
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBromide ion promoted practical synthesis of phosphinothioates of sulfinic acid derivatives and H-phosphine oxides†
Haoyuan Li‡
,
Wenjie Yan‡,
Peipei Ren,
Huimin Hu,
Runbo Sun,
Meixia Liu,
Zhengjiang Fu ,
Shengmei Guo* and
Hu Cai*
,
Shengmei Guo* and
Hu Cai*
Department of Chemistry, Nanchang University, No. 999, Xuefu Rd, Nanchang, 330031, P. R. China. E-mail: smguo@ncu.edu.cn; Caihu@ncu.edu.cn
First published on 15th November 2022
Abstract
A feasible method for the synthesis of phosphinothioates from sulfinic acid derivatives and phosphine oxides is described. This reaction can be carried out in an open flask at room temperature and in an aqueous medium. The scope of the sulfinic acid derivatives is extensive, with a wide range of sulfinate esters, sulfinic acids, and sodium sulfinates compatible with these conditions, with good to excellent yields of phosphinothioates. In addition, a gram-scale synthesis with this reaction is achieved. A mechanism of this procedure was proposed.
The S–P(O) structural unit is often found in pharmacologically active chemicals, organophosphorus insecticides, and natural products, and also serves as a building block in the preparation of a variety of compounds.1 Given its vast applications, numerous efforts have been made to develop efficient methods to approach the S–P(O) structurally contained compounds.2 Conventional methods for the construction of such compounds are involved in nucleophilic substitution of R2P(O)X or R–SX with nucleophiles.3 However, the toxicity and instability of R2P(O)X and R–SX limit their applications. As a result, green and efficient strategies for the synthesis of S–P(O) bonds attracted considerable attention. In recent years, catalysed oxidative coupling reactions utilizing inexpensive and readily available RS–H (RS–SR) and P(O)–H have emerged as appealing and potent techniques for the synthesis of these compounds.4 Working with thiol and sulfide reagents, however, may be a highly unpleasant experience, especially for large-scale synthesis and industrial processes.5 Alternatively, the reductive coupling reaction of RS(VI) or RS(IV) with P(O)–H has appeared as a new strategy for accessing these molecules. Among them, coupling reactions of RSO2Cl, RSO2NH2, and RSO2NH2NH2 with P(O)–H were well investigated.6 In these reactions, transition metals as catalysts are required. To overcome upon drawback, Hong and Yi separately devised elegant metal-free reductive coupling reactions using sulfinic acids or sodium sulfinates to construct S–P(O) bonds,7 while external reducer (PPh3) or H2SO4 were necessary. Despite significant progress, more practical synthetic methods for the formation of phosphinothioates are desirable. Based on our ongoing works on C–P and S–P bond formation,8 herein, we disclose a bromide ion accelerated reductive-coupling for the producing phosphinothioates from sulfinic acid derivatives and phosphine oxides.9 This method is suited for practical synthesis since it is free of metal reagents and external reducers, and allows to be carried out under an open flask and in an aqueous medium Scheme 1.
