mouse liver tissue fluids using kit-TiO2 enrichment strategy, while a total of 1801, 1629, and 1501 protein groups were isolated with ultracentrifugation, extraction kits, and TiO2-based isolation, respectively. In addition, the majority of the EV protein groups isolated using ultracentrifugation (56.8%), the extraction kit (60.2%), and TiO2-based enrichment (63%) were also isolated by the kit-TiO2 approach. Further analysis revealed that the kit-TiO2 strategy not only efficiently covered 62% proteins obtained by the other three methods, but also provided 321 extra proteins, indicating its higher capability in identifying EV proteins from tissue sample. Further comparison in Fig. S2† revealed lower protein abundance of the uniquely identified proteins by the kit-TiO2 method than the shared proteins with other methods, which indicated the higher sensitivity of the kit-TiO2 method to identify more EV proteins. Over 84.5% of the protein groups were identified in at least two of the three replicates by the kit-TiO2 approach (see Fig. 5C). Good reproducibility with Pearson correlation coefficients greater than 0.83 was achieved (see Fig. 5D). This result demonstrates the high reproducibility of the method for extracting EVs from tissues using kit-TiO2 approach. | ||
| Fig. 5 (A) Ven diagram of EV proteins isolated using ultracentrifugation, kit only, TiO2 only, and kit-TiO2 methods; (B) overlap of protein groups identified in the tissue EV isolated using ultracentrifugation, extraction kit, TiO2 only, and kit-TiO2 approaches; (C) overlap of proteins identified from biological triplicates using kit-TiO2 method; (D) reproducibility evaluation of the kit-TiO2 enrichment strategy. | ||
Furthermore, Gene Ontology (GO) analysis was performed to probe the subcellular location of the identified tissue EV proteins. As illustrated in Fig. S3,† liver EV proteins were highly enriched in the cellular component of extracellular exosome, suggesting the vesicular and extracellular nature of the proteins. Importantly, the identified proteins were irrelevant to nuclear, Golgi, endoplasmic reticulum or blood microparticles, indicating negligible contamination from cellular debris or other vesicles. In the biological process category, the identified protein groups were mainly relevant to metabolic, oxidation–reduction, and protein transport processes. Additionally, the most abundant molecular functions included poly(A) RNA binding, catalytic activity, and cadherin binding involved in cell–cell adhesion. We further performed gene ontology (GO) analysis of EVs proteins enriched by four methods. In Fig. S4,† the number of proteins classified as EVs in the cellular component were obviously higher in EVs extracted using kit-TiO2 than using the other three methods. This result demonstrates the reliability of the kit-TiO2 method and the higher enrichment efficiency.
3.4 Proteomic analysis of tissue EVs from hepatocellular carcinoma and healthy mice
Characterization of EV proteomic profiles may contribute to finding therapeutic targets for the treatment of hepatocellular cancer. We compared the EV proteins extracted from liver tissue of HCC and healthy mice using kit-TiO2 method. Three HCC mice and three healthy mice were analyzed. The proteomic profile showed that a total of 1176 and 1083 EV protein groups were isolated from healthy and HCC mice, respectively (Fig. 6A). Multivariate statistical analysis was performed on proteins identified more than two times to screen the proteomic difference between the two groups. Proteins with a p value <0.01 and log2|FC| > 1.5 were considered significantly differentiated. The volcano plot generated in Fig. 6B reveals 25 upregulated and 45 downregulated proteins in the HCC group relative to the normal group. The 70 differentially expressed proteins in HCC were further probed using unsupervised hierarchical clustering analysis (Table S1†). As shown in Fig. 6C, the HCC and healthy groups were clearly separated, indicating that the differentially expressed EV proteins may be useful as biomarkers or therapeutic targets for HCC. | ||
| Fig. 6 Proteomic analysis of tissue EV from the healthy and HCC mice. (A) Overlap of EV protein groups identified from the two groups; (B) volcano plot of the tissue EV proteins; (C) unsupervised clustering of the significantly changed proteins. | ||
Furthermore, the biological functions of the significantly changed proteins were investigated. Among the 25 upregulated proteins, Atic, Copa, Cont3, Me1, Anxa3, Fth1, Anxa5, Phb1, Acaa2, ATPD, and Glud1 were found to be highly relevant to HCC. For example, Atic aided in HCC growth and migration by functioning in the AMPK and mTOR signaling pathways.42 Secretory Anxa3 contributes to tumorigenic, metastatic, and self-renewal processes in HCC, and its blockade has been reported as a therapeutic target for hepatocellular carcinoma.43 In addition, several proteins, e.g., Ndufv1, Pygl, Sardh, and Ftl1, were previously associated with other cancers including breast cancer and colorectal cancer. Therefore, the significantly altered proteins in tissue EVs might be considered useful markers for the diagnosis of HCC.
