
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast transient absorption spectroelectrochemistry: femtosecond to nanosecond excited-state relaxation dynamics of the individual components of an anthraquinone redox couple†
Sofia
Goia
 ab,
Matthew A. P.
Turner
abc,
Jack M.
Woolley
a,
Michael D.
Horbury
ad,
Alexandra J.
Borrill
ae,
Joshua J.
Tully
ae,
Samuel J.
Cobb
aef,
Michael
Staniforth
a,
Nicholas D. M.
Hine
c,
Adam
Burriss
g,
Julie V.
Macpherson
*a,
Ben R.
Robinson
*g and
Vasilios G.
Stavros
*a
ab,
Matthew A. P.
Turner
abc,
Jack M.
Woolley
a,
Michael D.
Horbury
ad,
Alexandra J.
Borrill
ae,
Joshua J.
Tully
ae,
Samuel J.
Cobb
aef,
Michael
Staniforth
a,
Nicholas D. M.
Hine
c,
Adam
Burriss
g,
Julie V.
Macpherson
*a,
Ben R.
Robinson
*g and
Vasilios G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk; j.macpherson@warwick.ac.uk
bMolecular Analytical Science CDT, Senate House, University of Warwick, Coventry, CV4 7AL, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
dSchool of Electronic and Electrical Engineering, University of Leeds, LS2 9JT, UK
eDiamond Science and Technology CDT, University of Warwick, Coventry, CV4 7AL, UK
fDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
gSyngenta, Warfield, Bracknell, RG42 6EY, UK. E-mail: ben.robinson@syngenta.com
First published on 17th December 2021
Abstract
Many photoactivated processes involve a change in oxidation state during the reaction pathway and formation of highly reactive photoactivated species. Isolating these reactive species and studying their early-stage femtosecond to nanosecond (fs–ns) photodynamics can be challenging. Here we introduce a combined ultrafast transient absorption-spectroelectrochemistry (TA-SEC) approach using freestanding boron doped diamond (BDD) mesh electrodes, which also extends the time domain of conventional spectrochemical measurements. The BDD electrodes offer a wide solvent window, low background currents, and a tuneable mesh size which minimises light scattering from the electrode itself. Importantly, reactive intermediates are generated electrochemically, via oxidation/reduction of the starting stable species, enabling their dynamic interrogation using ultrafast TA-SEC, through which the early stages of the photoinduced relaxation mechanisms are elucidated. As a model system, we investigate the ultrafast spectroscopy of both anthraquinone-2-sulfonate (AQS) and its less stable counterpart, anthrahydroquinone-2-sulfonate (AH2QS). This is achieved by generating AH2QS in situ from AQS via electrochemical means, whilst simultaneously probing the associated early-stage photoinduced dynamical processes. Using this approach we unravel the relaxation mechanisms occurring in the first 2.5 ns, following absorption of ultraviolet radiation; for AQS as an extension to previous studies, and for the first time for AH2QS. AQS relaxation occurs via formation of triplet states, with some of these states interacting with the buffered solution to form a transient species within approximately 600 ps. In contrast, all AH2QS undergoes excited-state single proton transfer with the buffered solution, resulting in formation of ground state AHQS− within approximately 150 ps.
