
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous catalyst modifier for alkyne semi-hydrogenation: systematic screening in an automated flow reactor and computational study on mechanisms†
Shusaku
Asano
 a,
Samuel J.
Adams
b,
Yuta
Tsuji
a,
Kazunari
Yoshizawa
a,
Atsushi
Tahara
c,
Jun-ichiro
Hayashi
a and
Nikolay
Cherkasov
*bd
a,
Samuel J.
Adams
b,
Yuta
Tsuji
a,
Kazunari
Yoshizawa
a,
Atsushi
Tahara
c,
Jun-ichiro
Hayashi
a and
Nikolay
Cherkasov
*bd
aInstitute for Materials Chemistry and Engineering, Kyushu University, Kasuga 816-8580, Japan
bStoli Chem, Prince Phillip Building, Wellesbourne, CV35 9 EF, UK
cFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, Sendai 980-8578, Japan
dSchool of Engineering, University of Warwick, Coventry CV4 7AL, UK. E-mail: n.cherkasov@warwick.ac.uk
First published on 16th May 2022
Abstract
The selectivity of palladium catalysed hydrogenation can be improved by adding a homogeneous modifier (or poison) such as quinoline to the reaction mixture. Although such selectivity improvement by modifiers (selective catalyst poisoning) has been known for decades, we still know little about them. We, ultimately, cannot select a modifier to improve a particular process. In this study, 21 types of modifiers are screened for the semi-hydrogenation of alkynes with varying catalyst type, reaction time, and target substrate using an automated flow reactor system. All of the studied variables changed affected hydrogenation activity and selectivity confirming the effectiveness of a multi-parameter optimization. 1,10-phenanthroline marked the best selectivity beyond quinoline. The density functional theory (DFT) calculations suggest that 1,10-phenanthroline has a remarkable ability to adsorb on the irregular surface of the catalyst that effects undesirable reaction.
1. Introduction
Hydrogenation is one of the most important reactions across a variety of industries from fine chemicals and pharmaceuticals1 to food production.2 Hydrogenation has lower activation energy compared with reactions such as skeletal isomerization, dehydrogenation, and hydrogenolysis; hence, hydrogenation is the main pathway once hydrogen gas and solid catalysts are in contact with a liquid substrate.3 In the majority of hydrogenation processes, complete hydrogenation of all the functional groups is desired due to the reliability, simplicity, and robustness of the reaction. In some cases, however, chemoselective hydrogenation or semi-hydrogenation of only the targeted functional group is needed. Vitamins A and E as well as several fragrances involve several semi-hydrogenation steps in their synthesis reducing triple C–C bonds in alkynes and alkynols into double C–C bonds without affecting other groups such as internal double bonds.4,5 Moreover, having a reliable, efficient, and selective route for alkyne semi-hydrogenation without major upfront R&D development could open compelling synthetic instead of multi-step processes to reduce costs and environmental impact of the synthesis. Thus, the development of catalyst and catalytic processes for selective hydrogenation is a major problem.The main approach to improve hydrogenation selectivity is an optimization of catalytic composition for the target reaction system. For example, the addition of Rh dramatically increases the catalytic activity of Pd. Rh–Pd nanoparticles can catalyze hydrogenation of arenes which pure Pd nanoparticles cannot.6 On the other hand, Bi or Pb decreases the activity of Pd as catalyst poisons and minimizes hydrogenation of alkene double bonds.7,8 Although catalyst optimization by changing its composition is effective, catalyst synthesis is a time- and labour-consuming procedure. Moreover, any larger-scale application requires complex, lengthy, expensive considerations for scaling-up catalyst synthesis and supply chain management. These problems increase technical and commercial risks of the development projects often making them unviable. Not surprisingly, a lot of selective hydrogenation processes still rely on catalysts with limited optimization for a particular molecular system.
An alternative approach of catalyst modification could use existing catalysts instead of fully engineering new ones. Quinoline9 and pyridine,10 for example, are known to improve the selectivity of alkyne semi-hydrogenation to an alkene by interacting with the catalyst surface. A pioneering example of tuning catalytic performance of heterogeneous metal catalysts is asymmetric hydrogenation catalysed by Pt/C with quinoline-derived cinchona alkaloids.11,12 A recent innovative example is for activation of inert gold nanoparticles on silica support.13 They have no activity for the hydrogenation of alkynes but show remarkable activity with piperazine. Such kinds of molecules are called homogeneous modifiers,14 ligands,15 additives,16 or poisons. The working mechanism of a modifier is not simple because there are multiple interactions between catalyst, catalyst support, reactant, solvent, and modifier. Thus, a slight change in the modifier structure, catalyst type or reaction conditions can change the yields and selectivities dramatically.15
Yet, homogeneous catalyst modifiers are not ideal either due to the additional separation and purification processes with a possibility for side-reactions.17 However, utilization of homogeneous modifiers has many benefits for rapidly developing fields of small-scale continuous production of pharmaceuticals18 and automated chemical synthesis with the assistance of artificial intelligence.19,20 First, optimization of a catalytic process with homogeneous modifiers can be decoupled from the catalyst synthesis. Usual approaches to optimize the catalytic process require the repetition of catalyst synthesis and reaction tests. In the case of highly value-added pharmaceuticals and fine chemicals, shorter process lead time has a priority than process running cost compared to the conventional bulk chemicals. Second, modifiers are in a liquid state and easy to handle by machines. Automation of liquid handling is already an established technology and can be done with readily available equipment.21 On the other hand, automation of solid handling is a challenging (that is substantially more expensive) task especially for small-scale.22 Thus, automated optimization of modifiers would be much easier and more fruitful than automated optimization of catalyst synthesis.
