
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic deoxygenation of fatty acids via ketonization and α-carbon scissions over layered alkali titanate catalysts under N2†
Tosapol Maluangnont *ab,
Piyasan Praserthdamc and
Tawan Sooknoi*bd
*ab,
Piyasan Praserthdamc and
Tawan Sooknoi*bd
aCollege of Materials Innovation and Technology, King Mongkut's Institute of Technology Ladkrabang, Bangkok 10520, Thailand. E-mail: tosapol.ma@kmitl.ac.th
bCatalytic Chemistry Research Unit, School of Science, King Mongkut's Institute of Technology Ladkrabang, Bangkok 10520, Thailand. E-mail: kstawan@gmail.com
cCenter of Excellence on Catalysis and Catalytic Reaction Engineering, Chulalongkorn University, Bangkok 10330, Thailand
dDepartment of Chemistry, School of Science, King Mongkut's Institute of Technology Ladkrabang, Bangkok 10520, Thailand
First published on 30th November 2022
Abstract
The ketonization of fatty acid with subsequent McLafferty rearrangement of the fatty ketone allows the deoxygenation to hydrocarbons. Here, we report the cascade reaction of palmitic acid (C16) to hydrocarbons (≤C14) over lepidocrocite-type alkali titanate K0.8Zn0.4Ti1.6O4, K0.8Mg0.4Ti1.6O4, and K0.8Li0.27Ti1.73O4 and the reassembled TiO2 catalysts at ≤400 °C under atmospheric N2 in a continuous fixed-bed flow reactor. The C16 acid is coupled to C31 ketone prior to the scissions mostly to a C17 methyl ketone and C14 hydrocarbons (i.e., the McLafferty rearrangement). The hydrocarbons yield increases with temperature and is proportional to partial charge at the O atom, suggesting that basic sites are responsible for C31 ketone scissions. The layered alkali titanate catalysts with two-dimensional (2D) space inhibit diffusion of the ketone primarily formed and promote its scissions to hydrocarbons within the confined space. Otherwise, low hydrocarbons yield (but high ketone yield) is obtained over TiO2 and the Mg/Al mixed oxide catalysts possessing no interlayer space. Meanwhile, the semi-batch experiment with pre-intercalated palmitic acid favors a direct deoxygenation, demonstrating the essential role of reaction mode toward ketone scission reaction pathway. Over K0.8Li0.27Ti1.73O4, the complete palmitic acid conversion leads to ∼47% hydrocarbons yield, equivalent to ∼80% reduction of the oxygen content in the feed under N2.
Introduction
Fatty acids are an abundant, naturally occurring, renewable feedstock for the green synthesis of value-added chemicals by several pathways.1 For example, their deoxygenation leads to bio-fuel (decarboxylation) and olefins (decarbonylation).2–4 Meanwhile, ketonization produces fatty ketones and methyl ketones which have specialized applications.5–8 A selective and efficient fatty acid transformation can potentially contribute to energy security, economic gain, and global warming mitigation. This is better achieved via a cascade reaction9,10 in which one reaction occurs after another in a controlled manner, allowing chemical syntheses in a single step without extensive work up or products separation.In the absence of hydrogen, the cascade reaction of fatty acids typically starts with ketonization, where the new C–C bond is formed, CO2 and H2O are liberated, and one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group is preserved for further reactions. As observed by several authors,6,11–15 the primarily generated fatty ketone can undergo an asymmetrical scission into a methyl ketone and the olefin in the so-called McLafferty rearrangement. The formed olefins could be subsequently converted into saturated hydrocarbons via hydrogenation/H-transfer (or into alcohols via hydration).11,16–18 This cascade reaction is represented using palmitic acid in eqn (1)–(3).
O group is preserved for further reactions. As observed by several authors,6,11–15 the primarily generated fatty ketone can undergo an asymmetrical scission into a methyl ketone and the olefin in the so-called McLafferty rearrangement. The formed olefins could be subsequently converted into saturated hydrocarbons via hydrogenation/H-transfer (or into alcohols via hydration).11,16–18 This cascade reaction is represented using palmitic acid in eqn (1)–(3).
|
2C15H31COOH → C15H31(CO)C15H31 + CO2 + H2O
| (1) |
|
C15H31(CO)C15H31 → C15H31(CO)CH3 + C14H28
| (2) |
| C14H28 + H2 → C14H30 | (3) |
The above reaction network has been reported over several metal oxide catalysts including those potentially containing oxygen vacancies (TiO2,6 CeO2 (ref. 11 and 12)), predominantly acidic (Al2O3 (ref. 13–15)), or basic (MgO (ref. 16 and 17)). The reaction condition varies from the gaseous atmospheres (H2, N2 or supercritical water19) to pressure (ambient or high one). For example, Corma et al.17 employed a two-bed reactor consisting of MgO and M/MgO (M = Pt, Pd, Ru) catalysts and a high H2 pressure, producing saturated hydrocarbons from fatty acid. Meanwhile, we investigated20 the Pt/K2Ti6O13 catalyst for the fatty acid-to-olefins conversion under atmospheric H2. Lee et al.6 studied the McLafferty rearrangement of fatty acids with varying chain length over TiO2 and under N2, where the rate of McLafferty rearrangement increased with the carbon number. Still, there is a gap in our understanding of the factors controlling the scissions of formed ketones as in eqn (2), especially the nature of active sites and how to tune their selectivity.
Anisotropic layered metal oxides with active basic sites are an interesting catalyst candidate. They have strong bonding within the sheets and their structural integrity is preserved even above their synthetic temperature.21,22 Yet, they enable the intercalation of guest species/reactants23–25 which are subsequently confined at the two-dimensional (2D) interlayer space. Following the general classification of ketonization catalysts,5 layered metal oxides are therefore intermediate between the low-lattice energy catalyst (showing bulk transformation to carboxylate) and high-lattice energy one (with structural preservation and surface reaction). Among several families of layered alkali metal oxides, the lepidocrocite-type layered alkali titanate K0.8MyTi2−yO4 [Mn+ = Zn2+, Mg2+, Li+, and others; 0.8 = y(4 − n)]26 has received increasing attention for many potential applications. The negatively-charged sheets are interleaved with K+ ions at the 2D space, stacking repeatedly along the b-direction. The lattice oxygen atoms on the external surface/crystal edge or at the internal surface are the basic sites.24,25 In addition, these layered solids can be exfoliated into nanosheets and reassembled back into a simple metal oxide with larger specific surface area.27 While the photocatalytic activity of the lepidocrocite-type alkali titanate and its nanostructures is widely investigated,28–30 the (thermal) catalytic activity is underexplored.
As a continuation of our previous works,24,25 we report herein the cascade reaction of palmitic acid to palmitone, and the subsequent scissions of the latter into hydrocarbons over the lepidocrocite-type K0.8Zn0.4Ti1.6O4, K0.8Mg0.4Ti1.6O4, and K0.8Li0.27Ti1.73O4 catalysts. The as-made and spent catalysts were characterized by X-ray diffraction, Raman spectroscopy, scanning electron microscopy, and CO2 temperature-programmed desorption. The catalytic activities were evaluated in a flow reactor at 375–400 °C and compared to our previous results24 conducted in a semi-batch mode. This allows us to understand not only the electronic effect (i.e., electronegativity of the intralayer metal Zn, Mg, Li; and the basic strength of the oxygen atom), but also the role of palmitic acid proximity in this cascade deoxygenation. The catalytic activities of the reassembled TiO2 and the Mg/Al mixed oxide which lack the interlayer space were also evaluated, further highlighting the benefit of the 2D confinement toward hydrocarbon yields and stability even under N2.