Introduction
Photoinduced intra- or intermolecular reactions are ubiquitous in many chemical and biological processes,1,2 with the latter typically comprising proton transfer or electron transfer (or both, proton-coupled electron transfer)3 between the solvent or a second chemical species. Such reactions are of vital importance in many areas including photosynthesis,4 photoredox catalysis,5,6 photoelectrocatalysis,7 electrophotocatalysis,8 and photoacidity.9 Understanding the transient dynamics of these processes is not only critically important in elucidating mechanistic pathways but also in determining the photostability of different species.Transient absorption spectroscopy (TAS) is a technique used widely to follow the excited-state dynamics of light-activated complexes. With TAS, measurements at the (sub) ns timescales are possible,10 shedding further insight into the critical first step(s) in a reaction mechanism, from excited-state evolution, to either solvent rearrangement,11 or intramolecular rearrangement (typically fs to ps),12 or proton/electron transfer to solvent (typically ps to ns).1 With the use of electrochemical techniques it is possible to switch the oxidation state of a soluble species, simply by control of the electrode potential and direction of electron flow.13 Electrochemistry can also be used to access higher reducing and oxidising potentials than possible using chemical oxidants and reductants.14 Bringing the two techniques together is an area which is ripe for exploration.15–18
There are many processes where the starting species change oxidation state during a reaction pathway, resulting in formation of a less stable, more reactive photoactivated species.7,19 In the complementary, burgeoning area of electrophotocatalysis,8,20 a reactive radical intermediate is first generated electrochemically and then brought to a higher oxidising or reducing potential by the action of light. Isolating reactive intermediates via chemical means for the individual study of early-stage photodynamics is challenging, typically requiring the use of strong chemical oxidising or reducing agents.14 Furthermore, the presence of these chemical reagents in the system, unless removed, may complicate dynamic spectroscopic analysis. In contrast, electrochemistry offers an attractive ‘green’ method for the creation of reaction intermediates, via controlled oxidation/reduction of the starting species. Moreover, through electrochemical means the product of interest can be generated continuously and there is no need for excess chemical oxidants or reductants. Although it is often useful to consider the impact, if any, of the supporting electrolyte added.21 Combined ultrafast TAS (ultraviolet-visible (UV/Vis) or infrared (IR)) – electrochemical measurements thus not only provide routes into the investigation of the photodynamics of isolated reaction intermediates, but also add a new dynamic time dimension to the broad field of spectroelectrochemistry.16
To develop this area, a robust experimental set-up is required which can be universally applied. In this work, we introduce an experimental system based on boron doped diamond (BDD) freestanding mesh electrodes combined with TAS, which enable the ultrafast photochemistry and photophysics of the reduced/oxidised states of a redox couple to be investigated. Such electrodes can also find use in more conventional spectroelectrochemical (SEC) measurements. Freestanding BDD has the advantage that it can be laser micromachined22 with high accuracy into an appropriate geometry mesh, through which light can pass with minimal scattering. The laser micromachining process is both time efficient and extremely reproducible leading to ease of manufacture. Electrochemically, due to its significantly reduced electrocatalytic activity towards water and dissolved oxygen, it also offers the widest solvent window in aqueous solution, negligible interference from a dissolved oxygen reduction signal, and presents very low background currents.23,24 BDD electrodes thus enable discrimination of electrochemical oxidation/reduction processes, via voltammetric means, which are often obscured on other electrodes, especially metals. From complementary voltammetric data, the electrode potential for generation of e.g. the reaction intermediates, can be established for ultrafast TA-SEC studies.
Quinones are important electron acceptors (resulting in the reduced form of the quinone)25,26 and play a crucial role in many biological processes.27 In this study we investigate the photoactivated dynamics and stability of a model quinone redox couple, anthraquinone-2-sulfonate (AQS)/anthrahydroquinone-2-sulfonate (AH2QS). AQS is water-soluble with many applications in the biological as well as chemical fields including as an organo-photocatalyst in the oxidation of alcohols28 and nitration of phenol,29 a quantifier of reaction lifetimes in surface water bodies,30 and in photoelectrochemical cells.31 AQS can be electrochemically reduced via proton-coupled electron transfer to form its less stable counterpart, AH2QS,32,33Fig. 1. Using combined ultrafast TA-SEC measurements we showcase how both the oxidised and electrochemically reduced photoactivated states of the quinone can be interrogated at the fs–ns timescale, revealing important information on the photostability dynamics of the AQS/AH2QS redox system in aqueous buffer.