Understanding the reasons for catalyst modifier operation may open new ways for understanding the catalytic mechanisms and developing more efficient catalysts. The mechanistic understanding of selectivity improvement is still limited. Few examples on this topic involve a density functional theory (DFT) study on the role of quinoline to the acetylene semi-hydrogenation on the Lindlar catalyst23 and our direct liquid-phase adsorption study featuring selective sites and unselective sites of Pd catalyst.8 The former study revealed that quinoline isolates the adsorbed reactants to prevent oligomerization. The latter study proposed the model of two different types of active sites of selective sites and unselective sites. Quinoline is purported to block unselective sites similar to the Pb addition to Pd which weakens the reactivity of the unselective sites. The relationship between modifier structure and performance, the interaction between reactants and modifiers, and the rationalized strategy for using modifiers are still unclear.
In this study, we develop a system for automatic screening of catalyst modifiers and perform a systematic screening of N-containing base molecules as modifiers. An adsorption study with a DFT calculation was also conducted to elucidate the working mechanisms.
2. Methodology
2.1 Experimental
Alkyne semi-hydrogenation to a corresponding alkene with H2 gas was conducted using Pd catalysts. 3 alkynyl alcohols shown in Fig. 1 are used as substrates. These are excellent model compounds of multifunctional alkynes used in fine chemistry. These materials themselves, moreover, are important in the production of vitamins A and E.5 The reaction scheme for 2-methyl-3-butyn-2-ol (MBY) is shown in Scheme 1 as an example. | ||
| Fig. 1 Alkynyl alcohol substrates used in this study. | ||
 | ||
| Scheme 1 Example hydrogenation of MBY. | ||
Semi-hydrogenation produces the corresponding alkenyl alcohol 2-methyl-3-buten-2-ol (MBE). Further hydrogenation converts MBE to the undesirable 2-methyl-2-butanol (MBA). 21 nitrogen containing aromatic and aliphatic molecules shown in Fig. 2 were examined as modifiers. Alkynyl alcohols were provided from DSM (Heerlen, Netherlands). All the other reagents were purchased and used without additional purification.
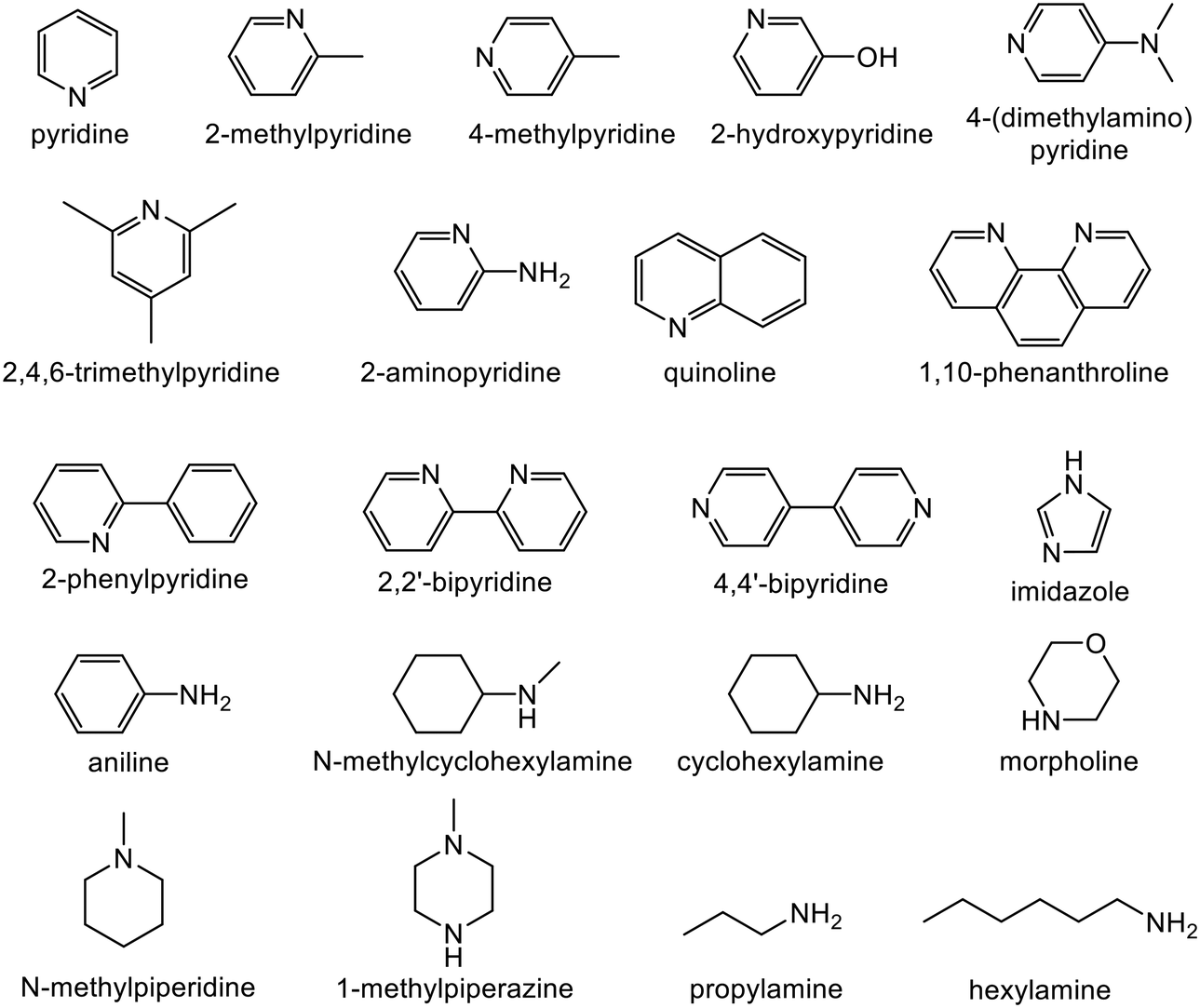 | ||
| Fig. 2 List of N-containing homogeneous modifiers studied. | ||
Hydrogenation was conducted using an bespoke automated flow reactor system based on the OpenFlowChem platform.24,25Fig. 3 illustrates the system configuration. A substrate solution was prepared at 1 mol L−1 concentration in isopropanol. There were 3 HPLC pumps that fed (i) substrate solution, (ii) solution of the modifier, and (iii) isopropanol solution. Hydrogen gas flow was supplied via a mass flow controller and was fed at a fixed substrate to hydrogen molar ratio of 1.10 (H2 excess) for MBY, and 1.20 for DLL and DIP to increase conversion hampered by decreased selectivity. A 12-way valve selected a desired catalyst modifier out of a small library of modifiers. Modifiers were prepared at the concentration of 1 mol L−1 in isopropanol and placed in vials. The system adjusted the flow rates of the components to maintain 0.1 mol L−1 substrate and 0.01 mol L−1 modifier concentrations. The total flow rate of liquid was varied from 3 mL min−1 to 6 mL min−1.