Experimental
Synthesis
The lepidocrocite titanate catalysts K0.8Zn0.4Ti1.6O4, K0.8Mg0.4Ti1.6O4, and K0.8Li0.27Ti1.73O4 were made via calcination of the stoichiometric mixture of K2CO3, (ZnO/MgO/Li2CO3), and TiO2 at 900 °C for 20 h twice with intermediate grinding.24–26 The reassembled TiO2 catalyst was prepared by proton exchange of K0.8Zn0.4Ti1.6O4, exfoliation with tetrabutylammonium hydroxide (TBAOH),31–33 reassembling with KOH, and calcination in air at 450 °C for 6 h, see (ESI†) for details. The layered double hydroxide with Mg/Al mol ratio of 2.5 was synthesized by a coprecipitation method25 and was calcined at 450 °C for 6 h, giving the Mg/Al mixed oxide.Characterization
Powder X-ray diffraction (XRD) measurements were performed on a Rigaku, DMAX 2200/Ultima+ diffractometer (Cu Kα radiation, 40 kV, 30 mA) covering the range 2θ = 5–55° at the rate of 0.02° per step and a scanning rate of 0.4 s per step. In the measurement of specific surface area SBET (Autosorb-1C, Quantachrome), ∼0.05 g of the catalyst was heated to 350 °C under vacuum prior to the introduction of N2 gas. Microscopic images of the catalysts were obtained from a Zeiss Scanning Electron Microscope (EVO/MA10). Bulk compositional analyses were performed using a Rigaku ZSX Primus IV wavelength dispersive X-ray fluorescence (WD-XRF) spectrometer. CO2 temperature-programmed desorption (CO2 TPD) experiments were conducted following the reported procedure,24,25 comprising of sample activation at 450 °C for 2 h, CO2 sorption at RT and purging, and the temperature ramp from 35 °C to 600 °C (5 °C min−1).Catalytic activity testing and products analysis
Catalytic activity testing was conducted in a continuous fixed-bed flow (glass) reactor (length, 50 cm; outside diameter, 8 mm; inner diameter, 6 mm) under atmospheric pressure of N2. The catalyst (sieved to the size of 600–850 μm) was packed into the reactor and activated by heating from room temperature to 800 °C (2 °C min−1) and held there for an hour under air stream (20 mL min−1). The reactor was next cooled to the reaction temperature (mostly 375 °C, but also 400 °C) under N2 gas. After that, 5% palmitic acid (Fluka, ≥98%) in p-xylene was fed into the reactor by an HPLC pump at the rate of 1.5 mL h−1. The contact time was fixed at 1500 g h mol−1. The reaction was operated for a total time on stream of 360 min under N2. The liquid products were trapped by an ice bath and collected hourly. They were analyzed by an HP 6890 gas chromatograph equipped with a flame ionization detector (GC-FID) and a capillary column DB-1 (length, 30 m; internal diameter, 0.32 mm; film thickness, 5.00 μm). The temperature program started with holding at 40 °C for 5 min, ramping to 280 °C (15 °C min−1), and holding for 24 min. An HP 5890 gas chromatograph coupled with a mass spectrometer (GC-MS) was also employed to identify/confirm the structure of the reaction products. The product distribution was compared to that in a semi-batch mode from the titanate pre-intercalated with the acid, as reported recently.24Results and discussion
Physicochemical properties of catalysts
Fig. 1a–d show the XRD patterns of the lepidocrocite-type catalysts, which are characteristics of the C-based centered potassium lepidocrocite titanate structure (Cm2c1).24–26 The highest-intensity peak at 2θ ∼11.3° represents the interlayer distance which is ∼7.8 Å. While K0.8Zn0.4Ti1.6O4 is highly crystalline, the structural ordering of other two samples is lower and they contain some additional peaks due to the impurity phase(s). The low SBET (3–13 m2 g−1, Table 1) of these samples indicates that their internal 2D space is inaccessible to N2 and that they are essentially non-porous. This finding is common for lepidocrocite titanate prepared by a high-temperature solid state method.20,21,25,34 | ||
| Fig. 1 The XRD patterns of catalysts. The hkl indices of the C-based center potassium lepidocrocite titanate structure with orthorhombic symmetry26 are shown as an example for K0.8Zn0.4Ti1.6O4. The ×, ■, and ○ symbols denote the impurity phase(s). The ▲ mark reflections are due to anatase-type TiO2. | ||
| Catalyst | SBETa (m2 g−1) | Db (Å) | Basicityc (μmol g−1) | Tpd (°C) | δOe |
|---|---|---|---|---|---|
| a Specific surface area.b Crystallite size calculated using Scherrer equation and the full width at half maximum of the strongest peak. The value in parenthesis is from the spent sample.c Basicity from CO2 TPD measurement in μmol CO2 per g catalyst.d NH3 desorption peak temperature (Tp)e Partial charge at the oxygen atom calculated from the nominal composition. The value in bracket [] is calculated from the XRF-derived composition. | |||||
| K0.8Zn0.4Ti1.6O4 | 3 | 570 (580) | 39 | 86 | −0.467 [−0.509] |
| K0.8Mg0.4Ti1.6O4 | 13 | 330 | 36 | 49, 59 | −0.492 |
| K0.8Li0.27Ti1.73O4 | 5 | 330 | 71 | 103 | −0.505 |
| Reassembled TiO2 | 27 | 50 (50) | 29 | 132 | −0.313 [−0.311] |
| Mg/Al mixed oxide | 122 | N.A. | 820 | 170 | N.A. |
K0.8Zn0.4Ti1.6O4 was converted to the protonic form which is accompanied by a significant Zn leaching as proven by XRF analysis. It is subsequently exfoliated into the Ti0.8O20.8− nanosheets colloid by reacting with tetrabutylammonium hydroxide,31–33 see Fig. S1 in ESI.† The addition of KOH destabilized the nanosheets which then electrostatically reassembled into precipitates. Fig. 1e shows that two phases were present after calcination at 450 °C. The majority is anatase, while the minor one is apparently the reassembled structure with larger (8.5 Å) repeating distance. Because XRF analysis indicates the small K and Zn content in the precursor (Ti0.99Zn0.01K0.004O2), this calcined sample will be referred to simply as the reassembled TiO2 catalyst. Consistently, its Raman spectrum displayed in Fig. S2† is the overlap of signals due to the dominating anatase and much smaller band close to the starting lepidocrocite titanate.35 The crystallite size of the reassembled TiO2 catalyst is ∼50 Å, smaller than the starting K0.8Zn0.4Ti1.6O4 by ∼100-fold. In a supporting manner, the SBET increases from 3 to 27 m2 g−1 after the reassembling. Fig. 2a shows the morphology of as-made K0.8Zn0.4Ti1.6O4 as the dense, platy microcrystals typical of layered materials with dimension ≥2 × 2 μm2. Meanwhile, Fig. 2b shows that the reassembled TiO2 is loosely-stacked and has even more irregular shape and smaller particle sizes, consistent with the increase in SBET.
