
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional organoboron–phosphonium catalysts for coupling reactions of CO2 and epoxides†
Mani Sengodena,
Gulzar A. Bhat *b and
Donald J. Darensbourg*a
*b and
Donald J. Darensbourg*a
aDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: djdarens@chem.tamu.edu
bCentre for Interdiciplinary Research and Innovations, University of Kashmir, Srinagar, Jammu and Kashmir 190006, India. E-mail: gulzarbhat@uok.edu.in
First published on 14th November 2022
Abstract
Recent years have witnessed intensive research activity in exploring novel metal-free organocatalysts for catalyzing the coupling reactions of CO2 and epoxides to afford cyclic or polymeric carbonates. In this direction, herein we report a series of boron–phosphonium organocatalysts for catalyzing the coupling reactions of CO2 and epoxides. These organophosphonium catalysts were synthesized in high yields by following a two step protocol involving Menschutkin and hydroboration reactions in succession. The purity of these organocatalysts was confirmed by spectroscopic techniques like 1H, 13C and 31P NMR, and molecular structures were confirmed by single crystal X-ray diffraction studies. We have also demonstrated that these bifunctional organoboron–phosphonium catalysts are comparatively much less hygroscopic compared to the analogus ammonium catalysts. These phosphonium organocatalysts were shown to catalyze the copolymerization of CO2 and cyclohexene oxide or vinyl cyclohexene oxide to provide polycarbonates with >99% polymer selectivity and carbonate linkages. The coupling reactions of aliphatic epoxides such as PO, having lower energy barrier to cycloaddition formation compared to alicyclic epoxides, preferentially provided cyclic carbonates in good yields. It was demonstrated that these organoboron–phosphonium catalysts are sensitive to chain transfer agents like water, and hence are deactivated in its presence. This is opposite to what is observed for metal based catalysts for these transformations, where water serves as a precursor to the chain-transfer agent diols.
Introduction
A major challenge our world faces today is the mitigation of CO2 emissions. In order to accomplish this requires two strategies, produce less carbon dioxide and capture more. Capturing and safely storing CO2 emissions are currently emerging technologies which must be partnered with CO2 utilization (CCU) in attempts to remain economically viable.1–4 Currently, approximately 250 Mt per year of CO2 are used as a raw material in the chemical industry out of 40 Gt per year of CO2 emissions.5 Therefore, as a means to achieve the necessary reduction in global CO2 emissions and address its impact on climate change, significant advancement in the valorization of carbon dioxide are needed. Among various efforts on the utilization of CO2 as a C-1 feedstock, the coupling of CO2 and epoxides to synthesize cyclic and polymeric carbonates is attractive and has drawn considerable attention from both academia and industrial communities because of its atom-economic and greener approach.6,7 Both the cycloaddition and polymeric products find diverse applications. For example, the cyclic carbonates have widely been utilized as polar aprotic solvents,8 electrolytes for lithium-ion battery,9 starting materials for polycarbonate synthesis via ring opening polymerization and pharmaceutical industry.10,11 The polycarbonates have been employed and proposed as alternatives to petro-based chemicals for use in the fields of automotive,12 medical,13 and electronic industries14,15 besides being used as starting materials for making polyurethane industry.16Metal-based homogeneous or heterogeneous catalytic systems for the coupling of CO2 and epoxides to afford either copolymers or cycloaddition products have seen tremendous developments since the first report by Inoue and coworkers in 1969 using poorly defined zinc catalysts.17–36 The exponential growth in exploring new catalysts is strongly aided by diverse applications of these cyclic or polycarbonate products. Many of these new metal catalysts have found to be very effective in terms of selectivity and regio-regularity in providing polycarbonate polymers.37,38 Despite the impressive advances made in the development of metal based catalysts for this coupling reaction, toxicity associated with the residual metal contaminants especially in medical applications of these polycarbonates and cost concerns strongly advocates for finding the alternative catalyst for this coupling reaction. In this direction, recent years have witnessed exponential progress in exploring various organocatalysts for different organic transformation reactions primarily motivated by low cost, versatile chemical structure, simple synthetic procedures etc.39–41
Recent evolution of organocatalysts for performing the coupling of epoxides and CO2 offers an appealing strategy.42–46 Numerous studies have been reported where these organocatalysts effectively catalyze the coupling of CO2 and epoxides to provide either five-membered cyclic carbonates or polymers selectively.27,47–59 These catalyst systems include imidazolium salts, quaternary ammonium salts, phosphonium salts, amidine-based, carbene-based, and hydrogen bond donor-systems. For example very recently Wu and coworkers reported a bifunctional organoboron catalyst system which effectively catalyzes the coupling of CO2 and CHO to selectively afford PCHC with excellent TOF.49 It has also been observed that the acidity of the Lewis acid, the nucleophilicity of the Lewis base, along with the steric effects of the Lewis pair are the deciding factors for effective metal-free catalysts.44,60
Thus keeping these results into consideration with their main limitation being air and moisture sensitivity, in this study we report a series of closely related boron–phosphonium organocatalysts for catalyzing the coupling of CO2 and epoxides.61 Compared with the previously developed organocatalytic systems, these catalysts show equally good activity, and are relatively more stable under air and moisture conditions.