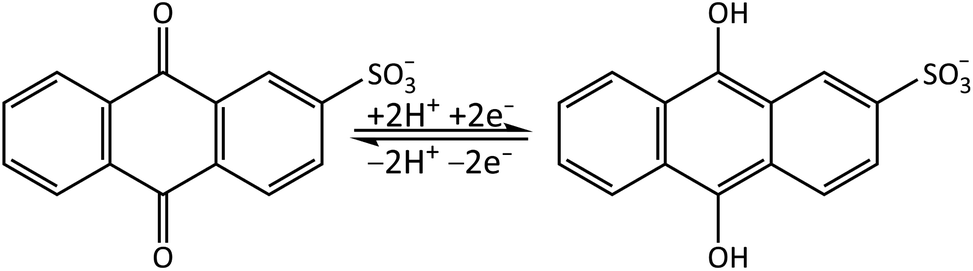 | ||
| Fig. 1 Reversible redox cycle of AQS/AH2QS through the acceptance/loss of two electrons (2e−) and two protons (2H+). | ||
Methods
Materials and equipment
Solutions were prepared using the following chemicals: sodium anthraquinone-2-sulfonate (≥98% (HPLC), Sigma-Aldrich), sodium acetate trihydrate (laboratory reagent grade, Fisher Scientific), acetic acid glacial (≥99.7% analytical reagent grade, Fisher Scientific), potassium chloride (analytical reagent grade, VWR Chemicals), phosphate buffered saline (PBS) tablet (Fisher Scientific BioReagents™, pH 7.4, 1 tablet/100 mL), sodium carbonate anhydrous (99.5% laboratory reagent grade, Fisher Scientific), sodium bicarbonate (≥99.7% ACS reagent grade, Honeywell Fluka™). Without any further purification, all reagents were used as received. For sample preparation, ultrapure water with a resistivity of 18.2 MΩ cm was used throughout. The solutions were freshly prepared before each measurement, and experiments were performed at room temperature, (20 ± 2 °C), unless otherwise stated.Steady-state absorption measurements were conducted using a Cary 60 UV/Vis Spectrophotometer (Agilent Technologies, US) and quartz fluorescence cell with a 1 cm path length (Hellma Analytics). Time-resolved measurements were conducted using the TAS instrumentation available at the Warwick Centre for Ultrafast Spectroscopy (https://warwick.ac.uk/fac/sci/wcus/about/). Electrochemical measurements were performed using a PalmSens EmStat3 potentiostat.
Spectroelectrochemical cell and BDD working mesh electrode fabrication
The 3D printed cell was designed using Autodesk Fusion 360 software (Autodesk, USA) and printed at a 200 μm layer height (Taz 6, Lulzbot, USA) from PET filament (Innofil, USA; green parts) and a FormLabs Form 3 in FormLabs Standard Clear (clear parts). The cell built on a panel holder was designed to fit into the steady-state and the ultrafast instruments (see ESI 1, Fig. S1† for details). A 1 mm path length quartz cuvette (Hellma Analytics) was then cut and adhered to the 3D printed cell using an epoxy resin (Araldite Rapid, USA).Electrodes were fabricated from freestanding, highly doped (∼3 × 1020 boron atoms per cm3) electrochemical processing grade polycrystalline BDD (Element Six Ltd, UK),34 polished to ∼ nm roughness on the growth face and lapped to ∼μm roughness on the nucleation face with a thickness of 0.25 mm. This material was laser-cut into mesh electrodes with the following dimensions: 6 × 7 mm with holes of 0.25 mm diameter and a 0.35 mm centre to centre space, using a 355 nm Nd:YAG laser micromachining system (E-355-ATHI-O system, Oxford Lasers Ltd. UK) with a nominal pulse-length of 34 ns. Cutting was performed in two passes with a fluence of 350 J cm−2. After laser cutting the electrodes were acid cleaned at ∼200 °C in concentrated H2SO4 saturated with KNO3 for 30 min, then rinsed with water and cleaned in concentrated H2SO4 at ∼200 °C for 30 min, to remove debris introduced during laser micromachining.35 After acid cleaning any residual sp2 bonded carbon from laser micromachining, was significantly reduced by annealing in air at 600 °C for 5 hours.35 A titanium (Ti: 10 nm)/gold (Au: 400 nm) contact was sputtered (Moorfield MiniLab 060 Platform Sputter system) onto the rectangular tail of the electrode and annealed in air (400 °C for 5 h) to create an ohmic contact.34 The BDD electrode was connected to a copper wire using a conductive epoxy (Silver Epoxy, Circuitworks, USA), and the connection waterproofed with a layer of epoxy resin (Araldite Rapid, USA).
Reduction of AQS
Electrochemical measurements were performed using the built cell and a three-electrode system (Pt wire counter electrode, CHI150 Saturated Calomel Electrode (SCE, CH Instruments, Inc) as reference electrode, and BDD mesh working electrode) under an inert atmosphere by bubbling the solution with N2. Cyclic voltammetry (CV) measurements were performed on a 1 mM AQS solution (0.1 M acetate buffer pH 4.9, 0.2 M KCl) at a 0.1 V s−1 scan rate between −1.0 V and 0.5 V vs. SCE (see ESI 1, Fig. S2† for details). A reduction potential of −0.65 V vs. SCE was chosen to drive the AQS reduction at a sufficient rate for a time period (30 min) sufficient to convert all the AQS to AH2QS. Steady-state spectroelectrochemical experiments were performed by mounting the SEC set-up inside the UV/Vis spectrophotometer. Stability measurements were taken on the electrochemically reduced species (0.2 mM AQS starting material, 0.1 M acetate buffer pH 4.9, 0.2 M KCl) by analysing the UV/Vis spectra over time (see ESI 2, Fig. S3†).pH dependence measurements
These measurements were taken using the SEC set-up mounted within the UV/Vis spectrophotometer, using an inert atmosphere and in the following environments: 1 mM AQS in either 0.1 M acetate buffer pH 4.9 (−0.65 V vs. SCE) or 0.1 M PBS buffer pH 7.4 (−0.70 V vs. SCE), and 0.2 mM AQS in 0.1 M carbonate–bicarbonate buffer pH 8.9 (−0.66 V vs. SCE), with all solutions containing 0.2 M KCl. The voltage used for reduction in the different pH solutions was chosen by recording a CV scan at a 0.1 V s−1 scan rate between −1.0 V and 0.5 V vs. SCE before each experiment. This voltage was applied for 30 minutes before each measurement was taken.Transient absorption spectroscopy (TAS)
TAS measurements were performed on 0.1 M acetate buffer pH 4.9, 0.2 M KCl solutions containing either 1.1 mM AQS or electrochemically reduced 1 mM AQS. For AQS, a diaphragm pump (SIMDOS 02) was employed to ensure a fresh flow of sample was delivered constantly to the demountable liquid cell (Harrick Scientific Products Inc.) and a sample thickness of 500 μm was used. For the electrochemically reduced species, the built SEC 1 mm path length static cell was used. Scans of the pH 4.9 buffer solution with no AQS present were taken using the same parameters chosen for the respective molecule of interest, in order to determine potential contributions from the solvent and windows of the Harrick cell or quartz cuvette.The TAS36 set-up has been described in detail in a previous study;37 briefly, a Ti-sapphire laser system (Spectra-Physics, Dual Ascend Pumped Spitfire Ace) was used to generate the fs 800 nm (12 W, 1 kHz) pulses, with a nominal pulse width of ≈40 fs. The different pump beams (330 nm at 1 mW and 382 nm at 0.5 mW for AQS and for AH2QS, respectively) were produced using an optical parametric amplifier (TOPAS-Prime with UV extension). Pump-probe delays (Δt) between −1 ps to 2.5 ns were used, with changes in optical densities being detected using an Avantes, AvaSpec-ULS1650F spectrometer. The retrieved data was further analysed using the Glotaran software38 for the R package TIMP.39 A global sequential fit (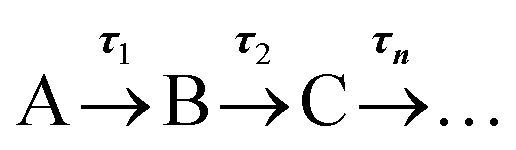 ) was employed throughout to extract the lifetimes contained in the TA spectra of our molecules. The standard error for each lifetime was acquired through the Glotaran fit, and the lifetime was reported with twice this