 | ||
| Fig. 2 The SEM images of (a) as-made K0.8Zn0.4Ti1.6O4, (b) reassembled TiO2, (c) spent K0.8Zn0.4Ti1.6O4, and (d) the spent, reassembled TiO2. | ||
The CO2 desorption profiles of the catalysts are shown in Fig. 3. For K0.8Zn0.4Ti1.6O4, the CO2 desorption spans from 50–300 °C. This is ascribed to a combination of weak/physisorbed CO2 at low desorption temperature, and CO2 interacting with the lattice oxygen atom at higher desorption temperature. While K0.8Mg0.4Ti1.6O4 behaves similarly, K0.8Li0.27Ti1.73O4 shows a tailing at high-temperature side. For the reassembled TiO2, a very broad desorption profile was evident with Tp = 132 °C (which is relatively close to Tp = 120 °C in TiO2 anatase25). In all cases, the observed Tps are lower than the basic Mg/Al mixed oxide where the desorption profile extends up to 400 °C with Tp = 170 °C.
 | ||
| Fig. 3 The CO2 desorption profiles from studied catalysts. The plots were arbitrarily normalized such that the peaks are of similar intensity. | ||
The basicity (μmol CO2 per gram of the catalyst, in parenthesis) is in the order: Mg/Al mixed oxide (820) ≫ K0.8Li0.27Ti1.73O4 (70.8) > K0.8Zn0.4Ti1.6O4 (38.9) ∼ K0.8Mg0.4Ti1.6O4 (36.1) > reassembled TiO2 (29.1). The higher basicity of the potassium titanate compared to TiO2 is understandable considering that K+ ion typically promotes basic sites in solids.36 Interestingly, the basicity of lepidocrocite titanate catalysts (∼36−70 μmol CO2 g−1) with a relatively small SBET (3–13 m2 g−1) is just slightly different from some of the values reported37 for alkali titanate nanotubes (SBET – a few hundred m2 g−1). Also, by comparing the basicity per surface area of K0.8Zn0.4Ti1.6O4 (13.0 μmol CO2 m−2) with that of reassembled TiO2 (1.1 μmol CO2 m−2), the essential role of the highly crystalline lepidocrocite layers toward basicity could be appreciated.
Deoxygenation of palmitic acid
The palmitic acid conversion and products yield as a function of time on stream over K0.8Zn0.4Ti1.6O4 under N2 are depicted in Fig. 4a. A high conversion (∼72–86%) with a moderate deactivation is evident. Palmitone C15H31(CO)C15H31 (a C31 ketone) is the major product at ∼47–59% yield. This is most likely produced via palmitic acid ketonization, well known to be catalyzed by several Ti-based oxides.20,21,25,38–40 While the catalyst pre-reduction or the use of H2 carrier gas is required for small acids,20,41 it is not necessary for fatty acids in the present work, consistent with previous reports.6,24,25,42
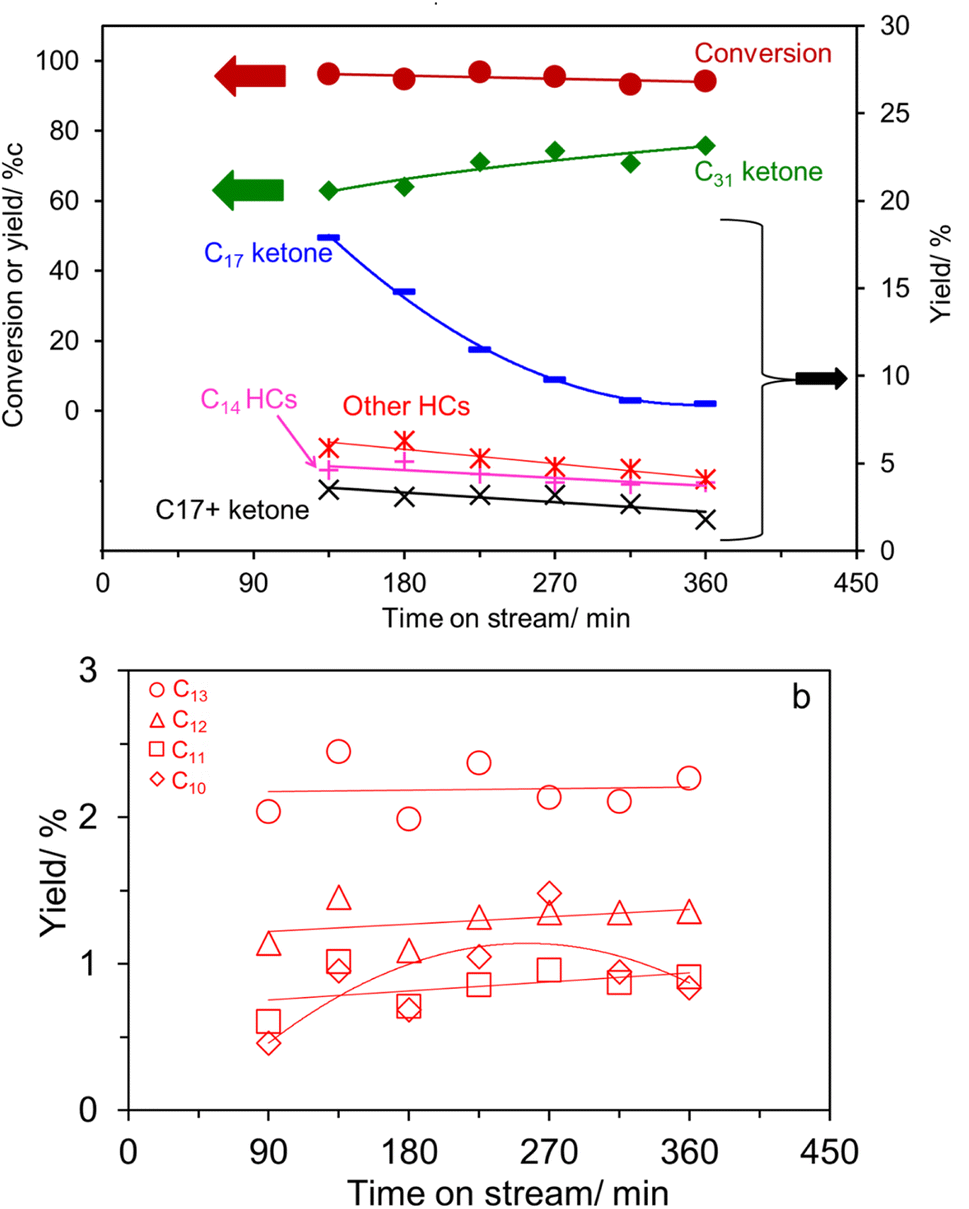 | ||
| Fig. 4 (a) The time on stream dependence of palmitic acid conversion and products yield over K0.8Zn0.4Ti1.6O4. The right axis is the zoom-in to products with smaller amount. (b) Detailed product distributions of other hydrocarbons ‘Other HCs’ in (a). Reaction condition: 375 °C, 15 mL min−1 of N2, 5% palmitic acid in p-xylene, contact time 1500 g h mol−1. | ||
The pronounced C31 ketone formation contrasts with our previous results from palmitic acid-intercalated K0.8Zn0.4Ti1.6O4 in a semi-batch experiment,24 giving mostly C15 hydrocarbons (eqn (4) and (5)) but none of the C31 ketone.
| C15H31COOH → C15H32 + CO2 | (4) |
| C15H31COOH → C15H30 + CO + H2O | (5) |
Due to the limited amount of intercalated palmitic acid in the previous work (18.6 wt% (ref. 24) or 0.23 g palmitic acid per g catalyst), excessive sites are available for every acid to interact. Consequently, the acid coupling to ketone is inhibited because ketonization is a bimolecular reaction,5,20 and the direct deoxygenation was favored. In contrast, there is a continuous supply of palmitic acid in the present flow system (contact time 1500 g h mol−1 for 6 h, or 1.02 g g−1), apparently allowing enough proximity of two acid molecules for ketonization.
