
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of fluorine bonding and nonbonding interactions on 19F chemical shifts†
Yang Lu a,
Mingming Sunc and
Ning Xi*ab
a,
Mingming Sunc and
Ning Xi*ab
aInstitute of Drug Discovery Technology, Ningbo University, Ningbo, Zhejiang 315211, P. R. China. E-mail: xining@nbu.edu.cn
bSchool of Medicine, Ningbo University, Ningbo, Zhejiang 315211, P. R. China
cDepartment of Chemistry, Nanchang University, 999 Xuefu Avenue, Nanchang, 330031, P. R. China
First published on 9th November 2022
Abstract
19F-NMR signals are sensitive to local electrostatic fields and are useful in probing protein structures and dynamics. Here, we used chemically identical ortho-F nuclei in N-phenyl γ-lactams to investigate the relationship between 19F NMR chemical shifts and local environments. By varying the structures at the C5- and C7-substituents, we demonstrated that 19F shifts and Hammett coefficients in Hammett plots follow typical relationships in bonding interactions, while manifesting reverse correlations in nonbonding contacts. Quantum mechanics calculations revealed that one of the ortho-F nuclei engages in n → π* orbital delocalization between F lone pair electrons (n) and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/ArN antibonding orbital (π*), and the other ortho-F nucleus exhibits n ↔ σ orbital polarization between the n electrons and the C–H σ bonding orbital. As 19F NMR spectroscopy find increasing use in molecular sensors and biological sciences, our findings are valuable for designing sensitive probes, elucidating molecular structures, and quantifying analytes.
O/ArN antibonding orbital (π*), and the other ortho-F nucleus exhibits n ↔ σ orbital polarization between the n electrons and the C–H σ bonding orbital. As 19F NMR spectroscopy find increasing use in molecular sensors and biological sciences, our findings are valuable for designing sensitive probes, elucidating molecular structures, and quantifying analytes.
1. Introduction
Fluorine is a particularly important element in the broader space of chemical biology. Many FDA approved drugs and commercial compounds such as pesticides and refrigerants are fluorinated.1 There is a high degree of interaction between fluorine and biomolecules, which plays an important role in the functioning of fluorinated molecules.2 Owing to the lack of naturally-occurring fluorinated organic molecules, fluorine is also a unique probe to investigate the structures and functions of biological molecules,3 and a feasible sensor to detect analytes of biological, medicinal, and environmental importance.4 Central to these studies is 19F NMR spectroscopy, which allows users to readily visualize 19F chemical shifts and splitting patterns depending on the fluorine local chemical environments.The 19F isotope is 100% naturally abundant and shows a large NMR chemical shift range (∼300 ppm). The high resolution, coupled with high detection sensitivity and absence of background signals, affords well-resolved 19F resonances. To date, 19F NMR spectroscopy has been extensively applied in diverse biological, pharmaceutical, and material researches as evidenced by new reporter molecules and detection strategies.3,4 Zhao and Swager,5 as well as our own work,6 showed that 19F signals are sensitive to chiral environments even though the stereogenic centers are several carbons away from the fluorine. Recently, Prof. Huang and Zhao confirmed that substituents adjacent and distal to F are both important to the resolving ability of 19F-labeled sensors.7 In fact, the rich information obtained from 19F NMR spectroscopy also allows us to monitor molecular interaction and dynamics. Related studies based on fluorinated ligands and/or fluorinated proteins have found their broad utilities in molecular recognition, protein/nucleic acid characterization,3 and fragment-based drug discovery.8 However, interpreting 19F NMR data from biomolecular mixtures can be challenging and is often complicated by the lack of knowledge in 19F chemical shift assignments, among others.
In order to better use of fluorinated molecules in biochemical applications, a comprehensive understanding of correlations between 19F chemical shift and the environment/structure is essential. It is well-recognized that 19F shielding is not only determined by magnetic anisotropies associated with the covalent bond system and with the circulation of π-electrons in aromatic rings, but also greatly influenced by van der Waals interactions, hydrogen bonding, and electric fields.9 Noncovalent interactions such as multipolar contacts in C–F⋯CO and C–F⋯CN pairs,10 hydrogen-bonding in C–F⋯H–N, C–F⋯H–O, and C–F⋯H–C pairs,11 and interactions found in C–F⋯S12a and C–F⋯π pairs,12b have been reported. Sporadic data disclosed in literature suggest that C–F⋯H–X interactions shield 19F signals.13 Prof. Lectka confirmed that 19F signals in “jousting” C–F⋯H–C interactions display remarkable downfield shifts, likely due to dominating repulsive forces between C–F⋯H–C.14 Yet, the effects of CO/CN groups on 19F chemical shifts are not reported in the public domain, and a full understanding of relations between 19F chemical shifts and chemical environments remains elusive.
We have employed a slow rotating (on 19F NMR time scale), di-ortho-F substituted N-phenyl γ-lactam scaffold to study through-space contacts of F with remote functional groups such as stereogenic C9-amides and C9-esters, as shown in Fig. 1.15 Compounds are generally used as enantiomeric mixtures, as the individual enantiomer behaves the same spectroscopically. Assuming γ-lactam ring as a flat ring, we define the ortho-F nucleus siting on the same side of C9-substituents as syn-F, and the other ortho-F as anti-F (atom number assigned according to crystal structures, vide infra). By varying the p-substituent on C9-phenylamide, we showed that downfield 19F shifts δdown display “reverse” a linear relationship with Hammett constants σpara of the p-substituents. There are only small changes at the upfield 19F shifts δup in various C9-amide derivatives. We therefore assigned the downfield 19F signal to syn-F, and the upfield 19F peak to anti-F.16 In this report, we examined correlations of 19F chemical shifts with different C7- and C5-groups in γ-lactam-derived structures. We also carried out quantum calculations to investigate fluorine through-space contacts with the adjacent functional groups. Effects of through-bond and through-space interactions on 19F chemical shifts, as well as the through-space orbital delocalization are discussed.
 | ||
| Fig. 1 Assignments of syn-F and anti-F nuclei on N-phenyl group. The atom numbers are assigned according to crystal structures. | ||
2. Results and discussion
In di-ortho-F substituted N-phenyl γ-lactam derivatives, structural modifications of γ-lactam ring initiate unequal perturbations on ortho-F nuclei regarding to their through-space interactions, but afford the same through-bond effects on syn-F and anti-F. Thus, converting C7-amide to C7-thioamide or C7-amidine moiety on the γ-lactam ring alters chemical environments around syn-F and anti-F differently, as exemplified in thioamides 2a–f and amidines 4a–q. Installing a substituent at C5 on N-phenyl group, as in compounds 6a–w, produces the same bonding and nonbonding effects on both ortho-F nuclei. Fig. 2 shows the chemical structures of compounds 1–6, where structures of 1a–f, 3a–e and 5a–i are used as references to the corresponding C9-esters/amides. | ||
| Fig. 2 Structures of compounds 1–6. | ||
2.1. Synthetic schemes
Illustrative synthetic schemes for compounds 2–6 are provided in Fig. 3–5. Compounds 2a and 2b were converted from 1a and 1b, respectively, with Lawesson's reagent under mild conditions.17 For C7-phenylamidine derivatives 3 and 4, two synthetic routes were employed. The initial procedure requires the presence of POCl3 in CH2Cl2 solution under refluxing conditions.18 This one-pot reaction was used to generate compounds 3a–e and 4a–e. A more versatile and productive approach was later developed using thioamide 2b as the starting material.19 As shown in Fig. 3, thioamide 2b was treated with mCPBA at 0 °C, followed by the addition of para-substituted phenylamines to give the desired amidines 4f–q. This process is compatible with basic para-substituents such as dimethylamine (NMe2) and generally provides better yields than the POCl3 route.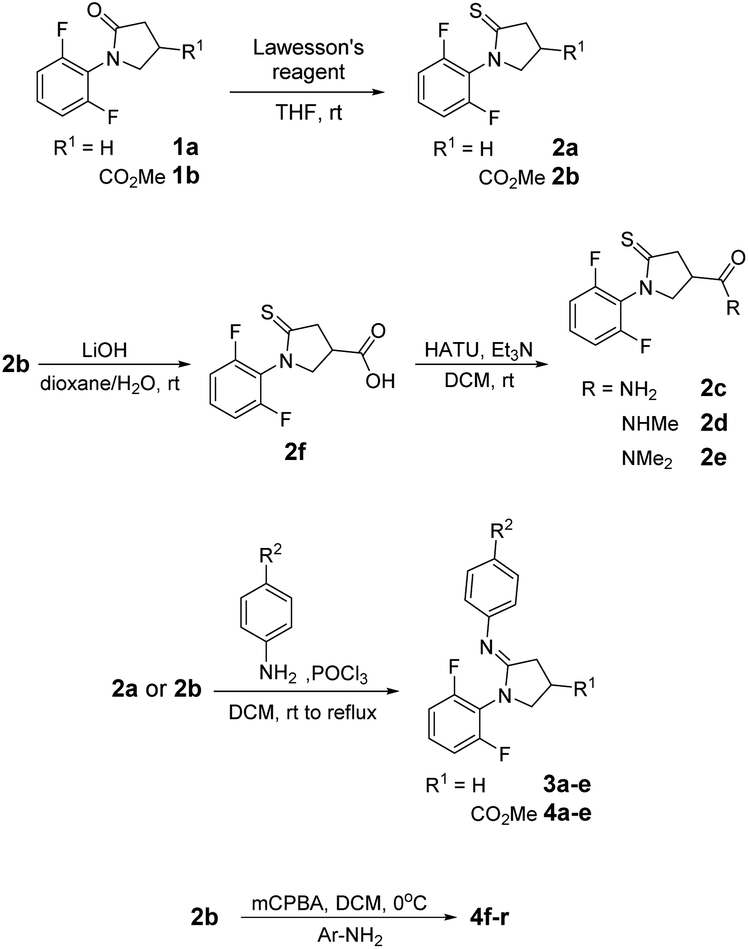 | ||
| Fig. 3 Syntheses of compounds 2–4. | ||
C5-substituted N-phenyl γ-lactams 6a–e, 6i–o and 6u were obtained through the synthetic routes as described previously, starting from appropriate 5-substituted anilines.15 The syntheses of 5f and 6f were attained by reducing the NO2 group in 5e or 6e. Reductive aminations of 5f and 6f with NaBH4 resulted in a mixture of anilines, which was separated by a column chromatography to give 5g/5h and 6g/6h. Acylation of 6f with different acylating agents (formal or acetic acid) afforded 6v and 6w in excellent yields, as illustrated in Fig. 4.