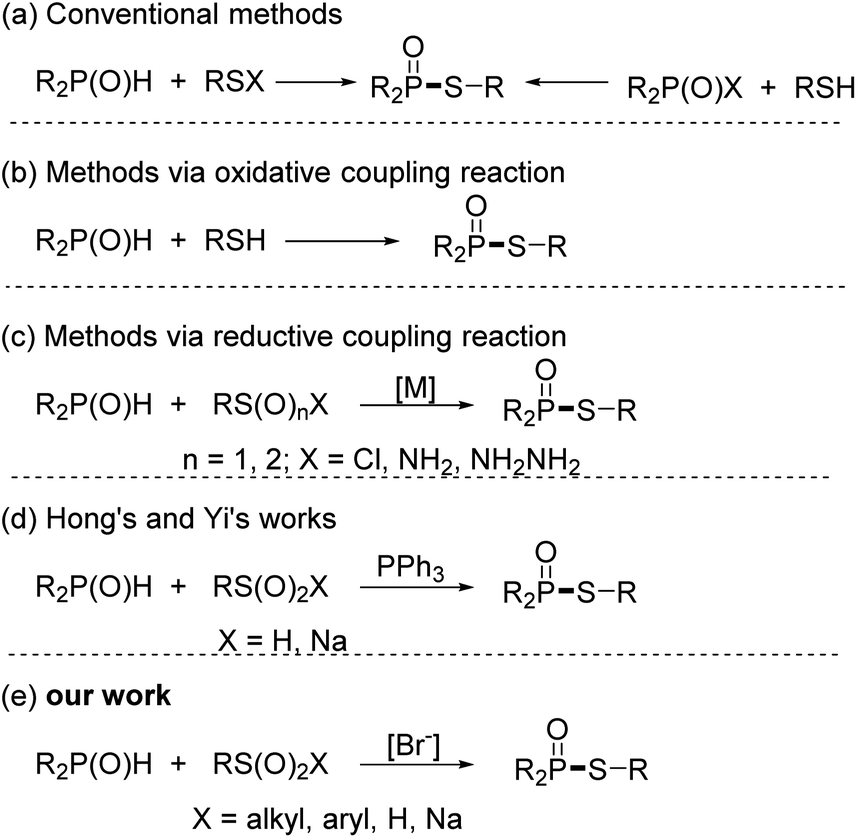 | ||
| Scheme 1 Methods for the formation of phosphinothioates. | ||
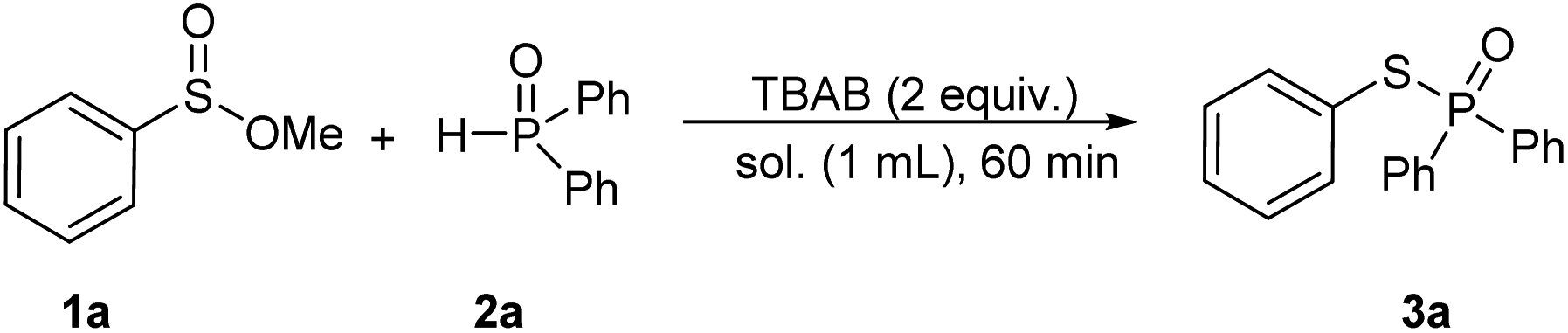
Initially, we chose methyl benzenesulfinate (1a, 0.2 mmol) and diphenylphosphine oxide (2a, 0.5 mmol) as model reactants to optimize the reaction conditions (Table 1). A tiny quantity of desired product was identified after 1 hour at room temperature without using any solvent (Table 1, entry 1). Then, TBAB (tetrabutylammonium bromide) was added to the reaction to increase the possibility of reactants exposure, and the product 3a was afforded in 30% yield (Table 1, entry 2). Inspired by this, we prolonged the reaction time to 4 h, but the yield of product had no significant increase (Table 1, entry 3). Interestingly, the product of 3a was isolated in 94% yield when CH3CN (1 mL) was employed as a solvent (Table 1, entry 4). In the upon solvent, TEAB (tetraethylammonium bromide) and HTAB (hexadecyl trimethyl ammonium bromide) were tested, and found that they could promote the reaction with the yields of 62% and 68%, respectively (Table 1, entries 5 and 6). Other quaternary ammonium salts with different anions like TBAF (tetrabutylammonium fluoride), TBAI (tetrabutylammonium iodide), or TBAC (tetrabutylammonium chloride) showed inefficient in this transformation (Table 1, entries 7–9). Considering the bromide anion has a positive effect on this coupling reaction, LiBr and KBr were independently applied to the reaction, and the 3a was obtained in 78% and 72% yields (Table 1, entries 10 and 11), whereas KI and K2CO3 delivered the desired product in 32% and 28% yields (Table 1, entries 13 and 14). Other solvents such as THF, dioxane, and CH3OH were studied, but lower yields were obtained in each case (Table 1, entries 14–16). It should be noted that the coupling product was isolated in 85% yield when H2O was utilized as a solvent after increasing the reaction duration to 4 h (Table 1, entry 17). Next, the ratio of reactants was examined. We found that decreasing the amount of phosphine oxide would reduce the yield; while increasing the ratio of 1a and 2a to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, the product has no improvement (Table 1, entries 18–20). Besides, 92% yield of product was obtained when the reaction was carried out under N2 (Table 1, entry 21). In addition, the reaction could deliver the desired product in 92% yield in 30 minutes (Table 1, entry 22).