 | ||
| Fig. 3 Automated flow reactor system used in this study. | ||
A catalyst-coated tube reactor (1.27 mm inner diameter, 1.6 mm outer diameter, 1 m length) provided by Stoli Chem Ltd was placed inside an oven at 70 °C. The reaction took place at the ambient pressure. In the reactor, 5 wt% loading Pd/C or Pd/ZnO was coated with 10 μm thickness. The catalyst coating had been obtained using an improved sol–gel procedure described in a reference.26 Detailed characterisations of these catalysts are reported in other references.27,28 The samples collected with a fraction collector were analysed offline analysis with a gas chromatograph (Shimadzu GC-2010) equipped with a flame ionization detector and a Stabilwax column. The yield of oligomers was negligible judging from obtained GC chromatograms. Oligomerization is a major side-reaction in the case reaction with neat alkyne17 or reaction in gas-phase23 but negligible with isopropanol solvent.8
We have previously developed flow reactor systems equipped with in-line GC24 and HPLC29 that take aliquots from the reactor outlet and directly inject them into the chromatography columns (note that in a strict classification,30 they are called at-line analysis). Such technic minimizes human operation and enables automated feedback to the design of experiment algorithms. However, reproducibility was not excellent for hydrogenation being ±10% likely because some gas droplets were inadvertently collected.24 Thus, off-line GC analysis was employed in the current study.
2.2 Computational
The surfaces of Pd(110) and Pd(210) were modelled by periodic (4 × 3) unit cell slabs consisting of six and ten atomic layers, respectively. A 20 Å thick vacuum space was added on the surfaces. The lower half atoms in the slabs were kept fixed at their optimized bulk positions. The optimized adsorption structures were visualized using VESTA.31The initial structures for geometry optimization at the DFT level were generated using the quench dynamics method with the Forcite module implemented in the Materials Studio software.32 The COMPASS force field33 was used with the conditions of an initial temperature of 300 K, NVE ensemble, the time step of 1 fs, and the simulation time of 1 ns. The quench-step number was set to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000: 101 optimized structures were obtained for each quench dynamics run, and then the most stable structure was selected.
000: 101 optimized structures were obtained for each quench dynamics run, and then the most stable structure was selected.
The Vienna ab initio simulation package (VASP 5.4.4)34–36 was used to perform the DFT calculation. The Perdew–Burke–Ernzerhof (PBE)37 exchange-correlation functional was used. The Kohn–Sham equations were solved with a plane-wave basis set using the projector-augmented wave method.38,39 The cutoff energy for the plane-wave basis set was set to 500 eV. The convergence threshold for self-consistent field iteration was set to 1.0 × 10−5 eV. The atomic coordinates were relaxed until the forces on all of the atoms were less than 0.03 eV Å−1. The Γ-centered k-point meshes with k spacing of 2π × 0.05 Å−1 were employed for sampling the Brillouin zone. Grimme's D3 dispersion correction formalism with Becke–Johnson damping was adopted.40
3. Results & discussion
3.1 General trend of modifier effect with varying flow rate
First, Pd/ZnO and Pd/C catalysts were compared with the semi-hydrogenation of MBY. The Pd/C catalyst is widely used in full hydrogenation but often has limited selectivity in semi-hydrogenation; the Pd/ZnO catalyst,41–43 on the contrary, typically has a significantly higher selectivity without any modifiers. When no catalyst modifier was added, Pd/ZnO catalyst achieved much higher selectivity with almost the same conversion. The superior performance of ZnO as support for semi-hydrogenation would be due to the intermetallic Pd-Zn phase formation.17,44,45 Pd–Zn alloy is considered to modify the adsorption strength of alkynes and alkenes.The full results for all the catalysts and catalyst modifiers are shown in Table S1–S4† in the ESI† with notable examples presented in Fig. 4. In all cases with the increasing MBY flow rate, the conversion decreased and selectivity increased to a plateau. This behaviour is well known and explained by the reduced residence time and simultaneously higher substrate to catalyst ratio at a higher flow rate that results in lower conversion. The conversion over the Pd/ZnO catalyst was slightly higher compared to Pd/C which may be a combination of PdZn (ref. 42, 44 and 46) alloying or purely extensive factors (larger Pd area) and is of no interest for the work. The selectivity at low conversion over many catalysts41,42,47 is typically high and decreases as full conversion is reached.