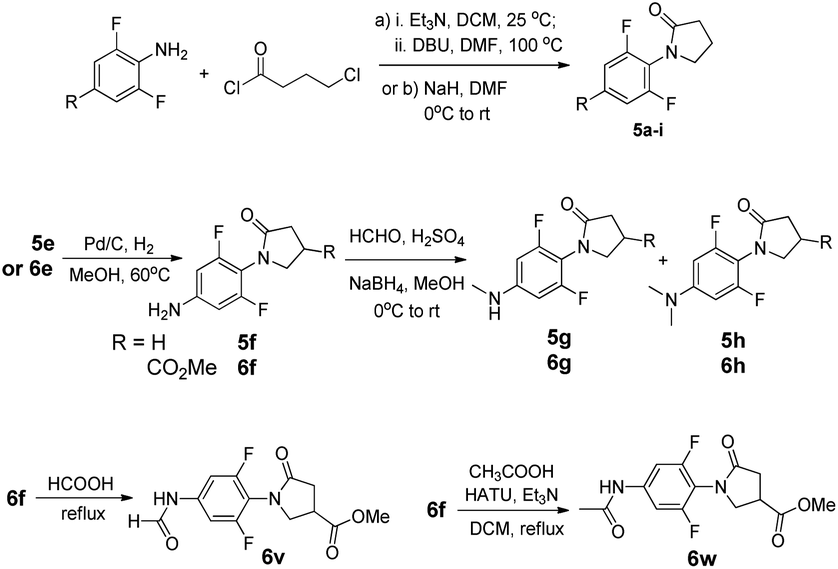 | ||
| Fig. 4 Syntheses of compounds 5–6. | ||
Fig. 5 shows the syntheses of 6p–t from 4-iodo 6l through Pd-catalyzed coupling reactions. Stille coupling was used to obtain 6p and 6q,20 while Suzuki–Miyaura coupling afforded 6r. Formylation of aryl halides from 6l using t-butyl isocyanide as a C1 source and formate salts as a hydride donor provided 6s.21 Similarly, vinylation of 6l with a vinyl ether followed by hydrolysis furnished ketone 6t.22
 | ||
| Fig. 5 Syntheses of compounds 6p–q from 6f and 6l. a dppe: 1,2-bis(diphenylphosphino)ethane; dppp: 1,3-bis(di-phenylphosphino)propane; dppf: 1,1′-bis(diphnylphosphino)-ferrocene. | ||
2.2. C7-substituent effects on ortho-19F chemical shifts
We surveyed 19F shift changes when the γ-lactam CO group was converted into CS and CNPh groups. All 19F NMR spectra were recorded in CDCl3 solution at 25 °C. Compound concentrations were ∼10 mg mL−1 and CFCl3 was used as an internal standard. As shown in Tables 1 and 2, 19F NMR signals become deshielded from −117.82 to −117.49 and −117.05 ppm, respectively, from compounds 1a to 2a and 3a, likely due to their different electron-withdrawing abilities and polarizabilities. In chiral C9-ester series, thioamide 2b and amidine 4a display larger shift differences Δδ (1.16 and 1.09 ppm) between syn-F and anti-F signals than γ-lactam 1b (Δδ = 0.79 ppm). Therefore, the chemical environment differences between syn-F or anti-F nuclei increase from γ-lactam to thioamide and amidine counterparts.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | δdown | δup | Δδ | δdown | δup | Δδ | ||
| H | 1a | −117.82 | −117.82 | 0 | 2a | −117.49 | −117.49 | 0 |
| CO2Me | 1b | −117.14 | −117.93 | 0.79 | 2b | −116.53 | −117.69 | 1.16 |
| CONH2 | 1c | −116.81 | −118.15 | 1.34 | 2c | −116.21 | −117.94 | 1.73 |
| CONHMe | 1d | −116.66 | −118.21 | 1.55 | 2d | −116.10 | −118.07 | 1.98 |
| CONMe2 | 1e | −116.25 | −118.23 | 1.98 | 2e | −115.59 | −118.16 | 2.56 |
|
|||||
|---|---|---|---|---|---|
| Cpd | R | δamidinedown | δamidineup | Δδamidine | σpara |
| a Δδamidine = δamidinedown − δamidineup; σpara is Hammett constants for para-substituents. | |||||
| 3a | H | −117.05 | — | 0 | 0 |
| 3b | Me | −117.06 | — | 0 | −0.17 |
| 3c | OMe | −117.07 | — | 0 | −0.27 |
| 3d | CF3 | −117.18 | — | 0 | 0.54 |
| 3e | NO2 | −117.30 | — | 0 | 0.78 |
| 4a | H | −116.31 | −117.41 | 1.09 | 0 |
| 4b | Me | −116.26 | −117.41 | 1.15 | −0.17 |
| 4c | OMe | −116.31 | −117.42 | 1.11 | −0.27 |
| 4d | CF3 | −116.49 | −117.47 | 0.98 | 0.54 |
| 4e | NO2 | −116.65 | −117.48 | 0.83 | 0.78 |
| 4f | NMe2 | −116.17 | −117.37 | 1.20 | −0.83 |
| 4g | OEt | −116.33 | −117.42 | 1.09 | −0.24 |
| 4h | F | −116.46 | −117.47 | 1.01 | 0.06 |
| 4i | Cl | −116.46 | −117.46 | 1.00 | 0.23 |
| 4j | Br | −116.46 | −117.46 | 1.00 | 0.23 |
| 4k | I | −116.44 | −117.44 | 1.00 | 0.18 |
| 4l | CN | −116.61 | −117.48 | 0.87 | 0.66 |
| 4m | CHO | −116.53 | −117.45 | 0.92 | 0.42 |
| 4n | COMe | −116.47 | −117.42 | 0.94 | 0.5 |
| 4o | CO2Me | −116.49 | −117.46 | 0.97 | 0.45 |
| 4p | CONMe2 | −116.40 | −117.42 | 1.01 | — |
| 4q | NHCOMe | −116.34 | −117.40 | 1.06 | 0 |
| 4r | CH2CH2 |
−116.33 | −117.41 | 1.08 | −0.02 |
In both γ-lactam and thioamide derivatives (i.e., 1b–e and 2b–e), 19F shift differences Δδ increase when the C9-substituent changes from CO2Me to CONH2, CONHMe and CONMe2 groups. Obviously, C9-substituents modulate ortho-F shielding in addition to C7-functional groups. The combination of the most polarizable groups at both C7- and C9-sites, i.e., C7–CS and C9–CONMe2, results in compound 2e displaying the largest chemical shift difference Δδ at 2.56 ppm. This result may be applicable to the design of molecular sensors, in which the purposely formed polarizable groups will expand 19F shift changes.
The C7-thioamide and the phenyl group in C7-amidines bring different electronic and steric influences on the pyrrolidine ring as compared to C7–CO group. The subtle conformational differences among these compounds are manifested in the crystal structures of 2a, 3a and 5a, which we determined by X-ray crystallography. As revealed in Fig. S1,† the pyrrolidine ring in 2a and 3a displays similar puckered conformations with C9 extruding out of the plane, in contrast to near flat γ-lactam ring in 5a.15 The torsion angles for 2a, 3a and 5a are 81.56°, 65.31° and 62.63°, respectively, correlating to the increasing bulkiness of thioamide, amidine and amide groups. Meanwhile, the two phenyl groups in 3a adopts trans-configuration. Hence, the effects of various para-substituted phenylamidines on 19F chemical shifts are mostly instigated by electrostatic rather than steric effects. To evaluate the influences of C7-amidines on 19F shielding, we prepared compounds 3b–e and 4b–q. Table 2 compiles their 19F chemical shifts and shift differences Δδ between the syn-F and anti-F, along with Hammett constants σpara.
As expected, achiral compounds 3a–e furnish convergent 19F signals for ortho-F nuclei. Electron-rich amidines 3b–c show deshielded 19F nuclei as compared to electron-deficient 3d–e. Close examination on divergent 19F chemical shifts in chiral compounds 4a–q shows that this “reverse” relationship between 19F shielding and electron-withdrawing abilities maintains.16 In fact, linear correlations of ortho-19F chemical shifts with Hammett constants σpara is seen in Hammett plots, as illustrated in Fig. 6. The negative slope coefficient κ indicates that amidine groups affect 19F shielding predominately through noncovalent interaction, not chemical bonds for both syn-F and anti-F nuclei. Notably, the slope coefficient (κ = −0.29) for δdown–σpara curve is bigger (in absolute value) than that for δup–σpara plot (κ = −0.06). Larger syn-19F chemical shift (δdown) changes are likely originated from cooperative effects of nonbonding interactions of syn-F nucleus with C9-ester and C7-amidine.
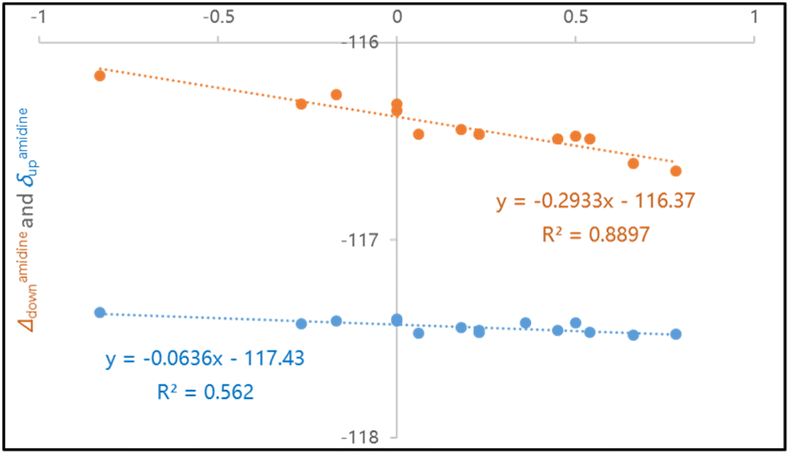 | ||
| Fig. 6 Hammett plots of 19F chemical shifts and shift differences versus σpara: (a) δamidinedown–σpara curve; (b) δamidineup–σpara curve (κ represents the slope coefficient). | ||
2.3. C5-substituent effects on 19F chemical shifts
In C7- and C9-substituted molecules, the electronic properties of F nuclei are little affected. Alternatively, substituent changes on the N-phenyl ring allow us to modulate fluorine electronic properties. Any different responses of syn-F and anti-F to C5-substituent variations reflect the electronic effects of fluorine on nonbonding interactions. To that end, we prepared a series of C5-substituted N-phenyl derivatives 5a–h and 6a–w (meta-substituent relative to ortho-F nuclei, see Fig. 1) and examined their 19F NMR spectra. Tables 3 and 4 provide detailed 19F chemical shifts for 5a–h and 6a–w.
|
|||||||
|---|---|---|---|---|---|---|---|
| Cpd | R | σmeta | δmetaref | Cpd | δdown | δup | Δδmeta |
| 1a | H | 0 | −117.82 | 1b | −117.14 | −117.93 | 0.79 |
| 5a | Me | −0.07 | −119.21 | 6a | −118.56 | −119.35 | 0.79 |
| 5b | OMe | 0.115 | −117.42 | 6b | −116.75 | −117.61 | 0.86 |
| 5c | CF3 | 0.43 | −113.36 | 6c | −112.73 | −113.43 | 0.70 |
| 5d | NO2 | 0.71 | −110.99 | 6d | −110.57 | −110.90 | 0.33 |
| 5e | CO2Me | 0.37 | −115.34 | 6e | −114.78 | −115.42 | 0.64 |
| 5f | NH2 | −0.16 | −119.53 | 6f | −118.89 | −119.70 | 0.81 |
| 5g | NHMe | −0.21 | −119.85 | 6g | −119.31 | −120.10 | 0.79 |
| 5h | NMe2 | −0.15 | −119.45 | 6h | −118.84 | −119.67 | 0.83 |
| 5i | F | 0.337 | −114.61 | 6i | −113.93 | −114.72 | 0.79 |

| Cpd | R | σmeta | δdown; δup | Δδmeta |
|---|---|---|---|---|
| 6j | Cl | 0.373 | −114.90, −115.58 | 0.68 |
| 6k | Br | 0.39 | −114.94, −115.73 | 0.788 |
| 6l | I | 0.35 | −115.44, −116.20 | 0.768 |
| 6m | Et | −0.07 | −118.28, −119.07 | 0.798 |
| 6n | n-Pr | −0.07 | −118.39, −119.16 | 0.77 |
| 6o | OEt | 0.10 | −117.02, −117.89 | 0.86 |
| 6p | CH2CH2 |
0.05 | −117.42, −118.16 | 0.74 |
| 6q | CH2CHMe |
0.05 | −117.98, −118.75 | 0.77 |
| 6r | Ph | 0.06 | −116.85, −117.62 | 0.77 |
| 6s | CHO | 0.35 | −113.13, −113.72 | 0.59 |
| 6t | COMe | 0.38 | −114.25, −114.86 | 0.61 |
| 6u | CN | 0.56 | −111.95, −112.30 | 0.35 |
| 6v | NHCHO | 0.19 | −117.46, −117.98 | 0.52 |
| 6w | NHCOMe | 0.21 | −118.54, −119.11 | 0.57 |
We expect that through-bond forces such as induction and resonance effects would be leading factors for C5-substituents to affect 19F chemical shifts. Indeed, plots of 19F chemical shifts δ from 5a–h, δdown and δup from 6a–u versus Hammett constants σmeta exhibit linear relationships, as demonstrated in 8 and S2.† The positive slope coefficients κ in the curves are in consistent with the conventional Hammett plots. Thus, 19F shielding variations in C5-substituted γ-lactams are predominately governed by either through-bond effects. It is worth to note that the chemical shifts of C5-amides 6v (R = NHCHO) and 6w (R = NHCOMe) are distinctly deviated from the linear curves, which we attribute to strong amide polarizable effects and their data are omitted from the plots in Fig. 7.
 | ||
| Fig. 7 Hammett plots of δdown–σmeta and δup–σmeta for compounds 6a–w. | ||
We envision that the magnitude of slope coefficient κ in a Hammett plot indicates the susceptibility of 19F shifts to substituent electronic characteristics. The slope coefficients for δdown–σmeta plots of C5-substituted γ-lactams (κ = 9.45 in Fig. 7) are much bigger than those for δdown–σpara plots of C7-amidines (κ = −0.29 in Fig. 6), proving that through-bond forces have greater impacts on 19F shielding than through-space interactions. In addition, the syn-F and anti-F nucleus behave differently in responding to their discrete surroundings, as the slope for δdown–σmeta plot (κ = 9.45) is smaller than that of δup–σmeta curve (κ = 9.86). The asymmetric environments around anti-F and syn-F nuclei are shaped by not only C9-ester but also neighboring C7-amidine. Anti-F shielding is more sensitive to the changes of fluorine electronic property than syn-F, suggesting anti-F is closer to C7-amidine that syn-F. Thus, for the first time, we demonstrate that the slope coefficients in Hammett plots are useful in detecting the type and strength of fluorine involved bonding and nonbonding interactions.