:3, the product has no improvement (Table 1, entries 18–20). Besides, 92% yield of product was obtained when the reaction was carried out under N2 (Table 1, entry 21). In addition, the reaction could deliver the desired product in 92% yield in 30 minutes (Table 1, entry 22).
|
||||
|---|---|---|---|---|
| Entry | Promoter | Solvent | 1a:2a |
Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.5 mmol), TBAB (0.4 mmol), CH3CN (1 mL) in an open flask at rt for 1 h.b Isolated yields.c 4 h.d Under N2.e 30 min. | ||||
| 1 | None | None | 1:2.5 |
Trace |
| 2 | TBAB | None | 1:2.5 |
30 |
| 3c | TBAB | None | 1:2.5 |
32 |
| 4 | TBAB | CH3CN | 1:2.5 |
94 |
| 5 | TEAB | CH3CN | 1:2.5 |
62 |
| 6 | HTAB | CH3CN | 1:2.5 |
68 |
| 7 | TBAF | CH3CN | 1:2.5 |
Trace |
| 8 | TBAI | CH3CN | 1:2.5 |
60 |
| 9 | TBAC | CH3CN | 1:2.5 |
45 |
| 10 | LiBr | CH3CN | 1:2.5 |
78 |
| 11 | KBr | CH3CN | 1:2.5 |
73 |
| 12 | KI | CH3CN | 1:2.5 |
32 |
| 13 | K2CO3 | CH3CN | 1:2.5 |
28 |
| 14 | TBAB | THF | 1:2.5 |
36 |
| 15 | TBAB | Dioxane | 1:2.5 |
31 |
| 16 | TBAB | CH3OH | 1:2.5 |
20 |
| 17c | TBAB | H2O | 1:2.5 |
85 |
| 18 | TBAB | CH3CN | 1:1 |
48 |
| 19 | TBAB | CH3CN | 1:2 |
79 |
| 20 | TBAB | CH3CN | 1:3 |
92 |
| 21d | TBAB | CH3CN | 1:2.5 |
92 |
| 22e | TBAB | CH3CN | 1:2.5 |
92 |
Ethyl, n-butyl, benzyl, and phenethyl benzenesulfinate (1b–1f) afforded the desired products (3a) above 80% yields but inefficient than OMe group. Besides, large steric hindrance group esters such as isopropyl, t-butyl, and cyclohexyl showed poor reactivity, which was probably due to the prohibition of the attack from phosphine oxide (please see ESI†).
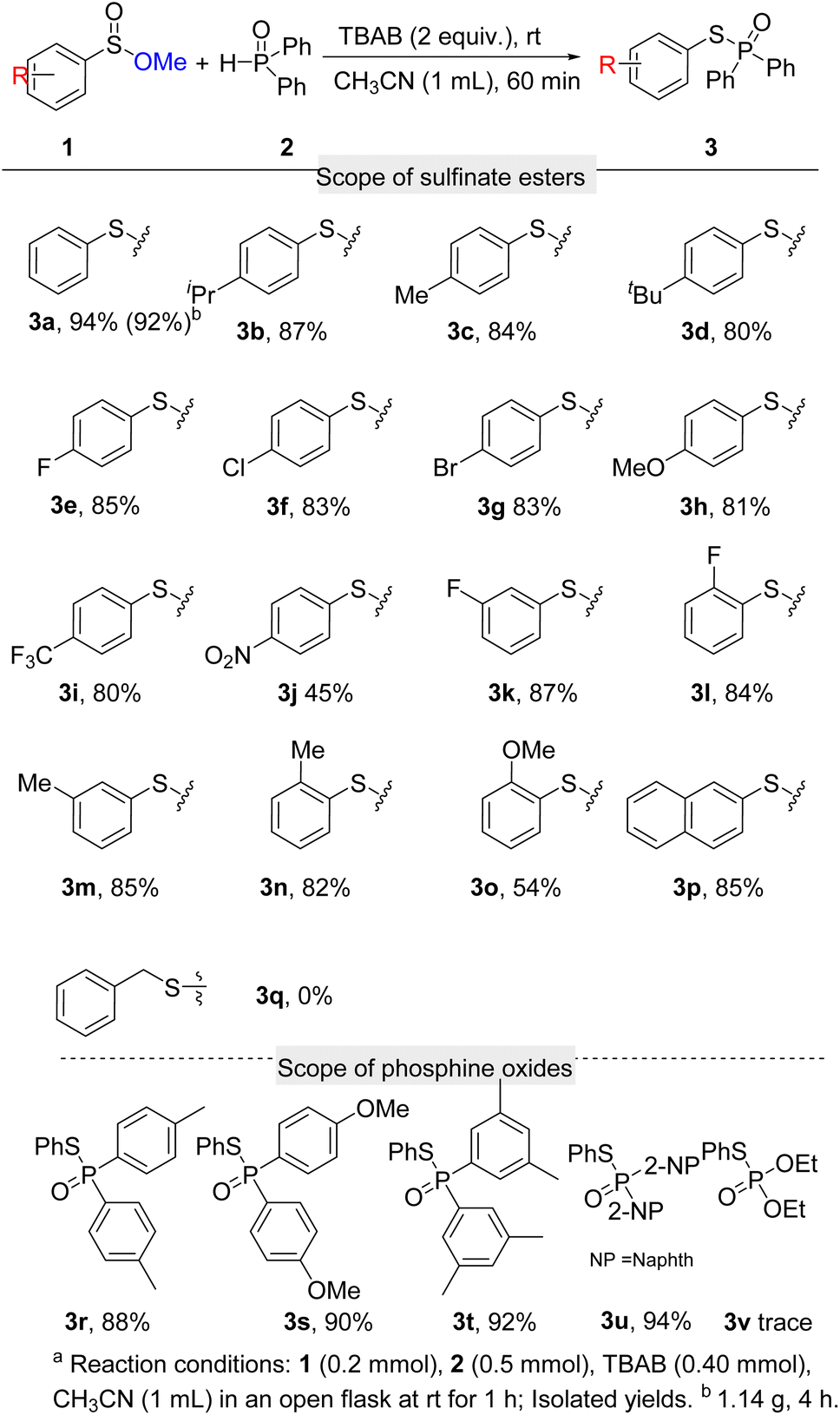
Having identified the optimal reaction conditions, the scope of sulfinate esters in this transformation was studied (Scheme 2). Firstly, we investigated the effect of the groups on the aryl ring on the reaction. Alkyl groups at the 4-position of aryl such as isopropyl, methyl, and tert-butyl reacted smoothly with diphenylphosphine oxide (2a) to afford 3b, 3c, and 3d in good yields. Halogens at the 4-position of the benzene ring have little effect on the reaction, which gave the desired products