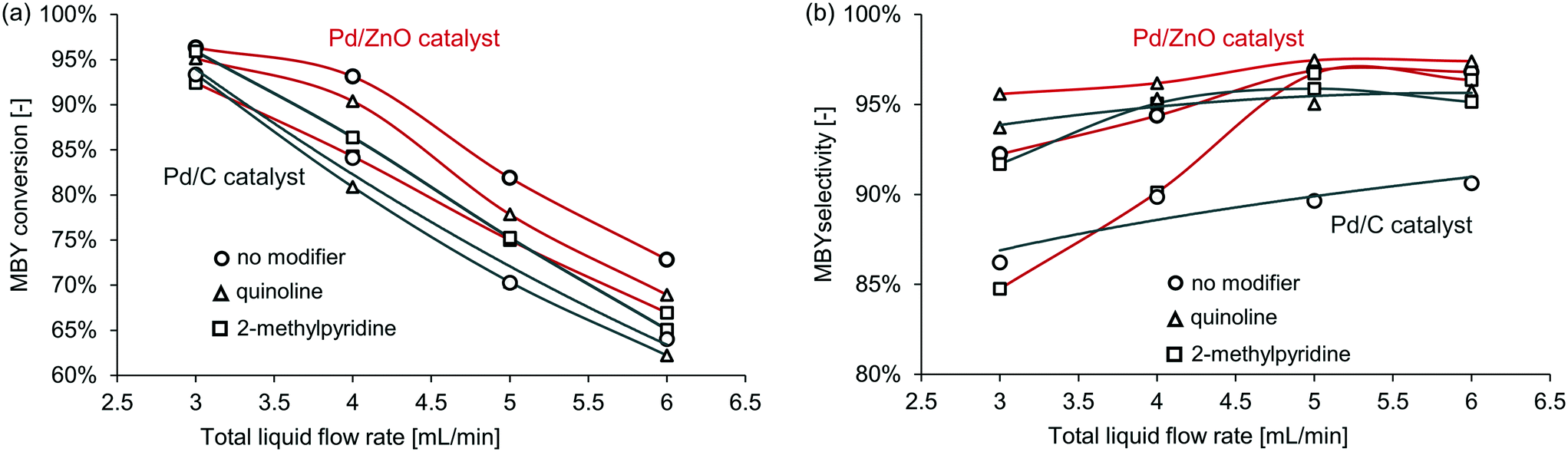 | ||
| Fig. 4 The effect of selected catalyst modifier on (a) conversion and (b) selectivity in MBY semi-hydrogenation over the (black) Pd/C and (red) Pd/ZnO catalysts. | ||
Fig. 4 shows the relative effect of the modifiers on the catalyst activity and selectivity – comparing the performance with the modifiers to that of the non-modified catalysts. In the case of the Pd/ZnO catalyst, the addition of the modifiers decreased conversion obviously due to blocking some of the active sites. In the case of the Pd/C catalyst, however, the addition of 2-methylpyridine consistently increased conversion – surprisingly, the catalysts became more active. The mechanistic origins of this effect require a separate study and are outside of the scope of this study.
Similarly, unpredictable behaviour of the modifiers was observed on the catalyst selectivity. The quinoline-modified catalysts showed the highest selectivity for both Pd/ZnO and Pd/C. Yet, the relative increase in selectivity was different. The selectivity with the Pd/ZnO catalyst and quinoline was higher than that of the Pd/C catalyst and quinoline. Interestingly, 2-methylpyridine behaved unexpectedly even here – the selectivity decreased in the case of the Pd/ZnO catalyst but not for Pd/C.
Therefore, the effect of catalyst modifiers on various heterogeneous catalysts (even similar catalysts) is very difficult to predict. The modifiers may increase or decrease both activity and selectivity. However, if the catalyst without modifiers shows high selectivity, its performance is often could be improved further with the homogeneous modifiers.
3.2 The effect of catalyst modifiers for semi-hydrogenation of similar molecules
Table 1 shows the effect of the catalyst modifiers on MBY semi-hydrogenation over Pd/C and Pd/ZnO as well as semi-hydrogenation of other substrates bearing the β-hydroxy alkyne motif over the more selective Pd/ZnO catalyst. The data presented as a difference with the corresponding non-modifier cases showing the percentage change in conversion and selectivity with the modifier addition. For example, the addition of quinoline to the Pd/C catalysts resulted in a conversion decrease of 1.8%; from 64.0% (non-modified case) to 62.2% (with quinoline). The results show 7 of the studied modifiers for the flow rates of 3 mL min−1 and 6 mL min−1. The other datasets are listed in the ESI.†| a Conversion or selectivity change compared to the case without a modifier. The “no modifier” cases provide absolute values. b Reactions are performed at 6 mL min−1 to highlight the case of low-conversion relevant for selectivity comparisons. c Reactions are performed at 3 mL min−1 for production-relevant selectivity comparisons at high conversion. |
|---|
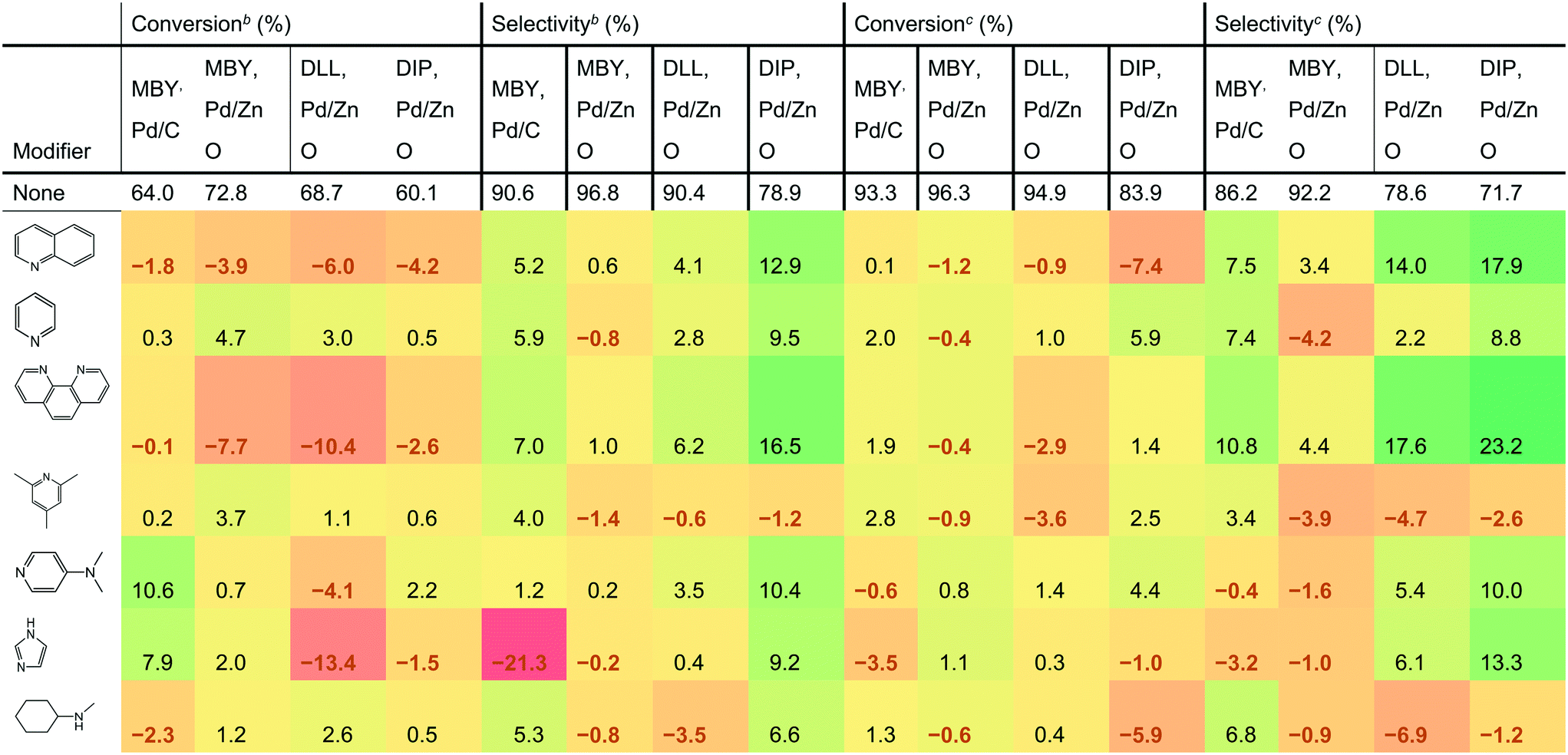
|