2.4. X-ray crystal structures of C5-substituted N-phenyl γ-lactams
In N-phenyl γ-lactam derivatives, the asymmetric chemical environments around ortho-F nuclei are mainly regulated by C9-ester and neighboring CO/CH2 groups. The conjugation effects of C5-substituents hinder rotations around central N–Ph bond, enlarging chemical differences between syn-F and anti-F. To inspect the structural details of C5-substituted N-phenyl γ-lactams, we determined single crystal structures of 6da (C9-acid derivative of 6d) and 6h by X-ray crystallography. Fig. 8 shows computer generated drawings with atomic numbering for S-6da and R-6h. Table 4 lists parameters that define important atom distances and functional group orientations. CCDC 1904410 and 1915197 contain the supplementary crystallographic data for 6da and 6h, respectively.†
 | ||
| Fig. 8 Computer-generated drawings of crystal structures 6da and 6h. 6da is a C9-acid analogue of 6d. | ||
As shown in Table 5, S-6da displays a small torsion angles ϕ at 57.59° due to C5–NO2 resonance effects, affording strong van der Waals forces between F2⋯CO (dF2–O1 = 2.83 Å) and F1⋯CH2 (dF1–H8 = 2.45 Å). F2 is anti-F relative to equatorial C9-acid in S-6da. On the contrary, C5NMe2 R-6h has larger torsion angle ϕ of 76.54° and longer distances from ortho-F nuclei to the nearby groups. None of the syn-F and anti-F nuclei approach to the CO/CH2 groups within the sum of van der Waals radius in R-6h. Despite marked differences in N-Ph torsion angles, C9-substituents in both S-6da and R-6h occupy similar pseudo-equatorial orientations, suggesting that there is no direct correlation between torsion angle ϕ and C9-substituent orientation. Significantly, strong nonbonding interactions as exemplified in structure S-6da correlates to small shift differences Δδ (Tables 3 and 4).
| Parameter | S-6da | R-6h | |
|---|---|---|---|
| a Sum of van der Waals radii: Rw(F–C) = 3.17 Å; Rw(F–O) = 2.99 Å; Rw(F–H) = 2.67 Å. | |||
| Dihedral angle β (carbonyl orientation) | β1[C7–C10–C9–C11] | −150.45 | −145.27 |
| β2[N1–C8–C9–C11] | 150.85 | 148.21 | |
| Torsion angle ϕ (between N–Ph and γ-lactam) | ϕ1[C1–C2–N1–C7] | −124.13 | 99.77 |
| ϕ2[C3–C2–N1–C7] | 57.59 | −76.54 | |
| Contact angle α (F to CO groups) |
α1[C3–F2–C(amide)] | 82.34 | 74.07 |
| α2[F2–CO] |
71.08 | 91.45 | |
| α1[C1–F1–C(amide)] | 66.91 | 70.44 | |
| α2[F1–CO] |
112.91 | 94.14 | |
| Distances d (F to CO and CH2 groups) |
dF1–C8(CH) | 2.903 | 3.609 |
| dF1–H8(CH) | 2.446 | 3.324 | |
| dF1–O1(amide) | 4.470 | 3.877 | |
| dF1–C7(amide) | 3.851 | 3.592 | |
| dF2–C8(CH) | 3.946 | 3.386 | |
| dF2–H7(CH) | 3.994 | 2.996 | |
| dF2−O1(amide) | 2.826 | 3.478 | |
| dF2–C7(amide) | 2.975 | 3.226 | |
| C–F bond lengths | dC1–F1 | 1.343 | 1.359 |
| dC3–F2 | 1.340 | 1.356 | |
| Distances of F to C9-carbonyl | dF2–O2 (F to axial carbonyl) | 6.663 | 5.536 |
| dF2–C11 (F to axial carbonyl) | 5.991 | 5.165 | |
| dF1–O3 (F to axial carbonyl) | 4.783 | 6.178 | |
| dF1–C11 (F to axial carbonyl) | 5.028 | 5.895 | |
2.5. Quantum mechanics calculation on nonbonding interactions between ortho-F nuclei and neighboring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O/CN/CH2 groups
O/CN/CH2 groups
Having a wealth of valuable geometric and 19F NMR spectroscopic information in hand, we set to explore the nature of the interactions between ortho-F nuclei and neighboring CO/CN/CH2 groups. We first carried out geometry optimizations using the Gaussian16 program with density functional theory (DFT) at the B3LYP/6-311G* level of theory.23 We analyzed LUMO, HOMO and HOMO−1 orbitals in amide 1b, thioamide 2b and amidine 4a, and their molecular orbitals are depicted inFig. 9 and S3.†
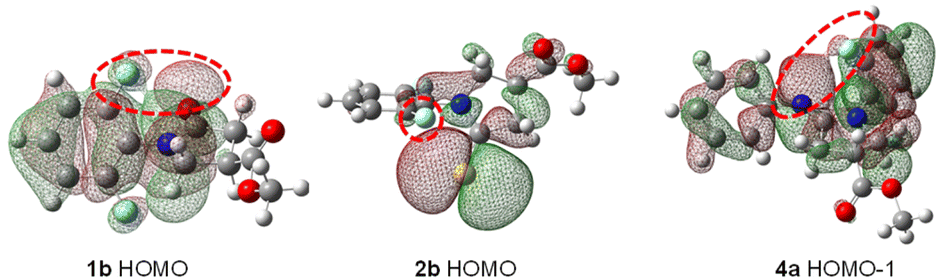 | ||
| Fig. 9 Molecular orbitals at HOMO energy levels in amide 1b, thioamide 2b, and HOMO−1 in amidine 4a (on 0.012 e au−3 electron density surface). | ||
Fluorine nonbonding interactions are multifaceted. In our model molecules 1–6, interactions such as van der Waals contacts, coulombic (electrostatic and polarization), and orbital delocalization play important roles in governing overall conformation, and affecting 19F NMR chemical shifts.24–26 It is known that lone pair electrons (n) on F can delocalize to the antibonding orbital (π*) of the adjacent CO group from the C end.25a However, ortho-F nucleus can only approach to CO group sideways in γ-lactam derivatives due to conformational confinement. In fact, HOMO and LUMO orbitals in 1b suggest that there is repulsive interaction between p-type lone pair electrons on F and O atoms (coulombic n ↔ n repulsive interactions). On the other hand, the same ortho-F nucleus also engages n → π* orbital overlaps through donation of n electrons on F into antibonding π* orbitals in NPh* tautomeric form (Ph* denotes the ortho-F substituted phenyl group). This attractive contact offsets F⋯O repulsion and favors small torsion angle.
The interaction of lone pair electrons between ortho-F and S, however, is not apparent in thioamide 2b, as the van der Waals repulsive force between the ortho-F and large S atom dominates, furnishing a large torsion angle (vide supra).
Notably, a π* orbital located between N and the attaching phenyl group (i.e., NAr, a tautomer of phenylamidine, see Fig. 9) exists. Unlike repelling C–F⋯OC interaction in γ-lactam 1b, the obvious n → π* orbital delocalization from lone pair electrons (n) on F to the antibonding orbital π* in NAr is deemed to stabilize C–F⋯NAr contact, although the attraction energy may be small.2,25,28 Accordingly, para-NO2 amidine 4e furnishes better n → π* orbital overlap than that of p-NMe2 amidine 4f, as 4e majorly adopts ArN form 4e(b), (see Fig. 10 and S3†).
 | ||
| Fig. 10 Tautomers of amidine 4e. | ||
C–F⋯H–C contacts in amidines 4 are little affected by the p-substituents, but are more likely influenced by C5-substituents in γ-lactams 6. Indeed, molecular orbital analysis of 4a indicates that lone pair electrons (n) on F clash sideway into the proximate C–H σ bonding orbital (n ↔ σ interaction, Fig. 11 and S4†).27 Since both orbitals are occupied, such a F⋯H interaction is repulsive, and the short F⋯H distance is a consequence of steric crowding (buttressing), rather than any meaningful hydrogen bonding interaction.14 Strong F⋯H van der Waals repulsion is anticipated in C5–NO2-γ-lactam 6d, where C5-conjugation effect shortens the F⋯H distance (Fig. 11and S4†). However, orbital polarization between n and σ orbitals in 6d seems insignificant, partly because fluorine is electron-deficient and less polarizable.
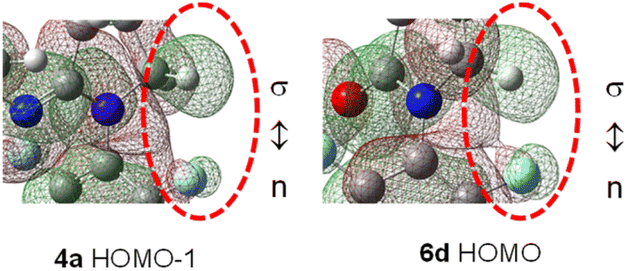 | ||
| Fig. 11 HOMO−1 orbital in amidine 4a and HOMO orbital in C5–NO2 γ-lactam 6d (on 0.012 e au−3 electron density surface, C–F⋯H–C portion). | ||
2.6. Fluorine nonbonding interactions and 19F NMR chemical shift changes
Through-space interactions between ortho-F nuclei and their surrounding groups may be measured by shift differences Δδdown (for syn-F) and Δδup (for anti-F) versus a corresponding reference chemical shift (δref). Here we use C9-unsubstituted γ-lactams 5a–i as references for 6a-I, and achiral amidines 3a–e as references for 4a–e. Values of Δδdown (=δdown − δref) and Δδup (= δup − δref) reflects substituent effects on 19F NMR chemical shifts for F⋯OC/NC and F⋯H–C interactions, respectively. Table 6 lists Δδdown and Δδup data for selected amidines and γ-lactams.
| Cpd | R | Δδdown | Δδup | dF–Ha (Å) |
|---|---|---|---|---|
| a F⋯H distances are obtained from optimized structures from QM calculations.b Data are from X-ray crystal structures. | ||||
| 4a | H | 0.74 | −0.36 | 2.550 |
| 4b | Me | 0.80 | −0.35 | 2.550 |
| 4c | OMe | 0.76 | −0.35 | 2.550 |
| 4d | CF3 | 0.69 | −0.29 | 2.538 |
| 4e | NO2 | 0.65 | −0.18 | 2.548 |
| 1b | H | 0.68 | −0.11 | 2.518 |
| 6a | Me | 0.65 | −0.14 | 2.585 |
| 6b | OMe | 0.67 | −0.19 | 2.639 |
| 6c | CF3 | 0.63 | −0.07 | 2.508 |
| 6d | NO2 | 0.42 | 0.09 | 2.446b |
| 6h | NMe2 | 0.61 | −0.22 | 3.324b |
Previously we established that 19F shielding variations correlate with dipole moments/polarizability in C–F⋯OC interactions.15,16 Our molecular orbital analysis confirms that 19F NMR signals are mostly affected by functional group polarity, where 19F chemical shifts are more deshielded when either F nucleus or CO/ArN groups become more polarizable. As detailed in Table 6, ortho-F nuclei are more polarizable in C5–NMe2 γ-lactam 6h than in C5–NO2 6d, leading to larger Δδdown for 6h (0.61 ppm) than for 6d (0.42 ppm).
In C–F⋯H–C interactions, short F⋯H distances are associated with deshielded 19F signals (Table 6). Thus, upfield 19F signal δup in 6d displays the most downfield shift value (Δδup = 0.09 ppm), which is associated with the shortest dF–H = 2.45 Å in γ-lactams. Similar correlation between Δδup and dF–H is observed in amidine series, and documented in literature.14
3. Conclusions
In this work, we investigated the relationship between 19F NMR shielding changes and chemical environments in rotameric N-phenyl γ-lactam analogues. 19F shift changes in C7-amide/thioamide/amidine derivatives 1–3 confirmed that the neighboring CO/CS/CN and CH2 groups, together with stereogenic C9-substituent on γ-lactam ring, synergistically enhance chemical environment differences between syn-F and anti-F. Substituent surveys among C7-phenylamidines 4a–r and C5-substituted γ-lactams 6a–u revealed that through-bond effects track conventional relationships between 19F chemical shift (δ) and Hammett coefficient (σ), while through-space interactions display “reverse” correlations in δ–σ Hammett plots. In fact, the signs (positive or negative) of the slope coefficients κ in the Hammett plots specify the types of fluorine-participated molecular recognitions.