above 80% yields (3e, 3f, 3g). When –OMe and –CF3 groups were donated on the phenyl ring, the desired products 3h and 3i were afforded in 81% and 80% yields, respectively. Substrate with a strong electron-withdrawing group such as –NO2 at the 4-position of aryl showed weaker activity under the standard conditions, which gave the corresponding product in 45% yield (3j). Group on the other positions of aryl rings were also studied. 3-F, 2-F, 3-Me, 2-Me, and 3-OMe substituted sulfinate esters were successfully carried out to contribute the desired products in 87%, 84%, 85%, 82%, and 54% yields respectively (3k, 3l, 3m, 3n, 3o). Naphthalenesulfinate ester was also compatible with the reaction, leading to 3p in 85% yield. Unfortunately, benzyl sulfinate ester was unsuitable for the conversion (3q). In addition, gram-scale synthesis of 3a was achieved with more reaction time (4 h), and excellent yield can be maintained.
 | ||
| Scheme 2 Scope of sulfinate esters and phosphine oxides.a | ||
Next, the compatibility of phosphine oxides was evaluated. Di-p-tolylphosphine oxide and bis(4-methoxyphenyl)phosphine oxide afforded the expected products in 88% and 90% yields (3r, 3s). Bis(3,5-dimethylphenyl)phosphine oxide delivered the coupling product 3t in 92% yield. Di(naphthalen-2-yl)phosphine oxide also displayed high reaction activity, which provided 3u in 94% yield. Despondently, diethyl phosphonate did not anticipate this reaction.
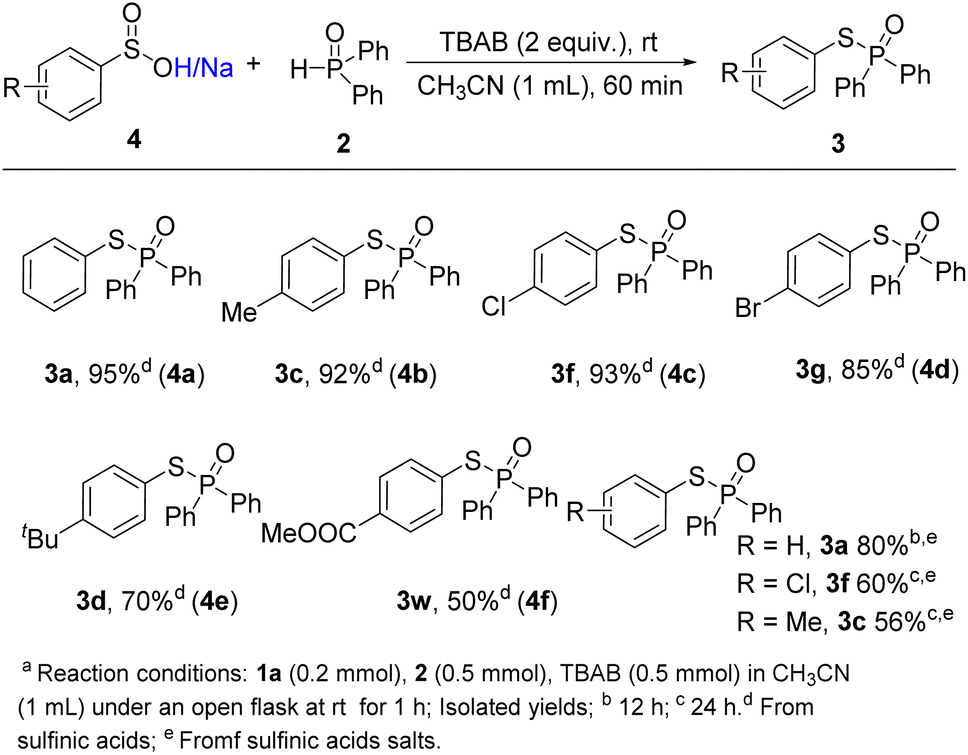
The compatibility of sulfinic acids and their salts were also tested under the standard conditions (Scheme 3). Benzenesulfinic acid delivered the reductive coupling product 3a in 95% yield. 4-Methylbenzenesulfinic acid 4b, 4-chlorobenzenesulfinic acid 4c and 4-bromobenzenesulfinic acid 4d also showed excellent reactive activities, leading to the desired products in 92%, 93% and 85% yields, respectively. Besides, 4e and 4f delivered the desired coupling products in moderate yields (3d, 3w). Although a longer reaction time was required (12 h), benzene sulfinic acid sodium could undergo the reductive coupling procedure, which gave the corresponding product 3a in 80% yield. When sodium 4-chlorobenzenesulfinate and sodium 4-methyl benzenesulfinate were subjected to the transformation, moderate yields of coupling products were obtained after prolonging the reaction time to 24 h (3f, 3c), which might be owing to the salts' solubility.