Other major products include a C17 ketone (specifically the methyl ketone, CH3(CO)C15H31) and C14 hydrocarbons. Their formation can be easily explained by scissions via the McLafferty rearrangement of the primarily produced C31 ketone6,11–17,19,25 (i.e., γ-hydrogen transfer, Scheme 1).
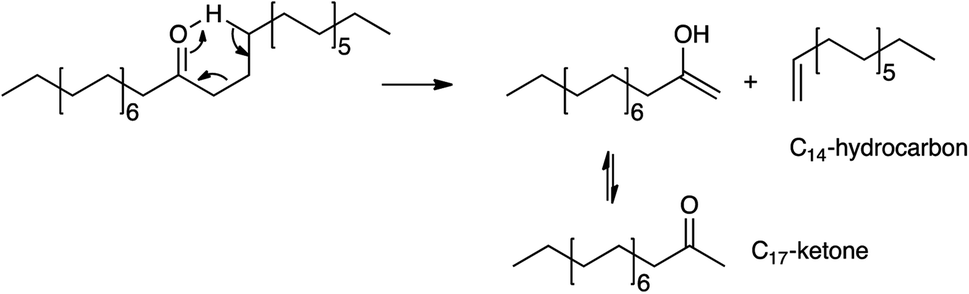 | ||
| Scheme 1 McLafferty rearrangement of C31 ketone. | ||
In agreement with eqn (2), the detected C14 hydrocarbons are mostly unsaturated (i.e., the unsaturated-to-saturated peak area ratio in GC analysis is ∼3). Because the reaction was performed under N2, the observed saturated hydrocarbon is likely due to the H-transfer during the coke formation.11 In a supporting manner, the presence of 10 wt% coke was deduced from TG analysis shown in Fig. S3.† While the hydrogen transfer activity of K0.8Zn0.4Ti1.6O4 has not yet been studied, TiO2 is an active catalyst for H-transfer of several feeds under atmospheric N2.42,43 Certainly, there is a trade-off between the use of hydrogen from coke precursor vs. catalyst deactivation, requiring an extended stability study. While the McLafferty rearrangement predicts the equimolar amount of the products, C14 hydrocarbons (11.8%) were in excess compared to C17 ketone (1.8–4.4%). This suggests an additional pathway for C14 hydrocarbons formation, possibly palmitic acid deacetylation, although this remains to be proven.
In addition, we observed a small amount (<8%) of ketones with more than 17 carbon atoms (C18, C28, and C29 ketones; shown as C17+ ketone in Fig. 4a), and ∼6% of other hydrocarbons, possessing less than 14 carbon atoms as shown in Fig. 4b. These two groups of products are likely originated from the C31 ketone scissions at other positions besides the C17/C18.11,25 Previously, Billuad et al.14,15 detected the hydrocarbon analog of the methyl ketone via reduction and dehydration over the acidic Al2O3 catalyst. However, the reductive dehydration of C17 methyl ketone to C17 olefin/alkane is not observed here. This finding suggests that K0.8Zn0.4Ti1.6O4 does not have active acidic sites for dehydration, consistent with the observed zero NH3 desorption in the NH3-TPD experiment shown in Fig. S4.† Similarly, the C31 hydrocarbon is not detected in the present work, suggesting that the H-transfer to C31 ketone must be slower than its scissions. In summary, the cascade reaction according to eqn (1)–(3) are dominated at this experimental condition.
While the as-made K0.8Zn0.4Ti1.6O4 is platy microcrystals, the spent K0.8Zn0.4Ti1.6O4 has a more irregular shape and rough surfaces (Fig. 2c). This finding suggests that palmitic acid vapor diffuses into the 2D space, consistent with the reported liquid-phase25 or melt23 intercalation. The intercalated species might exert appreciable mechanical stress to the layers such that they are deformed, and the microcrystals expanded. Yet, the integrity of the lepidocrocite structure was preserved as deduced from the preserved XRD pattern (Fig. 1b), the unchanged crystallite size D (Table 1), and the Raman spectrum (Fig. S2†). The stability of K0.8Zn0.4Ti1.6O4 at elevated temperature is clearly superior to the layered double hydroxides or layered hydroxy salt, undergoing irreversible structural transformation upon contact with liquid fatty acids at 100–140 °C.44–46 Considering the almost identical XRD and Raman spectra between as made and spent catalyst (Fig. 1 and S2†), it is assumed that all elements preserved their common oxidation state. This is also consistent with the reported stability of layered titanate with respect to high temperature,47 NH3,34 or H2 (at least up to 550 °C (ref. 20 and 48)).
Effect of intralayer metal and 2D confinement
The ketonization-McLafferty scission discussed above is influenced by catalyst composition, reaction temperature, and the confinement effect as summarized in Table 2. At 375 °C, the 77.2% palmitic acid conversion over K0.8Zn0.4Ti1.6O4 (entry 1) increased to 87.3% over K0.8Mg0.4Ti1.6O4 (entry 2) and 97.2% over K0.8Li0.27Ti1.73O4 (entry 3). These results are either due to the higher SBET or the higher basicity of the catalysts. Notably, the increased conversion is accompanied by a decreased C31 ketone yield but the increased yields of hydrocarbons. The C31 ketone yield at 375 °C is in the order: K0.8Zn0.4Ti1.6O4 (51.7%) > K0.8Mg0.4Ti1.6O4 (44.6%) > K0.8Li0.27Ti1.73O4 (36.6%), and the hydrocarbons yield is just the opposite. The increased hydrocarbons yield was similarly obtained at 400 °C, compare entry 3 and 4 for K0.8Li0.27Ti1.73O4. At this severe reaction condition, the scissions of the formed ketone are promoted, giving 46.8% hydrocarbons yield (vs. 33.4% at 375 °C).| Entry | Catalyst | T (°C) | Conv. | Yield, ketone | Yield, hydrocarbon | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C31b | C29 | C28 | C18 | C17c | C14 | C13 | C12 | C11 | C10 | C<10 | Total | ||||||
| a Reaction conditions: 5% palmitic acid in p-xylene; flow rate of feed plus carrier gas (N2) 30 mL min−1; reaction temperature 375 or 400 °C; atmospheric pressure; contact time 1500 g h mol−1; values were averaged from the time on stream from 90 to 360 min.b C15H31(CO)C15H31.c C15H31(CO)CH3. |
|||||||||||||||||
| 1 | K0.8Zn0.4Ti1.6O4 | 375 | 77.2 | 51.7 | 0 | 0 | 2.57 | 3.4 | 12.0 | 2.2 | 1.3 | 0.9 | 0.9 | 0 | 18.3 | ||
| 2 | K0.8Mg0.4Ti1.6O4 | 375 | 87.3 | 44.6 | 0 | 0 | 7.36 | 11.7 | 16.1 | 3.2 | 2.0 | 1.0 | 4.0 | 0 | 26.1 | ||
| 3 | K0.8Li0.27Ti1.73O4 | 375 | 97.2 | 36.6 | 0.34 | 0 | 3.35 | 23.1 | 21.0 | 4.5 | 2.6 | 1.7 | 2.7 | 0 | 33.4 | ||
| 4 | K0.8Li0.27Ti1.73O4 | 400 | 100 | 29.2 | 0.2 | 0 | 2.8 | 21.0 | 27.6 | 5.0 | 3.9 | 2.0 | 3.6 | 4.7 | 46.8 | ||
| 5 | Reassembled TiO2 | 375 | 95.4 | 51.7 | 1.1 | 1.3 | 0.5 | 11.3 | 4.4 | 1.7 | 1.3 | 0.7 | 1.6 | 0 | 9.7 | ||
| 6 | Mg/Al mixed oxide | 375 | 96.9 | 35.5 | 0.9 | 8.93 | 3.7 | 20.8 | 2.0 | 6.2 | 3.0 | 2.4 | 2.4 | 3.0 | 22.6 | ||
The hydrocarbons yield of 46.8% can be achieved at the complete palmitic acid conversion over K0.8Li0.27Ti1.73O4 at 400 °C, even under N2. As summarized in entry 4, the products also include 29.2% palmitone (C31H62O, 1.1 mol% O) and 24.0% C17 (or larger) ketones (which for simplicity will be represented solely by CH3(CO)C15H31, 1.9 mol% O). So, the nominal oxygen content in the products is (0.292 × 1.1%) + (0.240 × 1.9%) = 0.78 mol% O. Meanwhile, the palmitic acid feed (C16H32O2) contains 4.0 mol% O. This corresponds to the significant reduction of the nominal oxygen content by ∼80% relative to the feed. Notably, this high deoxygenation degree under N2 is comparable to some other catalysts for the cascade reaction of fatty acid under atmospheric H2.11,20 It is also worth emphasizing that palmitic acid is a solid while the products obtained are liquid at room temperature. In a separate experiment, CO2 was detected in the gaseous products, including other hydrocarbons which were not analyzed.
