DOI:
10.1039/D2RA06300J
(Paper)
RSC Adv., 2022,
12, 33825-33834
A novel bimetallic MOFs combined with gold nanoflakes in electrochemical sensor for measuring bisphenol A†
Received
7th October 2022
, Accepted 16th November 2022
First published on 25th November 2022
Abstract
In this paper, a novel bimetallic Fe–Cu metal–organic framework combined with 1,3,5-benzenetricarboxylic acid (Fe–Cu-BTC) are synthesized using hydrothermal reaction. The bimetallic Fe–Cu-BTC with high BET (1504 cm3 g−1) and high Langmuir surface area (1831 cm3 g−1) is composited by gold nanoparticles to improve the conductivity and to develop their synergistic effect. A novel bisphenol A (BPA) sensor was prepared by dropcasting Fe–Cu-BTC on glassy carbon electrodes (GCE) followed by AuNPs electrodeposition. The Fe–Cu-BTC framework were characterized by X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy studies (TEM), FT-IR, BET measurements and EDX spectra. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were carried out for surveying the electrochemical properties of the sensors and for the quantification of BPA. Two linear ranges of BPA concentrations 0.1–1.0 μM and 1.0–18 μM with 18 nM limit of detection were obtained. The developed sensor was used to measure the concentration of BPA in samples extracted from rain coat with the recovery ranging from 85.70 to 103.23%.
1. Introduction
Recently, much effort has been devoted to the integration of metal–organic frameworks (MOFs) based on transition metals onto electrochemical sensing platforms. The focal metals (Zn, Mn, Ni, Cu) in the framework act as active sites to accelerate electrocatalytic reactions whereas organic ligands (1,4-benzene dicarboxylate, 1,3,5-benzene tricarboxylate (BTC), 4,40,400-benzene-1,3,5-triyl-tribenzoate) act as adsorption sites towards the target organic molecules.1 MOFs have been used as modifiers in electrochemical sensors for the detection of organic compounds, e.g., Cu-BTC MOF platform for determining glyphosate,2 sunset yellow and tartrazine,3 dopamine and paracetamol using Cu-MOF/graphene composite,4 [Cu3(BTC)2(H2O)3]n for catechol,5 199-GO-n/GCE for catechol and hydroquinone,6 and [Cu3(BTC)2] in carbon-paste electrode for 2,4-dichlorophenol.7 Li et al.8 detected dihydroxyl benzene in water by using a Cu-MOF-GN-composite-based electrochemical sensor. ZIF-67-derived nickel–cobalt–manganese hydroxides on 2D V2CTx MXene for dual-functional orientation have been used as high-performance asymmetric supercapacitor and electrochemical sensor for hydroquinone detection.9
Bisphenol A (BPA) is one of the most widely used chemical raw materials in the world, which is commonly used as a monomer for the production of polymers (e.g. polycarbonate, polyphenylene ether resins, unsaturated polyester resins, etc.) or as an ingredient for the production of fine chemicals (e.g. plasticizers, flame retardants, antioxidants, heat stabilizers, rubber antioxidants, pesticides, coatings, etc.).10 BPA is also a typical endocrine disruptor in the environment which is commonly found in product packaging, thus gradually accumulating in human bodies through the ingestion of food and water from trace levels.11–28 For the detection of BPA, several MOFs have been utilized as functional materials to fabricate sensors and to improve sensing performance. For instance, a nano-sized iron MOF (Fe-MIL-88B-NH2) were used to detect BPA due to its adsorption and quenching of fluorescent modified DNA aptamer.29 Ce-MOF was employed to fabricate electrochemical sensors for the detection of BPA due to its excellent capacity and electrocatalytic activity.30 Monoclinic WO3 nanosheets-carbon nanotubes nanocomposite based electrochemical sensor for sensitive detection of bisphenol A reported by Tianyou Peng et al.31 with detection limit of 16.3 nM, good reproducibility, selectivity and stability. Despite the availability of various electrochemical methods for rapid and cost-effective detection of BPA,11–28,31 BMOF sensors has not been utilized popularly.
Bimetallic MOFs are novel form of MOFs that possess two types of metal ions, which have attracted attention very recently due to larger specific surface area, more catalytic sites, and stable compared to single-metal MOFs.32 The reaction of organic ligands with two different metal ions leads to bimetallic MOF as a pure compound instead of a mixture of two monometallic MOFs.33 Nevertheless, they display low electric conductivity which may range from 10−10 to 103 S cm−1.34,35 Several attempts have been made to improve the conductivity of MOFs such as the addition of carbon materials,36 graphene oxide (GO),37 reduced graphene oxides38 and gold nano particles.39,40 As mentioned in,41 because of the synergistic effect of MOF (Cu-BTC) and AuNP, the transmission signal of electrochemical sensor for BPA increases by about 2.5 times.
For BMOFs, it is possible to incorporate Fe2+ into the framework during synthesis to modify the surface morphology and pore formation of nanosized Cu-BTC via synergistic effect of the second metal ion.42 In the present paper, we propose a method for the synthesis of bimetallic Fe–Cu-BTC by adding two types of metal ions Cu and Fe to the organic ligand of 1,3,5-benzenetricarboxylic acid (H3BTC) combined with AuNPs by electrodeposition on modified electrodes. The synthesized BMOF exhibited high surface area, uniform pore size, extended surface morphology, pore width and enhanced adsorption affinity. The synergistic effect of both metal ions in the structure can result in greater adsorption performance.42 This combination of Fe–Cu-BTC, which is expected as an electrocatalyst with high adsorption performance, and AuNPs with extremely high electrical conductivity was developed to fabricate electrochemical sensors for sensitive and rapid detection of BPA. In this way, both synergistic effects for the detection of BPA in water may be enhanced.
2. Experimental
2.1. Materials and apparatus
CuCl2·2H2O and FeCl3·6H2O and trimesic acid (H3BTC) (Sigma-Aldrich) were used as precursors. Potassium ferri/ferro-cyanide (K3[Fe(CN)6])/(K4[Fe(CN)6]) (Sigma-Aldrich) were employed for electrochemical characterization of redox probes. XPS was obtained by a monochromic Al Kα source at 1486.6 eV using a Thermo VG-ESCALAB spectrometer (USA). XRD measurement was performed on a D8 advance diffractometer (Bruker, Germany) using CuKα as the radiation source (λ = 0.15406 nm) at scan range of 2θ = 5°–70°. SEM images were captured using a JEOL JSM 6500F system operating at 20 kV. TEM was performed using a JEOL JEM 1400 system at 100 kV acceleration voltage. FT-IR spectra were recorded using a Jacos 4700 spectrometer. The sample surface area was determined by BET measurement on a Tristar-3000 instrument using nitrogen as the adsorbate. EDX spectra were recorded using a JEOL JED-2300 spectrometer.
All electrochemical measurements (EIS, CV and DPV) were performed with an AUTOLAB potentiostat PGS302N using NOVA 2.0.1 software (Metrohm, Netherland) equipped with a three-electrode configuration, where Ag/AgCl/Sat. KCl and a Pt wire were used as the pseudo-reference and counter electrodes, respectively, and bare or modified Fe–Cu-BTC electrode was employed as the working electrode. All experiments in this study were performed at room temperature (25 ± 1 °C) and potentials cited herein are referenced versus Ag/AgCl.
2.2. Synthesis of bimetallic Fe–Cu-BTC
Twelve mmol of H3BTC were added to 60 mL of NaOH 1 M solution and stirred vigorously until a clear solution (solution A) was obtained. A volume of 9.6 mmol FeCl3·6H2O was added to 40 mL of distilled water to form solution B, and 2.4 mmol of CuCl2·2H2O was added to 20 mL of distilled water to form solution C. Solutions B and C were mixed (molar ratio Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu = 8:2) and sonicated for 30 min to form a homogeneous solution. Finally, the mixture (solution of A + (B + C)) was placed in a Teflon bottle and heated in a microwave oven (700 W) at 100 °C for 30 min. The solid product was filtered by centrifugation at 8000 rpm for 5 min and washed with water and finally with ethanol. The product was dried by heating in an oven at 80 °C overnight to obtain the Fe–Cu-BTC sample.
:Cu = 8:2) and sonicated for 30 min to form a homogeneous solution. Finally, the mixture (solution of A + (B + C)) was placed in a Teflon bottle and heated in a microwave oven (700 W) at 100 °C for 30 min. The solid product was filtered by centrifugation at 8000 rpm for 5 min and washed with water and finally with ethanol. The product was dried by heating in an oven at 80 °C overnight to obtain the Fe–Cu-BTC sample.