 | ||
| Scheme 3 Scope of sulfinic acids and their salts.a | ||
Green synthesis is one of the main themes in modern organic chemistry. Water is considered a green solvent in reaction. Interestingly, this reaction may also be carried out in aqueous medium (Scheme 4). When 1a and 2a were subjected to water instead of CH3CN and were stirring for 4 h, the coupling product 3a was obtained in 85% yield. Methyl 4-methylbenzenesulfinate and 4-chlorobenzenesulfinate were compatible with these reaction conditions, which delivered the desired products in 78% and 80% yields (3c, 3f), respectively. The other phosphine oxide like di-p-tolylphosphine oxide was also tested in water, which gave the product 3t in 65% yield.
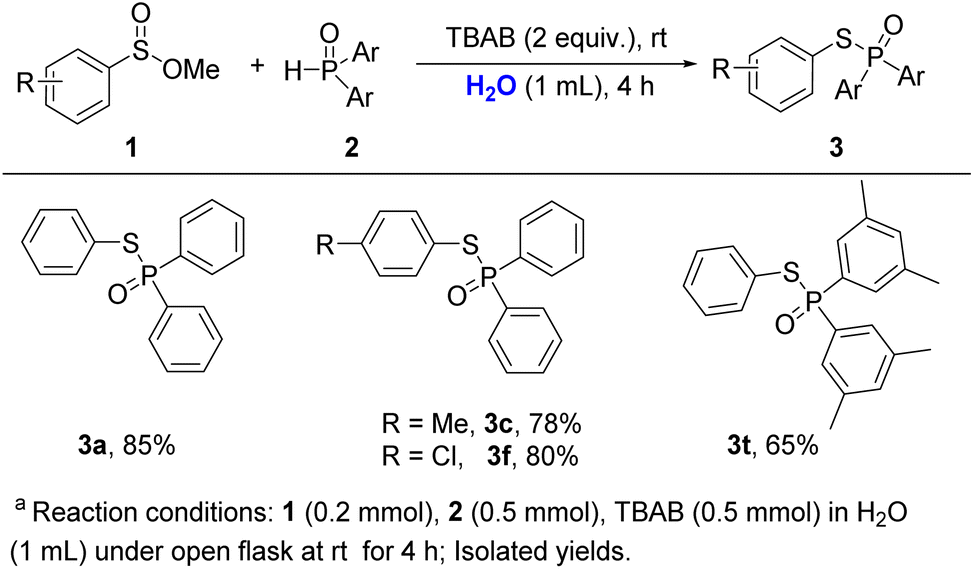 | ||
| Scheme 4 Water as a solvent.a | ||
To investigate the mechanism of this transformation, several control experiments were conducted (Scheme 5). When BHT (butylated hydroxytoluene) and 1,1-diphenylethylene (DPE) were added to the model reaction under the standard conditions, the yields of 3a were obtained in 85% and 87% yields, which suggested that the reaction does not undergo a radical route (Scheme 5a and b). However, when TEMPO was employed in the reaction, a 17% yield of the coupling product was obtained. This is because TEMPO is an oxidant, which will disrupt the reductive reaction conditions. When the ratio of 1a and 2a was lowered to 2:1, the corresponding product was obtained in 46% yield, this indicated that one molecule of 2a is served as a reducer (Scheme 5c). Moreover, lots of diphenylphosphinic acid was precipitated as a colorless solid from the reaction solution, and the crystal diffraction of this compound was obtained (Scheme 5d). The 1H NMR track experiments showed that the shift of 3.45–3.50 (–OMe group of 1a) appeared several peaks, suggested that 1a was affected in the conditions (Scheme 6, and please ESI†).