Meanwhile, palmitic acid conversion of 95.4% obtained over the reassembled TiO2 catalyst (entry 5) at 375 °C is likely due to a higher SBET compared to the K0.8Zn0.4Ti1.6O4. However, this is accompanied by a very high C31 ketone yield (70.3%) and the small hydrocarbons yield (13.0%), sharply contrasting with the product distribution over the lepidocrocite titanate catalysts. Interestingly, TGA indicates an equal coke content (∼10 wt%, Fig. S3†), even though the reassembled TiO2 has a higher SBET and exhibits a higher palmitic acid conversion than K0.8Zn0.4Ti1.6O4. This result can be explained assuming that, palmitone easily desorbs from the external surfaces of the reassembled TiO2 without being further converted or accumulated to form coke, consistent with its well-known ketonization activity.20,40 The absence of fragmented products is explained by the lack of confinement at the interlayer space. After use of the reassembled TiO2 as the catalyst, no significant change in the morphology (Fig. 2d), the crystal structure (Fig. 1f) and the local structure (Fig. S2†) was observed.
We also tested the catalytic activity of the mixed Mg/Al oxide which is known25,49–53 to catalyze carboxylic acid ketonization by basic sites. Generally, it shows similar catalytic activity (entry 6) to that lepidocrocite titanate catalysts especially K0.8Li0.27Ti1.73O4. The resemblance of catalytic activities for titanates and the Mg/Al-mixed oxide (but not TiO2) suggests that they could have the same type of active sites, as will be discussed in the next section. The high C17+ ketones yield over the mixed oxide (16.2% vs. 4.1%) suggests the scissions of palmitone at other positions, in addition to the typical C17/C18 characteristics of the McLafferty rearrangement. The lower hydrocarbons yield (22.6% vs. 33.4%) might be similarly explained assuming the lack of the interlayer space which facilitates the ketone scissions.
Basic-catalyzed scission and catalyst deactivation
Considering that the McLafferty rearrangement is the major pathway for the observed hydrocarbons formation, it is likely that the basic sites are the active sites for C31 ketone scissions. Here, we employ Sanderson's electronegativity equalization principle22,23 to rank the basic strength of each catalyst. Taking the nominal composition “K0.8Zn0.4Ti1.6O4” as an example, the intermediate electronegativity Sint is shown in eqn (6).| Sint(K0.8Zn0.4Ti1.6O4) = [S(K)0.8 × S(Zn)0.4 × S(Ti)1.6 × S(O)4]1/(0.8+0.4+1.6+4) | (6) |
Then, δO is given by eqn (7) based only on the chemical composition.
| δO = [Sint(K0.8Zn0.4Ti1.6O4) – S(O)]/[1.57S(O)1/2] | (7) |
The δO for the nominal “K0.8Zn0.4Ti1.6O4” is −0.467, compared to −0.509 for K0.74Zn0.37Ti1.45O4 according to XRF analysis. Accordingly, the deviation of ±0.04 will not significantly affect the trend of δO (Table 1) which are in the range −0.467 (“K0.8Zn0.4Ti1.6O4”) to −0.505 (“K0.8Li0.27Ti1.73O4”). These are more negative than −0.313 in TiO2, consistent with the fact that the lepidocrocite sheets are negatively charged (i.e., electron-rich), while TiO2 is neutral. The negative δO represents the more basic oxygen atom and vice versa.
Fig. 5 shows that palmitic acid conversion and several products selectivity are linear with respect to δO. Accordingly, the C31 ketone selectivity decreases with the increasing magnitude of δO, because its scissions to hydrocarbons and C17/C17+ ketone are promoted by basic sites. These results hint at the importance of acid-base chemistry over lepidocrocite titanate catalysts, complementing our previous works in the semi-batch reactor.24 The participation of other active sites is rather unlikely because these titanate catalysts (i) show no appreciable acidity (Fig. S4†), (ii) do not contain metal phase, and (iii) are not pre-reduced. Future work might investigate how this correlation will be modified in the presence of added functions. Fig. S5† show the expanded plot including the off-the-trend data from the reassembled TiO2 at the far left. Apparently, the correlation is valid for a catalyst with 2D confinement and a closely related composition only. Yet, the observed correlation based solely on the presumed composition could provide a simple predictive tool for catalysts selection by lepidocrocite titanates.
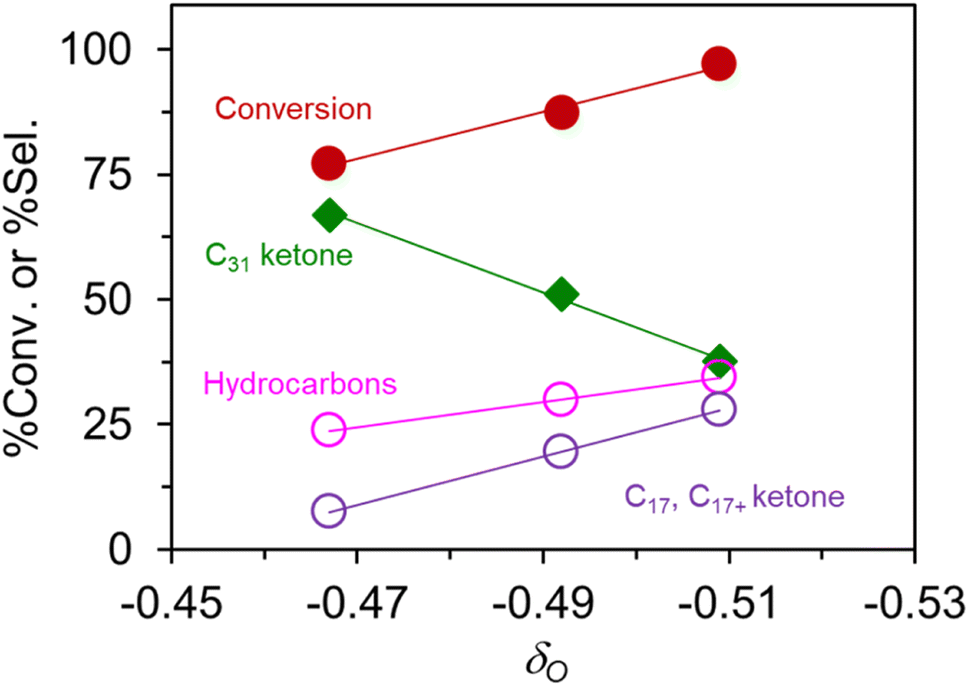 | ||
| Fig. 5 The apparent correlation between the partial charge at the O atom δO vs. the yield of C31 ketone, total hydrocarbons, and the fragmented ketones (i.e., C17 ketone and C17+ ketones). | ||
In addition to an improved activity, K0.8Li0.27Ti1.73O4 – the most active catalyst-exhibit a high stability as shown in Fig. 6a. This behavior is in sharp contrast to the reassembled TiO2 shown in Fig. 6b. Here, palmitic acid conversion slightly decreases which is parallel to the decreasing fragmented products yield (32% to 18%). Accordingly, it is deduced that the basic sites in the reassembled TiO2 become inactive with time on stream. This is presumably due to limited number of basic sites in the reassembled TiO2, as compared to that in the lepidocrocite titanate (Table 1). In addition, the lack of the 2D confinement could discourage the ketone scission, as discussed above. On the other hand, the active sites for ketonization remain active at least up to 360 min, leading to an increasing yield of C31 ketone. Notably, the liquid alkane yield obtained over K0.8Li0.27Ti1.73O4 with 2D confinement is comparable to the pioneering work on ketone scissions over Al2O3 (ref. 14) (Table S1†). Table S1† also suggests that the basic catalysts are superior to Al2O3 in that the liquid ketone yield is high (up to 63%, vs. 22%), as the ketone is not lost by reacting with acid sites.
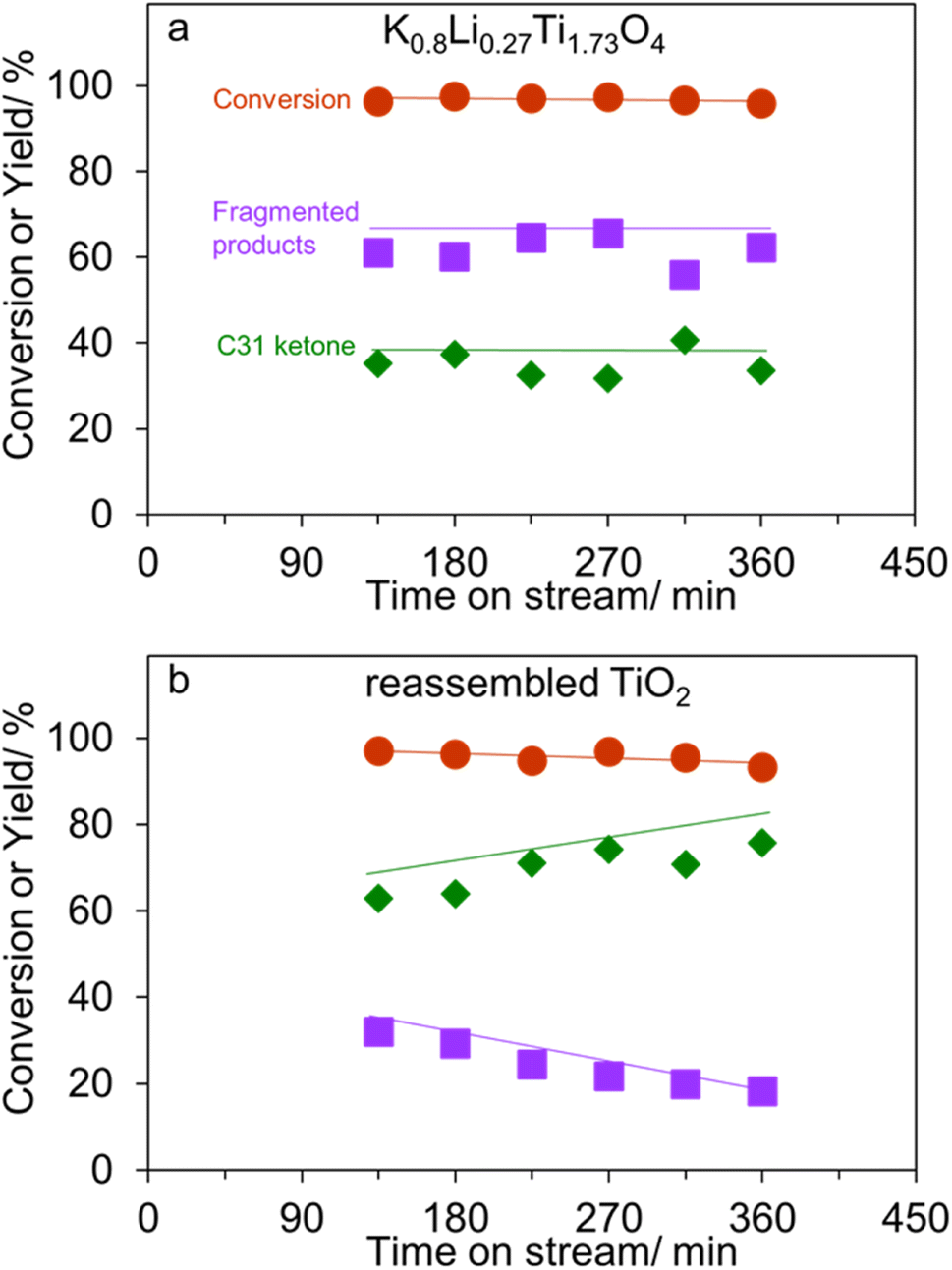 | ||
| Fig. 6 The time on stream dependences of palmitic acid conversion, C31 ketone yield, and fragmented products yield (the combination of C17 ketone, C17+ ketones, C14 and smaller hydrocarbons) over (a) K0.8Li0.27Ti1.73O4, and (b) reassembled TiO2. Reaction conditions: 5% palmitic acid in p-xylene; flow rate of feed plus carrier gas (N2) 30 mL min−1; reaction temperature 375 or 400 °C as indicated; atmospheric pressure; contact time 1500 g h mol−1. | ||
Conclusions
The cascade reaction of palmitic acid (C16) to hydrocarbons (≤C14) was investigated over lepidocrocite-type alkali titanate. This reaction proceeds via the C16 acid ketonization to C31 ketone, prior to the scissions mostly to a C17 methyl ketone and the unsaturated C14 hydrocarbon (i.e., the McLafferty rearrangement). The latter undergoes the H-transfer from the coke precursor to a saturated C14 analog. We propose basic sites as the active sites for the ketone scissions, as evidenced from the apparent correlation of secondary products yield with the calculated δO. In addition, the products distribution depends on the reaction temperature, catalyst compositions, confinement at the 2D space, and the proximity between palmitic acid molecules (in flow- vs. semi-batch experiments). Over K0.8Li0.27Ti1.73O4 at 400 °C and complete palmitic acid conversion, the obtained 46.8% hydrocarbons yield presents a significant deoxygenation with high stability under N2. Future works could be directed towards the conversion of other feeds such as ester, aldehyde, or more complex biomass-derived chemicals including triglycerides.Author contributions
Tosapol Maluangnont: conceptualization, data acquisition, writing, review, and editing. Piyasan Praserthdam: review. Tawan Sooknoi: conceptualization, review, editing, and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by the National Research Council of Thailand. We thank Kanokporn Limsakul, Songsit Juntarachairot, and Saithong Sangsan for assistance in catalytic activity measurements. The authors acknowledge the facilities and technical assistance from Nanotechnology and Materials Analytical Instrument Service Unit (NMIS) of College of Materials Innovation and Technology, KMITL.References
- D. R. Dodds and R. A. Gross, Chemicals from biomass, Science, 2007, 318, 1250–1251 CrossRef CAS PubMed.