DLL and DIP hydrogenation was examined over the Pd/ZnO catalyst to elucidate the substrate dependency on catalyst modification. DLL has more possible side-reactions than MBY due to another alkene group. DLL resulted in a conversion similar to MBY (94.9% for DLL and 96.3% for MBY, value at 3 mL min−1) but resulted in lower selectivity (78.6% for DLL and 94.2% for MBY) as expected. DIP is much larger molecule with (C20) than the others (C5 and C10). DIP hydrogenation showed both a lower conversion of 83.9% and selectivity of 71.7% at 3 mL min−1 probably owing to disordered adsorption on the Pd surface with bulky chain. Correlation between the DLL, DIP, and MBY hydrogenation performance was observed previously.25,48,49 In both cases, the addition of catalyst modifiers changed selectivity dramatically. The largest increase in DLL and DIP selectivity was 17.6% and 23.2%, respectively, observed at the flow rate of 3 mL min−1. The effect on modifier is dependent on the substrate. For example, the addition of imidazole decreased the selectivity of MBY both for Pd/C and Pd/ZnO cases but increased that of DLL and DIP. Clear dependency on the catalyst type was also confirmed. For example in the cases of pyridine and N-methylcyclohexylamine, the selectivity of MBY was improved with the Pd/C catalyst but decreased with Pd/ZnO catalyst.
Quinoline is the most common catalyst modifier used to improve semi-hydrogenation selectivity. However, its effect in improving selectivity was not the highest. Indeed, 1,10- phenanthroline was the best performing among the tested modifiers and remarkably improved the selectivity for all the substrates. It is noteworthy that the increased performance of 1,10-phenanthroline compared with quinoline can be explained neither by the number of N atoms. Other molecules having two N atoms such as bipyridine and N-methylcyclohexylamine resulted in a lower selectivity than quinoline.
The addition of 1,10-phenanthroline resulted in significantly decreased conversion at 6 mL min−1 (−10.4% compared with no modifier). However, with the higher conversion at 3 mL min−1, a positive effect on the conversion was confirmed except for the DLL case. The improvement in conversion is surprising considering that previous studies reported a strong poisoning effect of 1,10-phenanthroline. For example, Sajiki et al. reported complete deactivation of Pd/C catalyst with 1,10-phenanthroline for the hydrogenolysis of benzyl ether.50 Bayram et al. used the 1,10-phenanthroline as a quantitative catalyst poison that perfectly blocks active sites of Rh nanoparticles.51 The nature of the study required frequent return to the reference non-modified conditions and we observed no significant (above 1% relative activity change) deactivation over the course of the study with all the catalysts.
To visualize the general trends of the modifier effect, all the results are plotted in Fig. 5. A general tendency that higher selectivity brought lower conversion can be seen at 6 mL min−1 but, interestingly, was not observed at 3 mL min−1. At this flow rate, conversion was improved in most cases, especially for DIP. Basicity of the modifier is the first factor that can accelerate the reaction16 but no clear correlation was found in between the pKa value of the modifier and the conversion change as shown in the ESI.† Fiorio et al. revealed the catalysis of piperazine on the gold nanoparticle for hydrogenation by the formation of frustrated Lewis pairs with a target substrate.13 In the case of asymmetric hydrogenation, activation of substrates by hydrogen bonding network with cinchona alkaloid modifiers is reported.52 Such electronic or structural interaction between the modifier and the substrate on the catalyst surface may exist and will be studied in the future.