Results and discussion
Catalyst synthesis and characterization
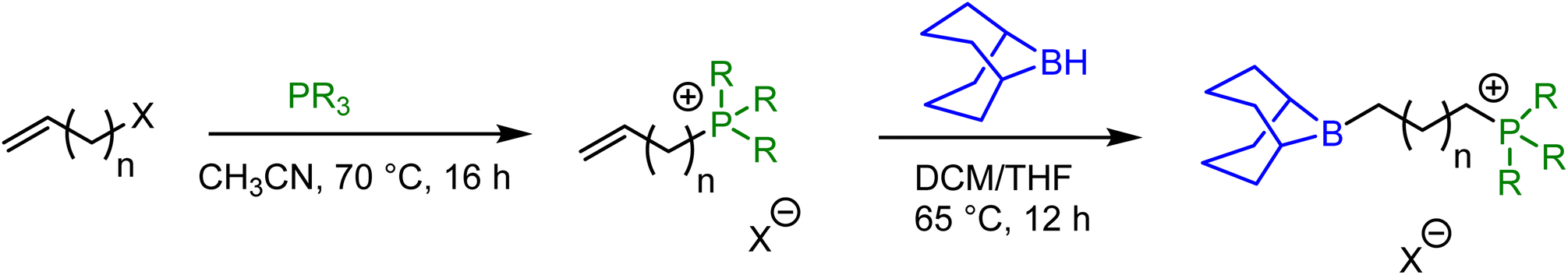
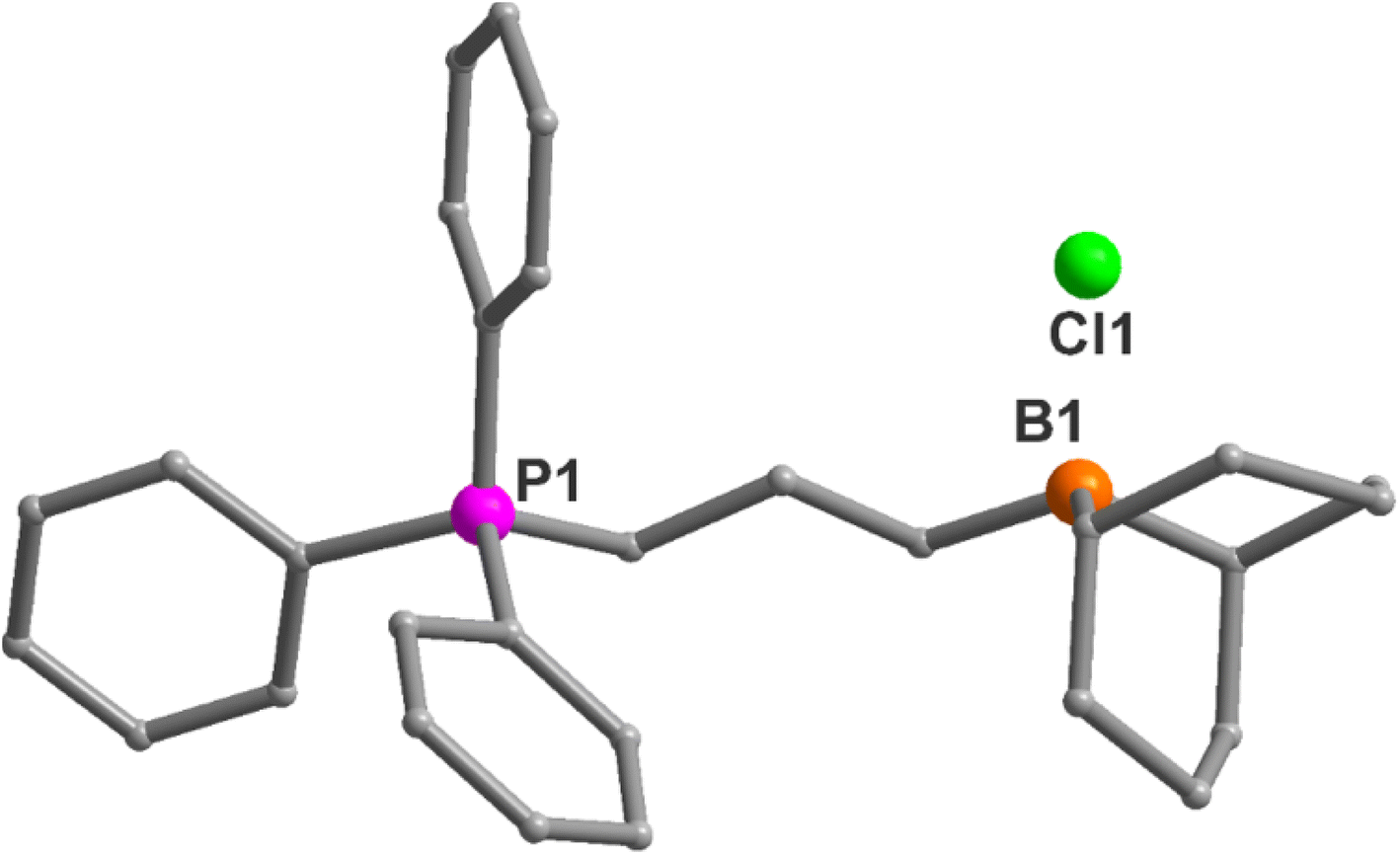
Bifunctional catalysts featuring Lewis acidic 9-borabicyclo[3.3.1]nonane (BBN) centers and a phosphonium halide were synthesized in high yields by following the two step route shown in Table 1. In the first step, the stoichiometric reaction of phosphines with alkenyl halides (i.e., Menschutkin reaction) in acetonitrile produced the corresponding phosphonium salts. In the second step, the desired bifunctional organocatalysts were achieved through the hydroboration reaction of the double bond of the phosphonium salts using 9-BBN in CH2Cl2 at 65 °C. All of these organocatalysts having different alkyl lengths between boron and phosphorus centers and variable –R groups on phosphonium salts were obtained as white solids by simply washing with n-pentane and subsequent drying in vacuum in high yields (95–99%). The catalysts were characterized by 1H and 31P NMR spectroscopy and their molecular structures were confirmed by single crystal X-ray diffraction studies as shown in Fig. 1 for entry 1 in Table 1. X-ray diffraction quality crystals were grown by slow evaporation at room temperature in DCM/toluene solvent mixture. Catalyst 1a was shown to crystallize in the triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The catalyst 1b also crystallizes in triclinic crystal system with P space group and has one water molecule in the crystal lattice (Fig. 2). The water molecule is involved in hydrogen bonding with the Br− which eventually result in the formation of a weakly bound dimer as shown in Fig. 2b. The synthetic protocol of these catalysts is simple and the starting materials employed are cheap and easily available, thus preparation of the catalyst in a large scale is practical, as we have demonstrated on a 10 g scale.
space group. The catalyst 1b also crystallizes in triclinic crystal system with P space group and has one water molecule in the crystal lattice (Fig. 2). The water molecule is involved in hydrogen bonding with the Br− which eventually result in the formation of a weakly bound dimer as shown in Fig. 2b. The synthetic protocol of these catalysts is simple and the starting materials employed are cheap and easily available, thus preparation of the catalyst in a large scale is practical, as we have demonstrated on a 10 g scale.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | n | R | X | Yield (%) |
| 1 | 1a | 1 | Ph | Cl | 96 |
| 2 | 1b | 1 | Ph | Br | 97 |