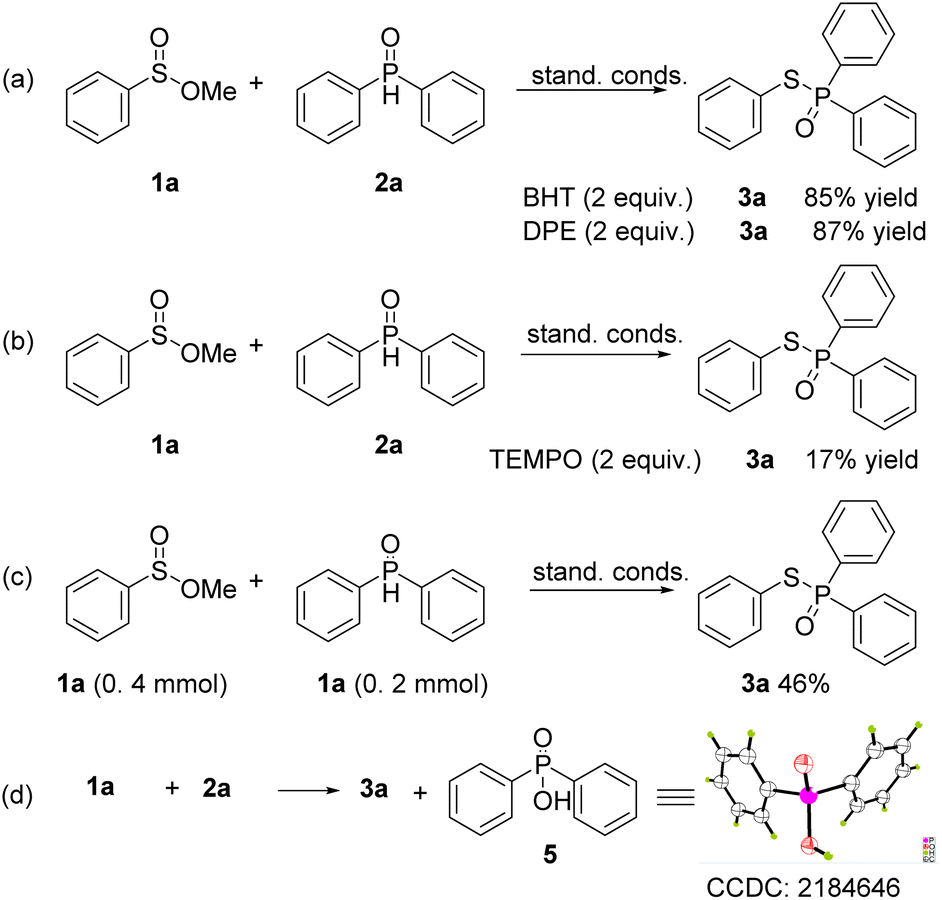 | ||
| Scheme 5 Control experiments (a–c) and investigation of byproduct (d). | ||
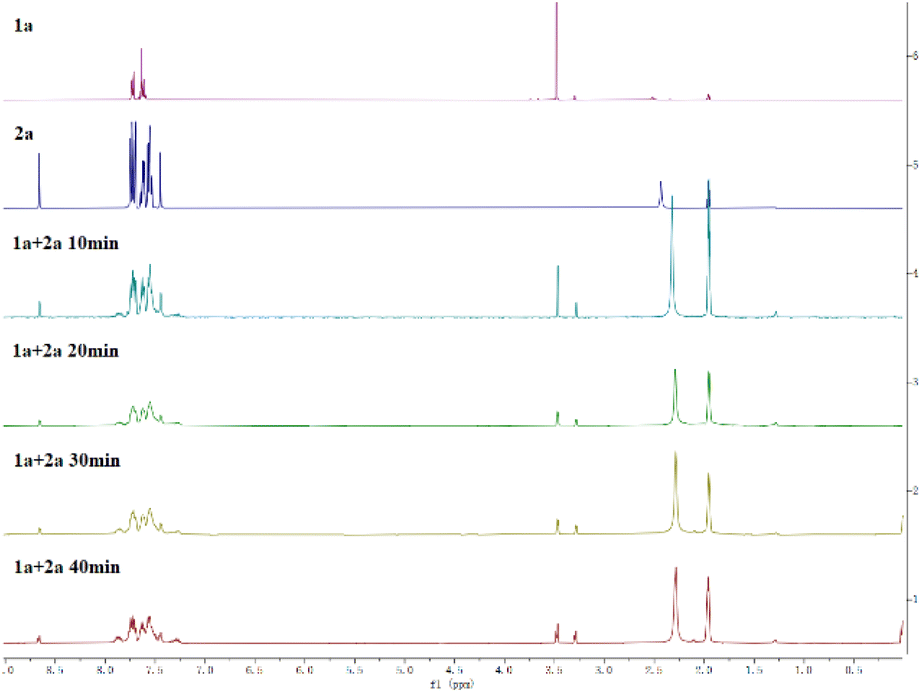 | ||
| Scheme 6 1H NMR experiments (CD3CN). | ||
According to literature and the results of our experiments,6,7,9 a plausible mechanism of this transformation is proposed (Scheme 7). Firstly, bromide anion acts on the sulfinate esters 1a to produce B. Then the P(V) in phosphine oxide maintains equilibrium with P(III) phosphorus anion to attacks B to obtain the intermediate C and release a molecular of HBr. After that, C is reduced by 2a to give the desired product 3a and D.