The absolute values of slope coefficients κ in δ–σ Hammett plots verified that nonbonding contacts (κ = −0.29 for F⋯NAr contacts) are much weaker than bonding interactions in amending 19F shielding (Fig. 6 and 7). Such results corroborated with our earlier observations from C9-amides series,16 where the nonbonding effects of C9-amides on 19F shielding afforded κ = −0.35 in the δdown–σpara Hammett plot. To the best of our knowledge, this is the first report to show that Hammett plots can be used to categorize fluorine nonbonding interactions, and to explicitly gauge the magnitude of these interactions using the slope coefficients κ from Hammett plots.
QM calculations established that repulsive interactions between F⋯OC and F⋯H–C are the main schemes in N-phenyl γ-lactams, while a favorable, n → π* orbital delocalization from lone pair electrons (n) on F into ArN antibonding orbital π* exists in amidine analogous. All the orbital overlaps are supposed to be weak interactions. It is evident that more polarizable functional groups link to more deshielded 19F signals. On the other hand, C–F⋯H–C interaction comprises orbital polarizations between lone pair electrons on F and C–H σ bonding orbital (n ↔ σ). As both n and σ orbitals are occupied, this contact is considered steric repulsive, and is associated with 19F signal deshielding.
4. Experimental section
4.1. General information
Unless otherwise stated, all the reactions were carried out under atmosphere conditions. All chemical reagents were purchased from commercial sources and used without further purification. Flash column chromatography was performed with Agilent Technologies Claricep FlashSilica. All 1H and 13C NMR experiments were carried out using a Bruker AVANCE 400 spectrometer or a Bruker AVANCE III 600 spectrometer. 19F NMR data was recorded using a Bruker AVANCE 400 spectrometer. Chemical shifts in 1H, 19F and 13C NMR spectra were reported in parts per million (ppm). The residual solvent signals were used as references and the chemical shifts converted to the TMS scale (CDCl3: δ (H) = 7.26 ppm, δ (C) = 77.16 ppm) for 1H and 13C NMR spectra. For 19F NMR experiments, CFCl3 was added as an internal reference (0.5% CFCl3). All coupling constants (J values) were reported in Hertz (Hz). Multiplicities were reported as follows: singlet (s), doublet (d), doublet of doublets (dd), doublet of doublet of doublets (ddd), doublet of triplets (dt), triplet (t), triplet of doublets (td), quartet (q), and multiplet (m). High resolution mass spectra (HRMS) were obtained from an Agilent 6200 series TOF/6500 series spectrometer, using electrospray ionization (ESI) as an ion source. The MS spectra were assessed with an Agilent 1260-6120 spectrometer with an electrospray ionization (ESI) source, equipped with an Agilent SB-C18 (2.1 × 30 mm, 3.5 μm) column.4.2. Compound characterization
Compound 2b: 133 mg, 58%, pale yellow oil. MS (ESI, pos. ion) m/z: 272.4 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.43–7.33 (m, 1H), 7.03 (t, J = 8.4 Hz, 2H), 4.23 (ABX, J = 10.7, 6.4, 8.4 Hz, 2H), 3.79 (s, 3H), 3.60–3.50 (m, 1H), 3.48–3.46 (m, 2H);19F NMR (376 MHz, CDCl3): δ (ppm) −116.53 (s), −117.69 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 203.1, 172.1, 158.3 (dt, 3JCF = 9.3, 5.0 Hz), 130.6 (t, 3JCF = 9.8 Hz), 116.8 (t, 3JCF = 16.1 Hz), 112.8–112.1 (m), 58.1, 52.8, 46.9, 39.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H12F2NO2S 272.0557; found: 272.0550.
To a solution of compound 2f (200 mg, 0.78 mmol), aniline (1.00 equiv.) and HATU (443 mg, 1.17 mmol) in dichloromethane (30 mL) was added Et3N (118 mg, 1.17 mmol). The mixture was stirred at room temperature for 8 h. The reaction was quenched with water (30 mL), then extracted with dichloromethane (30 mL × 3). The combined organic phases were washed with brine (30 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/1) to give compounds 2c, 2d and 2e.
Compound 2c (100 mg, 50%) as pale yellow oil. MS (ESI, pos. ion) m/z: 257.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.41–7.32 (m, 1H), 6.99 (t, J = 8.5 Hz, 2H), 5.79 (d, J = 57.9 Hz, 2H), 4.01 (dd, J = 9.4, 7.5 Hz, 1H), 3.88 (t, J = 9.0 Hz, 1H), 3.45–3.31 (m, 1H), 2.85 (ABX, J = 10.7, 6.4, 8.4 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.81 (s), −118.15 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 173.6, 172.4, 159.1 (dd, 1JCF = 253.0, 3JCF = 4.9 Hz), 129.5 (t, 3JCF = 9.9 Hz), 115.0 (t, 3JCF = 16.4 Hz), 112.3 (d, 2JCF = 21.8 Hz), 51.4, 38.6, 34.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H11F2N2OS 257.0560; found: 257.0547.
Compound 2d (120 mg, 57%) as pale yellow oil. MS (ESI, pos. ion) m/z: 271.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37 (dq, J = 8.5, 6.2 Hz, 1H), 7.01 (tt, J = 12.8, 6.4 Hz, 2H), 6.15 (brs, 1H), 4.35–4.26 (m, 1H), 4.11–4.02 (m, 1H), 3.46–3.33 (m, 3H), 2.83 (d, J = 4.8 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.10 (s), −118.07 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 203.5, 171.5, 158.2 (ddd, 2JCF = 22.6, 3JCF = 18.5, 4.9 Hz), 130.6 (t, 3JCF = 9.7 Hz), 116.8 (t, 3JCF = 16.1 Hz), 112.5 (ddd, 2JCF = 33.3, 3JCF = 19.5, 3.3 Hz), 59.0, 48.0, 41.2, 26.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H13F2N2OS 271.0717; found: 271.0704.
Compound 2e (140 mg, 63%) as pale yellow oil. MS (ESI, pos. ion) m/z: 285.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.34 (tt, J = 8.5, 6.2 Hz, 1H), 7.00 (ddd, J = 9.3, 3.6, 1.3 Hz, 2H), 4.45 (dd, J = 10.3, 7.7 Hz, 1H), 4.01 (dd, J = 10.2, 8.8 Hz, 1H), 3.82–3.70 (m, 1H), 3.47–3.29 (m, 2H), 3.05 (s, 3H), 2.97 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −115.59 (s), −118.16 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 202.9, 170.3, 158.2 (ddd, 3JCF = 17.3, 13.7, 4.8 Hz), 130.4 (t, 3JCF = 9.8 Hz), 116.9 (t, 3JCF = 16.2 Hz), 112.4 (ddd, 2JCF = 30.5, 3JCF = 19.6, 3.5 Hz), 58.64, 47.6, 38.6, 37.5, 37.2, 36.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H15F2N2OS 285.0873; found: 285.0864.
Compound 3b: 630 mg, 41%, yellow oil. MS (ESI, pos. ion) m/z: 287.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.25–7.15 (m, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.96 (t, J = 8.2 Hz, 2H), 6.72 (d, J = 8.1 Hz, 2H), 3.69 (t, J = 6.8 Hz, 2H), 2.51 (t, J = 7.8 Hz, 2H), 2.27 (s, 3H), 2.20–2.10 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.06 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 161.6, 159.7 (dd, 1JCF = 252.1, 3JCF = 5.8 Hz), 149.5, 131.5, 129.3, 128.0 (t, 3JCF = 10.0 Hz), 121.9, 118.1, 112.3 (dd, 3JCF = 18.4, 5.2 Hz), 50.8, 26.8, 21.4, 20.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H17F2N2 287.1360; found: 287.1355.
Compound 3c: 270 mg, 45%, colorless oil. MS (ESI, pos. ion) m/z: 303.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.25–7.15 (m, 1H), 6.96 (t, J = 8.4 Hz, 2H), 6.76 (q, J = 8.9 Hz, 4H), 3.76 (s, 3H), 3.68 (t, J = 6.8 Hz, 2H), 2.50 (t, J = 7.7 Hz, 2H), 2.19–2.09 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.07 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 161.9, 159.7 (dd, 1JCF = 252.0, 3JCF = 5.6 Hz), 155.2, 145.5, 128.0 (t, 3JCF = 10.0 Hz), 122.8, 114.1, 112.3 (dd, 3JCF = 19.8, 4.0 Hz), 55.6, 50.7, 26.9, 21.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H17F2N2O 303.1309; found: 303.1337.
Compound 3d: 201 mg, 13%, yellow oil. MS (ESI, pos. ion) m/z: 341.2([M + H]+); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.46 (d, J = 8.2 Hz, 2H), 7.28–7.19 (m, 1H), 6.98 (t, J = 8.2 Hz, 2H), 6.88 (d, J = 8.1 Hz, 2H), 3.73 (t, J = 6.8 Hz, 2H), 2.51 (t, J = 7.8 Hz, 2H), 2.23–2.13 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −62.09 (s), −117.18 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 161.6, 159.6 (dd, 3JCF = 252.1, 3JCF = 5.6 Hz), 155.6, 128.4 (t, 3JCF = 10.0 Hz), 125.9 (d, 3JCF = 3.6 Hz), 124.9–123.2 (m), 122.2, 117.6 (t, 3JCF = 16.1 Hz), 112.3 (dd, 3JCF = 18.4, 4.8 Hz), 50.8, 26.9, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H14F5N2 341.1077; found: 341.1086.
Compound 3e: 80 mg, 3%, yellow oil. MS (ESI, pos. ion) m/z: 318.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 8.09 (d, J = 8.4 Hz, 2H), 7.32–7.17 (m, 1H), 6.98 (t, J = 8.0 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 3.75 (t, J = 6.9 Hz, 2H), 2.56 (t, J = 7.7 Hz, 2H), 2.26–2.19 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.30 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 160.1 (d, 1JCF = 394.9 Hz), 159.4 (dd, 1JCF = 252.8, 3JCF = 4.9 Hz), 142.9, 128.8 (t, 3JCF = 10.0 Hz), 124.9, 122.5, 112.3 (dd, 3JCF = 19.7, 3.8 Hz), 51.0, 27.3, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H14F2N3O2 318.1054; found: 318.1045.
Compound 4a: 133 mg, 30%, colorless oil. MS (ESI, pos. ion) m/z: 331.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.29–7.17 (m, 3H), 6.97 (t, J = 8.3 Hz, 3H), 6.83–6.81 (m, 2H), 3.98–3.86 (m, 2H), 3.74 (s, 3H), 3.43–3.35 (m, 1H), 2.81 (ABX, J = 16.7, 8.2, 9.0 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.31 (s), −117.41 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.8, 159.6 (dd, 3JCF = 251.7, 1JCF = 5.4 Hz), 158.8, 151.5, 128.9, 128.5 (t, 3JCF = 10.0 Hz), 122.6, 121.9, 117.1 (t, 3JCF = 16.0 Hz), 112.5 (td, 3JCF = 12.2, 2.1 Hz), 52.5, 52.3, 39.3, 29.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H17F2N2O2 331.1258; found: 331.1269.
Compound 4b: 153 mg, 43%, yellow oil. MS (ESI, pos. ion) m/z: 345.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.21 (ddd, J = 14.6, 8.3, 6.3 Hz, 1H), 7.03 (d, J = 7.9 Hz, 2H), 6.95 (t, J = 8.8 Hz, 2H), 6.72 (d, J = 8.0 Hz, 2H), 3.95–3.86 (m, 2H), 3.73 (s, 3H), 3.37 (p, J = 8.1 Hz, 1H), 2.81 (ABX, J = 16.7, 8.2, 9.0 Hz, 2H), 2.27 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.26, −117.41 (s).13C NMR (101 MHz, CDCl3): δ (ppm) 172.9, 159.7 (dd, 1JCF = 251.4, 3JCF = 5.4 Hz), 159.1, 148.8, 131.9, 129.5, 128.5 (t, 3JCF = 10.0 Hz), 121.8, 117.2 (t, 3JCF = 16.0 Hz), 112.3 (dd, 3JCF = 18.5, 11.3 Hz), 52.5, 52.3, 39.3, 29.9, 20.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H19F2N2O2 345.1415; found: 345.1424.