| 3 | 2 | 1 | Me | Br | 99 |
| 4 | 3 | 1 | Cy | Br | 97 |
| 5 | 4 | 3 | Ph | Br | 95 |
| 6 | 5 | 3 | Me | Br | 98 |
| 7 | 6 | 3 | Cy | Br | 96 |
 | ||
| Fig. 1 X-ray crystal structure of the organocatalyst 1a (entry 1 in Table 1). B1⋯P1 distance is 5.551 Å. | ||
 | ||
| Fig. 2 (a) X-ray crystal structure of the organocatalyst 1b (entry 2 in Table 1) B1⋯P1 distance is 5.533 Å. (b) Dimer formation of 2b due to hydrogen bonding interactions between lattice water, Br− and the C–H hydrogen atoms. | ||
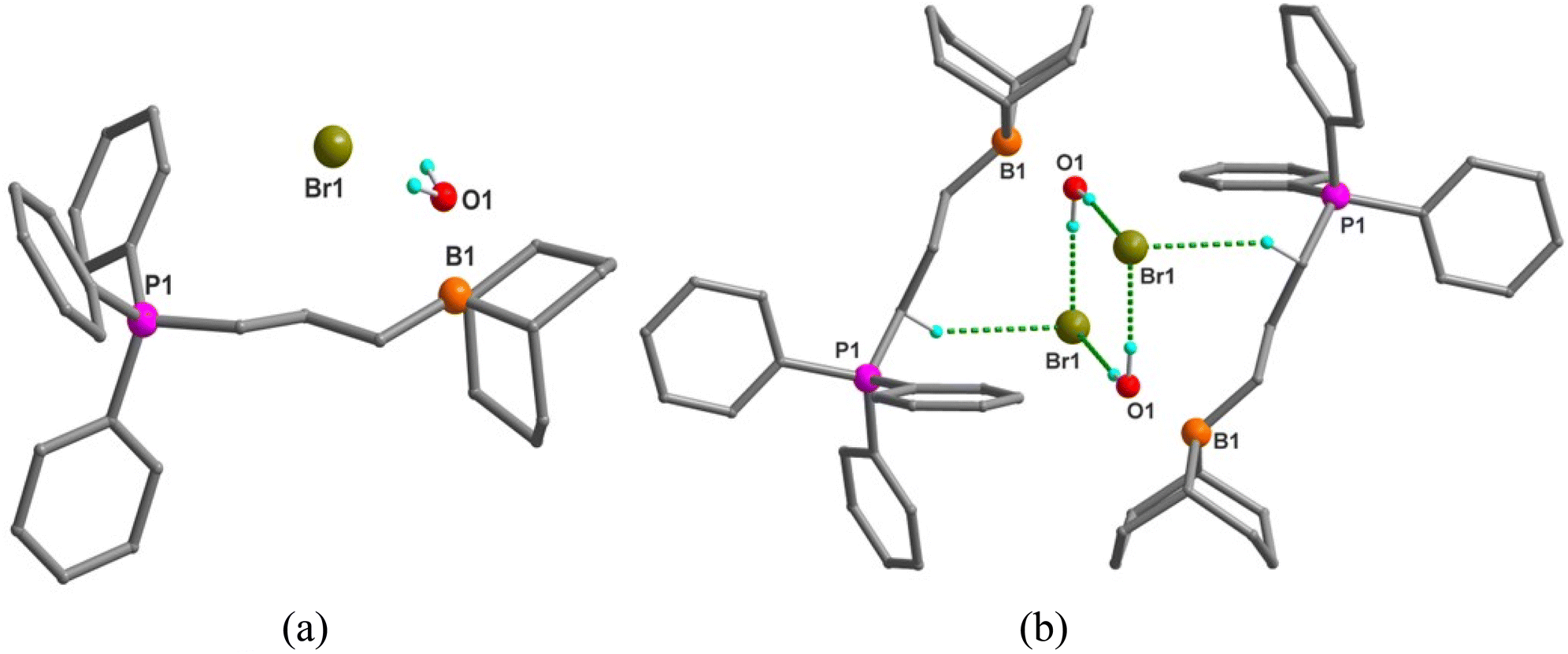
In order to test the moisture stability of these bifunctional organophosphonium catalysts, we have performed a “standing test in air” where we have maintained the newly synthesized organocatalysts at room temperature under aerobic conditions and compared it with the boron ammonium organocatalysts. We have observed that the boron ammonium organocatalysts under identical aerobic conditions rapidly absorbs the moisture, whereas the bifunctional organophosphonium catalysts are much less hygroscopic and does not lose their powdered morphology over this time period (Fig. 3). We have also recorded these events in a video which is provided in ESI.†
 | ||
| Fig. 3 Left: Comparison of the moisture stability standing test of boron phosphonium catalyst with the boron ammonium catalyst, both samples maintained at room temperature under aerobic conditions. Right: Quantitative comparison of the moisture stability standing test of boron ammonium catalyst (0.2 mmol) with the boron phosphonium catalyst (0.2 mmol). | ||
Catalyst screening and CHO/CO2 copolymerization studies
These organocatalysts were examined as catalysts for the copolymerization reactions of CO2 and cyclohexene oxide (CHO). A comprehensive study of the effects of catalyst structure, catalyst loading, reaction temperature, reaction time, and CO2 pressure on coupling of CHO and CO2 are summarized in Table 2. Firstly, we started our investigation by probing the influence of different anions on the catalytic activity of these organocatalysts for the CHO/CO2 copolymerization by choosing anions such as chloride and bromide. As shown in Table 2, entries 1 and 2, on using the bifunctional catalyst 1a and 1b having same –(CH2)3– linker length, catalyst having Cl− as counter anion gave 47% conversion in 5 h at 80 °C and 1.5 MPa CO2 pressure to afford PCHC with a Mn value of 10.5 kg mol−1 and polydispersity of 1.11. On the other hand, catalyst 1b having a bromide anion showed increased catalytic activity with a conversion of 61% and a Mn value of 13.8 kg mol−1 and narrow polydispersity. Next, we turned our attention to study the effect of different substituents on the phosphines and length of B⋯P linker on catalytic activities (Table 2, entries 3–7). Thus under similar conditions, and only varying the substituents on phosphines it was revealed that with an increase in bulky substituent on the phosphine a slight increase in the conversion was observed. That is, the catalyst derived from trimethylphosphine 2 showed 45% conversion with a decreased Mn value of 5.7 kg mol−1. Similarly, the tricyclohexylphosphine based catalyst 3 exhibited a slight increase in epoxide conversion (64%) over triphenylphosphine based 1b with a decreased Mn value of 9.1 kg mol−1. We have further also observed that by increasing the connecting methylene units between phosphorus and boron from 3 to 5, the catalytic activity of these organocatalysts for the copolymerization of CHO/CO2 decreased significantly. Thus, due to cost of starting materials and catalytic efficiency, we therefore choose 1b as a model catalyst to further investigate the effect of catalyst loadings, reaction temperature, and CO2 pressure on the catalytic activity of these organocatalysts. It was observed that on increasing the feed ratio CHO/1b from 2500 to 5000, the catalytic activity for polymerization decreased slightly from a conversion of 19 to 14%, respectively was observed (Table 2, entries 8 and 9). The influence of solvents on the catalytic performance of 1b was also probed, and it was observed that a CH2Cl2/toluene solvent system gave complete conversion for polymerization. Upon increasing the reaction time to 18 h, there was not much of an impact on the percentage conversion of the copolymerization reaction (Table 2, entry 10).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | Temp. (°C) | Time (h) | Conv.g (%) | TON | TOF (h−1) | Mn [kg mol−1] | PDI |
a The reaction was performed epoxide (9.9 mmol; 1.5 MPa CO2; catalyst/epoxides = 1/1000) in a 15 mL autoclave at 80 °C for 5 h.b Epoxide (2500 equiv.) used.c Epoxide (5000 equiv.) used.d Reaction was performed using toluene/CH2Cl2 (0.8 mL; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture).e H2O (5 equiv.) added.f CO2 (0.5 MPa) was applied.g Calculated by 1H NMR analysis. :1 mixture).e H2O (5 equiv.) added.f CO2 (0.5 MPa) was applied.g Calculated by 1H NMR analysis. |
||||||||
| 1 | 1a | 80 | 5 | 47 | 470 | 94 | 10.5 | 1.11 |
| 2 | 1b | 80 | 5 | 61 | 610 | 122 | 13.8 | 1.13 |
| 3 | 2 | 80 | 5 | 45 | 450 | 90 | 5.7 | 1.08 |
| 4 | 3 | 80 | 5 | 64 | 640 | 128 | 9.1 | 1.13 |
| 5 | 4 | 80 | 5 | 57 | 570 | 114 | 15.2 | 1.13 |
| 6 | 5 | 80 | 5 | 15 | 150 | 30 | — | — |
| 7 | 6 | 80 | 5 | 22 | 220 | 44 | 16.5 | 1.10 |
| 8b | 1b | 80 | 5 | 19 | 475 | 95 | 9.2 | 1.02 |
| 9c | 1b | 80 | 5 | 14 | 700 | 140 | 9.7 | 1.08 |
| 10d | 1b | 80 | 18 | 60 | 600 | 33 | 14.4 | 1.11 |
| 11e | 1b | 80 | 18 | — | — | — | — | — |
| 12 | 1b | 50 | 18 | 62 | 620 | 35 | 15.5 | 1.06 |
| 13f | 1b | 50 | 18 | 50 | 500 | 28 | 12.2 | 1.08 |
| 14f | 1b | rt | 18 | 19 | 190 | 11 | 5.6 | 1.07 |
With the optimized catalyst 1b, the influence of temperature and CO2 pressure was also examined for the coupling of CO2 and epoxides (Table 2, entries 12–14). It was observed that polymerization performed at 50–80 °C gave higher conversion compared to the reactions performed at ambient temperature. It was further observed that a decrease in CO2 pressure from 1.5 MPa to 0.5 MPa, resulted in a decrease in catalytic activity. When the reaction was carried out below 0.5 MPa CO2 at ambient temperature a significant decrease in monomer conversion (down to 19%) and molecular weight of the resulting polymer was observed.
To assess the influence of water on the catalytic activity of bifunctional catalyst 1b for the copolymerization of CO2 and CHO, we intentionally added five equivalents of water compared to the catalyst loadings. It was found that addition of excess water during the catalytic process resulted in complete deactivation of the catalyst (Table 2, entry 11). Hence, unlike most of the metal catalyzed copolymerization processes, these organocatalysts are not tolerant to an excess in the chain transfer agent, H2O.
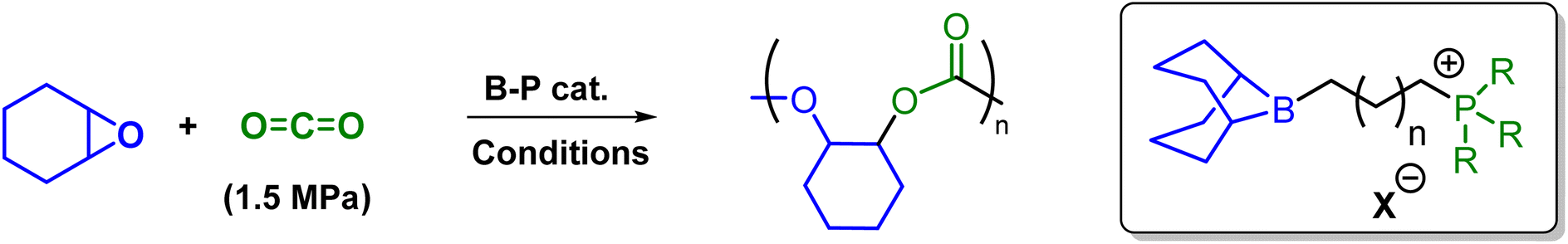
This organoboron–phosphonium catalyst (1b) catalyzed the copolymerization of CHO/CO2 to give PCHC with >99% polymer selectivity and carbonate linkage formation as detected by 1H NMR spectroscopy for all the above mentioned copolymerization reactions. Bimodal molecular weight distributions were observed as evidenced by MALDI-ToF spectrum and GPC traces (Fig. 4). The two series observed during this copolymerization are separated by 142 m/z, which correspond to the repeating unit (one unit of cyclohexene carbonate). The major population series (green circle) correspond to the potassium adduct of the PCHC copolymer which are initiated by bromide anion. The minor serious (red square) was assigned to the dihydroxy-end-capped PCHC copolymer. The bimodality of these processes presumably results from a trace amount of water present in the reaction system.
 | ||
| Fig. 4 GPC and MALDI-ToF spectra of the resulted PCHC. | ||
In further efforts of exploring the catalytic limits of these bifunctional boron–phosphonium organocatalysts, we selected 1b as a model catalyst for catalyzing the copolymerization of CO2 with a range of other commonly employed epoxides. It was observed that propylene oxide (PO), butylene oxide (BO), and styrene oxide (SO) lead only to the formation of five membered cyclic carbonates. We have later extended the substrate scope to epoxides similar to cyclohexene oxide, such as vinyl cyclohexene oxide and aziridine (Fig. 5). As expected, the copolymerization of vinylcyclohexene oxide (VCHO) and CO2 produced poly(vinylcyclohexene carbonate) with 81% conversion in 5 h at 80 °C and 1.5 MPa CO2 pressure. A number average molecular weight (Mn) and a polydispersity (PDI) of 17.1 kg mol−1 and 1.13 were obtained, respectively. Attempts of copolymerizing renewable epoxides such as monoterpenes, α-pinene, and limonene derived epoxides, which resemble CHO were unsuccessful. These results suggest that the coordination of the Lewis acidic boron center and epoxide is hampered by the methyl group in the cyclohexane epoxide scaffold. Furthermore, the N-benzyl cyclohexyl aziridine was tested under the optimized reaction conditions which only resulted in the formation of corresponding cyclic carbamate in 96% yield (Scheme 1).
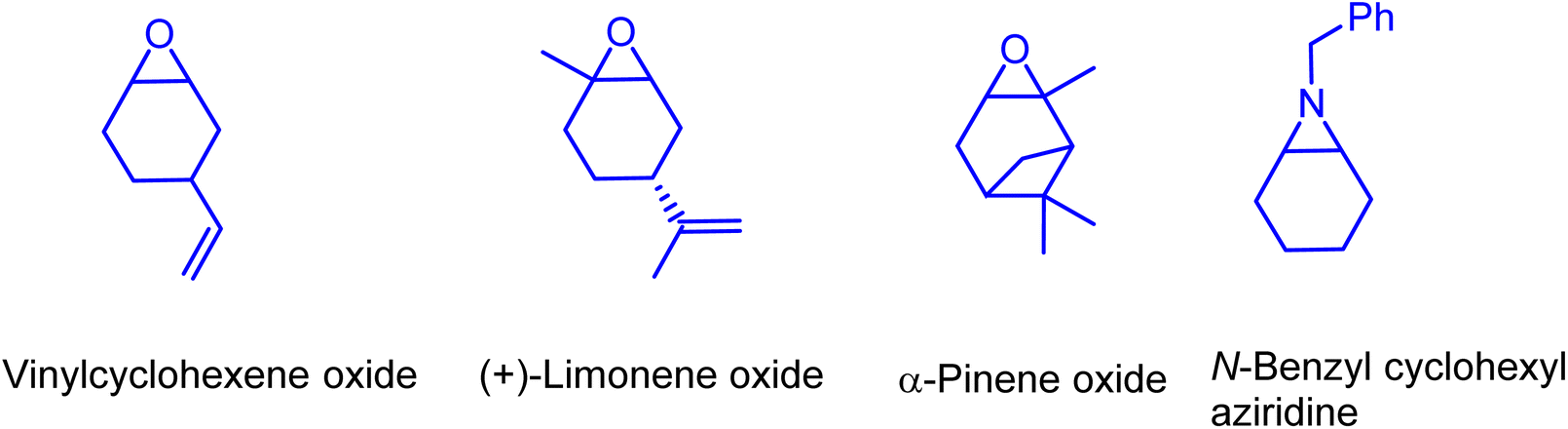 | ||
| Fig. 5 Employed cyclohexane based epoxide and aziridine monomers. | ||
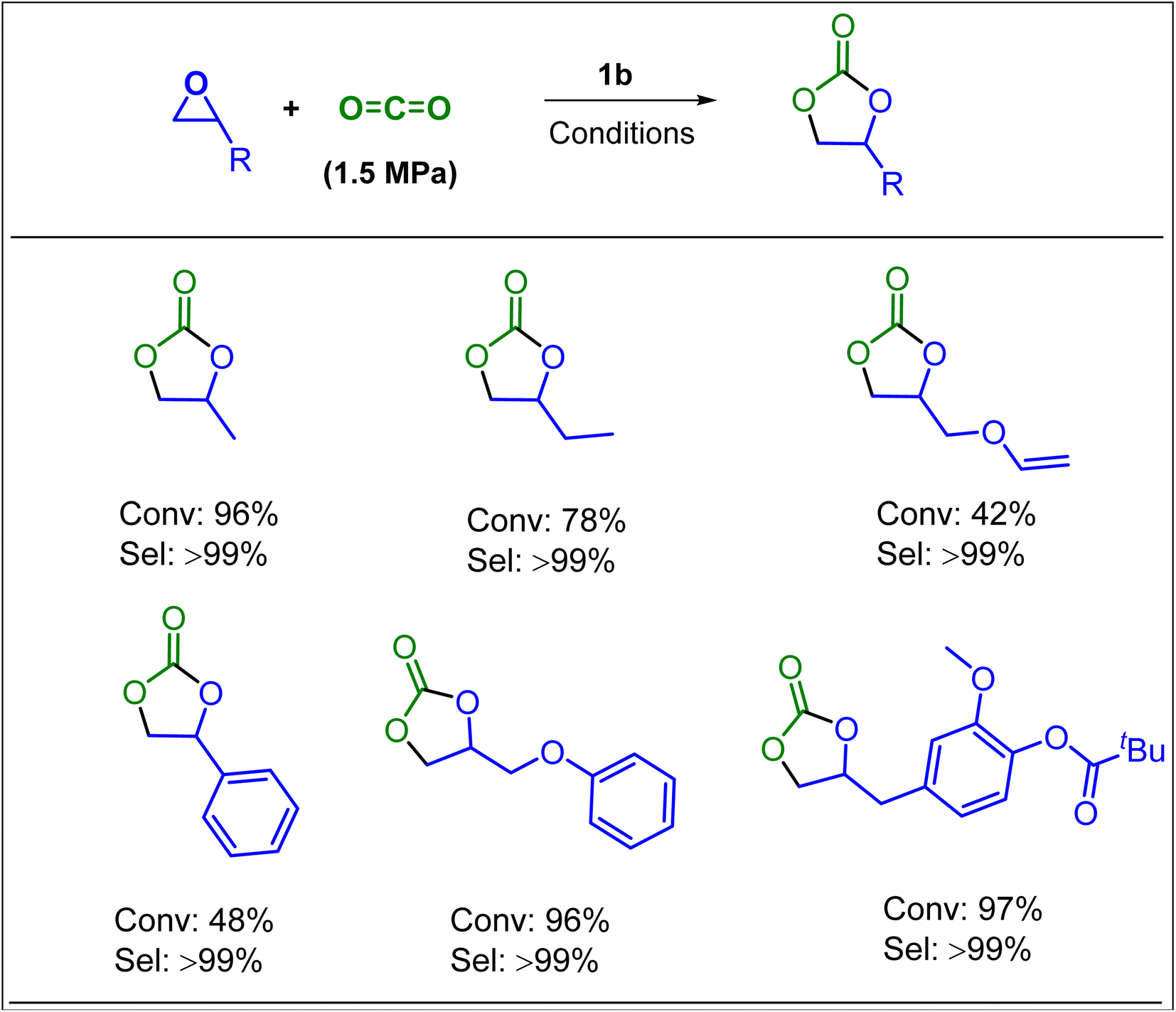 | ||
| Scheme 1 Synthesis of cyclic carbonates from various epoxides and CO2 using organocatalysts 1b. The reaction was performed epoxide (9.9 mmol; 1.5 MPa CO2; catalyst/epoxides = 1/1000) in a 15 mL autoclave using toluene/CH2Cl2 (0.8 mL) at 80 °C for 18 h. | ||
The role of these bifunctional organocatalysts is reminiscent to that proposed for bifunctional (salen)MX (M = Cr, Co) catalysts.27 That is, subsequent to epoxide binding to the boron center, ring opening by the anion, and CO2 insertion, displacement of the anion from boron by an epoxide is stabilized by an electrostatic interaction with the phosphonium cation. Hence, ring opening by the anionic polymer chain end is thought to be the rate determining step (Fig. 6). A more detailed analysis of the reaction mechanism consistent with this interpretation may be found in ref. 62.
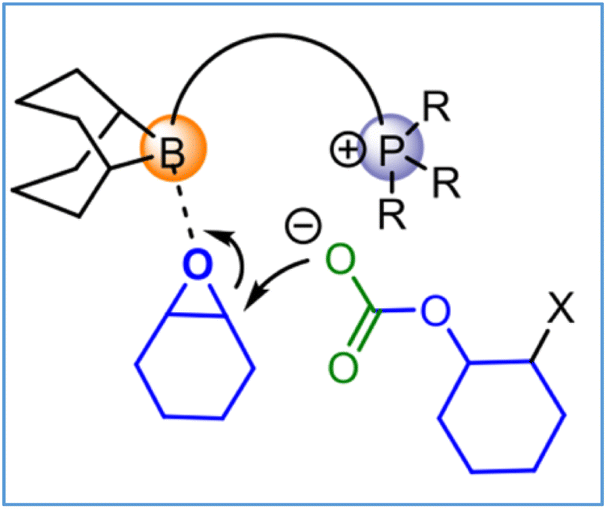 | ||
| Fig. 6 Rate-determining step in the copolymerization process. | ||
Conclusions
Herein, we report a series of bifunctional organoborane phosphonium catalysts for coupling reactions of epoxides and CO2 based on the novel contributions of Wu and coworkers on their ammonium analogs.49 Importantly, these phosphonium derivatives are much less sensitive to atmospheric moisture than their ammonium versions. Under very similar reaction conditions, these catalysts are more effective at copolymerizing the alicyclic epoxides, cyclohexene oxide and vinyl cyclohexene oxide and CO2, and at facilitating the cycloaddition reactions of aliphatic epoxides with CO2 as the organoboron ammonium catalysts. This strategy should also be applicable to synthesizing phosphonium derivatives of the multinuclear ammonium catalysts which have been shown to be very efficient at copolymerizing propylene oxide and epichlorohydrin with CO2.44 A significant disadvantage of these organocatalysts over commonly employed metal catalysts for these processes is their inability to carry out immortal copolymerization reaction in the presence of excess water. However, direct additions of diols are shown to be effective chain-transfer agents. Be that as it may, there are numerous applications where the absence of trace metals in CO2-based copolymers is beneficial.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJD gratefully acknowledge the financial support from Robert A. Welch Foundation (A-0923). GAB gratefully acknowledge DST-SERB for Ramanujan fellowship Grant (RJF 2020/000116). The authors also thank Dr Nattamai Bhuvanesh for helping in solving the X-ray Structures.References
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef PubMed.