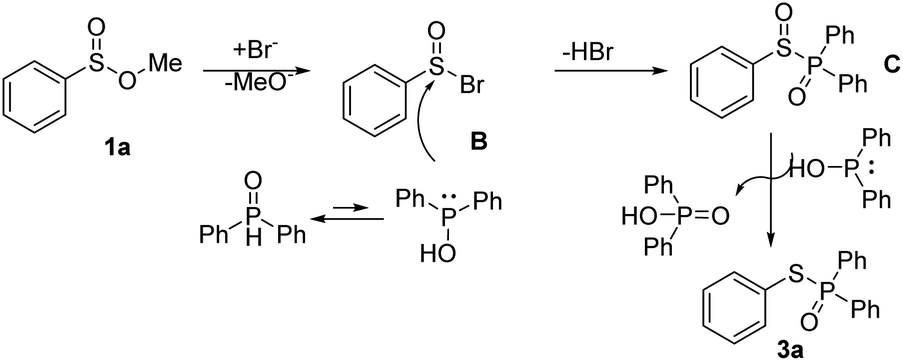 | ||
| Scheme 7 Proposed mechanism of the reaction. | ||
Conclusions
In conclusion, we have developed an efficient and practical method for the synthesis of phosphinothioates from sulfinate esters (sulfinic acids) and phosphine oxides. TBAB play an accelerator role this reaction in the absence of a metal catalyst an external reducer. The reaction may also be carried out in an open flask and aqueous conditions, and rapidly converted into phosphinothioate at room temperature, which can significantly reduce pollution and expense. A range of sulfinate esters and phosphine oxides may be compatible with the transformation. Furthermore, gram-scaled synthesis was achieved with excellent yield. Mechanism studies revealed that the reaction might go through a nucleophilic substitution and a reduction procedure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (21861024, 21861026, 21761021).Notes and references
- (a) N.-S. Li, J. K. Frederiksen and J. A. Piccirili, Acc. Chem. Res., 2011, 44, 1257 CrossRef CAS PubMed; (b) P. J. Murphy, Organophosphorus Reagents, Oxford University Press, Oxford, UK, 2004 Search PubMed; (c) R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278 CrossRef CAS; (d) J. H. Barnes, M. J. Fatome, E. L. Christopher and S. G. Murray, Eur. J. Med. Chem., 1988, 23, 211 CrossRef CAS; (e) T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J. B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902 CrossRef CAS PubMed; (f) B. Kaboudin, S. Emadi and A. Hadizadeh, Bioorg. Chem., 2009, 37, 101 CrossRef CAS PubMed.
- (a) L. Zhang, P. Zhang, X. Li, J. Xu, G. Tang and Y. Zhao, J. Org. Chem., 2016, 81, 5588 CrossRef CAS; (b) L. Wang, S. Yang, L. Chen, S. Yuan, Q. Chen, M.-Y. He and Z.-H. Zhang, Catal. Sci. Technol., 2017, 7, 2356 RSC; (c) C. Wen, Q. Chen, Y. Huang, X. Wang, X. Yan, J. Zeng, Y. Huo and K. Zhang, RSC Adv., 2017, 7, 45416 RSC; (d) Y. Chen, M. Li, Z. Gong and Z. Shen, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 196, 19 CrossRef; (e) S. Li, T. Chen, Y. Saga and L.-B. Han, RSC Adv., 2015, 5, 71544 RSC.