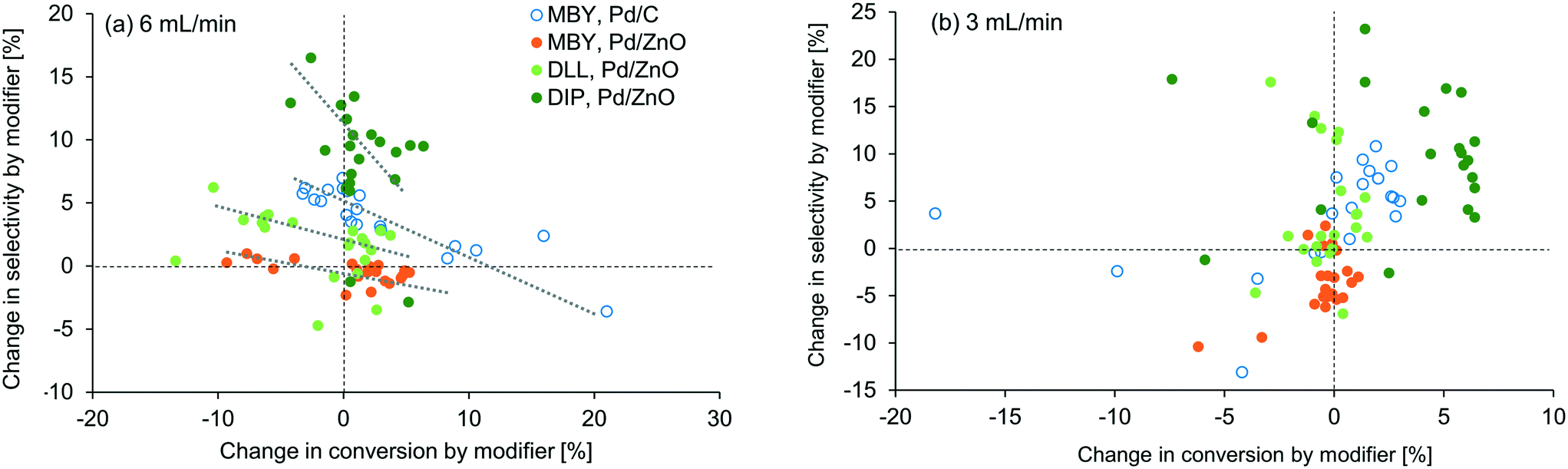 | ||
| Fig. 5 Effect of 21 modifiers in terms of alkyne conversion and alkene selectivity for (a) 6 mL min−1, and (b) 3 mL min−1 of total liquid flow rate. Dashed lines are displayed to show the reference value without any modifiers. Dotted lines are displayed to show the general tendency for the 6 mL min−1 case. | ||
In our examinations, types of modifiers, catalysts, and substrates are examined in the relatively narrow ranges (N-containing bases, Pd/C and Pd/ZnO, and alkynyl alcohols). Nevertheless, the modifiers changed the reaction conversion and selectivity substantially. In usual experimental investigations in literature, parameters of a catalytic reaction are examined one by one, e.g. modifier screening followed by catalyst support change13 or vice versa.16 However, the results in this study suggest that simultaneous investigation of modifier effect and other variables are important. For example, when taken alone, the effect of modifier addition appears to be minimal for MBY hydrogenation with Pd/ZnO due to the complex nature of the interaction between precious metal centres and support. However, multi-parameter investigation as shown in Table 1 shows that 1,10-phenanthroline can improve the selectivity dramatically with the same or even higher level of the conversion when paired with other catalyst systems and substrates, demonstrating the effectiveness of automated flow systems in achieving rigorous experimental work.
3.3 Mechanistic insights into the catalyst effect
As discussed earlier, experimental studies are required to find the best catalyst-modifier-substrate combination. Yet, it is a multi-variable optimization problem with its complexity increasing exponentially with the number of variables. Therefore, it is worthwhile to examine the mechanism of the catalyst modification to reduce the scope of experimental work. Previous studies focusing on Lindlar catalyst8,23 with quinoline proposed models to explain selectivity improvement. They feature the adsorption of quinoline on the catalyst surface to block undesired reactions. Many of the findings in this study such as catalyst type dependency, substrate dependency, and conversion improvement are out of those adsorption models. However, the better selectivity of the 1,10-phenanthroline than that of quinoline was universal in the conditions examined in this study and are examined on the basis of previously established theories.To elucidate the mechanism of selectivity improvement, the adsorption of modifiers to the catalyst surface was examined computationally. In our previous study with MBY hydrogenation over Pd catalyst, a mechanistic model with alkyne site and alkene site has been proposed.8 There are two types of active catalysts sites: (i) the terrace sites that strongly adsorb alkynes and produce 99+% alkene selectivity alone, and (ii) the low-coordination sites that hydrogenate alkene even in excess of alkyne – resulting in an average selectivity of 80–95%. Detailed adsorption studies and DFT calculations confirm this postulation. In the DFT calculations, the selective terrace site was represented by Pd(111) surface and the non-selective by Pd(210) and Pd(110) surface.
In this study, we also present DFT calculations of adsorption of catalyst modifiers over the selective and non-selective Pd surfaces. We have compared the adsorption energy of the selected modifiers taking the most efficient one (1,10-phenanthoroline), efficient conventional one (quinoline), and the least efficient one with a similar aromatic structure (2,4,6-trimethylpyridine).
Fig. 6 illustrates the adsorption structures of these three molecules optimized at the DFT level to the alkene sites. In both cases, the modifiers interacted with Pd horizontal configuration as clear in top views of Pd surfaces. Santarossa et al. examined the adsorption mode of quinoline and confirmed that vertical configurations are possible with Pt and Rh but only the horizontal direction is stable with Pd.53 The current results agree well with their conclusions. 1,10-Phenanthroline showed high affinity to Pd(110) surface so that 12 of 14 atoms (except hydrogen atoms) made bonding with Pd as shown in Fig. 6b. Pd(210) surface has a bumping structure and the adsorption affinity decreased its number of bonding to 6 as shown in Fig. 6d. In the case of quinoline, numbers of bonds were 5 to Pd(110) and 3 to Pd(210). 3 2,4,6-trimethylpyridine showed far less interaction to Pd surface compared to 1,10-phenanthoroline and quinoline. Three methyl groups caused severe steric hindrance so that the molecule needed to bend at the site of adsorption. In this instance, only 3 atoms can make bonds with Pd atoms as shown in Fig. 6j. In the case of Pd(210), there is only one bond with 2,4,6-trimethylpyridine and Pd as shown in Fig. 6l.
 | ||
| Fig. 6 DFT-optimized adsorption structures of modifiers: (a–d) 1,10-phenanthroline, (e–h) quinoline, (i–l) 2,4,6-trimethylpyridine; (a, e and i) top view of Pd(110) surface, (b, f and j) side view of Pd(110) surface, (c, g and k) top view of Pd(210) surface and (d, h and l) side view of Pd(210) surface. Pd–C and Pd–N bonds are drawn when the distance between atoms is shorter than 2.5 Å. | ||
Table 2 lists the adsorption energy of the modifiers calculated in this study and that of MBE reported in the previous study.8 The adsorption energy of MBE is reported to be similar to MBY on Pd(110) and Pd(210) indicating that adsorption of MBE is equally likely over these surfaces opening the possibility for MBE adsorption and subsequent reactions. Note that the adsorption structure of MBE (adsorbed via H atom, OH group, or CH3 group) little changed the adsorption energy.8
Hence, it is interesting to compare how the modifiers adsorb over these non-selective Pd(210) and Pd(110) surface and estimate the possibility of blocking the surface to increase the catalyst selectivity. The 1,10-phenanthroline had 1.5 times higher adsorption energy than that of MBE. The adsorption energy of quinoline was comparable than that of MBE, while 2,4,6-trimethylpyridine had half the adsorption energy compared with MBE. This comparison agrees with the general picture of quinoline and 1,10-phenanthroline blocking the non-selective sites, while 2,4,6-trimethylpyridine does not. 1,10-phenanthroline has a remarkable advantage to block both Pd(110) and Pd(210) surface compared to quinoline which cannot achieve complete blocking of MBE adsorption on Pd(210).
The DFT insights, therefore, could be used to decrease the experimental programme. A relatively low-cost (computationally) adsorption energy calculation could be performed over the candidate modifiers and the modifiers with low adsorption energy could be discarded while the other modifiers tested experimentally. The rejection threshold for the adsorption energy value should be comparable or below that of the substrate adsorption energy (−2.0 eV in this case).
Conclusions
We have developed a novel automated system for screening modifiers for heterogeneous catalysts and studied 21 various nitrogen-containing molecules in the alkyne semi-hydrogenation.The nitrogen modifiers have a different effect on various catalysts. The less selective Pd/C catalyst behaved differently to the modifiers compared to Pd/ZnO. The general trends were observed (for example, polyaromatic modifiers were the most efficient over these catalysts), the particular details vary. Hence, it is vital to perform experimental screening of modifiers for the selected catalysts. Moreover, it is beneficial to use modifiers with the best-performing catalysts to maximise selectivity.
The effect of catalyst modifiers on the semi-hydrogenation of similar alkynes is also different. Alkynes with various lengths behave differently to the modifiers. Similar to various catalysts, the general trends could be observed, but exact behaviour must also be determined experimentally.
The DFT study indicates that the catalyst modifiers adsorb on and block non-selective low-coordination Pd sites. The most efficient modifier, 1,10-phenanthroline, had the adsorption energy much higher than that of the target alkene molecule. Hence, the DFT adsorption energy calculations could be used as a relatively low-cost way to identify the promising catalyst modifiers.
Therefore, the optimal process for selective catalyst modifiers seems to contain the steps of
(i) Selecting the range of candidate modifiers available commercially at the required scale and compatible chemically with the target process and the catalysts. The compatibility includes the possibility of side-reactions, material compatibility, ease of separation.
(ii) Narrowing the range of modifiers with inexpensive DFT calculations. In the case of alkene semi-hydrogenation, the feasible way to reject inefficient modifiers seems the modified adsorption energy over Pd(210) above −1.5 eV.
(iii) Performing automated modifier screening of the process using the narrowed modifier list.
Our approach using homogeneous modifiers will be able to play an important role in future chemical processing by decoupling the catalyst synthesis and process optimization, and by shortening the process development time with the help of automated and AI-assisted flow chemistry systems. Some of the important factors of homogeneous modifiers such as optimum concentration and reaction acceleration effect were not focused on in this study and would be reported in future works.
Conflicts of interest
NC is the founder and director of Stoli Chem.Acknowledgements
Stoli Chem acknowledges the European Union (EU) Horizon 2020 funding (SME Instrument phase 2 project, 848926) indispensable for this research. NC also is grateful to IChemE for Andrew Fellowship and the Royal Society for the International Exchanges grant IEC\R3\193016. SA acknowledges the support from JSPS KAKENHI (Grant number 20 K15081, 22 K14536, and 21H05083) and Cooperative Research Program of Network Joint Research Center for Materials and Devices that has been supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The computations in this work were primarily performed using computer facilities at the Research Institute for Information Technology, Kyushu University. We thank DSM for providing substrates.References
- F. Roessler, Chimia, 1996, 50, 106–109 CrossRef CAS.
- J. W. Veldsink, M. J. Bouma, N. H. Schöön and A. A. C. M. Beenackers, Catal. Rev.: Sci. Eng., 1997, 39, 253–318 CrossRef CAS.
- G. A. Somorjai and A. S. Mujumdar, Introduction to surface chemistry and catalysis, 1995 Search PubMed.
- W. Bonrath, J. Medlock, J. Schütz, B. Wüstenberg and T. Netscher, Hydrogenation, ed. I. Karamé, 2012, pp. 69–90, DOI:10.5772/48751.
- M. Eggersdorfer, D. Laudert, U. Létinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960–12990 CrossRef CAS PubMed.
- H. Miyamura, A. Suzuki, T. Yasukawa and S. Kobayashi, J. Am. Chem. Soc., 2018, 140, 11325–11334 CrossRef CAS PubMed.
- N. Cherkasov, A. J. Expósito, M. S. Aw, J. Fernández-García, S. Huband, J. Sloan, L. Paniwnyk and E. V. Rebrov, Appl. Catal., A, 2019, 570, 183–191 CrossRef CAS.
- N. Cherkasov, D. Yu. Murzin, C. R. A. Catlow and A. Chutia, Catal. Sci. Technol., 2021, 11, 6205–6216 RSC.
- H. Lindlar and R. Dubuis, Org. Synth., 1966, 46, 89 CrossRef CAS.
- E. V. Rebrov, E. A. Klinger, A. Berenguer-Murcia, E. M. Sulman and J. C. Schouten, Org. Process Res. Dev., 2009, 13, 991–998 CrossRef CAS.
- Y. Orito, S. Imai and S. Niwa, Nippon Kagaku Kaishi, 1979, 1118–1120 CrossRef CAS.
- Y. Orito, S. Imai, S. Niwa and N.-G.- Hung, J. Synth. Org. Chem., Jpn., 1979, 37, 173–174 CrossRef CAS.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- C. Moreno-Marrodan, F. Liguori and P. Barbaro, Beilstein J. Org. Chem., 2017, 13, 734–754 CrossRef CAS PubMed.
- M. A. Ortunõ and N. López, Catal. Sci. Technol., 2019, 9, 5173–5185 RSC.
- J. Tu, L. Sang, H. Cheng, N. Ai and J. Zhang, Org. Process Res. Dev., 2020, 24, 59–66 CrossRef CAS.
- S. Vernuccio, R. Goy, P. Rudolf Von Rohr, J. Medlock and W. Bonrath, React. Chem. Eng., 2016, 1, 445–453 RSC.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. John Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, 6453 CrossRef PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- N. Hawbaker, E. Wittgrove, B. Christensen, N. Sach and D. G. Blackmond, Org. Process Res. Dev., 2016, 20, 465–473 CrossRef CAS.
- H. Wang, A. Mustaffar, A. N. Phan, V. Zivkovic, D. Reay, R. Law and K. Boodhoo, Chem. Eng. Process.: Process Intesif., 2017, 118, 78–107 CrossRef CAS.
- M. García-Mota, J. Gómez-Díaz, G. Novell-Leruth, C. Vargas-Fuentes, L. Bellarosa, B. Bridier, J. Pérez-Ramírez and N. López, Theor. Chem. Acc., 2011, 128, 663–673 Search PubMed.
- N. Cherkasov, Y. Bai, A. J. Expósito, E. V. Rebrov and A. Exposito, React. Chem. Eng., 2018, 3, 769–780 RSC.
- N. Cherkasov, A. J. Expósito, Y. Bai and E. V. Rebrov, React. Chem. Eng., 2019, 4, 112–121 RSC.
- N. Cherkasov, A. O. Ibhadon and E. V. Rebrov, Lab Chip, 2015, 15, 1952–1960 RSC.
- N. Cherkasov, Y. Bai and E. Rebrov, Catalysts, 2017, 7, 12 CrossRef.
- A. J. J. Exposito, Y. Bai, K. Tchabanenko, E. V. Rebrov and N. Cherkasov, Ind. Eng. Chem. Res., 2019, 58, 4433–4442 CrossRef CAS.
- K. Igawa, S. Asano, Y. Yoshida, Y. Kawasaki and K. Tomooka, J. Org. Chem., 2021, 86, 9651–9657 CrossRef CAS PubMed.
- M. A. Morin, W. Zhang, D. Mallik and M. G. Organ, Angew. Chem., 2021, 60, 20606–20626 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Materials Studio, Version 2020, Software for Materials Modeling, Dassault Systèmes, BIOVIA, San Diego, 2020, https://www.materialsstudio.com Search PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- N. Cherkasov, A. O. Ibhadon and E. V. Rebrov, Appl. Catal., A, 2016, 515, 108–115 CrossRef CAS.
- N. Cherkasov, M. 'moun Al-Rawashdeh, A. O. Ibhadon and E. V. Rebrov, Catal. Today, 2016, 273, 205–212 CrossRef CAS.
- N. Cherkasov, Y. Bai and E. Rebrov, Catalysts, 2017, 7, 358 CrossRef.
- N. Semagina, M. Grasemann, N. Xanthopoulos, A. Renken and L. Kiwi-Minsker, J. Catal., 2007, 251, 213–222 CrossRef CAS.
- M. Crespo-Quesada, M. Grasemann, N. Semagina, A. Renken and L. Kiwi-Minsker, Catal. Today, 2009, 147, 247–254 CrossRef CAS.
- M. W. Tew, H. Emerich and J. A. Van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465 CrossRef CAS.
- S. K. Johnston, N. Cherkasov, E. Pérez-Barrado, A. Aho, D. Y. Murzin, A. O. Ibhadon and M. G. Francesconi, Appl. Catal., A, 2017, 544, 40–45 CrossRef CAS.
- E. V. Rebrov, A. Berenguer-Murcia, H. E. Skelton, B. F. G. Johnson, A. E. H. Wheatley and J. C. Schouten, Lab Chip, 2009, 9, 503–506 RSC.
- N. V. Semagina, A. V. Bykov, E. M. Sulman, V. G. Matveeva, S. N. Sidorov, L. V. Dubrovina, P. M. Valetsky, O. I. Kiselyova, A. R. Khokhlov, B. Stein and L. M. Bronstein, J. Mol. Catal. A: Chem., 2004, 208, 273–284 CrossRef CAS.
- H. Sajiki, T. Ikawa and K. Hirota, Tetrahedron Lett., 2003, 44, 8437–8439 CrossRef CAS.
- E. Bayram and R. G. Finke, ACS Catal., 2012, 2, 1967–1975 CrossRef CAS.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- G. Santarossa, M. Iannuzzi, A. Vargas and A. Baiker, ChemPhysChem, 2008, 9, 401–413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00147k |
| This journal is © The Royal Society of Chemistry 2022 |