- K. A. Rogers and Y. Zheng, Selective deoxygenation of biomass-derived bio-oils within hydrogen-modest environments: a review and new insights, ChemSusChem, 2016, 9, 1750–1772 CrossRef CAS PubMed.
- E. Santillan-Jimenez and M. Crocker, Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation, J. Chem. Technol. Biotechnol., 2012, 87, 1041–1050 CrossRef CAS.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Heterogeneous catalytic deoxygenation of stearic acid for production of biodiesel, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, Ketonization of carboxylic acids: mechanisms, catalysts, and implications for biomass conversion, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- K. Lee, M. Y. Kim and M. Choi, Effects of fatty acid structures on ketonization selectivity and catalyst deactivation, ACS Sustainable Chem. Eng., 2018, 6, 13035–13044 CrossRef CAS.
- B. Oliver-Tomas, M. Renz and A. Corma, High quality biowaxes from fatty acids and fatty esters: catalyst and reaction mechanism for accompanying reactions, Ind. Eng. Chem. Res., 2017, 56, 12870–12877 CrossRef CAS.
- O. Marie, A. V. Ignatchenko and M. Renz, Methyl ketones from carboxylic acids as valuable target molecules in the biorefinery, Catal. Today, 2021, 367, 258–267 CrossRef CAS.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Cascade utilization of lignocellulosic biomass to high-value products, Green Chem., 2019, 21, 3499–3535 RSC.
- M. Jin, M. E. Lee, M. Seo, J.-K. Kim, S. Li and M. Choi, Coproduction of value-added lube base oil and green diesel from natural triglycerides via a simple two-step process, Ind. Eng. Chem. Res., 2020, 59, 8946–8954 CrossRef CAS.
- T. Maluangnont, C. Dararat, T. Kulrat, S. Soontontaweesub, T. Anothaiwalaikul, P. Bunprechawong, R. Chanda, J. Kanchanawarin, P. Kidkhunthod and T. Sooknoi, Production of liquid fuel from palmitic acid over nanocrystalline CeO2-based catalysts with minimal use of H2, Catal. Commun., 2017, 102, 123–126 CrossRef CAS.
- L. M. Orozco, M. Renz and A. Corma, Cerium oxide as a catalyst for the ketonization of aldehydes: mechanistic insights and a convenient way to alkanes without the consumption of external hydrogen, Green Chem., 2017, 19, 1555–1569 RSC.
- A. Leung, D. G. B. Boocock and S. K. Konar, Pathway for the catalytic conversion of carboxylic acids to hydrocarbons over activated alumina, Energy Fuels, 1995, 9, 913–920 CrossRef CAS.
- F. Billaud, A. K. T. Minh, P. Lazano and D. Pioch, Catalytic cracking of octanoic acid, J. Anal. Appl. Pyrolysis, 2001, 58–59, 605–616 CrossRef CAS.
- F. Billaud, Y. Guitard, A. K. T. Minh, O. Zahraa, P. Lozano and D. Pioch, Kinetic studies of catalytic cracking of octanoic acid, J. Mol. Catal. Chem., 2003, 192, 281–288 CrossRef CAS.
- A. L. Baylon, J. Sun, K. J. Martin, P. Venkitasubramanian and Y. Wang, Beyond ketonization: selective conversion of carboxylic acids to olefins over balanced Lewis acid-based pairs, Chem. Commun., 2016, 52, 4975–4978 RSC.
- A. Corma, M. Renz and C. Schaverien, Coupling fatty Acids by ketonic decarboxylation using solid catalysts for the direct production of diesel, lubricants, and chemicals, ChemSusChem, 2008, 1, 739–741 CrossRef CAS PubMed.
- A. Witsuthammakul and T. Sooknoi, Selective hydrodeoxygenation of bio-oil derived products: ketones to olefins, Catal. Sci. Technol., 2015, 5, 3639–3648 RSC.
- M. Watanabe, T. Iida and H. Inomata, Decomposition of a long chain saturated fatty acid with some additives in hot compressed water, Energy Convers. Manag., 2006, 47, 3344–3350 CrossRef CAS.
- P. Promchana, A. Boonchun, J. T-Thienprasert, T. Sooknoi and T. Maluangnont, Direct conversion of carboxylic acid to olefins over Pt-loaded, oxygen-deficient alkali hexatitanate catalysts with ketonization-hydrogenation-dehydration activity, Catal. Today, 2021, 375, 418–428 CrossRef CAS.
- T. Maluangnont, N. Chanlek, T. Suksawad, N. Tonket, P. Saikhamdee, U. Sukkha and N. Vittayakorn, Beyond soft chemistry - bulk and surface modifications of polycrystalline lepidocrocite titanate induced by post-synthesis thermal treatment, Dalton Trans., 2017, 46, 14277–14285 RSC.
- T. Charoonsuk, S. Sriphan, P. Pulphol, W. Vittayakorn, N. Vittayakorn and T. Maluangnont, AC conductivity and dielectric properties of lepidocrocite-type alkali titanate tunable by interlayer cation and intralayer metal, Inorg. Chem., 2020, 59, 15813–15823 CrossRef CAS PubMed.
- T. Maluangnont and T. Sooknoi, Inclusion of alkali carboxylate salts at the two-dimensional space of layered alkali titanate via carboxylic acids intercalation, J. Solid State Chem., 2020, 291, 121648 CrossRef CAS.
- T. Maluangnont, P. Arsa and T. Sooknoi, Extending the basic function of lattice oxygen in lepidocrocite titanate – the conversion of intercalated fatty acid to liquid hydrocarbon fuels, J. Solid State Chem., 2017, 256, 219–226 CrossRef CAS.
- T. Maluangnont, P. Arsa, K. Limsakul, S. Juntarachairot, S. Sangsan, K. Gotoh and T. Sooknoi, Surface and interlayer base-characters in lepidocrocite titanate: the adsorption and intercalation of fatty acid, J. Solid State Chem., 2016, 238, 175–181 CrossRef CAS.
- D. Groult, C. Mercey and B. Raveau, Nouveaux oxydes à structure en feuillets: Les titanates de potassium non-stoechiométriques Kx(MyTi2-y)O4, J. Solid State Chem., 1980, 32, 289–296 CrossRef CAS.
- T. Maluangnont, K. Matsuba, F. Geng, R. Ma, Y. Yamauchi and T. Sasaki, Osmotic swelling of layered compounds as a route to producing high-quality two-dimensional materials. A comparative study of tetramethylammonium versus tetrabutylammonium cation in a lepidocrocite-type titanate, Chem. Mater., 2013, 25, 3137–3146 CrossRef CAS.