- (a) D. C. Morrison, J. Am. Chem. Soc., 1955, 77, 181 CrossRef CAS; (b) R. G. Harvey, H. I. Jacobson and E. V. Jensen, J. Am. Chem. Soc., 1963, 85, 1623 CrossRef CAS; (c) M. Arisawa, T. Ono and M. Yamaguchi, Tetrahedron Lett., 2005, 46, 5669 CrossRef CAS; (d) Y.-J. Ouyang, Y.-Y. Li, N.-B. Li and X.-H. Xu, Chin. Chem. Lett., 2013, 24, 1103 CrossRef CAS; (e) T.-L. Au-Yeung, K.-Y. Chan, W.-K. Chan, R. K. Haynes, I. D. Williams and L. L. Yeung, Tetrahedron Lett., 2001, 42, 453 CrossRef CAS; (f) J. Xu, L. Zhang, X. Li, Y. Gao, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1266 CrossRef CAS; (g) Y.-F. Zhao, G. Tang, Y.-X. Gao and Y. Cao, Synthesis, 2009, 2009, 1081 CrossRef; (h) D. S. Panmand, A. D. Tiwari, S. S. Panda, J.-C. M. Monbaliu, L. K. Beagle, A. M. Asiri, C. V. Stevens, P. J. Steel, C. D. Hall and A. R. Katritzky, Tetrahedron Lett., 2014, 55, 5898 CrossRef CAS; (i) P. Carta, N. Puljic, C. Robert, A.-L. Dhimane, L. Fensterbank, E. Lacôte and M. Malacria, Org. Lett., 2007, 9, 1061 CrossRef CAS PubMed; (j) W. M. Wang, L. J. Liu, L. Yao, F. J. Meng, Y. M. Sun, C. Q. Zhao, Q. Xu and L. B. Han, J. Org. Chem., 2016, 81, 6843 CrossRef CAS PubMed.
- (a) Y. Zhu, T. Chen, S. Li, S. Shimada and L. B. Han, J. Am. Chem. Soc., 2016, 138, 5825 CrossRef CAS PubMed; (b) J. G. Sun, H. Yang, P. Li and B. Zhang, Org. Lett., 2016, 18, 5114 CrossRef CAS; (c) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487 CrossRef CAS; (d) C. Y. Li, Y. C. Liu, Y. X. Li, D. M. Reddy and C. F. Lee, Org. Lett., 2019, 21, 7833 CrossRef CAS PubMed; (e) J. Wang, X. Huang, Z. Ni, S. Wang, J. Wu and Y. Pan, Green Chem., 2015, 17, 314 RSC; (f) Y.-C. Liu and C.-F. Lee, Green Chem., 2014, 16, 357 RSC; (g) J. Wang, X. Huang, Z. Ni, S. Wang, Y. Pan and J. Wu, Tetrahedron, 2015, 71, 7853 CrossRef CAS; (h) R. Choudhary, P. Singh, R. Bai, M. C. Sharma and S. S. Badsara, Org. Biomol. Chem., 2019, 17, 9757 RSC.
- K. Nishide, P. K. Patra, M. Matoba, K. Shanmugasundaram and M. Node, Green Chem., 2004, 6, 142 RSC.
- (a) G. Kumaraswamy and R. Raju, Adv. Synth. Catal., 2014, 356, 2591 CrossRef CAS; (b) J. Bai, X. Cui, H. Wang and Y. Wu, Chem. Commun., 2014, 50, 8860 RSC; (c) X. Zhang, D. Wang, D. An, B. Han, X. Song, L. Li, G. Zhang and L. Wang, J. Org. Chem., 2018, 83, 1532 CrossRef CAS PubMed; (d) F. M. Moghaddam, M. Daneshfar and R. Azaryan, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 196, 311 CrossRef; (e) B. Kaboudin, Y. Abedi, J.-y. Kato and T. Yokomatsu, Synthesis, 2013, 45, 2323 CrossRef CAS; (f) T. Liu, Y. Zhang, R. Yu, J. Liu and F. Cheng, Synthesis, 2020, 52, 253 CrossRef CAS.
- (a) Y. Moon, Y. Moon, H. Choi and S. Hong, Green Chem., 2017, 19, 1005 RSC; (b) Y. M. Lin, G. P. Lu, G. X. Wang and W. B. Yi, J. Org. Chem., 2017, 82, 382 CrossRef CAS.
- (a) L. Huang, J. Gong, Z. Zhu, Y. Wang, S. Guo and H. Cai, Org. Lett., 2017, 19, 2242 CrossRef CAS PubMed; (b) L. Huang, Z. Zhang, K. Jie, Y. Wang, Z. Fu, S. Guo and H. Cai, Org. Chem. Front., 2018, 5, 3548 RSC; (c) S. Guo, W. Yan, Z. Zhang, Z. Huang, Y. Guo, Z. Liang, S. Li, Z. Fu and H. Cai, J. Org. Chem., 2022, 87, 5522 CrossRef CAS PubMed; (d) S. Guo, S. Li, W. Yan, Z. Liang, Z. Fu and H. Cai, Green Chem., 2020, 22, 7343 RSC.
- H. Xiang, J. Liu, J. Wang, L. Jiang and W. Yi, Org. Lett., 2022, 24, 181 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2184647 and 2184646. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06351d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |