
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructurally diverse biflavonoids from Dysosma versipellis and their bioactivity†
Yan-Jun Sun *abc,
Rui-Jie Hanab,
Hong-Yun Baiab,
Hao-jie Wangab,
Meng Liab,
Ying-Ying Siab,
Jun-Min Wangab,
Jian-Hong Gongab,
Hui Chenab and
Wei-Sheng Feng*ab
*abc,
Rui-Jie Hanab,
Hong-Yun Baiab,
Hao-jie Wangab,
Meng Liab,
Ying-Ying Siab,
Jun-Min Wangab,
Jian-Hong Gongab,
Hui Chenab and
Wei-Sheng Feng*ab
aCo-construction Collaborative Innovation Center for Chinese Medicine and Respiratory Diseases by Henan & Education Ministry of P. R. China, Henan University of Chinese Medicine, Zhengzhou 450046, P. R. China. E-mail: sunyanjunily@126.com; fwsh@hactcm.edu.cn
bSchool of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, P. R. China
cHenan Research Center for Special Processing Technology of Chinese Medicine, Zhengzhou, 450046, P. R. China
First published on 7th December 2022
Abstract
Five pairs of new biflavonoid enantiomers, (±)-dysosmabiflavonoids A–E (1–5), two new biflavonoids, dysosmabiflavonoids F–G (6–7), and four biosynthetically related precursors (8–11) were isolated from the roots and rhizomes of Dysosma versipellis. Their structures were elucidated by extensive spectroscopic analysis, including HR-ESI-MS and 2D NMR. Their absolute configurations were determined by comparison of the calculated and experimental ECD spectra. All isolated compounds were evaluated for AChE inhibitory activity. Compounds 6 and 7 exhibited more potent inhibitory activities with IC50 values of 1.42 and 0.73 µM, respectively, than their biosynthetically related precursors kaempferol (8, 17.90 µM) and quercetin (9, 3.96 µM). The preliminary structure–activity relationship study indicated that the connection mode of biflavonoid subunits, oxidation degree of the C ring, and 3,4-dihydroxy group of the B ring were important structural factors for AChE inhibitory activity. Racemates 1–5 and their corresponding levorotatory and dextrorotatory enantiomers were tested for their potential to impede the generation of NO in lipopolysaccharide-stimulated RAW264.7 cells, and their mushroom tyrosinase inhibitory effect. Racemate 1 displayed more potent mushroom tyrosinase inhibitory activity (IC50, 28.27 µM) than the positive control kojic acid (IC50, 32.59 µM). D. versipellis may have therapeutic potential for melanogenesis disorders and neurodegenerative diseases.
Introduction
Neurodegenerative diseases are an internationally faced public health problem. Acetylcholinesterase (AChE) catalyzes the hydrolysis of the neurotransmitter acetylcholine, which plays a key role in memory and cognition.1 It is also the key enzyme target for treatments of myasthenia gravis, glaucoma, Alzheimer's disease, Parkinson's disease, and other neurodegenerative diseases.2 Although a few cholinesterase inhibitors from synthetic and natural origins are available in the drug market, they are used only for symptomatic treatment. Moreover, side effects and relatively low bioavailability limit their clinical applications.3 There is still a great demand to discover new cholinesterase inhibitors, which can improve cholinergic transmission in neurodegenerative diseases. Traditional Chinese medicine has become a promising source of structurally novel cholinesterase inhibitors.A perennial herb of the Berberidaceae family, Dysosma versipellis (Hance) M. Cheng ex Ying, is widely distributed in the central/south regions of China.4 Its roots and rhizomes, called “Bajiaolian” in Chinese, have the functions of eliminating phlegm, detoxification, removing blood stasis and dispersing knots.5 As a common folk medicine, it has a satisfactory effect on the treatment of parotitis,4 cough, sore throat, scrofula, nameless swollen venom/carbuncles, snake bite, fall injury, rheumatoid arthritis,5 epidemic encephalitis B, parasites, tumor,6 lumbago, skelalgia,7 ulcer,8 weakness, condyloma accuminata, lymphadenopathy,9 pyogenic infection,10 hemiplegia, joint pain,11 and syphilis12 since the Han dynasty in China. Its previous phytochemical and pharmacological investigations revealed the presense of bioactive biflavonoids,13–16 and their neuraminidase inhibitory13 and cytotoxic properties.14 In our search for bioactive natural products, five pairs of new biflavonoid enantiomers, (±)-dysosmabiflavonoids A–E (1–5), two new biflavonoids, dysosmabiflavonoids F–G (6–7), along with four biosynthetically related precursors (8–11) (Fig. 1) were isolated from the roots and rhizomes of D. versipellis. Reported herein are their isolation, structure elucidation, and bioactive evaluation.
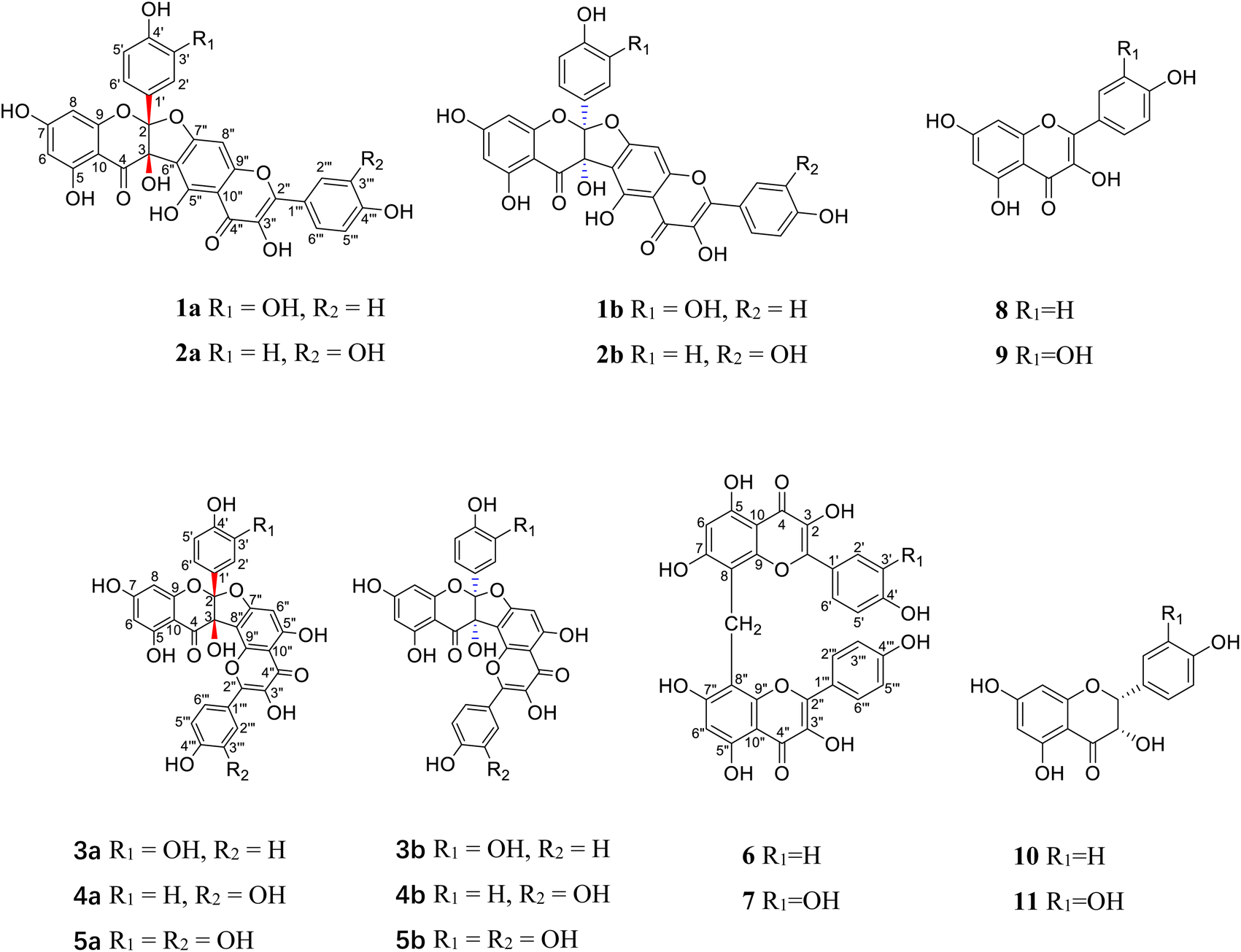 | ||
| Fig. 1 The structures of compounds 1–11. | ||
Results and discussion
Compound 1 was obtained as a yellow amorphous powder and possessed a molecular formula C30H18O13 with twenty-two degrees of unsaturation, as revealed from its HR-ESI-MS analysis (m/z 587.0806 [M + H]+, calcd for C30H19O13, 587.0826). The IR spectrum displayed two carbonyl groups (1726, 1635 cm−1), hydroxy groups (3374 cm−1), aromatic rings (1607, 1512 cm−1) and tertiary alcohol (1179 cm−1). The UV spectrum exhibited absorption maxima characteristic for flavonol at 275 and 360 nm. The 13C NMR (Table 1) and HSQC spectra revealed one biflavone skeleton including two carbonyl groups δC 176.6, 190.1 (characteristic of a flavonol and a flavanonol, respectively), four benzene rings, two oxygen-bearing olefinic carbons δC 147.8, 136.1, two aliphatic quaternary carbons δC 117.6, 79.1. The 1H NMR spectrum (Table 2) showed four sets of aromatic systems including one para-disubstituted benzene ring δH 8.08 (2H, d, J = 8.5 Hz), 6.94 (2H, d, J = 8.5 Hz), one penta-substituted benzene ring δH 7.01 (1H, s), one 1,3,4-tri-substituted benzene ring δH 6.89 (1H, s), 6.72 (1H, d, J = 8.6 Hz), 6.75 (1H, d, J = 8.6 Hz), and one 1,2,3,5-tetra-substituted benzene ring δH 5.86 (1H, s) and 5.78 (1H, s). The HMBC correlations (Fig. 2) of the aromatic protons δH 8.08 (2H, d, J = 8.5 Hz, H-2‴, 6‴) with C-2″ (δC 147.8), in combination with one penta-substituted benzene ring at δH 7.01 (1H, s), indicated that compound 1 contained kaempferol as a subunit.17 Another subunit was identified as dihydroquercetin by the HMBC cross peak of the aromatic protons δH 6.89 (1H, s, H-2′) and 6.75 (1H, d, J = 8.6 Hz, H-6′) with C-2 (δC 117.6), one 1,2,3,5-tetra-substituted benzene ring δH 5.86 (1H, s), 5.78 (1H, s), and two aliphatic quaternary carbons δC 117.6, 79.1. A careful comparison of the 13C NMR spectra of 1 with kaempferol indicated that the subunit of kaempferol was substituted at C-6 and C-7, which was confirmed by the chemical shift change from δC 156.8 (C-5″), 110.3 (C-6″), 164.3 (C-7″), 91.1 (C-8″), and δH 7.01 (1H, s, H-8″) in 1 to δC 160.7 (C-5), 98.2 (C-6), 163.9 (C-7), 93.5 (C-8), and δH 6.48 (1H, d, J = 2.0 Hz, H-8) in kaempferol.17 An ether bridge C-2-O-C-7″ and a C-3-C-6″ bond between the two flavonoid subunits were determined by two aliphatic quaternary carbons [one di-oxygenated δC 117.6 (C-2), one mono-oxygenated δC 79.1 (C-3)].18 The planar structure of 1 (Fig. 1) was determined by detailed elucidation on 1D and 2D NMR data. Despite repeated experiments, suitable single crystals of 1 for X-ray diffraction were not obtained successfully. NOESY experiments failed to provide any correlations between the two flavonoid subunits. Compound 1 showed no distinct Cotton effects (200–400 nm) at all examined concentrations, indicating it was a racemic mixture. This result is also consistent with the observation that 1 was optically inactive. Racemate 1 was resolved into 1a and 1b in a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by a chiral Daicel PAK AD-H column. As expected, the ECD spectra of 1a and 1b showed opposite Cotton effects. The absolute configurations of C-2 and C-3 were extrapolated by comparing the experimental with calculated ECD spectra, the latter performed by density functional theory. A comparison of the observed ECD spectra for 1a and 1b with the calculated ECD spectra for the (2S, 3S)-1 and (2R, 3R)-1 is shown in Fig. 3. The overall ECD spectra for (2S, 3S)-1 and (2R, 3R)-1 are in good accordance with the experimental ECD for 1a and 1b, respectively. Therefore, the absolute configurations of 1a and 1b were respectively deduced to be 2S, 3S and 2R, 3R. Thus, compounds 1a and 1b were determined as (2S,3S)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7′′:3-6″)-3″,5″,4‴-trihydroxyflavone and (2R,3R)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3-6″)-3″,5″,4‴-trihydroxyflavone, and named as (+)-dysosmabiflavonoid A and (−)-dysosmabiflavonoid A, respectively.
:1 by a chiral Daicel PAK AD-H column. As expected, the ECD spectra of 1a and 1b showed opposite Cotton effects. The absolute configurations of C-2 and C-3 were extrapolated by comparing the experimental with calculated ECD spectra, the latter performed by density functional theory. A comparison of the observed ECD spectra for 1a and 1b with the calculated ECD spectra for the (2S, 3S)-1 and (2R, 3R)-1 is shown in Fig. 3. The overall ECD spectra for (2S, 3S)-1 and (2R, 3R)-1 are in good accordance with the experimental ECD for 1a and 1b, respectively. Therefore, the absolute configurations of 1a and 1b were respectively deduced to be 2S, 3S and 2R, 3R. Thus, compounds 1a and 1b were determined as (2S,3S)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7′′:3-6″)-3″,5″,4‴-trihydroxyflavone and (2R,3R)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3-6″)-3″,5″,4‴-trihydroxyflavone, and named as (+)-dysosmabiflavonoid A and (−)-dysosmabiflavonoid A, respectively.
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 2 | 117.6 C | 117.8 C | 117.8 C | 117.7 C | 117.8 C | 146.9 C | 146.7 C |
| 3 | 79.1 C | 79.2 C | 79.9 C | 79.9 C | 79.9 C | 135.4 C | 135.4 C |
| 4 | 190.1 C | 190.7 C | 191.0 C | 190.5 C | 191.0 C | 176.2 C | 176.1 C |
| 5 | 163.3 C | 163.4 C | 163.5 C | 163.5 C | 163.5 C | 158.4 C | 158.4 C |
| 6 | 97.5 CH | 96.9 CH | 97.0 CH | 97.1 CH | 97.0 CH | 97.9 CH | 98.0 CH |
| 7 | 168.9 C | 167.8 C | 167.9 C | 168.1 C | 167.9 C | 162.1 C | 162.1 C |
| 8 | 95.4 CH | 95.1 CH | 95.3 CH | 95.3 CH | 95.3 CH | 104.6 C | 104.7 C |
| 9 | 160.4 C | 160.4 C | 160.6 C | 160.3 C | 160.6 C | 153.8 C | 153.8 C |
| 10 | 97.8 C | 98.4 C | 98.6 C | 98.3 C | 98.6 C | 102.8 C | 102.6 C |
| 1′ | 123.9 C | 123.7 C | 123.7 C | 123.4 C | 123.7 C | 122.1 C | 122.5 C |
| 2′ | 114.5 CH | 128.8 CH | 114.5 CH | 128.3 CH | 114.5 CH | 129.6 CH | 115.2 CH |
| 3′ | 144.6 C | 115.2 CH | 144.7 C | 114.8 CH | 144.7 C | 115.4 CH | 145.0 C |
| 4′ | 146.8 C | 159.1 C | 146.9 C | 158.7 C | 146.9 C | 159.1 C | 147.6 C |
| 5′ | 114.9 CH | 115.2 CH | 115.5 CH | 114.8 CH | 115.1 CH | 115.4 CH | 115.6 CH |
| 6′ | 118.1 CH | 128.8 CH | 118.2 CH | 128.3 CH | 118.2 CH | 129.6 CH | 120.0 CH |
| 2″ | 147.8 C | 148.0 C | 147.5 C | 147.5 C | 147.5 C | 146.9 C | 146.6 C |
| 3″ | 136.1 C | 136.2 C | 136.1 C | 136.0 C | 136.1 C | 135.4 C | 135.3 C |
| 4″ | 176.6 C | 176.6 C | 176.3 C | 176.2 C | 176.2 C | 176.2 C | 176.1 C |
| 5″ | 156.8 C | 156.9 C | 163.9 C | 163.8 C | 163.8 C | 158.4 C | 158.3 C |
| 6″ | 110.3 C | 110.0 C | 94.8 CH | 94.7 CH | 94.7 CH | 97.9 CH | 97.9 CH |
| 7″ | 164.3 C | 164.3 C | 164.9 C | 164.8 C | 164.8 C | 162.1 C | 162.1 C |
| 8″ | 91.1 CH | 91.1 CH | 106.3 C | 106.1 C | 106.1 C | 104.6 C | 104.6 C |
| 9″ | 157.2 C | 157.2 C | 151.2 C | 151.5 C | 151.2 C | 153.8 C | 153.7 C |
| 10″ | 105.6 C | 105.7 C | 105.2 C | 105.2 C | 105.2 C | 102.8 C | 102.6 C |
| 1‴ | 121.3 C | 121.6 C | 121.4 C | 121.7 C | 121.7 C | 122.1 C | 122.1 C |
| 2‴ | 129.7 CH | 115.3 CH | 130.2 CH | 116.6 CH | 116.2 CH | 129.6 CH | 129.5 CH |
| 3‴ | 115.5 CH | 145.2 C | 115.3 CH | 145.4 C | 144.9 C | 115.4 CH | 115.4 CH |
| 4‴ | 159.6 C | 148.2 C | 159.6 C | 148.1 C | 148.1 C | 159.1 C | 159.0 C |
| 5‴ | 115.5 CH | 115.7 CH | 115.3 CH | 115.3 CH | 115.3 CH | 115.4 CH | 115.4 CH |
| 6‴ | 129.7 CH | 120.2 CH | 130.2 CH | 120.5 CH | 120.5 CH | 129.6 CH | 129.5 CH |
| 8-CH2-8″ | 16.4 CH2 | 16.5 CH2 |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 6 | 5.86, s | 5.93, d (1.6) | 5.90, d (1.6) | 5.88, d (1.9) | 5.76, s | 6.17, s | 6.12, s |
| 8 | 5.78, s | 5.86, d (1.6) | 5.86, d (1.6) | 5.86, d (1.9) | 5.73, s | ||
| 2′ | 6.89, s | 7.28, d (8.7) | 6.90, d (1.6) | 7.29, d (8.8) | 6.89, d (2.0) | 7.97, d (8.7) | 7.82, d (2.0) |
| 3′ | 6.79, d (8.7) | 6.79, d (8.8) | 6.84, d (8.7) | ||||
| 5′ | 6.72, d (8.6) | 6.79, d (8.7) | 6.74, d (8.4) | 6.79, d (8.8) | 6.73, d (8.3) | 6.84, d (8.7) | 6.80, d (8.5) |
| 6′ | 6.75, d (8.6) | 7.28, d (8.7) | 6.76, dd (8.4, 1.6) | 7.29, d (8.8) | 6.76, dd (8.3, 2.0) | 7.97, d (8.7) | 7.54, dd (8.5, 2.0) |
| 6″ | 6.71, s | 6.69, s | 6.67, s | 6.16, s | |||
| 8″ | 7.01, s | 7.00, s | |||||
| 2‴ | 8.08, d (8.5) | 7.72, d (1.9) | 8.16, d (8.8) | 7.88, d (2.2) | 7.89, d (2.0) | 7.97, d (8.7) | 8.01, d (8.8) |
| 3‴ | 6.94, d (8.5) | 6.94, d (8.8) | 6.84, d (8.7) | 6.83, d (9.0) | |||
| 5‴ | 6.94, d (8.5) | 6.91, d (8.5) | 6.94, d (8.8) | 6.90, d (8.5) | 6.90, d (8.6) | 6.84, d (8.7) | 6.83, d (9.0) |
| 6‴ | 8.08, d (8.5) | 7.58, dd (8.5, 1.9) | 8.16, d (8.8) | 7.64, dd (8.5, 2.2) | 7.65, dd (8.6, 2.0) | 7.97, d (8.7) | 8.01, d (8.8) |
| 8-CH2-8″ | 4.22, s | 4.24, s | |||||
| 5-OH | 12.53, s | 12.52, s | |||||
| 5″-OH | 12.53, s |
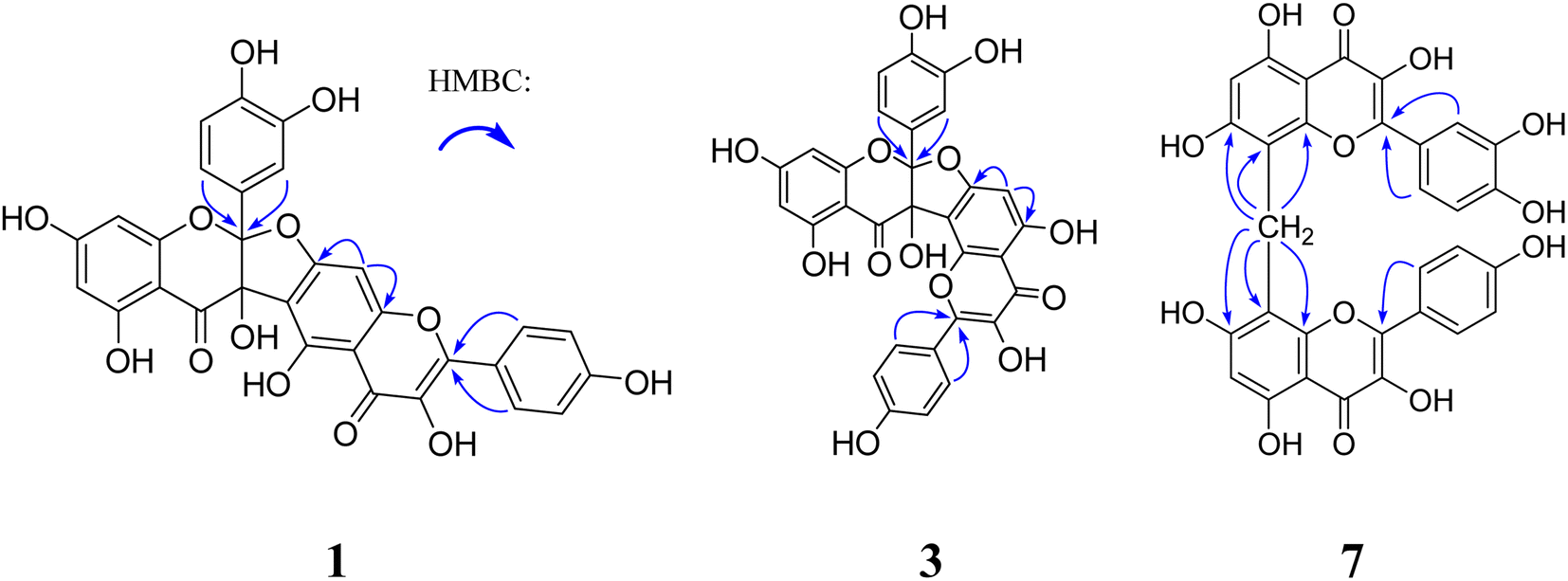 | ||
| Fig. 2 Key HMBC correlations of compounds 1, 3 and 7. | ||
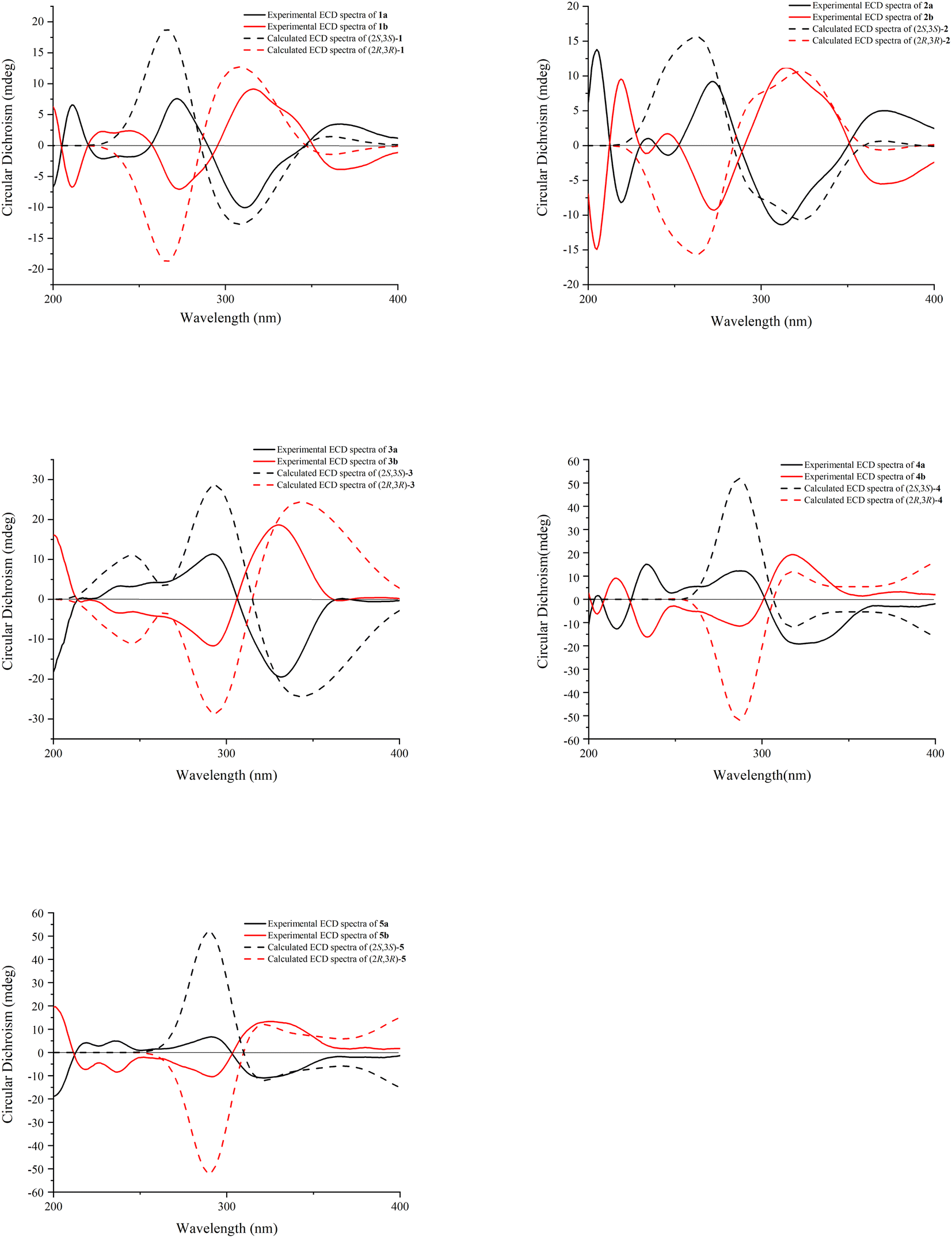 | ||
| Fig. 3 Experimental and calculated ECD spectra of compounds 1a–5a and 1b–5b. | ||
Compound 2 was obtained as a yellow amorphous powder. It gave the same molecular formula C30H18O13 as that of 1, based on the positive HR-ESI-MS ion observed at m/z 587.0824 [M + H]+ (Calcd 587.0826). Its 13C and 1H NMR spectra (Tables 1 and 2) were quite similar to those of 1, except that quercetin and dihydrokaemferol subunit in 2 were observed instead of kaempferol and dihydroquercetin subunit respectively in 1. Those were further supported by HMBC cross peak of the aromatic protons δH 7.72 (1H, d, J = 1.9 Hz, H-2‴) and 7.58 (1H, dd, J = 8.5, 1.9 Hz, H-6‴) with C-2″ (δC 148.0), of the aromatic proton δH 7.28 (2H, d, J = 8.7 Hz, H-2′, 6′) with C-2 (δC 117.8). The lack of optical activity and Cotton effect in the ECD spectrum indicated that 2 was a racemic mixture. Chiral HPLC resolution of 2 gave 2a and 2b in a ratio of approximately 1:1. Comparison between the experimental and calculated ECD spectra of two enantiomers allowed the absolute configurations of 2a and 2b to be established as (2S,3S) and (2R, 3R), respectively. Thus, compounds 2a and 2b were determined as (2S,3S)-3,5,7,4′-tetrahydroxyflavanone-(2-O-7″:3-6″)-3″,5″,3‴,4‴-tetrahydroxyflavone and (2R,3R)-3,5,7,4′-tetrahydroxyflavanone-(2-O-7″:3-6″)-3″,5″,3‴,4‴-tetrahydroxyflavone, and named as (+)-dysosmabiflavonoid B and (−)-dysosmabiflavonoid B, respectively.
Compounds 3 and 4 were obtained as yellow, amorphous powders. They possessed the same molecular formula C30H18O13 as that of 1 and 2, as revealed from their HR-ESI-MS analyses (m/z 587.0808 [M + H]+ in 3; m/z 587.0800 [M + H]+ in 4). Their 13C and 1H NMR data (Tables 1 and 2) were quite similar to those of 1 and 2, except that the chemical shift changes of an aromatic carbon and an aromatic proton [from δC 94.8 (C-6″), δH 6.71 (1H, s, H-6″) in 3 to δC 91.1 (C-8″), δH 7.01 (1H, s, H-8″) in 1, from δC 94.7 (C-6″), δH 6.69 (1H, s, H-8″) in 4 to δC 91.1 (C-8″), δH 7.00 (1H, s, H-8″) in 2, indicating the subunits of kaempferol in 3 and quercetin in 4 were substituted at C-7 and C-8. Those were also supported by chemical shift change from δC 94.8 (C-6″), 164.9 (C-7″), and 106.3 (C-8″) in 3 to δC 98.2 (C-6), 163.9 (C-7), and 93.5 (C-8) in kaempferol [17], and from δC 94.7 (C-6″), 164.8 (C-7″), and 106.1 (C-8″) in 4 to δC 98.6 (C-6), 164.3 (C-7), and 93.8 (C-8) in quercetin.19 Compounds 3 and 4 were also racemic mixtures, and separated into 3a and 3b, and 4a and 4b, respectively. The absolute configurations of 3a, 3b, 4a, 4b were determined as (2S, 3S), (2R, 3R), (2S, 3S), (2R, 3R) by comparison of their experimental and calculated ECD spectra. Thus, compounds 3a, 3b, 4a and 4b were deduced as (2S,3S)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3-8″)-3″,5″,4‴-trihydroxyflavone, (2R,3R)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3→8″)-3″,5″,4′′′-trihydroxyflavone, (2S,3S)-3,5,7,4′-tetrahydroxyflavanone-(2-O-7″:3-8″)-3″,5″,3,4‴-tetrahydroxyflavone, and (2R,3R)-3,5,7,4′-tetrahydroxyflavanone-(2-O-7″:3-8″)-3″,5″,3‴,4‴-tetrahydroxyflavone, and named as (−)-dysosmabiflavonoid C, (+)-dysosmabiflavonoid C, (−)-dysosmabiflavonoid D, and (+)-dysosmabiflavonoid D, respectively.
Compound 5 was obtained as a yellow amorphous powder. Its 1H and 13C NMR spectra data (Tables 1 and 2) were quite analogous to those of 4, except that 1,3,4-trisubstituted benzene ring δH 6.89 (1H, d, J = 2.0 Hz), 6.73 (1H, d, J = 8.3 Hz), 6.76 (1H, dd, J = 8.3, 2.0 Hz) was observed in 5 instead of 1,4-disubstituted benzene ring in 4. This was further supported by their HR-ESI-MS, which gave an [M + H]+ quasi-molecular ion peak m/z 603.0752 (Calcd 603.0775) in 5, being 16 mass-units more than that of 4. The dihydroquercetin subunit was identified as by the HMBC cross peak of the aromatic protons δH 6.89 (1H, d, J = 2.0 Hz, H-2′) and 6.76 (1H, dd, J = 8.3, 2.0 Hz, H-6′) with C-2 (δC 117.8), one 1,2,3,5-tetra-substituted benzene ring δH 5.76 (1H, s), 5.73 (1H, s), and two oxygenated aliphatic quaternary carbons δC 117.8, 79.9. The apparent lack of a Cotton effect in ECD spectrum indicated that 5 was a racemic mixture. Compounds 5a and 5b were obtained as a pair of enantiomers by chiral-phase HPLC isolation of 5. The absolute configurations of 5a and 5b were determined as (2S, 3S) and (2R, 3R) by comparison of calculated ECD and experimental data. Thus, compound 5 was deduced as (2S,3S)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3-8″)-3″,5″,3‴,4‴-tetrahydroxyflavone and (2R,3R)-3,5,7,3′,4′-pentahydroxyflavanone-(2-O-7″:3-8″)-3″,5″,3‴,4‴-tetrahydroxyflavone, (−)-dysosmabiflavonoid E, and (+)-dysosmabiflavonoid E, respectively.
Compound 6 was obtained as a yellow amorphous powder and possessed a molecular formula C31H20O12 with twenty-two degrees of unsaturation, as revealed from its HR-ESI-MS analysis (m/z 585.1023 [M + H]+, calcd for C31H21O12, 585.1033). The IR spectrum displayed the conjugated carbonyl group (1647 cm−1), hydroxy groups (3361 cm−1), and aromatic ring (1510 cm−1). The UV spectrum exhibited absorption maxima characteristic for flavonol at 271 and 369 nm. The 1H NMR spectrum showed two sets of aromatic systems including one para-disubstituted benzene ring δH 7.97 (2H, d, J = 8.7 Hz), 6.84 (2H, d, J = 8.7 Hz), and one penta-substituted benzene ring δH 6.17 (1H, s), and one isolated aliphatic proton δH 4.22 (1H, s). The 13C NMR and HSQC spectrum revealed one aliphatic carbon δC 16.4 and one flavonol skeleton including one carbonyl group δ 176.2, two benzene rings, two oxygen-bearing olefinic carbons δC 146.9, 135.4. Its 1H NMR spectrum was almost similar to that of kaempferol, except that an extra proton signal at δH 4.22 (1H, s) was observed in 6 instead of one aromatic proton signal at δH 6.48 (1H, br s) in kaempferol.17 In combination with the molecular formula C31H20O12, two symmetrical kaempferol moieties were linked at C-8 and C-8′ by one methylene group. This was also supported by the HMBC correlations of the methylene group δH 4.22 (2H, s) with C-7 and C-7″ (δC 162.1), C-8 and C-8″ (δC 104.6), and C-9 and C-9″ (δC 153.8). Thus, compound 6 was deduced as 3,5,7,4′,3″,5″,7″,4‴-octahydroxyl-[8-CH2-8′]-biflavone, and named dysosmabiflavonoid F.
Compound 7 was obtained as a yellow amorphous powder. Its 1H and 13C NMR (Tables 1 and 2) were quite analogous to those of 6, except that one 1,3,4-trisubstituted benzene ring δH 7.82 (1H, d, J = 2.0 Hz), 6.80 (1H, d, J = 8.5 Hz), 7.54 (1H, dd, J = 8.5 2.0 Hz) was observed in 7 instead of 1,4-disubstituted benzene ring in 6. This was further supported by their HR-ESI-MS, which gave an [M + Na]+ quasi-molecular ion peak m/z 623.0799 (Calcd 623.0802) in 7, being 16 mass-units more than that of 6. The quercetin subunit was identified as by the HMBC cross peak of the aromatic protons δH 7.82 (1H, d, J = 2.0 Hz, H-2′) and 7.54 (1H, dd, J = 8.5, 2.0 Hz, H-6′) with C-2 (δ 146.7), one 1,2,3,4,5-tetra-substituted benzene ring δH 6.12 (1H, s), and two oxygenated olefinic carbons δC 146.7, 135.4. Thus, compound 7 was 3,5,7,3′,4′,3″,5″,7″,4‴-nonahydroxyl-[8-CH2-8′]-biflavone, and named dysosmabiflavonoid G.
By comparing their physical and spectroscopic data with literature values, the known metabolites were identified as kaempferol (8),17 quercetin (9),19 dihydrokaempferol (10),20 dihydroquercetin (11).20
All the isolates (1–11) were evaluated for their acetylcholinesterase inhibitory activity, using a modified spectrophotometric method.21 Tacrine was utilized as the positive control. The results are listed in Table 3 as IC50 values. Based on the IC50 values, all racemates 1–5 exhibited satisfactory AChE inhibitory activity in the range of micromolar concentrations (IC50 values 2.72–6.74 µM), and more potent inhibitory activity than their corresponding levorotatory or dextrorotatory enantiomers. Among all of biosynthetically related precursors (8–11), only quercetin (9) are more active than racemates 2–5. The combination between quercetin and dihydrokaempferol or between quercetin and dihydroquercetin by a furan ring reduced the inhibitory activity, for example, compared 2, 4 and 5 to 9. However, this phenomenon is inverse the combination between kaempferol and dihydrokaempferol or between kaempferol and dihydroquercetin, for example, compared 1 and 3 to 8. Hydrogenation (10 and 11) at C-2 and C-3 drastically reduced the inhibitory activity of the flavonol derivatives (8 and 9). The hydroxy group at C-3′ is important, as 3′,4′-dihydroxy derivative 9 showed more potent activity than its corresponding 4′-hydroxy analogue 8. The methylene linkage bridge between flavonol skeletons plays a very important role in maintaining inhibitory activity for this series of biflavonoids, for example, compared 8 and 9 to 7, and 8 to 6. In terms of the structure, racemates 1–5 are different completely from general biflavonoids. Moreover, due to the similarity in AChE inhibitory activity, they can be used as a template for the development of new drugs against neurodegenerative diseases.
| No. | IC50 (µM) | No. | IC50 (µM) |
|---|---|---|---|
| 1 | 2.72 ± 0.25 | 5 | 6.74 ± 0.04 |
| 1a | 32.95 ± 2.90 | 6 | 1.42 ± 0.14 |
| 1b | 6.25 ± 0.57 | 7 | 0.73 ± 0.07 |
| 2 | 4.55 ± 0.36 | 8 | 17.90 ± 0.18 |
| 2a | 4.97 ± 1.80 | 9 | 3.96 ± 0.27 |
| 3 | 6.39 ± 0.48 | 11 | 43.95 ± 2.43 |
| 3a | 10.65 ± 1.43 | 2b, 4a, 4b, 5a, 5b, 10 | >50 |
| 3b | 9.89 ± 0.71 | Tacrine | 0.21 ± 0.01 |
| 4 | 6.33 ± 0.45 |
Racemates 1–5 and their corresponding levorotatory and dextrorotatory enantiomers were measured for the potential to impede the generation of NO in lipopolysaccharide-stimulated RAW264.7 cells,22 and mushroom tyrosinase inhibitory effect.23 Unfortunately, they were inactive (IC50 > 50 µM) against the inhibition of NO production. Only racemates 1 and 2 were found to display potent mushroom tyrosinase inhibitory activity with IC50 values of 28.27 µM and 39.75 µM (the positive control, kojic acid, 32.59 µM), respectively, whereas racemates 3–5 displayed no inhibitory effect against tyrosinase activity. The variation in inhibitory activity between them indicates the linkage bridges of C-2-O-C-7″:C-3-C-6″ were structurally required for anti-tyrosinase activity. Racemate 1, as the most potent tyrosinase inhibitor, has the therapeutic potential for skin hyperpigmentation and depigmentation in cosmetics.
Conclusions
So far, more than 600 natural biflavonoids have been found, and the vast majority of them are isolated from the plants of families Clusiaceae, Thymelaeaceae, Ochnaceae, and Selaginellaceae, etc.24 Only eleven biflavonoids have been obtained from plants of the genus Dysosma.13–16 Our phytochemical investigations on D. versipellis resulted in the isolation of five pairs of new biflavonoid enantiomers, (±)-dysosmabiflavonoids A–E (1–5), two new biflavonoids, dysosmabiflavonoids F–G (6–7), along with four biosynthetically related precursors (8–11). Racemates 1–2 represent the first report of natural C-2-O-C-7″:C3-C6″ biflavonoids. Based upon the IC50 values, compound 7 was the most potent AChE inhibitor among all isolated compounds. Further studies on compound 7 are necessary to action mechanism and in vivo efficacy in neurodegenerative diseases. Above all, the present investigation will broaden the application field of D. versipellis.Experimental method
General experimental procedures
Optical rotations and ECD spectra were measured by a Rudolph AP-IV polarimeter and an Applied Photophysics Chirascanq CD spectropolarimeter, respectively. UV and IR spectra were acquired using a ThermoEVO 300 spectrometer and a ThermoNicolet IS 10 spectrometer, respectively. NMR and mass spectra were recorded on a Bruker Avance III 500 spectrometer and a Bruker maXisHD mass spectrometer, respectively. Preparative HPLC separations were performed on a SEP system (Beijing Sepuruisi scientific Co., Ltd, China) equipped with a variable-wavelength UV detector, using a YMC-Pack ODS-A column (250 × 20 mm, 5 µm) and a Daicel chiral PAK AD-H column (250 × 10 mm, 5 µm). Sephadex LH-20 (40–70 µm), and silica gel (160–200 mesh) were acquired from Amersham Pharmacia Biotech AB, Uppsala, Sweden, and Marine Chemical Industry, Qingdao, China, respectively. Chemical reagents for isolation were of analytical grade and purchased from Tianjin Siyou Co., Ltd, China. Biological reagents were from Sigma Company.Plant material
The roots and rhizomes of D. versipellis were collected in Qingzhen, Guizhou Province, China, in July 2019, and identified by Prof. Cheng-Ming Dong at School of Pharmacy, Henan University of Chinese Medicine, where a voucher specimen (DV 20190706) was deposited.Extraction and isolation
The powder of air-dried roots and rhizomes of D. versipellis (40 kg) were refluxed with 95% EtOH (v/v 120 L × 3, 1.5 h each) and 50% EtOH (v/v 120 L × 1, 1.5 h each) at 95 °C, respectively. The filtrate was concentrated under reduced pressure to yield a dark brown residue (5.4 kg). The residue was adsorbed by silicious earth and fractioned by CH2Cl2, EtOAc, and MeOH. The MeOH extract (3.4 kg) was separated into nine fractions M1–M9 by silica gel column chromatography (CC) with a gradient of CH2Cl2–MeOH (v/v 100:0, 100:1, 100:3, 100:5, 100:7, 100:10, 100:30, 100:50, 0:100) based on TLC monitoring results. Fraction M5 (110.0 g) was subjected to sephadex LH-20 CC eluted by methanol to yield subfractions M5-1–M5-8. Subfraction M5-8 (42.5 g) was submitted to silica gel CC eluted by PE–acetone (100:10, 100:20, 100:30, 100:40, 100:50, 100:70, 1:1, 1:2, 1:3) to afford nine subfractions (M5-8-1–M5-8-9). Subfraction M5-8-4 (0.5 g) was isolated by preparative HPLC (MeOH:H2O, 60:40) at a flow rate of 3 mL min−1 to give compound 8 (tR 40.5 min, 10.0 mg) and 6 (tR 47.2 min, 6.0 mg). Subfraction M5-8-5 (1.07 g) was separated by preparative HPLC (MeOH:H2O, 55:45) at a flow rate of 3 mL min−1 to afford subfractions [M5-8-5-1, (±) 2, tR 37.4 min, 8.5 mg], (M5-8-5-2, tR 46.0 min, 78.2 mg), (M5-8-5-3, tR 72.2 min, 90.1 mg), [M5-8-5-4, (±) 3, tR 77.0 min, 12.9 mg]. Subfraction M5-8-5-1 was isolated by preparative chiral HPLC (cyclohexane:isopropanol, 60:40) at a flow rate of 2 mL min−1 to give 2a (tR 7.9 min, 3.6 mg) and 2b (tR 18.0 min, 3.6 mg). Subfraction M5-8-5-2 was chromatographed by preparative HPLC (CH3CN:H2O, 45:55) at a flow rate of 3 mL min−1 to give subfractions [M5-8-5-2-1, (±) 5, tR 25.7 min, 6 mg], [M5-8-5-2-2, (±) 1, tR 29.1 min, 9.4 mg] and compound 11 (tR 20.6 min, 10.9 mg). Subfraction M5-8-5-2-1 was isolated by preparative chiral HPLC (cyclohexane:isopropanol, 81:19) at a flow rate of 2 mL min−1 to give 5a (tR 31.8 min, 2.4 mg) and 5b (tR 43.8 min, 2.4 mg). Subfraction M5-8-5-2-2 was subjected to preparative chiral HPLC (cyclohexane:isopropanol, 60:40) at a flow rate of 2 mL min−1 to give 1a (tR 7.8 min, 4.2 mg) and 1b (tR 21.4 min, 4.2 mg). Subfraction M5-8-5-3 was isolated by preparative HPLC (CH3CN:H2O, 43:57) at a flow rate of 3 mL min−1 to give subfractions [M5-8-5-3-1, (±) 4, tR 26.9 min, 5.5 mg] and 10 (tR 22.5 min, 5.5 mg). Subfraction M5-8-5-3-1 was isolated by preparative chiral HPLC (cyclohexane: isopropanol, 81:19) at a flow rate of 2 mL min−1 to give 4a (tR 20.8 min, 2.5 mg) and 4b (tR 30.2 min, 2.5 mg). Subfraction M5-8-5-4 was submitted to preparative chiral HPLC (cyclohexane:isopropanol, 70:30) at a flow rate of 2 mL min−1 to give 3a (tR 9.7 min, 5.0 mg) and 3b (tR 12.7 min, 5.0 mg). Fraction M6 (130.0 g) was subjected to sephadex LH-20 CC eluted by methanol to yield subfractions M6-1, M6-2 and 11 (5 mg). The subfraction M6-2 (60.5 g) was applied to silica gel CC with a CH2Cl2–MeOH (100:1, 100:3, 100:5, 100:7, 100:10, 100:30) gradient to give six subfractions (M6-2-1–M6-2-6). Further separation of M6-2-4 (1.69 g) using sephadex LH-20 CC eluted by MeOH resulted in six subfractions M6-2-4-1–M6-2-4-6. Subfractions M6-2-4-4 (0.59 g) was purified by preparative HPLC (MeOH:H2O, 60:40) at a flow rate of 3 mL min−1 to afford compounds 7 (tR 24.1 min, 3.0 mg) and 9 (tR 20.4 min, 8.0 mg).