- H. Wang, Y. Song, J. Xiong, J. Bi, L. Li, Y. Yu, S. Liang and L. Wu, Highly selective oxidation of furfuryl alcohol over monolayer titanate nanosheet under visible light irradiation, Appl. Catal., B, 2018, 224, 394–403 CrossRef CAS.
- Y. Song, H. Wang, J. Xiong, B. Guo, S. Liang and L. Wu, Photocatalytic hydrogen evolution over monolayer H1.07Ti1.73O4·H2O nanosheets: roles of metal defects and greatly enhanced performances, Appl. Catal., B, 2018, 221, 473–481 CrossRef CAS.
- M. Pilarski, R. Marschall, S. Gross and M. Wark, Layered cesium copper titanate for photocatalytic hydrogen production, Appl. Catal., B, 2018, 227, 349–355 CrossRef CAS.
- S. Sriphan, T. Charoonsuk, T. Maluangnont, P. Pakawanit, C. Rojviriya and N. Vittayakorn, Multifunctional nanomaterials modification of cellulose paper for efficient triboelectric nanogenerators, Adv. Mater. Technol., 2020, 5, 2000001 CrossRef CAS.
- N. Petpiroon, N. Bhummaphan, R. Soonnarong, W. Chantarawong, T. Maluangnont, V. Pongrakhananon and P. Chanvorachote, Ti0.8O2 nanosheets inhibit lung cancer stem cells by inducing production of superoxide anion, Mol. Pharmacol., 2019, 95, 418–432 CrossRef CAS PubMed.
- S. Sriphan, T. Charoonsuk, T. Maluangnont and N. Vittayakorn, High-performance hybridized composited-based piezoelectric and triboelectric nanogenerators based on BaTiO3/PDMS composite film modified with Ti0.8O2 nanosheets and silver nanopowders cofillers, ACS Appl. Energy Mater., 2019, 2, 3840–3850 CrossRef CAS.
- T. Maluangnont, B. Wuttitham, P. Hongklai, P. Khunmee, S. Tippayasukho, N. Chanlek and T. Sooknoi, An unusually acidic and thermally stable cesium titanate CsxTi2-yMyO4 (x = 0.67 or 0.70; M = vacancy or Zn), Inorg. Chem., 2019, 58, 6885–6892 CrossRef CAS.
- T. Gao, H. Fjellvag and P. Norby, Crystal structures of titanate nanotubes: a Raman scattering study, Inorg. Chem., 2009, 48, 1423–1432 CrossRef CAS PubMed.
- H. Hattori, Solid base catalysts: generation, characterization, and catalytic behavior of basic sites, J. Jpn. Petrol. Inst., 2004, 47, 67–81 CrossRef CAS.
- A. V. Grigorieva, V. Y. Yuschenko, I. I. Ivanova, E. A. Goodilin and Y. D. Tretyakov, Chemical tuning of adsorption properties of titanate nanotubes, J. Nanomater., 2012, 920483 Search PubMed.
- M. Gliński, J. Kijeński and A. Jakubowski, Ketones from monocarboxylic acids: catalytic ketonization over oxide systems, Appl. Catal., A, 1995, 128, 209–217 CrossRef.
- T. N. Pham, D. Shi, T. Sooknoi and D. E. Resasco, Aqueous-phase ketonization of acetic acid over Ru/TiO2/carbon catalysts, J. Catal., 2012, 295, 169–178 CrossRef CAS.
- G. Pacchioni, Ketonization of carboxylic acids in biomass conversion over TiO2 and ZrO2 surfaces: a DFT perspective, ACS Catal., 2014, 4, 2874–2888 CrossRef CAS.
- S. Tosoni, H.-Y. T. Chen, R. Puigdollers and G. Pacchioni, TiO2 and ZrO2 in biomass conversion: Why catalyst reduction helps, Philos. Trans. R. Soc., A, 2018, 376, 20170056 CrossRef PubMed.
- B. Oliver-Tomas, M. Renz and A. Corma, Direct conversion of carboxylic acids (Cn) to alkenes (C2n-1) over titanium oxide in absence of noble metals, J. Mol. Catal. Chem., 2016, 415, 1–8 CrossRef CAS.
- L. E. Oi, M.-Y. Choo, H. V. Lee, Y. H. Taufiq-Yap, C. K. Cheng and J. C. Juan, Catalytic deoxygenation of triolein to green fuel over mesoporous TiO2 aided by in situ hydrogen production, Int. J. Hydrogen Energy, 2020, 45, 11605–11614 CrossRef CAS.
- C. S. Cordeiro, F. R. da Silva, R. Marangoni, F. Wypych and L. P. Ramos, LDHs instability in esterification reactions and their conversion to catalytically active layered carboxylates, Catal. Lett., 2012, 142, 763–770 CrossRef CAS.
- C. S. Cordeiro, G. G. C. Arizaga, L. P. Ramos and F. Wypych, A new zinc hydroxide nitrate heterogeneous catalyst for the esterification of free fatty acids and the transesterification of vegetable oils, Catal. Commun., 2008, 9, 2140–2143 CrossRef CAS.
- L. P. Dill, D. M. Kochepka, L. L. Lima, A. A. Leitão, F. Wypych and C. S. Cordeiro, Brazilian mineral clays: classification, acid activation and application as catalysts for methyl esterification reactions, J. Braz. Chem. Soc., 2021, 32, 145–157 CAS.
- T. Maluangnont, P. Pulphol and W. Vittayakorn, Interlayer alkali ion governs robustness, reactivity, and dielectric properties of sintered lepidocrocite titanate, J. Solid State Chem., 2022, 305, 122713 CrossRef CAS.
- J. Kanchanawarin, W. Limphirat, P. Promchana, T. Sooknoi, T. Maluangnont, K. Simalaotao, A. Boonchun, P. Reunchan, S. Limpijumnong and J. T-Thienprasert, Local structure of stoichiometric and oxygen-deficient A2Ti6O13 (A = Li, Na, and K) studied by X-ray absorption spectroscopy and first-principles calculations, J. Appl. Phys., 2018, 124, 155101 CrossRef.
- K. Yan, Y. Liu, Y. Lu, J. Chai and L. Sun, Catalytic application of layered double hydroxide-derived catalysts for the conversion of biomass-derived molecules, Catal. Sci. Technol., 2017, 7, 1622–1645 RSC.
- Y. Song, S. K. Beaumont, X. Zhang, K. Wilson and A. F. Lee, Catalytic applications of layered double hydroxides in biomass valorisation, Curr. Opin. Green Sustain. Chem., 2020, 22, 29–38 CrossRef.
- E. Kandare and J. M. Hossenlopp, Thermal degradation of acetate-intercalated hydroxy double and layered hydroxy salts, Inorg. Chem., 2006, 45, 3766–3773 CrossRef CAS PubMed.
- K. Parida and J. Das, Mg/Al hydrotalcites: preparation, characterization and ketonisation of acetic acid, J. Mol. Catal. Chem., 2000, 151, 185–192 CrossRef CAS.
- J. Das and K. Parida, Catalytic ketonization of acetic acid on Zn/Al layered double hydroxides, React. Kinet. Catal. Lett., 2000, 69, 223–229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06530d |
| This journal is © The Royal Society of Chemistry 2022 |