Compound 4c: 167 mg, 40%, pale yellow oil. MS (ESI, pos. ion) m/z: 361.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.26–7.19 (m, 1H), 6.97 (t, J = 8.6 Hz, 2H), 6.80–6.73 (m, 4H), 3.96–3.84 (m, 2H), 3.76 (s, 3H), 3.74 (s, 3H), 3.42–3.36 (m, 1H), 2.80 (ABX, J = 16.7, 8.3, 9.0 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.31 (s), −117.42 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.9, 159.6 (d, 1JCF = 251.7 Hz), 159.3, 155.4, 144.8, 128.4 (t, 3JCF = 10.0 Hz), 122.7, 117.2 (t, 3JCF = 15.9 Hz), 114.2, 112.3 (td, 3JCF = 19.9, 3.2 Hz), 55.57, 52.53, 52.26, 39.35, 29.92; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H19F2N2O3 361.1364; found: 361.1361.
Compound 4d: 166 mg, 21%, colorless oil. MS (ESI, pos. ion) m/z: 399.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.48 (d, J = 8.2 Hz, 2H), 7.26 (dt, J = 8.3, 6.3 Hz, 1H), 6.99 (t, J = 8.6 Hz, 2H), 6.90 (d, J = 8.1 Hz, 2H), 3.94 (d, J = 7.7 Hz, 2H), 3.46–3.35 (m, 1H), 2.80 (ABX, J = 16.8, 7.9, 9.0 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −62.20 (s), −116.49 (s), −117.47 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.7, 159.6 (dd, 1JCF = 252.6, 3JCF = 5.3 Hz), 159.1, 154.9, 128.9 (t, 3JCF = 9.9 Hz), 126.2 (d, 3JCF = 3.6 Hz), 122.1, 116.7, 112.4 (dd, 2JCF = 21.8, 3JCF 10.2 Hz), 52.7, 52.5, 39.3, 30.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H16F5N2O2 399.1132; found: 399.1125.
Compound 4e: 15 mg, 4%, pale yellow oil. MS (ESI, pos. ion) m/z: 376.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 8.13 (d, J = 8.2 Hz, 2H), 7.30–7.23 (m, 1H), 6.98 (t, J = 8.0 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 3.97 (d, J = 7.5 Hz, 2H), 3.76 (s, 3H), 3.48–3.40 (m, 1H), 2.56 (ABX, J = 16.7, 7.6, 8.9 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.65 (s), −117.47 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.5, 159.3 (dd, 1JCF = 252.9, 3JCF = 3.6 Hz), 158.9, 158.1, 143.1, 129.1 (t, 3JCF = 9.9 Hz), 124.9, 122.3, 116.3, 112.5–112.1 (m), 52.7, 52.6, 39.1, 30.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16F2N3O4 376.1109; found: 376.1108.
Compound 4g: 168 mg, 65%, pale brown oil. MS (ESI, pos. ion) m/z: 349.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.28–7.19 (m, 1H, Ph–F), 6.97 (t, J = 8.5 Hz, 2H, Ph–F), 6.92 (d, J = 8.7 Hz, 2H, Ph–NC), 6.79–6.73 (m, 2H, Ph–NC), 3.92 (d, J = 7.8 Hz, 2H, CH2N), 3.74 (s, 3H, Me), 3.44–3.32 (m, 1H, CH), 2.79 (ABX, J = 16.7, 8.1, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.46 (s), −117.47 (s), −122.76 (s); 13C NMR (150 MHz, CDCl3): δ (ppm) 172.8, 160.4 (d, 3JCF = 3.6 Hz), 159.9, 159.5, 158.8, 158.3, 147.6 (d, 3JCF = 2.6 Hz), 128.6 (t, 3JCF = 10.0 Hz), 123.0 (d, 3JCF = 7.9 Hz), 117.0 (t, 3JCF = 16.0 Hz), 115.5, 115.4, 112.5–112.2 (m), 52.6, 52.4, 39.3, 29.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16F3N2O2 349.1164; found: 349.1155.
Compound 4h: 554 mg, 82%, pale brown oil. MS (ESI, pos. ion) m/z: 365.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.28–7.19 (m, 1H, Ph–F), 7.18 (d, J = 8.6 Hz, 2H, Ph–Cl), 6.97 (t, J = 8.5 Hz, 2H, Ph–F), 6.75 (d, J = 8.6 Hz, 2H, Ph–Cl), 3.92 (d, J = 7.7 Hz, 2H, CH2N), 3.74 (s, 3H, Me), 3.45–3.33 (m, 1H, CH), 2.79 (ABX, J = 16.7, 8.0, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.46 (s), −117.46 (s); 13C NMR (150 MHz, CDCl3): δ (ppm) 172.7, 160.4 (d, 3JCF = 5.5 Hz), 159.3, 158.7, 150.2, 128.9, 128.7 (t, 3JCF = 10.0 Hz), 127.8, 123.3, 116.9 (t, 3JCF = 16.0 Hz), 112.6–112.2 (m), 52.6, 52.4, 39.3, 29.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16ClF2N2O2 365.0868; found: 365.0851.
Compound 4i: 700 mg, 91%, pale brown oil. MS (ESI, pos. ion) m/z: 409.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.33 (d, J = 8.5 Hz, 2H, Ph–Br), 7.29–7.19 (m, 1H, Ph–F), 6.97 (t, J = 8.5 Hz, 2H, Ph–F), 6.70 (d, J = 8.5 Hz, 2H, Ph–Br), 3.92 (d, J = 7.7 Hz, 2H, CH2N), 3.74 (s, 3H, Me), 3.45–3.33 (m, 1H, CH), 2.79 (ABX, J = 16.7, 8.0, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.46 (s), −117.46 (s); 13C NMR (150 MHz, CDCl3): δ (ppm) 172.7, 160.4 (d, 3JCF = 5.2 Hz), 159.2, 158.7, 150.7, 131.8, 128.7 (t, 3JCF = 10.0 Hz), 123.8, 116.8 (t, 3JCF = 16.0 Hz), 115.5, 112.6–112.2 (m), 52.6, 52.4, 39.3, 29.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16BrF2N2O2 409.0363; found: 409.0345.
Compound 4j: 433 mg, 52%, pale brown oil. MS (ESI, pos. ion) m/z: 457.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.51 (d, J = 8.5 Hz, 2H, Ph–I), 7.31–7.18 (m, 1H, Ph–F), 6.97 (t, J = 8.6 Hz, 2H, Ph–F), 6.59 (d, J = 8.5 Hz, 2H, Ph–I), 3.92 (d, J = 7.7 Hz, 2H, CH2N), 3.74 (s, 3H, Me), 3.45–3.32 (m, 1H, CH), 2.79 (ABX, J = 16.7, 8.0, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.44 (s), −117.44 (s); 13C NMR (150 MHz, CDCl3): δ (ppm) 172.7, 160.4 (d, 3JCF = 5.4 Hz), 159.1, 158.7 (d, 3JCF = 4.5 Hz), 151.4, 137.8, 128.7 (t, 3JCF = 10.0 Hz), 124.3, 117.4, 116.8 (t, 3JCF = 16.0 Hz), 112.5–112.2 (m), 85.9, 52.6, 52.4, 39.2, 29.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16F2IN2O2 457.0225; found: 457.0212.
Compound 4k: 96 mg, 28%, yellow oil. MS (ESI, pos. ion) m/z: 356.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.52 (d, J = 7.7 Hz, 2H, Ph–CN), 7.32–7.21 (m, 1H, Ph–F), 6.99 (t, J = 8.4 Hz, 2H, Ph–F), 6.88 (d, J = 8.0 Hz, 2H, Ph–CN), 3.95 (d, J = 7.6 Hz, 2H, CH2N), 3.76 (s, 3H, Me), 3.48–3.36 (m, 1H, CH), 2.81 (ABX, J = 16.8, 7.6, 8.8 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.61 (s), −117.48 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 172.5, 160.7, 159.0, 158.2, 156.0, 133.2, 129.0 (t, 3JCF = 10.0 Hz), 122.8, 119.7, 112.4 (dd, 3JCF = 17.0, 10.9 Hz), 105.7, 52.7, 52.5, 39.2, 30.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H16F2N3O2 356.1211; found: 356.1204.
Compound 4l: 123 mg, 62%, yellow oil. MS (ESI, pos. ion) m/z: 389.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.93 (d, J = 8.3 Hz, 2H, Ph–CO2Me), 7.29–7.21 (m, 1H, Ph–F), 6.98 (t, J = 8.7 Hz, 2H, Ph–F), 6.87 (d, J = 8.3 Hz, 2H, Ph–CO2Me), 3.95 (d, J = 7.7 Hz, 2H, CH2N), 3.88 (s, 3H, Ph–CO2Me), 3.75 (s, 3H, Me), 3.47–3.36 (m, 1H, CH), 2.82 (ABX, J = 16.8, 7.9, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.49 (s), −117.46 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 172.7, 167.3, 160.9, 158.8, 158.3, 156.2, 130.8, 128.9 (t, 3JCF = 10.0 Hz), 124.5, 121.9, 116.7, 112.6–112.1 (m), 52.6, 52.5, 51.9, 39.3, 30.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H19F2N2O4 389.1313; found: 389.1307.
Compound 4m: 150 mg, 70%, yellow oil. MS (ESI, pos. ion) m/z: 388.5 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.33 (d, J = 8.6 Hz, 2H, Ph–NH), 7.25–7.18 (m, 1H, Ph–F), 7.17 (br. s, 1H, NH), 6.96 (t, J = 8.7 Hz, 2H, Ph–F), 6.77 (d, J = 8.5 Hz, 2H, Ph–NH), 3.96–3.85 (m, 2H, CH2N), 3.74 (s, 3H, Me), 2.80 (ABX, J = 16.8, 8.0, 9.0 Hz, 2H, CNCH2), 2.13 (s, 3H, COMe); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.34 (s), −117.40 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 172.8, 168.4, 160.8 (d, 3JCF = 5.3 Hz), 159.6, 158.3, 147.6, 133.3, 128.7 (t, 3JCF = 9.9 Hz), 122.2, 120.9, 117.0 (t, 3JCF = 16.0 Hz), 112.3 (dd, 3JCF = 18.5, 9.8 Hz), 52.6, 52.4, 39.3, 30.1, 24.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H20F2N3O3 388.1473; found: 388.1469.
Compound 4n: 150 mg, 22%, pale yellow oil. MS (ESI, pos. ion) m/z: 373.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.88 (d, J = 8.1 Hz, 2H, Ph–COMe), 7.29–7.22 (m, 1H, Ph–F), 6.97 (t, J = 8.6 Hz, 2H, Ph–F), 6.89 (d, J = 8.1 Hz, 2H, Ph–COMe), 3.96 (d, J = 7.7 Hz, 2H, CH2N), 3.75 (s, 3H, Me), 3.46–3.38 (m, 1H, CH), 2.77 (ABX, J = 16.8, 7.9, 9.0 Hz, 2H, CNCH2), 2.55 (s, 3H, COMe); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.47 (s), −117.42 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 197.3, 172.5, 160.7, 158.7, 158.1, 156.3, 131.9, 129.6, 128.8 (t, 3JCF = 9.9 Hz), 121.9, 116.6 (t, 3JCF = 15.8 Hz), 112.2 (dd, 3JCF = 19.7, 7.8 Hz), 52.5, 52.4, 39.1, 29.9, 26.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H19F2N2O3 373.1364; found: 373.1356.
Compound 4o: 150 mg, 57%, yellow oil. MS (ESI, pos. ion) m/z: 356.9 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.29 (d, J = 8.1 Hz, 2H, Ph–CHCH2), 7.25–7.19 (m, 1H, Ph–F), 6.98 (t, J = 8.7 Hz, 2H, Ph–F), 6.78 (d, J = 8.2 Hz, 2H, Ph–CHCH2), 6.66 (dd, J = 17.6, 10.9 Hz, 1H, Ph–CHCH2), 5.63 (d, J = 17.6 Hz, 1H, Ph–CHCH2), 5.11 (d, J = 10.9 Hz, 1H, Ph–CHCH2), 3.97–3.87 (m, 2H, CH2N), 3.75 (s, 3H, Me), 3.46–3.38 (m, 1H, CH), 2.82 (ABX, J = 16.7, 8.1, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.33 (s), −117.41 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 172.8, 160.9 (d, 3JCF = 5.5 Hz), 158.8, 158.4 (d, 3JCF = 5.4 Hz), 151.4, 136.9, 132.2), 128.5 (t, 3JCF = 10.0 Hz), 126.9, 122.1, 117.1 (t, 3JCF = 16.0 Hz), 112.4 (dd, 3JCF = 16.9, 12.8 Hz), 111.7, 52.5, 52.3, 39.4, 30.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H19F2N2O2 357.1415; found: 357.1438.