4. Conclusions
In this work, we presented a novel approach to enrich EVs from animal tissue using kit-TiO2 method. High-quality and high-purity EVs were obtained as demonstrated by western blot and proteomic analysis. Successful application of this method led to the identification of dozens of differentially expressed EV proteins in the HCC samples. This new isolation strategy provides a powerful tool for enriching EVs from animal tissues and may support biomarker discovery and the diagnosis of hepatic tumors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key R&D Program of China (No. 2021YFA1302604 and 2021YFA1301601), the National Natural Science Foundation of China (No. 32088101, 22074158 and 21904008), and the National Center for Protein Science (Beijing) Grant 2021-NCPSB-003.References
- B. György, T. G. Szabó, M. Pásztói, Z. Pál, P. Misják, B. Aradi, V. László, É. Pállinger, E. Pap, Á. Kittel, G. Nagy, A. Falus and E. I. Buzás, Cell. Mol. Life Sci., 2011, 68, 2667–2688 CrossRef PubMed.
- New Technologies for Analysis of Extracellular Vesicles – PMC, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6029891/, accessed 26 May, 2022 Search PubMed.
- Y. Sun, S. Liu, Z. Qiao, Z. Shang, Z. Xia, X. Niu, L. Qian, Y. Zhang, L. Fan, C.-X. Cao and H. Xiao, Anal. Chim. Acta, 2017, 982, 84–95 CrossRef CAS PubMed.
- S. A. Melo, L. B. Luecke, C. Kahlert, A. F. Fernandez, S. T. Gammon, J. Kaye, V. S. LeBleu, E. A. Mittendorf, J. Weitz, N. Rahbari, C. Reissfelder, C. Pilarsky, M. F. Fraga, D. Piwnica-Worms and R. Kalluri, Nature, 2015, 523, 177–182 CrossRef CAS.
- T. Chen, Q.-Y. Xi, R.-S. Ye, X. Cheng, Q.-E. Qi, S.-B. Wang, G. Shu, L.-N. Wang, X.-T. Zhu, Q.-Y. Jiang and Y.-L. Zhang, BMC Genomics, 2014, 15, 100 CrossRef.
- T. Pisitkun, R.-F. Shen and M. A. Knepper, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13368–13373 CrossRef CAS.
- M. Simons and G. Raposo, Curr. Opin. Cell Biol., 2009, 21, 575–581 CrossRef CAS.
- A. Clayton, A. Turkes, S. Dewitt, R. Steadman, M. D. Mason and M. B. Hallett, FASEB J., 2004, 18, 977–979 CrossRef CAS.
- H. Valadi, K. Ekström, A. Bossios, M. Sjöstrand, J. J. Lee and J. O. Lötvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- R. Kalluri, J. Clin. Invest., 2016, 126, 1208–1215 CrossRef PubMed.
- L. Belov, K. J. Matic, S. Hallal, O. G. Best, S. P. Mulligan and R. I. Christopherson, J. Extracell. Vesicles, 2016, 5, 25355 CrossRef PubMed.
- S. Li, Q. Man, X. Gao, H. Lin, J. Wang, F. Su, H. Wang, L. Bu, B. Liu and G. Chen, J. Extracell. Vesicles, 2021, 10, e12175 CAS.
- L. L. Leung, M. K. Riaz, X. Qu, J. Chan and K. Meehan, Semin. Cancer Biol., 2021, 74, 3–23 CrossRef.
- V. Luga, L. Zhang, A. M. Viloria-Petit, A. A. Ogunjimi, M. R. Inanlou, E. Chiu, M. Buchanan, A. N. Hosein, M. Basik and J. L. Wrana, Cell, 2012, 151, 1542–1556 CrossRef CAS.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent, A. Jemal and C. A. Cancer, J. Clin., 2015, 65, 87–108 Search PubMed.
- Z. Qu, J. Wu, J. Wu, D. Luo, C. Jiang and Y. Ding, J. Exp. Clin. Cancer Res., 2016, 35, 159 CrossRef.
- M. Grohmann, F. Wiede, G. T. Dodd, E. N. Gurzov, G. J. Ooi, T. Butt, A. A. Rasmiena, S. Kaur, T. Gulati, P. K. Goh, A. E. Treloar, S. Archer, W. A. Brown, M. Muller, M. J. Watt, O. Ohara, C. A. McLean and T. Tiganis, Cell, 2018, 175, 1289–1306 CrossRef CAS.
- D. W. Greening, S. K. Gopal, R. A. Mathias, L. Liu, J. Sheng, H.-J. Zhu and R. J. Simpson, Semin. Cell Dev. Biol., 2015, 40, 60–71 CrossRef CAS.
- N. Ludwig and T. L. Whiteside, Expert Opin. Ther. Targets, 2018, 22, 409–417 CrossRef CAS PubMed.
- C. Shao, F. Yang, S. Miao, W. Liu, C. Wang, Y. Shu and H. Shen, Mol. Cancer, 2018, 17, 120 CrossRef PubMed.
- X. Xue, X. Wang, Y. Zhao, R. Hu and L. Qin, Biochem. Biophys. Res. Commun., 2018, 502, 515–521 CrossRef CAS.