AChE inhibitory assay in vitro
The AChE inhibitory activity was measured by a classical spectrophotometric method with minor modifications.21 A 200 µL volume reaction system containing phosphate buffer (pH 8.0), tested compounds at different concentrations, and AChE (0.02 U mL−1), was incubated for 20 min at 37 °C. Then, the reaction was initiated by the addition of 40 µL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 30 seconds for one hour. All the reactions were performed in triplicate. The IC50 value of each compound was calculated by the Reed-Muench's method.Assay for LPS-induced NO production
Test protocols were carried out as previously described.22 RAW 264.7 macrophages cells (2 × 105 cells per well) were precultured in 96-well microplates for 24 h. The tested compounds (50 µM) and L-NMMA with 1 µg mL−1 LPS were added and incubated at 37 °C for another 18 h. Nitric oxide production was determined by the Griess Reagent System. L-NG-monomethyl arginine (L-NMMA) was used as a positive control.Assay for tyrosinase inhibitory activity
Tyrosinase inhibitory activity was measured using an established assay protocol, with slight modification.23 The tested compound was dissolved phosphate buffer solution containing 0.2% DMSO. The 60 µL of phosphate buffer (20 mM, pH 6.8) and 50 µL of the tested compounds at different concentrations, and 50 µL of mushroom tyrosinase solution (20 µg mL−1) were placed into each well of a 96-well cell culture plate. After incubation for 10 min at 25 °C, 40 µL of L-dopa (2.0 mM) was added to the reaction system. The reaction mixture was incubated for another 5 min, and the absorbance was measured at 490 nm. Kojic acid was used as standard agent. The IC50 value of each compound was calculated as the concentration of the sample when the residual enzyme activity is 50%.ECD computational method
All the quantum chemical ECDs were calculated by the literature method.25 Conformational searches of compounds 1a–5a and 1b–5b were investigated by Spartan software using the MMFF94S force field, which gave four stable conformers, respectively. Then, those selected conformers were further optimized by density functional theory method at the B3LYP/6-31G* level. The ECD of these optimized conformers were calculated by the TDDFT method at the B3LYP/6-31+G* level with a CPCM model in MeOH solvent.Author contributions
R. J. Han, H. Y. Bai and H. J. Wang performed the experiments, data analysis, and experimental planning. M. Li, Y. Y. Si, J. M. Wang, J. H. Gong and H. Chen screened the biological activities. The project was conceived and supervised by Y. J. Sun and W. S. Feng. The manuscript was written by Y. J. Sun. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Basic Science Foundation of Henan University of Chinese Medicine (No. 2014KYYWF-QN26), Science and Technology Innovation Talent Support Scheme of Henan University of Chinese Medicine (No. 2016XCXRC01), Scientific and Technological Key Project in Henan Province (No. 192102310438), and Research Project on Chinese Medicine Science in Henan Province (No. 20-21ZY1039).Notes and references
- S. H. Lu, J. W. Wu, H. L. Liu, J. H. Zhao, K. T. Liu, C. K. Chuang, H. Y. Lin, W. B. Tsai and Y. Ho, J. Biomed. Sci., 2011, 18, 8 CrossRef CAS PubMed.
- M. Rosini, V. Andrisano, M. Bartolini, M. L. Bolognesi, P. Hreli, A. Minarini, A. Tarozzi and C. Melchiorre, J. Med. Chem., 2005, 48, 360–363 CrossRef CAS PubMed.
- M. Przybylowska, K. Dzierzbicka, S. Kowalski, K. Chmielewska and I. Inkielewicz-Stepniak, Curr. Neuropharmacol., 2021, 19, 1323–1344 CrossRef CAS PubMed.
- B. C. Guan, C. X. Fu, Y. X. Qiu, S. L. Zhou and H. P. Comes, Am. J. Bot., 2010, 97, 111–122 CrossRef CAS PubMed.
- C. X. Liu, C. N. Zhang, T. He, L. Sun, Q. Wang, S. Han, W. X. Wang, J. Kong, F. L. Yuan and J. M. Huang, Ecotoxicol. Environ. Saf., 2020, 190, 110073 CrossRef CAS PubMed.
- X. Q. Xu, X. H. Gao, L. H. Jin, P. S. Bhadury, K. Yuan, D. Y. Hu, B. A. Song and S. Yang, Cell Div., 2011, 6, 14 CrossRef CAS PubMed.
- L. Man, Y. Liu, S. Ananda, Z. Luo and L. Liu, J. Med. Plants Res., 2010, 4, 717–721 Search PubMed.
- N. W. Lu, Q. An, N. Li and Y. M. Dong, J. Chromatogr. Sci., 2014, 52, 514–519 CAS.
- Y. C. Shi, H. P. Yuan, R. Zou and B. B. Liu, Mitochondrial DNA, 2019, 4, 4218–4219 CrossRef PubMed.
- H. L. Fan, J. B. Xuan, K. X. Zhang and J. L. Jiang, Anal. Methods, 2018, 10, 371–380 RSC.
- B. He, Y. Chen, H. Zhang, C. Y. Xia, Q. Zhang and W. Li, Hortic. Environ. Biotechnol., 2018, 59, 519–528 CrossRef CAS.
- X. M. Tan, Y. Q. Zhou, X. L. Zhou, X. H. Xia, Y. Wei, L. L. He, H. Z. Tang and L. Y. Yu, Sci. Rep., 2018, 8, 5929 CrossRef PubMed.
- R. D. Chen, R. G. Duan, Y. N. Wei, J. H. Zou, J. W. Li, X. Y. Liu, H. Y. Wang, Y. Guo, Q. H. Li and J. G. Dai, Fitoterapia, 2015, 107, 77–84 CrossRef CAS PubMed.
- Z. Yang, Y. Q. Wu, H. Zhou, X. J. Cao, X. H. Jiang, K. W. Wang and S. H. Wu, RSC Adv., 2015, 5, 77553–77564 RSC.
- Z. Yang, Y. Q. Wu and S. H. Wu, J. Chromatogr. A, 2016, 1431, 184–196 CrossRef CAS PubMed.
- Z. Yang, P. P. Guo, R. Han and J. M. Gao, J. Sep. Sci., 2018, 41, 3631–3643 CrossRef CAS PubMed.
- D. D. Zheng, J. Y. Ruan, Y. Zhang, J. J. Yan, T. Wang and Y. Zhang, Chin. J. Med. Chem., 2020, 30, 542–548 Search PubMed.
- K. M. Canuto, L. K. A. M. Leal, A. A. Lopes, C. M. Coleman, D. Ferreira and E. R. Silveira, J. Braz. Chem. Soc., 2014, 25, 639–647 CAS.
- M. Q. An, L. R. Li, C. R. Li, S. Y. Zhong, H. Y. Jiang, L. P. Wu and J. Huang, West China J. Pharm. Sci., 2019, 34, 577–582 Search PubMed.
- L. Chen, J. F. Xu and L. C. Sun, Chin. Med. Mater., 2016, 39, 90–93 CAS.
- C. L. Yang, H. M. Wu, C. L. Liu, X. Zhang, Z. K. Guo, Y. Chen, F. Liu, Y. Liang, R. H. Jiao, R. X. Tan and H. M. Ge, J. Nat. Prod., 2019, 82, 792–797 CrossRef CAS PubMed.
- Z. Z. Zhao, Q. L. Zhao, W. S. Feng, H. R. He, M. Li, G. M. Xue, H. P. Chen and J. K. Liu, RSC Adv., 2021, 11, 18693–18701 RSC.
- Y. J. Zhai, G. M. Huo, Q. Zhang, D. Li, D. C. Wang, J. Z. Qi, W. B. Han and J. M. Gao, J. Nat. Prod., 2020, 83, 1592–1597 CrossRef CAS PubMed.
- X. Q. He, F. Yang and X. A. Huang, Molecules, 2021, 26, 6088 CrossRef CAS PubMed.
- Y. Y. Si, W. W. Wang, Q. M. Feng, Z. Z. Zhao, G. M. Xue, Y. J. Sun, W. S. Feng, J. I. Yong and X. S. Wang, RSC Adv., 2021, 11, 27085–27091 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, HR-ESI-MS, ECD, 1D and 2D NMR spectra for compounds 1–7. See DOI: https://doi.org/10.1039/d2ra06961j |
| This journal is © The Royal Society of Chemistry 2022 |