Compound 4p: 126 mg, 43%, yellow oil. MS (ESI, pos. ion) m/z: 375.3 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.21 (td, J = 8.4, 4.2 Hz,1H, Ph–F), 6.96 (t, J = 8.6 Hz, 2H, Ph–F), 6.75 (q, J = 8.9 Hz, 4H, Ph–OCH2CH3), 3.97 (q, J = 7.0 Hz, 2H, OCH2), 3.93–3.86 (m, 2H, CH2N), 3.73 (s, 3H, OMe), 3.43–3.31 (m, 1H, CH), 2.80 (ABX, J = 16.7, 8.2, 9.0 Hz, 2H, CNCH2), 1.38 (t, J = 7.0 Hz, 3H, OCH2CH3); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.33 (s), −117.42 (s); 13C NMR (150 MHz, CDCl3): δ (ppm) 172.9, 160.9 (d, 3JCF = 5.5 Hz), 159.1, 158.4, 154.8, 144.8, 128.4 (t, 3JCF = 10.0 Hz), 122.6, 117.3 (t, 3JCF = 16.0 Hz), 114.9, 112.5–112.1 (m), 63.8, 52.5, 52.2, 39.4, 29.9, 15.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H21F2N2O3 375.1520; found:375.1540.
Compound 4q: 180 mg, 81%, pale yellow oil. MS (ESI, pos. ion) m/z: 402.5 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.32 (d, J = 8.2 Hz, 2H, Ph–CO), 7.29–7.18 (m, 1H, Ph–F), 6.98 (t, J = 8.6 Hz, 2H, Ph–F), 6.84 (d, J = 8.1 Hz, 2H, Ph–CO), 3.99–3.87 (m, 2H, CH2N), 3.75 (s, 3H, Me), 3.47–3.34 (m, 1H, CH), 3.16–2.92 (m, 6H, NMe2), 2.80 (ABX, J = 16.8, 7.9, 9.0 Hz, 2H, CNCH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.40 (s), −117.42 (s); 13C NMR (100 MHz, CDCl3): δ (ppm) 172.6, 172.0, 171.1 (d, 3JCF = 7.4 Hz), 160.7 (d, 3JCF = 5.4 Hz), 158.9, 158.2 (d, 3JCF = 5.4 Hz), 152.9, 130.3, 128.6 (t, 3JCF = 10.0 Hz), 128.2, 121.6, 116.8 (t, 3JCF = 16.0 Hz), 112.2 (ddd, 2JCF = 20.2, 3JCF = 11.2, 3.1 Hz), 60.4, 52.5, 52.3, 39.2, 29.9, 21.0, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H22F2N3O3 402.1629; found: 402.1620.
To a solution of compound 5bc (300 mg, 1.14 mmol) in DMF (2 mL) was added DBU (260 mg, 1.71 mmol) at room temperature. The reaction mixture was heated at 100 °C and stirred overnight. The reaction was quenched with water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic phases were washed with brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/5) to give compound 5b (220 mg, 85%) as pale yellow oil; MS (ESI, pos. ion) m/z: 228.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.51 (d, J = 9.5 Hz, 2H), 3.77 (s, 3H), 3.70 (t, J = 7.0 Hz, 2H), 2.54 (t, J = 8.1 Hz, 2H), 2.31–2.14 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.42 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 175.3, 159.6 (dd, 1JCF = 250.2, 3JCF = 8.2 Hz), 160.2 (t, 3JCF = 13.5 Hz), 108.3 (t, 3JCF = 17.2 Hz), 98.7 (dd, 2JCF = 23.0, 3JCF = 4.3 Hz), 56.1, 49.6, 30.4, 19.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H12F2NO2 228.0836; found: 228.0831.
Compound 5ec: 322 mg, 88%, pale yellow oil. MS (ESI, pos. ion) m/z: 292.0 ([M + H]+).1H NMR (400 MHz, CDCl3): δ (ppm) 7.60 (d, J = 8.0 Hz, 2H), 7.24 (brs, 1H), 3.92 (s, 3H), 3.66 (t, J = 6.1 Hz, 2H), 2.64 (t, J = 7.0 Hz, 2H), 2.25–2.15 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.47 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 170.4, 164.7, 157.1 (dd, 1JCF = 251.8, 3JCF = 4.7 Hz), 129.7, 118.3 (t, 3JCF = 16.5 Hz), 113.2 (dd, 2JCF = 21.6, 3JCF = 4.3 Hz), 52.9, 44.3, 27.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H13ClF2NO3 292.0552, found: 292.0548.
Compound 5e: 133 mg, 82%, pale yellow oil. MS (ESI, pos. ion) m/z: 256.3 [M + H]+.1H NMR (400 MHz, CDCl3): δ (ppm) 7.63 (d, J = 8.2 Hz, 2H), 3.92 (s, 3H), 3.79 (t, J = 7.0 Hz, 2H), 2.57 (t, J = 8.1 Hz, 2H), 2.28 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −115.34 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 174.6, 164.6 (t, 3JCF = 3.2 Hz), 159.8 (d, 3JCF = 5.3 Hz), 157.3 (d, 3JCF = 5.4 Hz), 130.9 (t, 3JCF = 9.1 Hz), 120.1 (t, 3JCF = 16.5 Hz), 113.5 (m), 52.9, 49.3, 30.4, 19.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H12F2NO3 256.0785, found: 256.0785.
Compound 5d: 175 mg, 13%, yellow oil. MS (ESI, pos. ion) m/z: 243.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.94–7.86 (m, 2H, Ph), 3.83 (t, J = 7.0 Hz, 2H, CH2N), 2.60 (t, J = 8.1 Hz, 2H, COCH2), 2.38–2.27 (m, 2H, CH2); 19F NMR (376 MHz, CDCl3): δ (ppm) −110.99 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 174.5, 159.6 (d, 3JCF = 5.9 Hz), 157.0 (d, 3JCF = 5.9 Hz), 146.7 (t, 3JCF = 10.4 Hz), 122.4 (t, 3JCF = 16.3 Hz), 108.8–108.3 (m), 49.1 (t, 3JCF = 2.1 Hz), 30.3, 19.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H9F2N2O3 243.0581, found: 243.0572.
Compound 5i: 66 mg, 62%, yellow oil. MS (ESI, pos. ion) m/z: 216.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.80–6.72 (m, 2H), 3.72 (t, J = 7.0 Hz, 2H), 2.56 (t, J = 8.1 Hz, 2H), 2.33–2.17 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −108.04 (t, J = 5.6 Hz), −114.61 (d, J = 7.1 Hz); 13C NMR (151 MHz, CDCl3): δ (ppm) 175.0, 162.5 (t, 3JCF = 14.9 Hz), 160.8 (t, 3JCF = 14.9 Hz), 159.3 (ddd, 1JCF = 253.3, 3JCF = 15.3, 7.7 Hz), 112.4 (td, 3JCF = 16.8, 5.0 Hz), 101.5–100.5 (m), 49.4, 30.3, 19.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H9F3NO 216.0636, found: 216.0631.
Compound 5f: To a solution of compound 5d (500 mg, 2.0 mmol) in MeOH (10 mL) was added Pd/C (50 mg, 10%) and charged with H2. The reaction mixture was heated to 60 °C and stirred for 5 h, then cooled to room temperature. The mixture was filtered and the filter was concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/hexanes (v/v) = 1/10 to 2/3) to give compound 5f (360 mg, 80%) as pale yellow oil; MS (ESI, pos. ion) m/z: 213.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.27–6.08 (m, 2H), 4.22 (brs, 2H), 3.66 (t, J = 7.0 Hz, 2H), 2.52 (t, J = 8.1 Hz, 2H), 2.33–2.08 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −119.53 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 175.5, 159.8 (dd, 1JCF = 248.1, 3JCF = 8.0 Hz), 148.3 (t, 3JCF = 13.4 Hz), 105.0 (t, 3JCF = 17.3 Hz), 98.4–98.0 (m), 49.9, 30.5, 19.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H11F2N2O 213.0839, found: 213.0840.
Compound 5g and Compound 5h: To a solution of compound 5f (246 mg, 1.16 mmol), sulfuric acid (1.5 mL) and formaldehyde (261 mg, 3.48 mmol, 40% in water) in methanol (10 mL) was added NaBH4 (226 mg, 5.79 mmol) at 0 °C. The reaction mixture was warmed to room temperature and stirred overnight. The solvent was evaporated and the residue was dissolved with EtOAc (100 mL). The organic phase was washed with brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/1) to give compound 5g (31 mg, 12%) as pale yellow oil and compound 5h (66 mg, 24%) as pale yellow oil.
Compound 5g: MS (ESI, pos. ion) m/z: 227.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.16–6.02 (m, 2H), 4.39 (brs, 1H), 3.66 (t, J = 7.0 Hz, 2H), 2.75 (s, 3H), 2.53 (t, J = 8.1 Hz, 2H), 2.28–2.12 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −119.85 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 175.5, 159.9 (dd, 1JCF = 247.3, 3JCF = 8.4 Hz), 150.4 (t, 3JCF = 12.9 Hz), 103.7 (t, 3JCF = 17.5 Hz), 95.4 (d, 2JCF = 26.3 Hz), 50.0, 30.5 (d, 3JCF = 3.6 Hz), 19.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H13F2N2O 227.0996, found: 227.0995.
Compound 5h: MS (ESI, pos. ion) m/z: 241.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.29–6.11 (m, 2H), 3.67 (t, J = 7.0 Hz, 2H), 2.92 (s, 6H), 2.53 (t, J = 8.1 Hz, 2H), 2.28–2.10 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −119.45 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 175.4, 159.8 (dd, 1JCF = 246.5, 3JCF = 8.8 Hz), 150.9 (t, 3JCF = 13.1 Hz), 103.2 (t, 3JCF = 17.3 Hz), 95.8–95.0 (m), 49.9, 40.3, 30.5, 19.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H15F2N2O 241.1152, found: 241.1168.
Compound 6a: 368 mg, 43%, pale yellow oil. MS (ESI, pos. ion) m/z: 270.3 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.78 (d, J = 9.3 Hz, 2H), 3.93 (m, 2H), 3.77 (s, 3H), 3.47 (m, 1H), 2.85 (ABX, J = 17.2, 7.8, 9.6 Hz, 2H), 2.34 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.56 (s), −119.35 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.7, 172.3, 158.6 (dd, 1JCF = 251.9, 3JCF = 5.8 Hz), 140.8 (t, 3JCF = 9.5 Hz), 112.8 (dd, 3JCF = 19.9, 2.7 Hz), 112.0 (t, 3JCF = 16.7 Hz), 52.7, 50.9, 37.4, 33.4, 21.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H14F2NO3 270.0942; found: 270.0936.
Compound 6b: 657 mg, 75%, pale yellow oil. MS (ESI, pos. ion) m/z: 286.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.52 (m, 2H), 3.93 (m, 2H), 3.77 (s, 3H), 3.47 (m, 1H), 2.85 (ABX, J = 17.2, 7.8, 9.6 Hz, 2H), 2.34 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.75 (s), −117.60 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.8, 172.4, 159.6 (dd, 1JCF = 250.7, 3JCF = 8.0 Hz), 160.5 (t, 3JCF = 13.5 Hz), 107.6 (t, 3JCF = 17.1 Hz), 98.7 (dd, 2JCF = 23.9, 3JCF = 3.2 Hz), 56.1, 52.6, 51.1, 37, 33.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H14F2NO4 286.0891; found: 286.0859.
Compound 6c: 365 mg, 54%, pale yellow oil. MS (ESI, pos. ion) m/z: 314.0 ([M + H]+); 1H NMR (600 MHz, CDCl3): δ (ppm) 7.28 (d, J = 8.2 Hz, 2H), 4.02 (dd, J = 9.6, 6.6 Hz, 1H), 3.97 (t, J = 9.1 Hz, 1H), 3.79 (s, 3H), 3.51 (dt, J = 16.1, 8.0 Hz, 1H), 2.88 (ABX, J = 17.5, 7.6, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −63.69 (s), −112.71 (s), −113.42 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.5, 172.0, 158.8 (dd, 1JCF = 255.9, 3JCF = 5.2 Hz), 131.9–131.8 (m), 131.7–131.4 (m), 123.4, 121.6, 118.6–118.1 (m), 110.2 (d, 2JCF = 23.5 Hz), 52.9, 50.6, 37.5, 33.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H11F5NO3 314.0840; found: 314.0795.