- K. Takahashi, I. K. Yan, J. Wood, H. Haga and T. Patel, Mol. Cancer Res., 2014, 12, 1377–1387 CrossRef CAS PubMed.
- Z. Fang, X. Zhang, H. Huang and J. Wu, Chin. Chem. Lett., 2022, 33, 1693–1704 CrossRef CAS.
- Y. Huang, L. Cheng, A. Turchinovich, V. Mahairaki, J. C. Troncoso, O. Pletniková, N. J. Haughey, L. J. Vella, A. F. Hill, L. Zheng and K. W. Witwer, J. Extracell. Vesicles, 2020, 9, 1785746 CrossRef CAS.
- R. Perez-Gonzalez, S. A. Gauthier, A. Kumar and E. Levy, J. Biol. Chem., 2012, 287, 43108–43115 CrossRef CAS.
- Enrichment of extracellular vesicles from tissues of the central nervous system by PROSPR – PubMed, https://pubmed.ncbi.nlm.nih.gov/27216497/, accessed 27 May, 2022 Search PubMed.
- B.-Y. Chen, C. W.-H. Sung, C. Chen, C.-M. Cheng, D. P.-C. Lin, C.-T. Huang and M.-Y. Hsu, Clin. Chim. Acta, 2019, 493, 14–19 CrossRef CAS PubMed.
- J. Caradec, G. Kharmate, E. Hosseini-Beheshti, H. Adomat, M. Gleave and E. Guns, Clin. Biochem., 2014, 47, 1286–1292 CrossRef CAS.
- E. A. Mol, M.-J. Goumans, P. A. Doevendans, J. P. G. Sluijter and P. Vader, Nanomedicine Nanotechnol. Biol. Med., 2017, 13, 2061–2065 CrossRef CAS.
- T. Soares Martins, J. Catita, I. Martins Rosa, O. A. B. da Cruz E Silva and A. G. Henriques, PloS One, 2018, 13, e0198820 CrossRef.
- F. Cao, Y. Gao, Q. Chu, Q. Wu, L. Zhao, T. Lan and L. Zhao, Electrophoresis, 2019, 40, 3092–3098 CrossRef CAS.
- T. Uehara, I. P. Pogribny and I. Rusyn, Curr. Protoc. Pharmacol., 2014, 66, 14.30.1–14.30.10 Search PubMed.
- C. Coughlan, K. D. Bruce, O. Burgy, T. D. Boyd, C. R. Michel, J. E. Garcia-Perez, V. Adame, P. Anton, B. M. Bettcher, H. J. Chial, M. Königshoff, E. W. Y. Hsieh, M. Graner and H. Potter, Curr. Protoc. Cell Biol., 2020, 88, e110 CAS.
- R. Crescitelli, C. Lässer and J. Lötvall, Nat. Protoc., 2021, 16, 1548–1580 CrossRef CAS PubMed.
- M. D. Robinson, D. J. McCarthy and G. K. Smyth, Bioinformatics, 2010, 26, 139–140 CrossRef CAS PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef CAS.
- X. Fang, C. Chen, B. Liu, Z. Ma, F. Hu, H. Li, H. Gu and H. Xu, Acta Biomater., 2021, 124, 336–347 CrossRef CAS.
- R. J. Lobb, M. Becker, S. Wen Wen, C. S. F. Wong, A. P. Wiegmans, A. Leimgruber and A. Möller, J. Extracell. Vesicles, 2015, 4, 27031 CrossRef.
- Q. U. A. Reshi, M. M. Hasan, K. Dissanayake and A. Fazeli, in Next Generation Culture Platforms for Reliable In Vitro Models: Methods and Protocols, ed. T. A. L. Brevini, A. Fazeli and K. Turksen, Springer US, New York, NY, 2021, pp. 201–206 Search PubMed.
- J. A. Paulo, L. S. Lee, B. Wu, K. Repas, P. A. Banks, D. L. Conwell and H. Steen, Electrophoresis, 2010, 31, 2377–2387 CrossRef CAS PubMed.
- S. Keerthikumar, D. Chisanga, D. Ariyaratne, H. A. Saffar, S. Anand, K. Zhao, M. Samuel, M. Pathan, M. Jois, N. Chilamkurti, L. Gangoda and S. Mathivanan, J. Mol. Biol., 2016, 428, 688–692 CrossRef CAS.
- M. Li, C. Jin, M. Xu, L. Zhou, D. Li and Y. Yin, Cell Commun. Signal, 2017, 15, 52 CrossRef CAS PubMed.
- M. Tong, T.-M. Fung, S. T. Luk, K.-Y. Ng, T. K. Lee, C.-H. Lin, J. W. Yam, K. W. Chan, F. Ng, B.-J. Zheng, Y.-F. Yuan, D. Xie, C.-M. Lo, K. Man, X.-Y. Guan and S. Ma, Stem Cell Rep., 2015, 5, 45–59 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06185f |
| ‡ Present address: School of Medical Technology, Beijing Institute of Technology, Beijing 100081, PR China. |
| This journal is © The Royal Society of Chemistry 2022 |