- F. Nocito and A. Dibenedetto, Curr. Opin. Green Sustain. Chem., 2020, 21, 34–43 CrossRef.
- Z. Zhang, T. Wang, M. J. Blunt, E. J. Anthony, A.-H. A. Park, R. W. Hughes, P. A. Webley and J. Yan, Appl. Energy, 2020, 278, 115627 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustain. Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- C.-C. Su, M. He, R. Amine, Z. Chen, R. Sahore, N. D. Rago and K. Amine, Energy Storage Mater., 2019, 17, 284–292 CrossRef.
- D. J. Darensbourg and A. I. Moncada, Macromolecules, 2010, 43, 5996–6003 CrossRef CAS.
- S. R. Kosuru, Y.-L. Chang, P.-Y. Chen, W. Lee, Y.-C. Lai, S. Ding, H.-Y. Chen, H.-Y. Chen and Y.-C. Chang, Inorg. Chem., 2022, 61, 3997–4008 CrossRef CAS PubMed.
- B. J. Tuazon, N. A. V. Custodio, R. B. Basuel, L. A. Delos Reyes and J. R. C. Dizon, Key Eng. Mater., 2022, 913, 3–16 Search PubMed.
- K. Ghosal, S. Pal, D. Ghosh, K. Jana and K. Sarkar, Biomater. Adv., 2022, 212961 CrossRef CAS PubMed.
- A. Kausar, J. Plast. Film Sheeting, 2018, 34, 60–97 CrossRef CAS.
- H. Cao and X. Wang, SusMat, 2021, 1, 88–104 CrossRef.
- M. DeBolt, A. Kiziltas, D. Mielewski, S. Waddington and M. J. Nagridge, J. Appl. Polym. Sci., 2016, 133, 44086 CrossRef.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS PubMed.
- M. H. Chisholm and Z. Zhou, J. Mater. Chem., 2004, 14, 3081–3092 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- G. Trott, P. Saini and C. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- A. C. Deacy, A. F. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- Z. Chen, J.-L. Yang, X.-Y. Lu, L.-F. Hu, X.-H. Cao, G.-P. Wu and X.-H. Zhang, Polym. Chem., 2019, 10, 3621–3628 RSC.
- M. Alves, B. Grignard, R. Méreau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- M. Jia, D. Zhang, G. W. de Kort, C. H. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 5297–5307 CrossRef CAS PubMed.
- P. Alagi, G. Zapsas, N. Hadjichristidis, S. C. Hong, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 6144–6152 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS.
- J. L. Yang, H. L. Wu, Y. Li, X. H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed. Engl., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- Y. Tong, R. Cheng, H. Dong, Z. Liu, J. Ye and B. Liu, J. CO2 Util., 2022, 60, 101979 CrossRef CAS.
- C.-J. Zhang, S.-Q. Wu, S. Boopathi, X.-H. Zhang, X. Hong, Y. Gnanou and X.-S. Feng, ACS Sustain. Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS.
- M. Alves, B. Grignard, A. Boyaval, R. Méreau, J. De Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jérôme, ChemSusChem, 2017, 10, 1128–1138 CrossRef CAS PubMed.
- J. Huang, J. De Winter, A. P. Dove and O. Coulembier, Green Chem., 2019, 21, 472–477 RSC.
- J. Huang, C. Jehanno, J. C. Worch, F. Ruipérez, H. Sardon, A. P. Dove and O. Coulembier, ACS Catal., 2020, 10, 5399–5404 CrossRef CAS.
- L. Červenková Št'astná, A. Krupkova, R. Petrickovic, M. Mullerova, J. Matousek, M. Kostejn, P. Curinova, V. Jandova, S. Sabata and T. Strasak, ACS Sustain. Chem. Eng., 2020, 8, 11692–11703 CrossRef.
- Z. Wang, Y. Wang, Q. Xie, Z. Fan and Y. Shen, New J. Chem., 2021, 45, 9403–9408 RSC.
- X.-F. Liu, Q.-W. Song, S. Zhang and L.-N. He, Catal. Today, 2016, 263, 69–74 CrossRef CAS.
- Y. Hao, D. Yuan and Y. Yao, ChemCatChem, 2020, 12, 4346–4351 CrossRef CAS.
- C. Calabrese, L. F. Liotta, F. Giacalone, M. Gruttadauria and C. Aprile, ChemCatChem, 2019, 11, 560–567 CrossRef CAS.
- Y. Y. Zhang, G. W. Yang, R. Xie, L. Yang, B. Li and G. P. Wu, Angew. Chem., 2020, 132, 23491–23498 CrossRef.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 CrossRef CAS.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2191004 and 2191005. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06358a |
| This journal is © The Royal Society of Chemistry 2022 |