Compound 6e: 429 mg, 43%, pale yellow oil. MS (ESI, pos. ion) m/z: 314.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.66 (d, J = 8.6 Hz, 2H), 4.05 (m, 2H), 3.93 (s, 3H), 3.79 (s, 3H), 3.53 (m, 1H), 2.96 (m, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −114.76 (s), −115.37 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 173.8, 172.4, 164.5 (t, 3JCF = 3.0 Hz), 158.5 (dd, 1JCF = 254.3, 3JCF = 5.1 Hz), 131.9 (t, 3JCF = 9.2 Hz), 118.5 (t, 3JCF = 16.4 Hz), 113.6 (dd, 2JCF = 22.1, 3JCF = 3.3 Hz), 53.0 (d, 3JCF = 13.1 Hz), 51.2, 37.4, 33.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H14F2NO5 314.0840; found: 314.0795.
Compound 6f: the method as compound 5f. 390 mg, 93%, yellow oil. MS (ESI, pos. ion) m/z: 271.3 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.18 (d, J = 10.2 Hz, 2H), 4.04 (brs, 2H), 3.92–3.80 (m, 2H), 3.77 (s, 3H), 3.50–3.34 (m, 1H), 2.82 (ABX, J = 17.2, 7.8, 9.7 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.89 (s), −119.70 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.9 (s), 172.7 (s), 159.8 (dd, 1JCF = 248.6, 3JCF = 7.7 Hz), 148.5 (t, 3JCF = 13.5 Hz), 104.3 (t, 3JCF = 16.3 Hz), 98.2 (d, 2JCF = 23.8 Hz), 52.6, 51.34, 37.2, 33.; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H13F2N2O3 271.0894, found: 271.0809.
Compound 6g: the method as compound 5g. 36 mg, 8%, yellow oil. MS (ESI, pos. ion) m/z: 285.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.08 (d, J = 10.9 Hz, 2H), 4.33 (d, J = 4.3 Hz, 1H), 3.86 (p, J = 9.6 Hz, 2H), 3.76 (s, 3H), 3.49–3.36 (m, 1H), 2.83 (ABX, J = 17.2, 7.9, 9.7 Hz, 2H), 2.74 (d, J = 5.0 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −119.31 (s), −120.0988 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.9, 172.9, 159.9 (dd, 1JCF = 247.6, 3JCF = 8.2 Hz), 150.7 (t, 3JCF = 13.5 Hz), 102.6 (t, 3JCF = 17.5 Hz), 95.3 (dd, 2JCF = 23.5, 3JCF = 7.5 Hz), 52.6, 51.4, 37.1, 33.4, 30.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H15F2N2O3 285.1051, found: 285.1050.
Compound 6h: the method as compound 5h. 58 mg, 13%, yellow oil. MS (ESI, pos. ion) m/z: 299.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.21 (d, J = 12.0 Hz, 2H), 3.88 (m, 2H), 3.77 (s, 3H), 3.44 (m, 1H), 2.93 (s, 6H), 2.83 (ABX, J = 17.2, 8.0, 9.7 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.84 (s), −119.67 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 173.0, 172.7, 159.8 (dd, 1JCF = 246.9, 3JCF = 8.4 Hz), 151.2 (t, 3JCF = 13.1 Hz), 102.2 (t, 3JCF = 17.6 Hz), 95.4 (dd, 2JCF = 24.0, 3JCF = 3.7 Hz), 52.6, 51.3, 40.3, 37.2, 33.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H17F2N2O3 299.1207, found: 299.1218.
Compound 6i: 85 mg, 61%, yellow oil. MS (ESI, pos. ion) m/z: 274.2 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.74 (p, J = 3.2 Hz, 2H), 3.97–3.84 (m, 2H), 3.76 (s, 3H), 3.46 (dt, J = 16.1, 8.0 Hz, 1H), 2.83 (ABX, J = 17.3, 7.7, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −107.20 (t, J = 6.0 Hz), −113.92 (s), −114.72 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.6, 172.3, 162.7 (t, 3JCF = 14.8 Hz), 161.0 (t, 3JCF = 14.8 Hz), 159.2 (ddd, 1JCF = 254.0, 3JCF = 15.3, 7.4 Hz), 111.5 (td, 3JCF = 16.6, 5.1 Hz), 101.2 (t, 2JCF = 25.1 Hz), 52.7, 50.7, 37.2, 33.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H11F3NO3 274.0691, found: 274.0647.
Compound 6j: 132 mg, 59%, yellow oil. MS (ESI, pos. ion) m/z: 290.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.97 (d, J = 8.1 Hz, 2H), 3.88 (p, J = 9.5 Hz, 2H), 3.71 (s, 3H), 3.43 (dt, J = 16.0, 7.8 Hz, 1H), 2.79 (ABX, J = 17.4, 7.6, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −114.90 (s), −115.58 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.4, 172.1, 158.6 (dd, 1JCF = 255.7, 3JCF = 6.3 Hz), 134.4 (t, 3JCF = 12.7 Hz), 113.8 (t, 3JCF = 16.5 Hz), 113.3 (dd, 2JCF = 23.1, 3JCF = 1.5 Hz), 52.6, 50.5, 37.1, 33.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H11ClF2NO3 290.0396, found: 290.0349.
Compound 6k: 112 mg, 42%, yellow oil. MS (ESI, pos. ion) m/z: 334.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.18 (d, J = 7.8 Hz, 2H), 4.00–3.87 (m, 2H), 3.78 (s, 3H), 3.53–3.40 (m, 1H), 2.85 (ABX, J = 17.4, 7.7, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −114.94 (s), −115.72 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.6, 172.1, 158.8 (dd, 1JCF = 256.8, 3JCF = 6.0 Hz), 121.5 (t, 3JCF = 11.8 Hz), 116.4 (d, 2JCF = 22.9 Hz), 114.4 (t, 3JCF = 16.2 Hz), 52.8, 50.7, 37.4, 33.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H11BrF2NO3 333.9890, found: 333.9885.
Compound 6l: 65 mg, 37%, yellow oil. MS (ESI, pos. ion) m/z: 382.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (d, J = 7.6 Hz, 2H), 4.01–3.84 (m, 2H), 3.78 (s, 3H), 3.53–3.39 (m, 1H), 2.84 (ABX, J = 17.4, 7.7, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −115.44 (s), −116.20 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.6, 172.1, 158.5 (dd, 1JCF = 258.2, 3JCF = 5.4 Hz), 122.1 (d, 2JCF = 22.2 Hz), 115.2 (t, 3JCF = 16.3 Hz), 91.0 (t, 3JCF = 10.0 Hz), 52.8, 50.6, 37.4, 33.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H11F2INO3 381.9752, found: 381.9997.
Compound 6o: 154 mg, 73%, yellow oil. MS (ESI, pos. ion) m/z: 300.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.50 (d, J = 10.1 Hz, 2H), 3.98 (q, J = 7.0 Hz, 2H), 3.95–3.83 (m, 2H), 3.77 (s, 3H), 3.52–3.38 (m, 1H), 2.84 (ABX, J = 17.2, 7.8, 9.6 Hz, 2H), 1.40 (t, J = 7.0 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.02 (s), −117.89 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.8, 172.5, 159.5 (dd, 1JCF = 250.2, 3JCF = 8.0 Hz), 159.8 (t, 3JCF = 13.3 Hz), 107.2 (t, 3JCF = 17.1 Hz), 99.1 (dd, 2JCF = 23.5, 3JCF = 2.9 Hz), 64.6, 52.7, 51.1, 37.2, 33.4, 14.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H16F2NO4 300.1047, found: 300.1048.
Compound 6u: 65 mg, 33%, yellow oil. MS (ESI, pos. ion) m/z: 281.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.30 (d, J = 7.8 Hz, 2H), 4.06–3.89 (m, 2H), 3.77 (s, 3H), 3.55–3.41 (m, 1H), 2.85 (ABX, J = 17.5, 7.5, 9.5 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −111.95 (s), −112.30 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.3, 171.8, 158.7 (dd, 1JCF = 257.0, 3JCF = 5.7 Hz), 120.3 (t, 3JCF = 16.0 Hz), 116.6 (dd, 2JCF = 23.9, 3JCF = 3.8 Hz), 116.2 (t, 3JCF = 3.2 Hz), 112.6 (t, 3JCF = 11.6 Hz), 52.8, 50.5, 37.5, 33.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H11F2N2O3 281.0738, found: 281.0728.
Compound 6p: To a solution of compound 6l (260 mg, 0.68 mmol), tributyl(vinyl)stannane (221 mg, 0.70 mmol) and chlorolithium (95 mg, 2.25 mmol) in DMF (5 mL) was added PdCl2(PPh3)2 (62 mg, 0.1 mmol) and charged with N2. The reaction mixture was heated to 80 °C and stirred overnight, then concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/5) to give compound 6p (140 mg, 73%) as colorless oil; MS (ESI, pos. ion) m/z: 282.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.00 (d, J = 9.5 Hz, 2H), 6.60 (dd, J = 17.5, 10.8 Hz, 1H), 5.75 (d, J = 17.5 Hz, 1H), 5.38 (d, J = 10.8 Hz, 1H), 4.03–3.86 (m, 2H), 3.78 (s, 3H), 3.54–3.41 (m, 1H), 2.86 (ABX, J = 17.3, 7.8, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.42 (s), −118.16 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.7, 172.2, 158.9 (dd, 1JCF = 252.2, 3JCF = 6.0 Hz), 139.7 (t, 3JCF = 9.4 Hz), 134.6, 117.4, 113.8 (t, 3JCF = 16.9 Hz), 109.8 (d, 2JCF = 20.7 Hz), 52.7, 50.9, 37.4, 33.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H14F2NO3 282.0942, found: 282.0931.
Compound 6q: the method as compound 6p. 337 mg, 81%, colorless oil. MS (ESI, pos. ion) m/z: 296.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.91 (d, J = 9.8 Hz, 2H), 6.35–6.18 (m, 2H), 4.01–3.85 (m, 2H), 3.78 (s, 3H), 3.54–3.40 (m, 1H), 2.85 (ABX, J = 17.3, 7.8, 9.6 Hz, 2H), 1.87 (d, J = 5.0 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.98 (s), −118.75 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.7, 172.3, 158.9 (dd, 1JCF = 251.5, 3JCF = 6.0 Hz), 140.2 (t, 3JCF = 9.7 Hz), 129.7, 128.9, 112.7 (t, 3JCF = 16.8 Hz), 109.3 (d, 2JCF = 20.4 Hz), 52.7, 50.9, 37.4, 33.4, 18.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H16F2NO3 296.1098, found: 296.1099.
Compound 6m: the method as compound 5f. 82 mg, 91%, colorless oil. MS (ESI, pos. ion) m/z: 284.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.81 (d, J = 9.3 Hz, 2H), 4.00–3.85 (m, 2H), 3.77 (s, 3H), 3.47 (dt, J = 16.4, 8.1 Hz, 1H), 2.85 (ABX, J = 17.2, 7.8, 9.6 Hz, 2H), 2.63 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.28 (s), −119.07 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.7, 172.3, 158.7 (dd, 1JCF = 252.0, 3JCF = 5.8 Hz), 147.0 (t, 3JCF = 8.8 Hz), 112.0 (t, 3JCF = 16.7 Hz), 111.6 (d, 3JCF = 19.8 Hz), 52.6, 50.9, 37.3, 33.4, 28.8, 14.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H16F2NO3 284.1098, found: 284.1092.
Compound 6n: the method as compound 5f. 77 mg, 84%, colorless oil. MS (ESI, pos. ion) m/z: 298.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 6.78 (d, J = 9.4 Hz, 2H), 4.01–3.84 (m, 2H), 3.76 (s, 3H), 3.52–3.39 (m, 1H), 2.84 (ABX, J = 17.2, 7.8, 9.6 Hz, 2H), 2.55 (t, J = 7.6 Hz, 2H), 1.68–1.54 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.39 (s), −119.16 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.7, 172.3, 158.6 (dd, 1JCF = 252.0, 3JCF = 5.6 Hz), 145.5 (t, 3JCF = 8.8 Hz), 112.0 (t, 3JCF = 17.0 Hz), 52.7, 50.9, 37.8, 37.3, 33.4, 24.0, 13.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H18F2NO3 298.1255, found: 298.1252.