2.3. The fabrication of the modified electrode
The GCE (BAS) surface was polished in a 3 μm alumina slurry and washed with ultrapure water in an ultrasonic bath. Five μL of Fe–Cu-BTC (in ethanol 1 mg mL−1) was drop-cast on the prepared GCE, left dry under ambient condition in 1 h and put into a solution containing HAuCl4 1 mM and H2SO4 0.5 M.39 This electrode was denoted as Fe–Cu-BTC/AuNPs/GCE. Chronoamperometry techniques were used by applying a potential of +0.5 V (vs. Ag/AgCl). AuNP electrodeposition time was varied from 60 to 300 s to evaluate the impact of AuNPs on the analyte signals. The oxidation current of BPA was determined by cyclic voltammetry (CV) in PBS 0.1 M (pH 7.0). CV measurements were performed by scanning from 0.2 to 0.8 V at 100 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were conducted in the frequency range of 0.1 Hz to 100 MHz using a single sinusoidal excitation signal at 300 mV open circuit potential in KCl 0.1 M containing Fe(CN)63−/4− 5 mM.43
2.4. Optimization of sensor operating conditions
The detection of BPA was carried out in aqueous media using differential pulse voltammetry (DPV) method with a differential step of 0.005 V, a sampling time of 0.04 s, a pulse width of 0.08 s and a pulse amplitude of 0.05 V. To improve the effectiveness of sensing performance, the experimental conditions were optimized at following conditions: pH 5.0–9.0 and the accumulation conditions of BPA onto Fe–Cu-BTC/AuNPs/GCE (60–300 s with steps of 60 s each).
2.5. Sample preparation
Rain coat samples were collected and analyzed for BPA using the developed sensor. The samples were treated and adjusted to a pH of 7.0. BPA with certain concentrations were spiked to these prepared samples and their concentrations were determined using the standard addition method.
3. Results and discussion
3.1. Chemical composition and structural behaviors of Fe–Cu-BTC
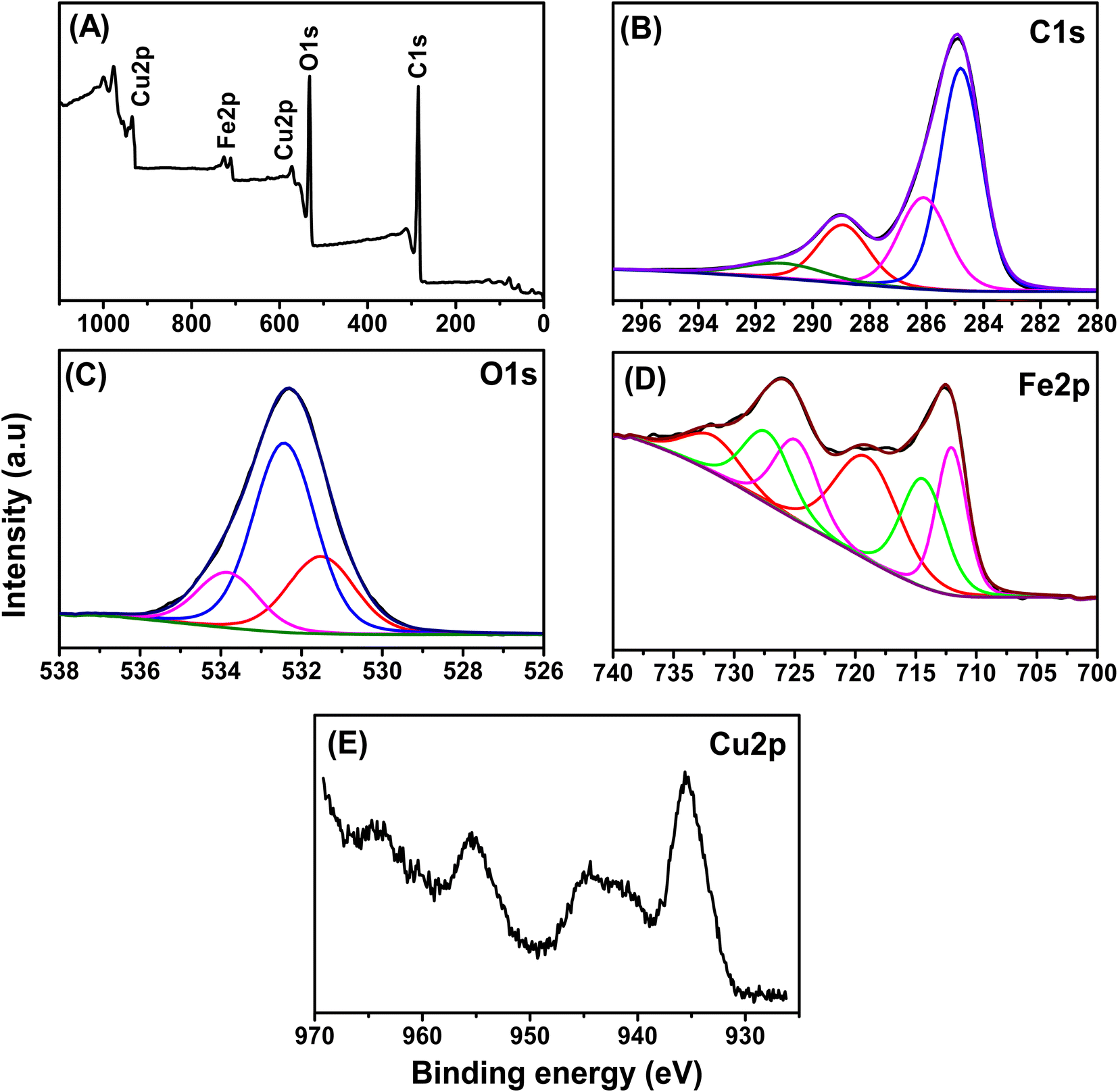
The chemical states and surface compositions of the prepared Fe–Cu-BTC sample were confirmed by XPS analysis (Fig. 1). The full-scan XPS spectra of Fe–Cu-BTC (Fig. 1A) confirm the existence of C (284.42 eV), O (532.82 eV), Cu (575.38 and 935.14 eV) and Fe (713.33 and 728.62 eV) elements. Four peaks observed at 284.79, 286.10, 288.93 and 291.00 eV in the high-resolution C1s-XPS spectra of (Fig. 1B) characterized for the bond energies of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C, C– O, C= O and O–CO, respectively.42 Three peaks at 531.51 eV, 532.43 eV and 533.84 eV observed in the high-resolution O1s-XPS spectra (Fig. 1C) were classified as lattice oxygen in the metal–oxygen bond (Fe–O or Cu–O), chemisorbed oxygen species, and oxygen in carboxyl groups, respectively.44 The binding energies at 712.05 eV and 724.87 eV in the Fe2p-XPS spectra (Fig. 1D) were assigned to Fe2+, while the peaks at 714.39 and 727.29 eV were attributed to Fe3+ and two satellites rocking at 719.04 and 731.73 eV (denoted sat.).45 The proportions of Fe2+ and Fe3+ in the Fe–Cu-BTC sample accounted for 49.48% and 50.42%, respectively. The Cu2p-XPS spectra (Fig. 1E) displayed major peaks of Cu2p (935.8 eV for Cu 2p3/2 and 955.8 eV for Cu 2p1/2) and their satellites (944.6 eV for Cu 2p3/2 satellites and 964.2 eV for Cu 2p1/2 satellites).
C/C–C, C– O, C= O and O–CO, respectively.42 Three peaks at 531.51 eV, 532.43 eV and 533.84 eV observed in the high-resolution O1s-XPS spectra (Fig. 1C) were classified as lattice oxygen in the metal–oxygen bond (Fe–O or Cu–O), chemisorbed oxygen species, and oxygen in carboxyl groups, respectively.44 The binding energies at 712.05 eV and 724.87 eV in the Fe2p-XPS spectra (Fig. 1D) were assigned to Fe2+, while the peaks at 714.39 and 727.29 eV were attributed to Fe3+ and two satellites rocking at 719.04 and 731.73 eV (denoted sat.).45 The proportions of Fe2+ and Fe3+ in the Fe–Cu-BTC sample accounted for 49.48% and 50.42%, respectively. The Cu2p-XPS spectra (Fig. 1E) displayed major peaks of Cu2p (935.8 eV for Cu 2p3/2 and 955.8 eV for Cu 2p1/2) and their satellites (944.6 eV for Cu 2p3/2 satellites and 964.2 eV for Cu 2p1/2 satellites).
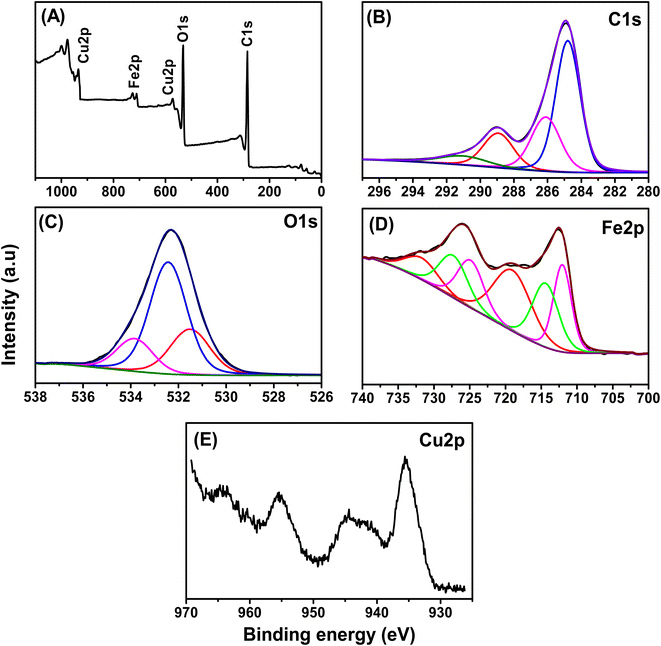 |
| | Fig. 1 The XPS spectra of the Fe–Cu-BTC sample, (A) survey scan, (B) C1s, (C) O1s, (D) Fe2p and (E) Cu2p. | |
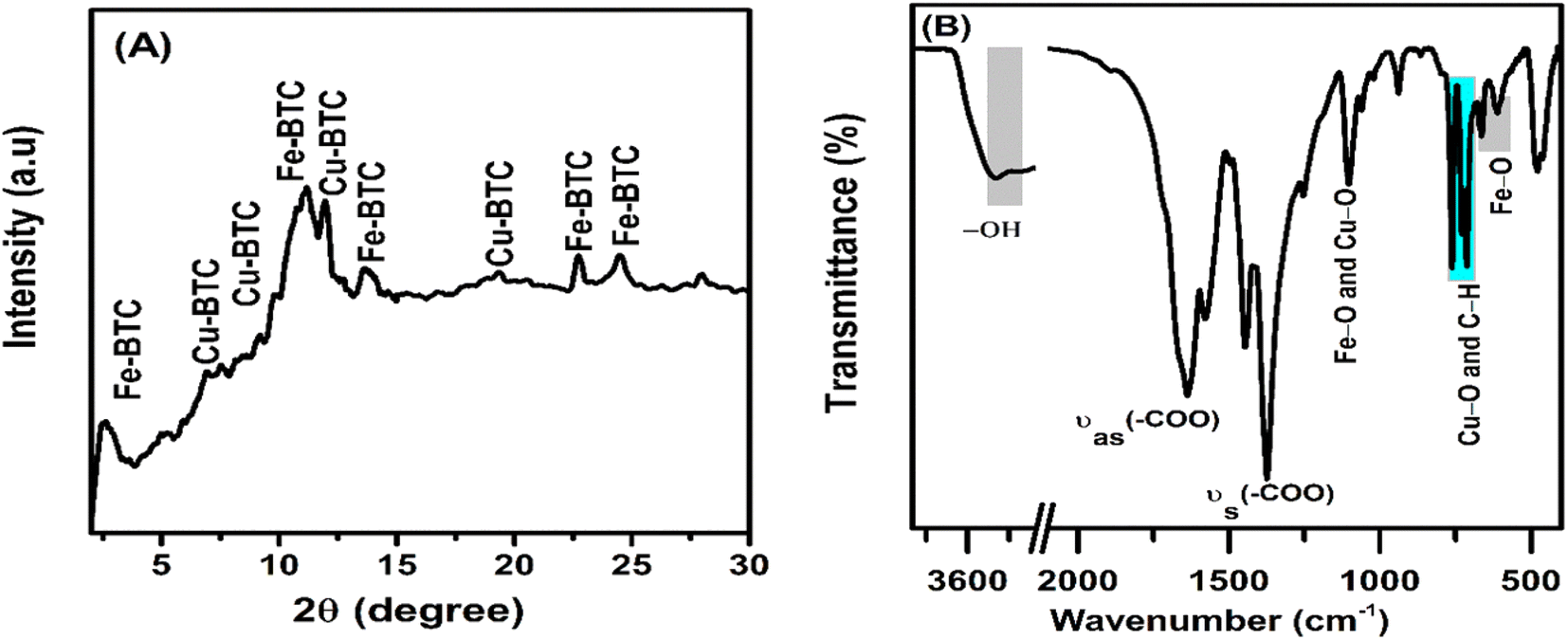
In the XRD pattern (Fig. 2), the 2θ-diffraction peaks at 2.53°, 11.18°, 13.55°, 22.70°, 24.52° and 28.02° mainly characterized for the Fe-BTC crystal structure.46–48 Meanwhile, the diffraction peaks at 6.99°, 9.22°, 11.99°, and 19.31° characterized for the Cu-BTC crystal structure. The X-ray diffraction patterns had relatively wide or overlapping peaks ranging from 10 to 12° due to the small crystal size and flawed material structure.49 In Fig. 2B, the oscillations at 3600 to 3300 cm−1 were attributed to the binding of loosely bound water molecules. The strong peaks at 1632 and 1372 cm−1 were assigned to the asymmetric stretching transformation and symmetric stretching variation of carboxyl groups, respectively.50 In addition, the peak oscillation at 1108 cm−1 was associated with the metal functional group C–O–Fe or C–O–Cu.51 The peak oscillation at 609 cm−1 was associated to the central oxygen in the single Fe3O. The peaks at 710 and 758 cm−1 were assigned to the C–C ring out-of-plane bending mode.52
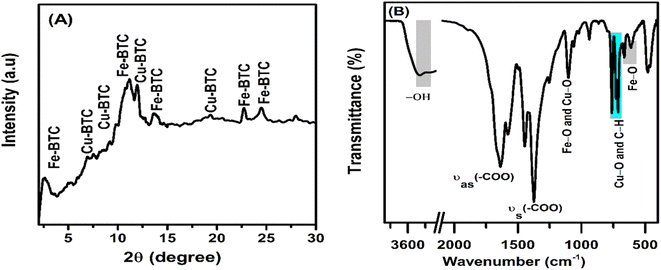 |
| | Fig. 2 The XRD pattern (A) and FT-IR spectra (B) of the Fe–Cu-BTC sample. | |
The SEM images (Fig. 3A) detected a grain size of about 50–100 nm in which clusters of those grains were likely to combine into larger particles. The TEM images (Fig. 3B) show that the Fe–Cu-BTC nanoparticles with a uniform particle size of 20–30 nm stacked on top of each other. The EDS elemental classification and EDS spectra (Fig. 4 and S1†) indicate the presence of C, O, Cu, and Fe elements uniformly distributed on the sample surface. The atomic elemental composition of C, O, Cu and Fe were 55.90, 32.56, 1.73 and 9.8%, respectively.
 |
| | Fig. 3 SEM (A) and TEM (B) images of the Fe–Cu-BTC sample. | |
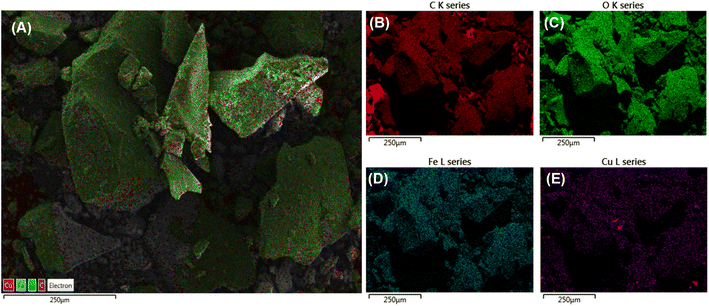 |
| | Fig. 4 The EDS layered image (A), EDS element mapping images of C (B), O (C), Fe (D) and Cu (E) for Fe–Cu-BTC sample. | |
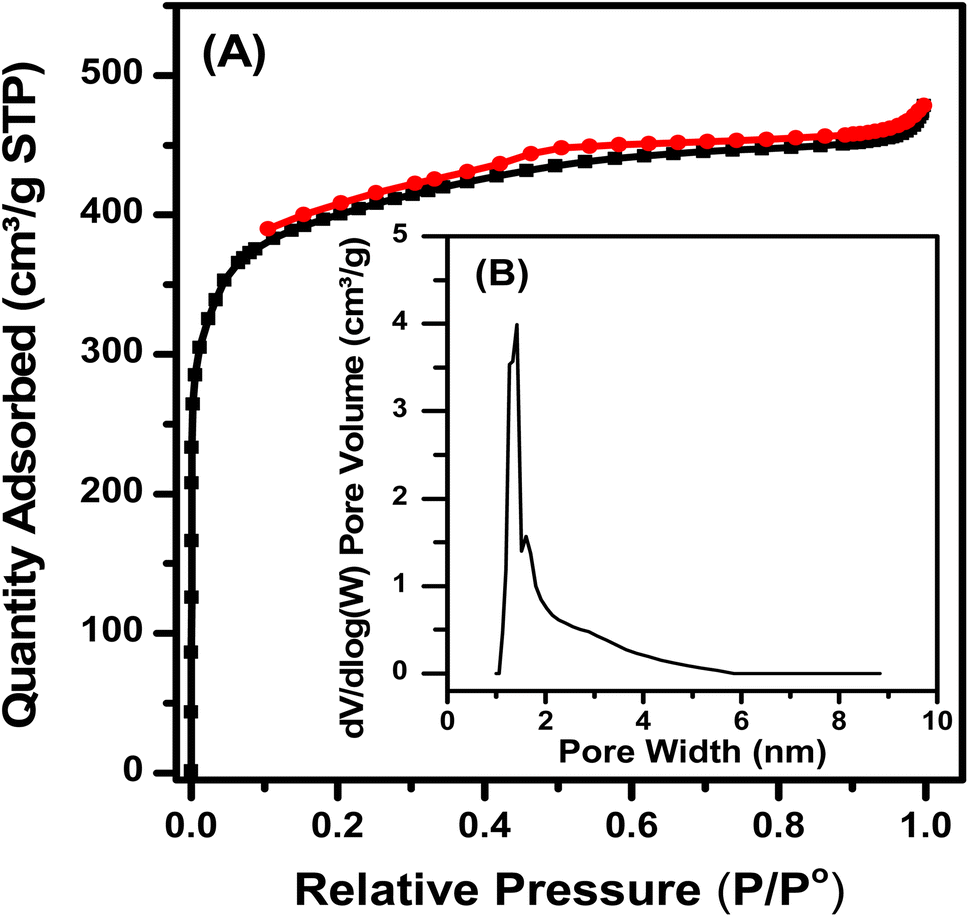
3.2. N2 adsorption–desorption isotherms of Fe–Cu-BTC
The N2 adsorption and desorption isotherm (Fig. 5) shows that the Fe–Cu-BTC sample belonged to type I, with no hysteresis loops, as the pores with microporous diameters mainly exist. The pore size distribution obtained from the calculation of non-local density functional theory (NL-DFT) was mainly micro-capillary. The sample has an average pore diameter of 1.40 nm, a large pore volume of 0.924 cm3 g−1, a BET surface area of 1504 cm3 g−1 and a high Langmuir surface area of 1831 cm3 g−1.
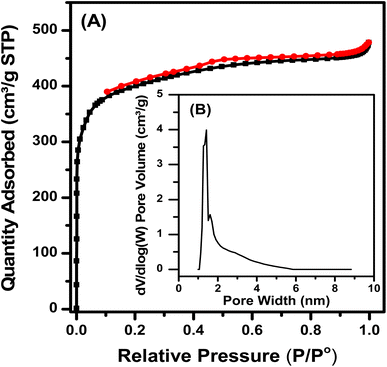 |
| | Fig. 5 N2 adsorption–desorption isotherm (A) and pore size distribution (B) of the Fe–Cu-BTC sample. | |
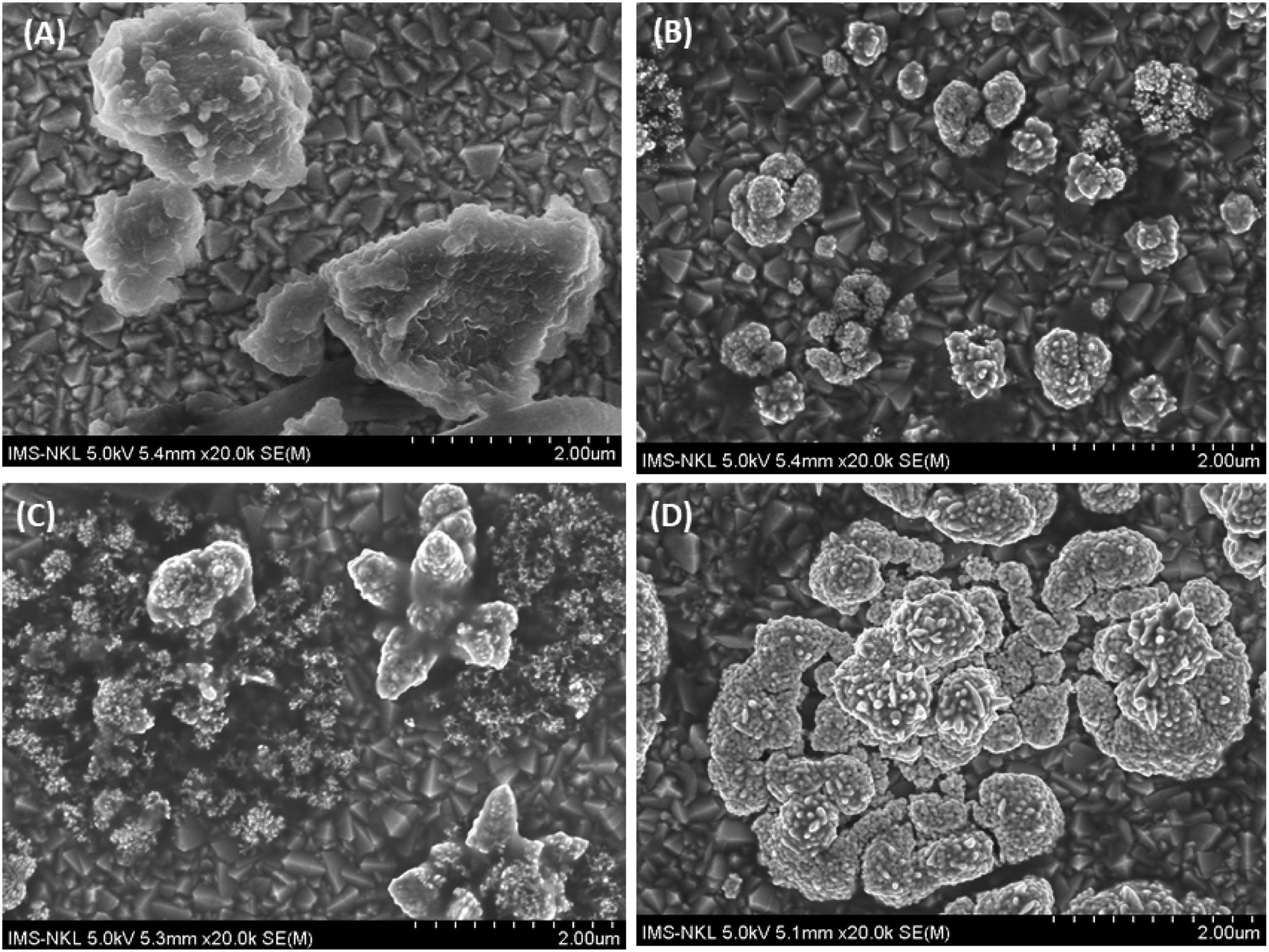
3.3. Modification of the Fe–Cu-BTC morphology after electrochemical deposition of AuNPs
The AuNPs was deposited on the sample to enhance the signals of BPA by both synergistic effects of them. The effects of AuNPs were investigated by increasing the deposition time from 60 to 300 s. The SEM images of AuNPs constructed on Fe–Cu-BTC/FTO at different deposition times are shown in Fig. 6. Initially, the Fe–Cu-BTC had an average size of 50–100 nm and an aggregation shape (Fig. 6A). Under 60 s deposition time in solution containing 1 mM HAuCl4 and 0.5 M H2SO4 with a potential of +0.5 V, Au particles appeared at low density next to the Fe–Cu-BTC clusters (Fig. 6B). When deposition time was increased to 180 s, the size of the AuNP flakes were also increased, and only a few Fe–Cu-BTC clusters were observed (Fig. 6C). Eventually, at 300 s deposition time, the Fe–Cu-BTC/GCE was fully coated with AuNPs, and Fe–Cu-BTC became invisible (Fig. 6D). This observation is in agreement with the EDS data (Fig. S2†).
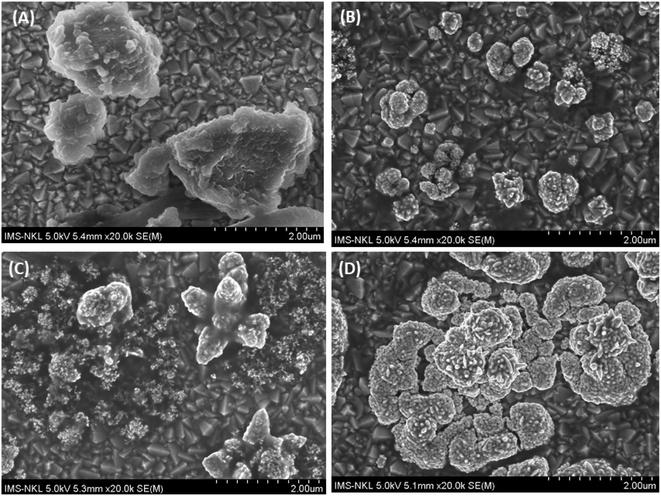 |
| | Fig. 6 SEM images of AuNPs deposited on the Fe–Cu-BTC/FTO sample at different deposition times: (A) intact Fe–Cu-BTC/FTO; (B) after 60 s; (C) after 180 s and (D) after 300 s. | |
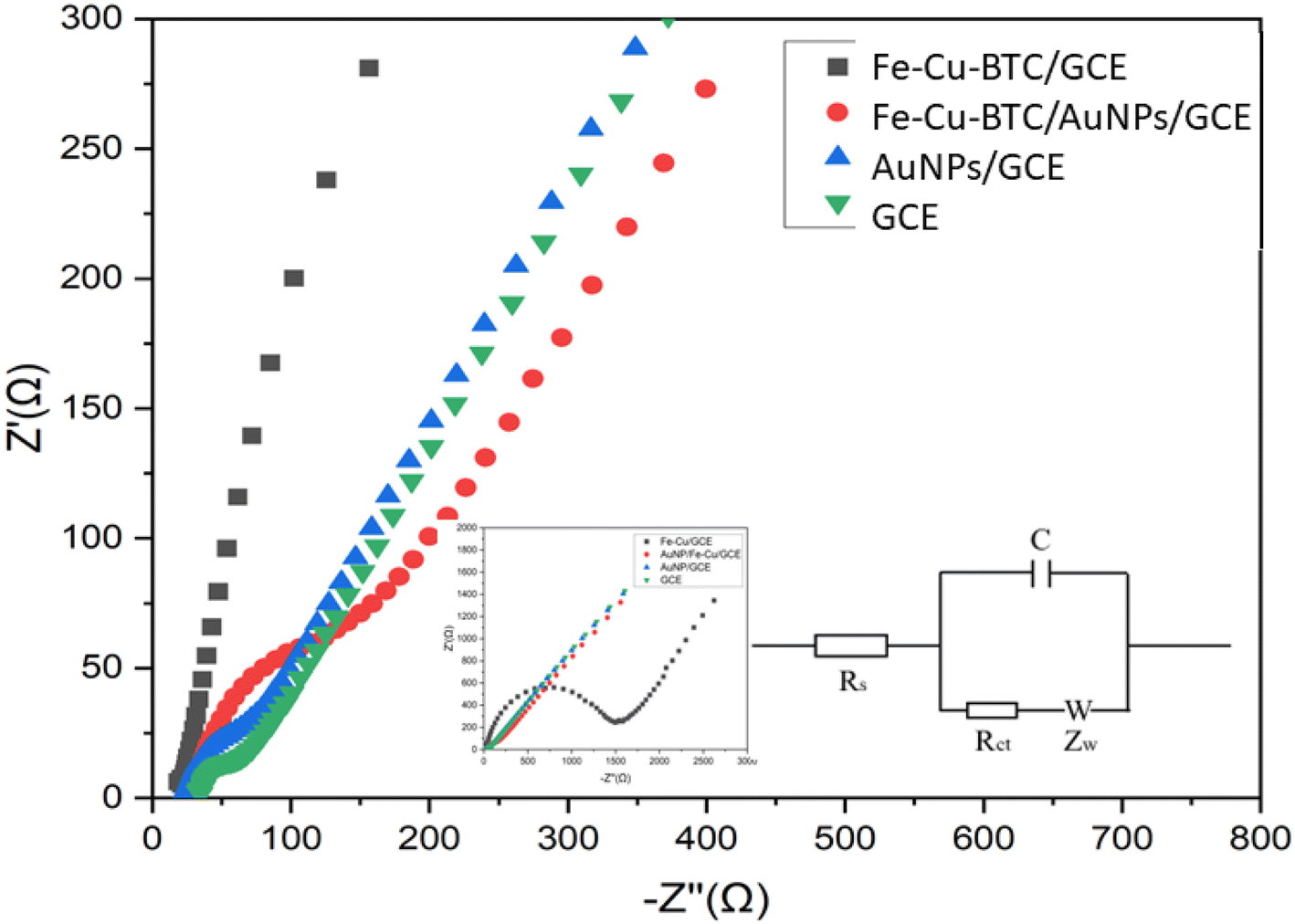
3.4. Electrochemical performance of Fe–Cu-BTC/AuNPs/GCE
Electrochemical behavior of the bare GCE, Fe–Cu-BTC/GCE, AuNPs/GCE and Fe–Cu-BTC/AuNPs/GCE samples were gradually tested with electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CVs) using Fe(CN)63−/4− couple as the electrochemical redox probes in solution containing 5 mM Fe(CN)63−/4− and 0.1 M KCl. In the Nyquist diagram (Fig. 7), the bare GCE exhibited a relatively low electron transfer resistance (Rct = 146.5 Ω) with a small semicircle. The electron transfer rate was dramatically dropped after the deposition of Fe–Cu-BTC, which resulted from an increase of the charge transfer resistance (Rct = 1420 Ω) due to the low conductivity of Fe–Cu-BTC. Meanwhile, for AuNPs/GCE, the charge transfer resistance was only at 102 Ω. As the results, the electrical conductivity of Fe–Cu-BTC/AuNPs attached on GCE was relatively improved (with Rct = 126 Ω), indicating a success fabrication of the modified electrode. Therefore this sensor was used for futher study. This result was also in line with the CV tests (Fig. S3†) which examined the electron transfer properties of those four samples in the presence of an inner sphere redox probe (i.e., Fe(CN)63−). From an electrochemical standpoint, the electron transfer in the inner sphere systems requires a strong interaction between the redox probe and the electrode surface. While bare GCE depicted a well-defined reversible redox system with a peak-to-peak separation (ΔEp) of 60 mV, the Fe–Cu-BTC/GCE exhibited extremely low electron transfer rate with a full blocking effect, which resulted from the absence of a faradaic signal of Fe(CN)63−/4− in the studied electrochemical window (ΔEp ∼60 mV). After the deposition AuNPs onto Fe–Cu-BTC/GCE sample, the obtained electrode quickly recovered the excellent electron transfer rate (ΔEp = 60 mV), showing an increase of electron transfer compared to the solely bimetallic MOF (Fig. S3†).
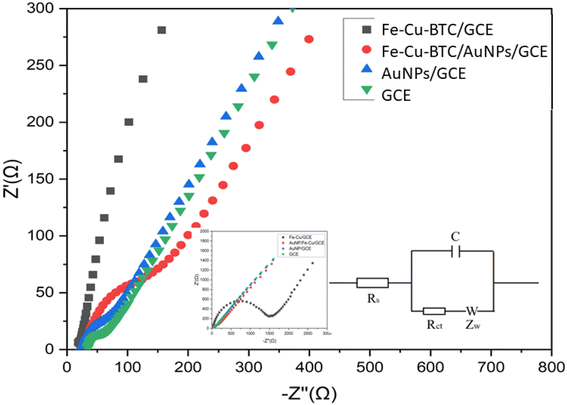 |
| | Fig. 7 Nyquist diagram of the GCE, Fe–Cu-BTC/GCE, AuNPs/GCE and Fe–Cu-BTC/AuNPs/GCE samples denoted by black squares, red circles, blue triangles and green inversed triangles, respectively, in the solution containing 5 mM Fe(CN)63−/4− and 0.1 M KCl. The parameter settings were as follows: frequency range from 0.01 Hz to 10000 Hz, an initiative potential of 0.23 V, an amplitude of 10 mV and a quiet time of 5 s. Its equivalent circuit is in inset. | |
3.4.1. Effects of scan rate (ν). The electroactive surface area (ECSA) of the electrodes were tested by scanning CV curves at different scan rates from 10 to 300 mV s−1. A bare GCE was used for comparison (Fig. S4†). Both anodic and cathodic peak heights were proportional to the square root of the scan rate (ip,a = 0.2442 Sqrt (ν) + 0.0011 with R2 = 0.9900; ip,c = −0.2211 Sqrt (ν) − 0.0311 with R2 = 0.9960; ip,a = 0.2222 Sqrt (ν) + 0.0015 with R2 = 0.9840; and ip,c = −0.2203 Sqrt (ν) − 0.0044 with R2 = 0.9940) for GCE and Fe–Cu-BTC/AuNPs/GCE, respectively. These peak heights were used for further calculations of effective surface areas. At 100 mV s−1, the ECSA of the Fe–Cu-BTC/AuNPs/GCE sample was estimated using the Randle–Sevcik equation as 0.0707 and 0.0721 cm2 for bare and modified electrodes, respectively.
3.5. Optimization of the electrochemical experiment parameters
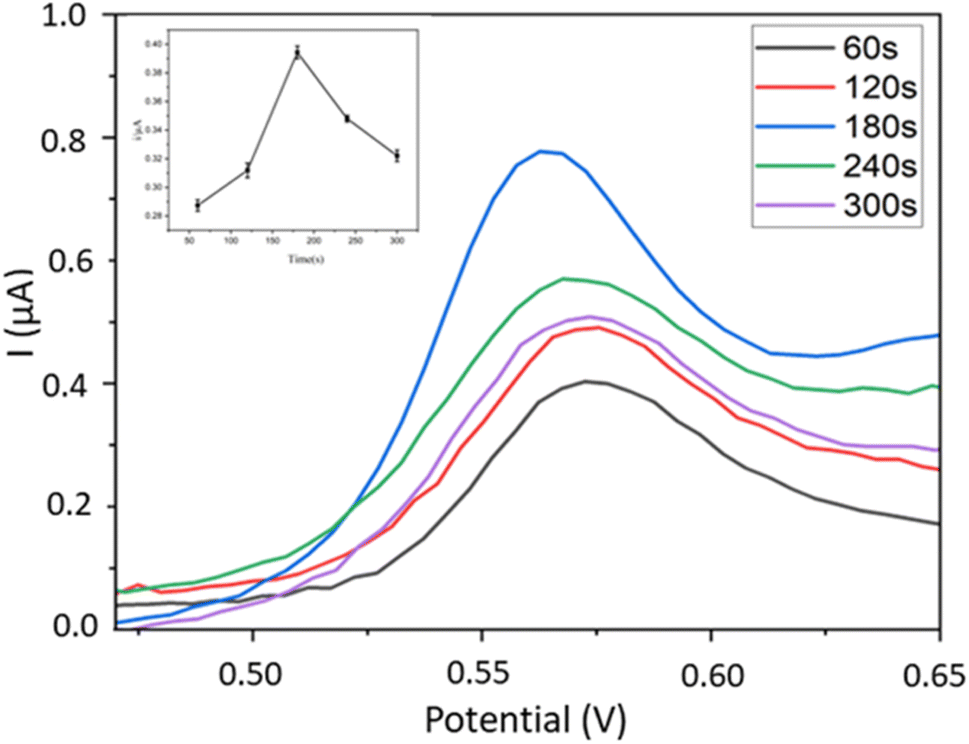
3.5.1. AuNPs deposition time. Here we investigated the effect of AuNPs deposition time on the DPV signals of BPA detected by the Fe–Cu-BTC/AuNPs/GCE electrodes optimized beforehand. The deposition time of AuNPs ranged from 60 to 300 s with 60 s interval. The results show that the highest electrochemical signal of BPA was recorded on the sensor with 180 s deposition time of AuNPs (Fig. 8). This phenomenon can be explained by the improvement of sensor conductivity by the Au layer39 and the electrocatalytic property of bimetallic MOFs,32 which enhance their synergistic effects for detecting BPA signals.
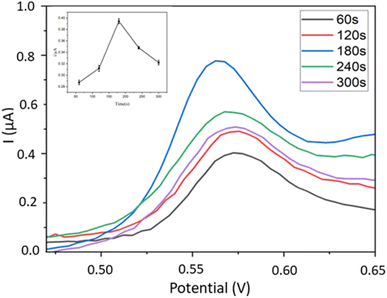 |
| | Fig. 8 Influence of Au deposition time on DPV signals of BPA using Fe–Cu-BTC/AuNPs/GCE. | |
3.5.2. Effect of pH. DPV signals of 0.4 mM BPA were recorded by sweeping the potential from 0.3 V to 0.8 V in a PBS buffer with pH level ranging from 6.0 to 9.0. The highest current was recorded at pH of 7, after which, the peak potential was negatively shifted as the pH of the solution continued to increase (Fig. 9). The peak potential was positively correlated to the buffer pH, Ep,a = −0.0615 pH + 0.725, R2 = 0.9933. Based on this equation, the electrochemical oxidation of BPA at Fe–Cu-BTC/AuNPs/GCE is a two-proton process as previously reported.10–13,32,39–41
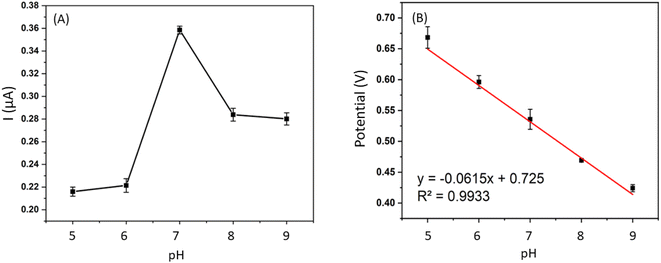 |
| | Fig. 9 Influence of pH on signals of BPA at different pH from 6.0 to 9.0. (A) Anodic peak currents varied by pH and (B) relationship between anodic peak (pa) potentials and pH (c). | |
3.5.3. Effects of accumulation time. The DPV voltammograms ranged from 0.35 V to 0.65 V of interested analyte at 0.4 mM were recorded at various accumulation time (i.e., 60–360 s) at 60 s interval in PBS electrolyte (pH 7.0). As indicating in this Fig. S5,† the saturation of BPA on modified and was reached at the accumulation time of 240 s. This adsorption time was used as the optimized duration to achieve maximum electrochemical signals.
3.6. Detection of BPA using Fe–Cu-BTC/AuNPs/GCE
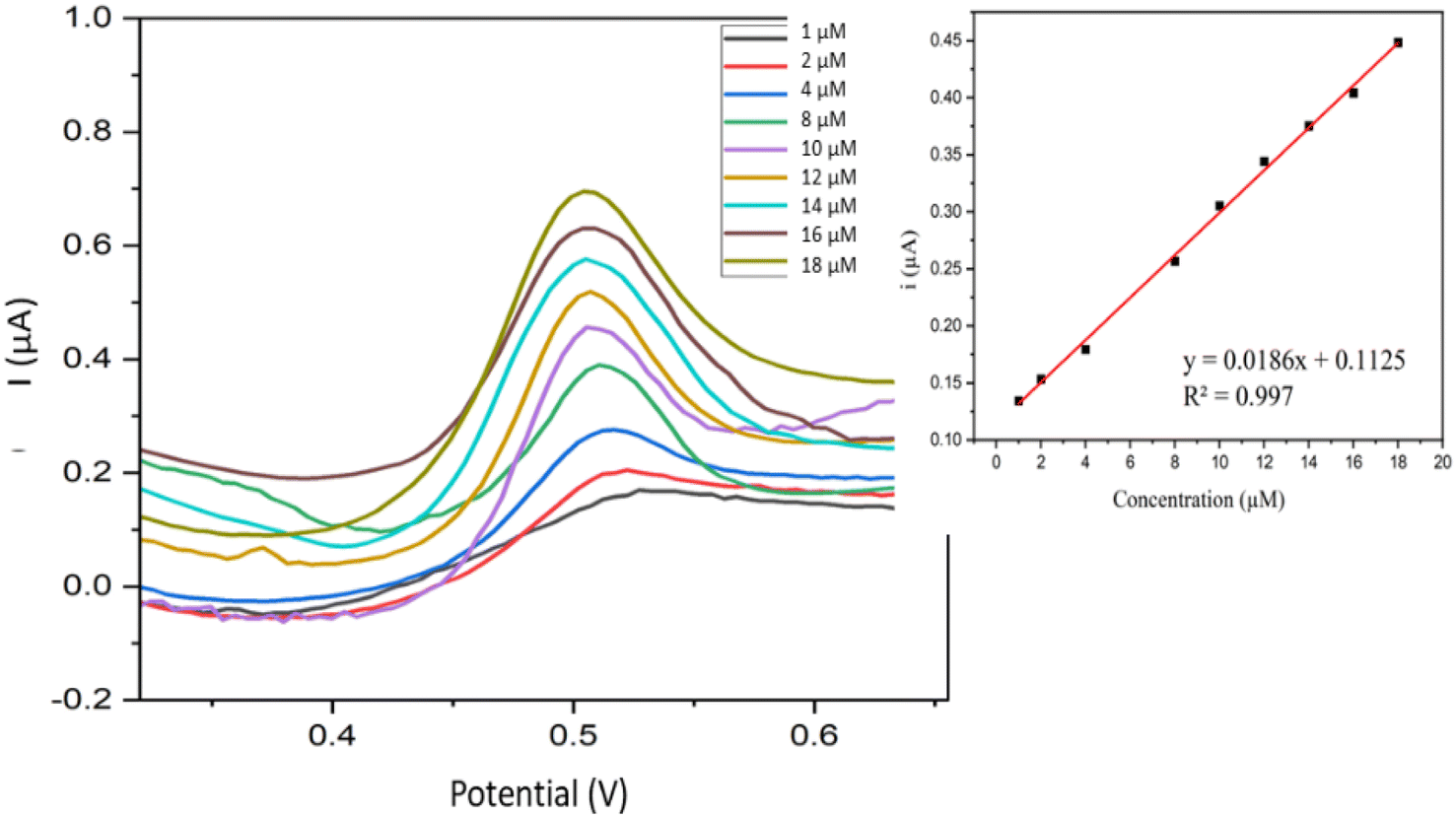
3.6.1. Calibration curve. Fig. 10 shows voltammograms of all the samples measured using Fe–Cu-BTC/AuNPs/GCE under optimized conditions (pH of 7, AuNPs deposition time of 180 s, and accumulation time of 240 s). The oxidation peak current of BPA exhibited a positive association with BPA concentration, i(μA) = 0.186C (μM) + 0.0125 (R2 = 0.9970) and the relative standard deviation (RSD) was 3.52%. The current on the Fe–Cu-BTC/AuNPs/GCE increased significantly (Fig. S6†), which indicated the effective electrocatalytic activity of the electrode and the improvement of the catalytic activity of BPA oxidation on the electrode surface. The electron transfer rate was facilitated between BPA and the Fe–Cu-BTC/AuNPs/GCE because of the extra high conductivity of AuNPs and the electrocatalytic activity on produced bimetallic Fe–Cu-BTC. The presence of Fe2+ as the metal center also enhanced the electrostatic force of the absorbent surface and exerted stronger attraction force towards the target,47 which consequently increased the oxidation response signal of BPA on the electrode.
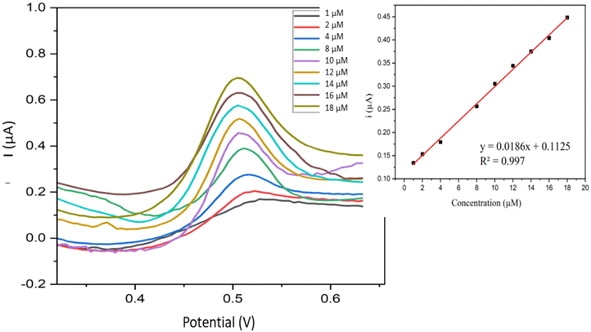 |
| | Fig. 10 Voltammograms acquired from BPA in the concentration range from 1.0 to 18 μM and the variations of peak intensities according to the concentration change (inset). | |
The limit of detection (LOD) of BPA was tested in a lower range (0.1–1.0 μM). LOD of was calculated as 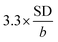 , in which SD was the standard deviation and b was the slope of the regression line. From the obtained regression curve y = 0.1915CBPA − 0.0158, R2 = 0.9989, LOD was determined as 18 nM which was more or less to the reported LOD (Table 1).
, in which SD was the standard deviation and b was the slope of the regression line. From the obtained regression curve y = 0.1915CBPA − 0.0158, R2 = 0.9989, LOD was determined as 18 nM which was more or less to the reported LOD (Table 1).
Table 1 Comparison of sensing performances of electrochemical sensor for detection of BPAa
| Electrode configuration |
Method |
Linear range (μM) |
LOD |
Ref. |
| Note: NPs – nanoparticles, CB – carbon black, MWNT – multi-walled carbon nanotubes, rGO – reduced graphene oxide, CTAB – cetyltrimethylammonium bromide, MOF – metal organic frames, BTC – benzene tricarboxylic, AuNDs – gold nano dendrites, Cd(TMPP) – (5,10,15,20-tetrakis[(4-methoxyphenyl)]porphyrinato)cadmium(II) complex, GO – graphene oxide, βCD – β-cyclodextrin, MIP – molecularly imprinted polymer, LSG – laser scribed graphene, screen-printed carbon electrode (SPCE), GCE – glassy carbon electrode, CAS – CB – casein (CAS) and carbon black (CB), CPE – carbon paste electrode, CS – chitosan, NiNP/NCN – nickel, nanoparticle/nitrogen-doped carbon nanosheet, PGE = pencil graphite electrode. |
| CB/f-MWCNTs |
CV |
0.1–130 |
0.08 μM |
12 |
| rGO-NiS |
DPV |
4.38 × 10−3–0.438 |
1.75 nM |
13 |
| Cu-BTC film/GCE |
DPV |
5 × 10−3–2.0 |
0.72 nM |
14 |
| AuNDs/CTAB/GCE |
DPV |
0.025–10 |
22 nM |
15 |
| x ([Cd(TMPP)]) |
DPV |
15–15 × 103 |
13.5 pM |
16 |
| 0.0015–15 |
| GO-MWCNT-βCD/SPE |
LSV |
0.05–5 |
6 nM |
17 |
| 5–30 |
| MCNTs/βCD |
DPV |
0.125–2 |
13.76 nM |
18 |
| 2–30 |
| MIP/PPy@LSG – tap |
DPV |
0.05–20 |
8 nM |
19 |
| NFO/SPCE |
DPV |
0.02–12.5 |
6 nM |
22 |
| PEN sensor AgNP 2% |
DPV |
2.5–15 |
0.24 μM |
23 |
| CAS-CB/GCE |
LSV |
0.49–24 |
0.25 μM |
25 |
| 3 wt% La–Yb2O3/CPE |
Amperometric |
0.04–2.66 |
13 nM |
26 |
| 2.6–15 |
| NiNP/NCN/CS/GCE |
DPV |
0.1–2.5 |
45 nM |
27 |
| 2.5–15 |
| WO3-CNT/GCE |
DPV |
0.03–3.0 μM and 3.0–100 μM |
16.3 nM |
31 |
| Ce–Ni-MOF/MWCNTs |
DPV |
0.1–100 |
7.8 nM |
32 |
| AuNPs/MWCNTs/GCE |
DPV |
0.01–0.7 |
4 nM |
39 |
| Fe–Cu-BTC/AuNPs/GCE |
DPV |
0.1–1.0 |
18 nM |
This work |
| 1.0–18 |
3.6.2. Reproducibility, repeatability and durability. The reproducibility was tested using the seven separate sensors. The small magnitudes of error bars as well as the average RSDs 3.7% in the measurements of BPA, shows the reproducible formation of Fe–Cu-BTC/AuNPs on GCE. The repeatability was evaluated by consecutively measuring a sample containing 10 μM BPA three times using one sensor. After three measurements, the peak intensities remain about 70%. The SEM and XRD of the Fe–Cu-BTC/AuNPs composite before and after BPA measurement are shown in Fig. S7.†
3.6.3. Interference study. Finally, the selectivity of measurements was evaluated by observing the electrochemical signals of 2,4-DCP samples in presence of Cd2+/Pb2+/Cu2+ or 4-nitrophenol as interferants. The concentrations of interferants were designedly fixed at 50 μM which is five times greater than BPA concentration. No significant changes in current intensity were observed, suggesting the absence of interference effect due to other compounds. The Fe–Cu-BTC/AuNPs/GCE exhibited good selective and effective property for BPA determination.
3.6.4. Rain coat real sample analysis. Fe–Cu-BTC/AuNPs/GCE was further used to determine BPA quantities in real samples prepared from rain coat. Sample preparation from plastic pieces was treated as previous procedure.16 Briefly, the plastic rain coat was cut into small pieces and washed several times with double distilled water. After vacuum drying, 1.0 g of the sample and 50 mL of purified water were added to a flask, sealed and heated at 70 °C for 48 h. The mixture was filtered through a 45 μm filter. The prepared solution was divided into two parts, one for fluorescence measurement and the other for electrochemical measurement in PBS solution of pH at 7. DPV voltammograms are shown in Fig. S8.† Table 2 shows BPA concentrations spiked in the treated solution using prepared Fe–Cu-BTC/AuNPs/GCE and the calculated recoveries.
Table 2 Determined BPA concentrations spiked in real sample using developed sensor its recoveries
| Sample |
BPA (μM) |
| Added |
Found |
Recovery (%) |
| Solution extracted from rain coat |
0 |
Not found |
— |
| 5.00 |
4.65 ± 0.044 |
93.00 |
| 10.00 |
8.57 ± 0.028 |
85.70 |
| 15.00 |
15.48 ± 0.091 |
103.23 |
4. Conclusion
In the present work, a bimetallic Fe–Cu-BTC material was prepared using hydrothermal reaction. Adding Fe2+ into the framework of MOF during synthesis to modify the surface morphology and pore formation of nanosized Cu-BTC was used for the synergistic effect of the second metal ion. The Fe–Cu-BTC sample has an average pore diameter of 1.40 nm, large pore volume (0.924 cm3 g−1), BET surface area (1504 cm3 g−1) and high Langmuir surface area (1831 cm3 g−1). Sensors developed by drop casting this porous material on GCE followed by AuNPs electrochemical deposition were used for detection of BPA with detection limits of 18 nM and two linear ranges from 0.1 to 1.0 μM and 1.0 to 18 μM. Sensor-to-sensor reproducibility was estimated by measuring seven separate prepared Fe–Cu-BTC/AuNPs/GCE sensors and RSDs of the peak intensities of BPA were 3.70%, thereby indicating the superior sensor-to-sensor reproducibility. Finally, Fe–Cu-BTC/AuNPs/GCE was applied to the real sample from rain coat with satisfactory recovery values from 85.70 to 103.23%.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by Vietnam Academy of Science and Technology under the grant number NCVCC06.09/22-23.
References
- W. Liu and X.-B. Yin, TrAC, Trends Anal. Chem., 2016, 75, 86–96 CrossRef CAS.
- Y. Cao, L. Wang, C. Shen, C. Wang, X. Hu and G. Wang, Sens. Actuators, B, 2019, 283, 487–494 CrossRef CAS.
- L. Ji, Q. Cheng and K. Wu, Sens. Actuators, B, 2016, 231, 12–17 CrossRef CAS.
- B. Ma, H. Guo, M. Wang, L. Li, X. Jia, H. Chen, R. Xue and W. Yang, Electroanalysis, 2019, 31, 1002–1008 CrossRef CAS.
- D. Brondani, E. Zapp, R. S. Heying, B. Souza and I. C. Vieira, Electroanalysis, 2017, 29, 2810–2817 CrossRef CAS.
- Q. Chen, X. Li, X. Min, D. Cheng, J. Zhou, Y. Li, Z. Xie, P. Liu and W. Cai, Electroanal. Chem., 2017, 789, 114–122 CrossRef CAS.
- S. Dong, G. Suo, N. Li, Z. Chen, L. Peng, Y. Fu, Q. Yang and T. Huang, Sens. Actuators, B, 2016, 222, 972–979 CrossRef CAS.
- J. Li, J. Xia, F. Zhang, Z. Wang and Q. Liu, Talanta, 2018, 181, 80–86 CrossRef CAS PubMed.
- T. Yu, S. Li, L. Zhang, F. Li, J. Wang, H. Pan and D. Zhang, J. Colloid Interface Sci., 2023, 629, 546–558 CrossRef CAS PubMed.
- F. S. vom Saal and C. Hughes, Environ. Health Perspect., 2015, 11, 926–933 Search PubMed.
- J. Huang, T. Zhang, G. Dong, S. Zhu, F. Yan and J. Liu, Front. Chem., 2022, 10, 900282 CrossRef CAS PubMed.
- A. Thamilselvan, V. Rajagopal and V. Suryanarayanan, J. Alloys Compd., 2019, 786, 698–706 CrossRef CAS.
- T. D. Vu, K. D. Pham, H. T. Bui, S.-H. Han and H. Chung, Sens. Actuators, B, 2019, 281, 320–325 CrossRef CAS.
- P. Hu, X. Zhu, X. Luo, X. Hu and L. Ji, Microchim. Acta, 2020, 187, 145 CrossRef CAS PubMed.
- J. Zhang, X. Xu and L. Chen, Sens. Actuators, B, 2018, 261(15), 425–433 CrossRef CAS.
- N. T. Lien, L. Q. Hung, N. T. Hoang, V. T. Thu, D. T. N. Nga, P. T. H. Yen, P. H. Phong and V. T. T. Ha, J. Anal. Methods Chem., 2020, 6693595, DOI:10.1155/2020/6693595.
- D. Jemmeli, C. Mchiri, C. Dridi, H. Nasri and E. Dempsey, RSC Adv., 2020, 10, 31740–31747 RSC.
- A. U. Alam and M. J. Deen, Anal. Chem., 2020, 92, 5532–5539 CrossRef CAS PubMed.
- A. Arif, U. Alama and Matiar M. R. Howlader, Sens. Actuators, B, 2020, 320, 128319 CrossRef.
- T. Beduk, A. Ait Lahcen, N. Tashkandi and K. N. Salama, Sens. Actuators, B, 2020, 314, 128026 CrossRef CAS.
- D. Jemmeli, E. Marcoccio, D. Moscone, C. Dridi and F. Arduini, Talanta, 2020, 216, 120924 CrossRef CAS PubMed.
- J. Cai, B. Sun, W. Li, X. Gou, Y. Gou, D. Li and F. Hu, J. Electroanal. Chem., 2019, 835, 1–9 CrossRef CAS.
- G. Kesavan, N. Nataraj, S. M. Chen and L. H. Lin, New J. Chem., 2020, 44, 7698–7707 RSC.
- M. Baccarin, M. A. Ciciliati, O. N. Oliveira, E. T. G. Cavalheiro and P. A. RaymundoPereira, Mater. Sci. Eng. C, 2020, 114, 110989 CrossRef CAS PubMed.
- S. Z. Bas, N. Yuncu, K. Atacan and M. Ozmen, Electrochim. Acta, 2021, 386, 138519 CrossRef CAS.
- L. V. Jodar, L. O. Orzari, T. S. Ortolani, M. H. M. T. Assumpção, F. C. Vicentini and B. C. Janegitz, Electroanalysis, 2019, 31, 2162–2170 CrossRef.
- X. Wang, Y. Li, Y. Zhou and Y. He, Anal. Methods, 2019, 11, 5613–5622 RSC.
- Y. Wang, C. Yin and Q. Zhuang, J. Alloys Compd., 2020, 827, 154335 CrossRef CAS.
- M. L. Chen, J. H. Chen, L. Ding, Z. Xu, L. Wen, L. B. Wang and Y. H. Cheng, Anal. Methods, 2017, 9(6), 906–909 RSC.
- J. Zhang, X. Xu and L. Chen, Sens. Actuators, B, 2018, 261(15), 425–433 CrossRef CAS.
- Y. Zhou, X. She, Q. Wu, J. Xiao and T. Peng, J. Electroanal. Chem., 2022, 915, 116355 CrossRef CAS.
- X. Huang, D. Huang, J. Y. Chen, R. Ye, Q. Lin and S. Chen, Anal. Bioanal. Chem., 2020, 412, 849–860 CrossRef CAS PubMed.
- D. M. Schubert, M. Z. Visi and C. B. Knobler, Main Group Chem., 2018, 7(4), 311–322 CrossRef.
- H. Xing, S. Peng, T. Zeyi, F. Zhang, J. Wang, H. Geng, D. Zou, C. Di, Y. Yi, Y. Sun and W. Xu, Nat. Commun., 2015, 6(1), 7408 CrossRef PubMed.
- S. Lei, G. M. Campbell and D. Mircea, Angew. Chem., Int., 2016, 55(11), 3566–3579 CrossRef PubMed.
- X. Wang, Q. Wang, Q. Wang, F. Gao, F. Gao, Y. Yang and H. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 11573–11580 CrossRef CAS PubMed.
- Q. Wang, Y. Yang, F. Gao, J. Ni, Y. Zhang and Z. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 32477–32487 CrossRef CAS PubMed.
- B. N. Manh, V. T. H. Nhung, V. T. Thu, D. T. N. Nga, N. P. T. Thuan, H. T. Giang, P. T. H. Yen, P. H. Phong, A. V. Tuan and V. T. T. Ha, RSC Adv., 2020, 10, 42212–42220 RSC.
- V. T. T. Ha, B. N. Manh, T. N. Tam, V. T. Thu, P. T. H. Yen, P. H. Phong, D. N. Nhiem, L. Q. Hung and T. Q. Hai, J. Appl. Electrochem., 2022, 52, 607–616 CrossRef CAS.
- N. B. Messaoud, M. E. Ghica, C. Dridi, M. B. Ali and C. M. A. Brett, Sens. Actuators, B, 2017, 253, 513–522 CrossRef.
- Y. Zhang, W. Zhang, L. Zhang, G. Song, N. Wang and W. Xu, et al., Microchem. J., 2021, 170, 106737–106746 CrossRef CAS.
- M. B. Nguyen, G. H. Le, T. Duy, Q. K. Nguyen, T. Trang, T. Pham, T. Lee and T. A. Vu, J. Hazard. Mater., 2021, 420, 126560 CrossRef PubMed.
- D. T. N. Nga, V. V. Hung, C. T. Thanh, N. V. Chuc, L. C. Thanh, T. D. Lam, P. N. T. Thuan, N. T. Loc, B. Piro and V. T. Thu, Sens. Actuators, B, 2019, 283, 52–60 CrossRef.
- W. Y. Siew, N. H. H. Abu Bakar, M. Abu Bakar and A. Zainal Abidin, J. Hazard. Mater., 2021, 416, 125846 CrossRef CAS PubMed.
- N. T. Dung, N. H. Duc, V. T. Binh, V. D. Thao, M. B. Nguyen, L. V. Ngan and N. N. Huy, Sep. Purif. Technol., 2021, 285, 120358 CrossRef.
- H. T. Vu, M. B. Nguyen, T. M. Vu, G. H. Le, T. T. T. Pham, T. D. Nguyen and T. A. Vu, Top. Catal., 2020, 63, 1046–1055 CrossRef CAS.
- X. Hu, X. Lou, C. Li, Y. Ning, Y. Liao, Q. Chen, E. S. Mananga, M. Shen and B. Hu, RSC Adv., 2016, 6, 114483–114490 RSC.
- A. R. Oveisi, A. Khorramabadizad and S. Daliran, RSC Adv., 2016, 6(2), 1136–1142 RSC.
- A. R. Oveisi, A. Khorramabadizad and S. Daliran, RSC Adv., 2016, 6, 1136–1142 RSC.
- Y. Li, J. Miao, X. Sun, J. Xiao, Y. Li, H. Wang, Q. Xia and Z. Li, Chem. Eng. J., 2016, 298, 191–197 CrossRef CAS.
- F. Dorosti and A. Alizadehdakhel, Chem. Eng. Res. Des., 2018, 136, 119–128 CrossRef CAS.
- A. Yurduşen, A. Yürüm and Y. Yürüm, Cryst. Eng. Comm., 2020, 22, 932–944 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Pham Thi Hai Yena,
Pham Hong Phonga,
Le Quoc Hunga,
Nguyen Thi Thanh Nganc,
Tran Quang Haib and
Vu Thi Thu Ha*a
c,
Pham Thi Hai Yena,
Pham Hong Phonga,
Le Quoc Hunga,
Nguyen Thi Thanh Nganc,
Tran Quang Haib and
Vu Thi Thu Ha*a



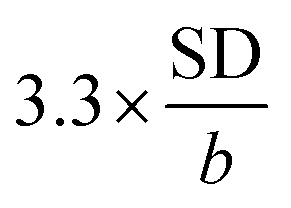 , in which SD was the standard deviation and b was the slope of the regression line. From the obtained regression curve y = 0.1915CBPA − 0.0158, R2 = 0.9989, LOD was determined as 18 nM which was more or less to the reported LOD (Table 1).
, in which SD was the standard deviation and b was the slope of the regression line. From the obtained regression curve y = 0.1915CBPA − 0.0158, R2 = 0.9989, LOD was determined as 18 nM which was more or less to the reported LOD (Table 1).