Compound 6r: to a solution of compound 6l (383 mg, 1.00 mmol), phenylboronic acid (247 mg, 2.03 mmol) and sodium carbonate (233 mg, 2.2 mmol) in 1,4-dioxane (2 mL) and water (2 mL) was added Pd(dppf)Cl2·CH2Cl2 (165 mg, 0.20 mmol) and charged with N2. The reaction mixture was heated to 100 °C and stirred overnight, then concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/1) to give compound 6r (270 mg, 82%) as colorless oil; MS (ESI, pos. ion) m/z: 332.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.49 (d, J = 7.4 Hz, 2H), 7.40 (m, 3H), 7.18 (d, J = 9.7 Hz, 2H), 3.97 (p, J = 9.6 Hz, 2H), 3.76 (s, 3H), 3.55–3.42 (m, 1H), 2.86 (ABX, J = 17.4, 7.6, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −116.85 (s), −117.62 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 172.8, 172.7, 158.9 (dd, 1JCF = 252.4, 3JCF = 5.9 Hz), 143.4 (t, 3JCF = 9.6 Hz), 138.3, 129.2, 128.8, 127.0, 113.3 (t, 3JCF = 16.8 Hz), 110.8 (dd, 2JCF = 20.9, 3JCF = 2.7 Hz), 52.7, 51.0, 37.3, 33.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16F2NO3 332.1098, found: 332.1089.
Compound 6s: to a solution of compound 6l (439 mg, 1.15 mmol), 1,2-bis(diphenylphosphino)ethane (50 mg, 0.12 mmol), diacetylpalladium (14 mg, 0.07 mmol) and formyloxysodium (165 mg, 2.43 mmol) in DMSO (4 mL) was added 2,2-dimethylpropanenitrile (1.5 mL) and charged with N2. The reaction mixture was heated to 120 °C and stirred overnight, then concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/1) to give compound 6s (160 mg, 49%) as colorless oil; MS (ESI, pos. ion) m/z: 284.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 9.92 (s, 1H), 7.51 (d, J = 8.2 Hz, 2H), 4.12–3.91 (m, 2H), 3.79 (s, 3H), 3.57–3.44 (m, 1H), 2.89 (ABX, J = 17.4, 7.6, 9.6 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −113.13 (s), −113.72 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 188.8, 172.4, 171.9, 159.2 (dd, 1JCF = 256.7, 3JCF = 4.6 Hz), 136.7 (t, 3JCF = 7.2 Hz), 130.0 (d, 3JCF = 4.4 Hz), 120.6 (t, 3JCF = 16.5 Hz), 113.0 (d, 2JCF = 20.7 Hz), 52.8, 50.6, 37.5, 33.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H12F2NO4 284.0734, found: 284.0750.
Compound 6t: to a solution of compound 6l (382 mg, 1.00 mmol), 1,3-bis(diphenylphosphino)propane (87 mg, 0.20 mmol), diacetylpalladium (27 mg, 0.14 mmol) and Na2CO3 (298 mg, 2.81 mmol) in n-butanol (3 mL) was added 1-vinyloxybutane (504 mg, 5.03 mmol) and charged with N2. The reaction mixture was heated to 120 °C and stirred overnight, then concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/2) to give compound 6t (122 mg, 41%) as colorless oil; MS (ESI, pos. ion) m/z: 298.4 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.56 (d, J = 8.8 Hz, 2H), 4.08–3.91 (m, 2H), 3.79 (s, 3H), 3.50 (dt, J = 16.0, 8.0 Hz, 1H), 2.89 (ABX, J = 17.4, 7.7, 9.5 Hz, 2H), 2.58 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −114.25, 114.86 (s); 13C NMR (101 MHz, CDCl3) δ 194.8, 172.5, 171.9, 158.8 (dd, 1JCF = 255.3, 3JCF = 5.2 Hz), 137.8 (t, 3JCF = 7.2 Hz), 130.1 (d, 3JCF = 3.4 Hz), 119.6–119.0 (m), 112.2 (d, 3JCF = 18.4 Hz), 52.8, 50.7, 37.5, 33.4, 26.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H14F2NO4 298.0891, found: 298.0902.
Compound 6v: a solution of compound 6f (568 mg, 2.10 mmol) in formic acid (7 mL) was heated to reflux and stirred for 1 h. The reaction was quenched with saturated Na2CO3 solution (100 mL), then extracted with dichloromethane (100 mL × 3). The combined organic phases were washed with brine (100 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 3/1) to give compound 6v (322 mg, 51%) as yellow oil; MS (ESI, pos. ion) m/z: 299.0 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 9.04 (s, 1H), 8.20 (s, 1H), 7.08 (d, J = 12.6 Hz, 2H), 3.99–3.83 (m, 2H), 3.78 (s, 3H), 3.55–3.43 (m, 1H), 2.89 (ABX, J = 17.4, 7.4, 9.7 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ (ppm) −117.46 (s), −117.98 (s); 13C NMR (101 MHz, CDCl3): δ (ppm) 13C NMR (101 MHz, CDCl3) δ 173.8, 172.5, 159.6, 158.7 (dd, 1JCF = 250.3, 3JCF = 6.6 Hz), 139.1 (t, 3JCF = 13.4 Hz), 109.6 (t, 3JCF = 17.0 Hz), 103.9 (dd, 2JCF = 24.7, 3JCF = 3.0 Hz), 52.8, 51.4, 37.2, 33.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H13F2N2O4 299.0843, found: 299.0841.
Compound 6w: to a solution of compound 6f (139 mg, 0.51 mmol), acetic acid (163 mg, 2.72 mmol), and Et3N (298 mg, 2.81 mmol) in CH2Cl2 (10 mL) was added HATU (383 mg, 1.01 mmol). The reaction mixture was heated to reflux and stirred overnight, then concentrated in vacuo. The residue was purified by a flash column chromatography (EtOAc/PE (v/v) = 1/10 to 1/0) to give compound 6w (139 mg, 87%) as yellow oil; MS (ESI, pos. ion) m/z: 313.1 ([M + H]+); 1H NMR (400 MHz, CDCl3): δ (ppm) 9.09 (s, 1H), 7.04 (d, J = 10.8 Hz, 2H), 3.96–3.84 (m, 2H), 3.78 (s, 3H), 3.55–3.43 (m, 1H), 2.91 (ABX, J = 17.4, 7.4, 9.7 Hz, 2H), 2.05 (s, 3H); 19F NMR (376 MHz, CDCl3): δ (ppm) −118.54 (s), −119.11 (s); 13C NMR (151 MHz, CDCl3): δ (ppm) 13C NMR (101 MHz, CDCl3) δ 174.0, 172.6, 169.4, 158.7 (dd, 1JCF = 249.3, 3JCF = 6.6 Hz), 140.4 (t, 3JCF = 13.4 Hz), 108.6 (t, 3JCF = 17.1 Hz), 103.3 (d, 2JCF = 25.8 Hz), 52.9, 51.6, 37.1, 33.6, 24.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H15F2N2O4 313.1000, found: 313.0999.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- X. Li, K. Christopher and F. B. Matthew, J. Fluorine Chem., 2017, 198, 47–53 CrossRef.
- D. Diana Gimenez, A. Aoife Phelan, C. D. Murphy and S. L. Cobb, Beilstein J. Org. Chem., 2021, 17, 293–318 CrossRef PubMed.
- Z. Xu, C. Liu, S. Zhao, S. Chen and Y. Zhao, Chem. Rev., 2019, 119, 195–230 CrossRef CAS PubMed.
- (a) Y. Zhao and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 3221–3224 CrossRef CAS PubMed; (b) Y. Zhao, G. Markopoulos and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 10683–10690 CrossRef CAS PubMed.
- M. Sun, W. Chen, T. Zhang, Z. Liu, J. Wei and N. Xi, Tetrahedron, 2020, 76, 131679 CrossRef CAS.
- (a) B. Huang, L. Xu, N. Wang, J. Ying, Y. Zhao and S. Huang, Anal. Chem., 2022, 94, 1867–1873 CrossRef CAS PubMed; (b) C. Dong, Z. Xu, L. Wen, S. He, J. Wu, Q.-H. Deng and Y. Zhao, Anal. Chem., 2021, 93, 2968–2973 CrossRef CAS PubMed.
- C. R. Buchholz and W. C. K. Pomerantz, RSC Chem. Biol., 2021, 2, 1312–1330 RSC.
- J. X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Nucl. Magn. Reson. Spectrosc., 2013, 70, 25–49 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- (a) C. Dalvit and A. Vulpetti, Chem.–Eur. J., 2016, 22, 7592–7601 CrossRef CAS PubMed; (b) R. Taylor, Acta Crystallogr, Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 474–488 CrossRef CAS PubMed.
- (a) M. R. Bauer, R. N. Jones, M. G. J. Baud, R. Wilcken, F. M. Boeckler, A. R. Fersht, A. C. Joerger and J. Spencer, ACS Chem. Bio., 2016, 11, 2265–2274 CrossRef CAS PubMed; (b) P. Li, J. M. Maier, E. C. Vik, C. J. B. Yehl, E. Dial, A. E. Rickher, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem.Int. Ed., 2017, 56, 7209–7212 CrossRef CAS PubMed.
- (a) J. Shang, N. M. Gallagher, F. Bie, Q. Li, Y. Che, Y. Wang and H. Jiang, J. Org. Chem., 2014, 79, 5134–5144 CrossRef CAS PubMed; (b) C. Dalvit, C. Invernizzi and A. Vulpetti, Chem.–Eur. J., 2014, 20, 11058–11068 CrossRef CAS PubMed.
- (a) M. D. Struble, J. Strull, K. Patel, M. A. Siegler and T. Lectka, J. Org. Chem., 2014, 79, 1–6 CrossRef CAS PubMed; (b) M. D. Struble, M. T. Scerba, M. Siegler and T. Lectka, Science, 2013, 340, 57–60 CrossRef CAS PubMed; (c) C. R. Pitts, M. G. Holl and T. Lectka, Angew. Chem., Int. Ed., 2018, 57, 1924–1927 CrossRef CAS PubMed; (d) M. G. Holl, C. R. Pitts and T. Lectka, Angew. Chem.Int. Ed., 2018, 57, 2–11 CrossRef PubMed.
- N. Xi, X. H. Sun, M. X. Li, M. M. Sun, M. A. Xi, Z. P. Zhan, J. Yao, X. Bai, Y. J. Wu and M. Liao, J. Org. Chem., 2018, 83, 11586–11594 CrossRef CAS PubMed.
- N. Xi, M. Sun, Y. Lu and B. Bai, Tetrahedron, 2022, 132733 CrossRef CAS.
- M. J. Milewska, T. Bytner and T. Polonski, Synthesis, 1996, 12, 1485–1488 CrossRef.
- C. L. Lopez, S. Patterson, T. Blum, A. F. Straight, J. Toth, A. M. Z. Slawin, T. J. Mitchison, J. R. Sellers and N. J. Westwood, Eur. J. Org. Chem., 2005, 2005, 1736–1740 CrossRef.
- F. K. Woolard, US pat., US4909834, Richmond, Calif, 1990 Search PubMed.
- G. A. Grasa, P. Steven and S. P. Nolan, Org. Lett., 2001, 3, 119–122 CrossRef CAS PubMed.
- Y. Zhang, X. Jiang, J.-M. Wang, J.-L. Chen and Y.-M. Zhu, RSC Adv., 2015, 5, 17060–17063 RSC.
- J. Mo, L. Xu and J. Xiao, J. Am. Chem. Soc., 2005, 127, 751–760 CrossRef CAS PubMed.
- Optimized structures and FMO orbitals were depicted with the GaussView 06 program.
- P. Politze and J. S. Murray, ChemPhysChem, 2020, 21, 579–588 CrossRef PubMed.
- (a) A. Rodrigo Cormanich, R. Rittner, D. O, Hagan and M. Bühl, J. Comput. Chem., 2016, 37, 25–33 CrossRef PubMed; (b) K. J. Kamer, A. Choudhary and R. T. Raines, J. Org. Chem., 2013, 78, 2099–2103 CrossRef CAS PubMed; (c) R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- N. Lu, R. M. Ley, C. E. Cotton, W.-C. Chung, J. S. Francisco and E. Negishi, J. Phys. Chem. A, 2013, 117, 8256–8262 CrossRef CAS PubMed.
- S. Hanessian, O. M. Saavedra, M. A. Vilchis-Reyes, J. P. Maianti, H. Kanazawa, P. Dozzo, R. D. Matias, A. Serio and J. Kondo, Chem. Sci., 2014, 5, 4621–4632 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1904410 and 1915197. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06660b |
| This journal is © The Royal Society of Chemistry 2022 |