standard error. The collected TA spectra were also chirp corrected using the KOALA package40 before plotting, for visual clarity. The instrument response duration calculated using the method devised by Kovalenko et al.,41 retrieved full width half maximum values of ≈130 fs for both the pH 4.9 buffer solution within the Harrick cell (pump at 330 nm) and for the pH 4.9 buffer solution within the 1 mm path length cuvette (pump at 382 nm). Pump power dependence measurements were also performed by varying the output power of the TOPAS. The range of powers used in the power dependence measurements for AQS was 0.15 mW to 1.5 mW and 0.1 mW to 1.0 mW for AH2QS, with five-points measurements taken at varying powers. A 10 nm integration window was then taken across a certain spectral window at a given Δt and the log(power) was plotted against the log(signal).
) was employed throughout to extract the lifetimes contained in the TA spectra of our molecules. The standard error for each lifetime was acquired through the Glotaran fit, and the lifetime was reported with twice this standard error. The collected TA spectra were also chirp corrected using the KOALA package40 before plotting, for visual clarity. The instrument response duration calculated using the method devised by Kovalenko et al.,41 retrieved full width half maximum values of ≈130 fs for both the pH 4.9 buffer solution within the Harrick cell (pump at 330 nm) and for the pH 4.9 buffer solution within the 1 mm path length cuvette (pump at 382 nm). Pump power dependence measurements were also performed by varying the output power of the TOPAS. The range of powers used in the power dependence measurements for AQS was 0.15 mW to 1.5 mW and 0.1 mW to 1.0 mW for AH2QS, with five-points measurements taken at varying powers. A 10 nm integration window was then taken across a certain spectral window at a given Δt and the log(power) was plotted against the log(signal).
Theoretical spectroscopy
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44–46 level of theory. The continuum solvation COnductor-like Screening MOdel (COSMO)47,48 was used in all calculations to implement implicit solvent (water) effects. The first 8 excited states (Sn) were then calculated at the Franck–Condon geometry, using the same level of theory.
000 steps, NVT ensemble), density equilibration (50000 steps, NPT ensemble) and equilibration (50000 steps, NVT ensemble) procedures as detailed in the ESTEEM documentation, with T = 300 K applied by a Langevin thermostat with a collision frequency of 1 ps, resulting in boxes of sizes around 44 Å cubed. 200 snapshots each spaced by 2000 timesteps were then extracted, separated by 4 ps. Of these, 50 equally-spaced cluster snapshots were extracted from the trajectory. The extracted cluster incorporated the solute and solvent molecules, with any atom of the solvent within 6 Å of any atom of the solute (typically around 400 atoms). The explicit solvent is intended to be sufficient to well-converge the solvatochromic effect of the solvent. Calculations in Fig. S8† compare results for a subset of the results at 3 Å, 6 Å, and 9 Å cluster sizes, and indicate that this has been achieved by 6 Å. The average number of atoms in models of AQS and AH2QS in water for R = 6 is 385 and 395, respectively; for R = 9, for AQS it is 914, and for AH2QS it is 917.
44–46 level of theory. The continuum solvation COnductor-like Screening MOdel (COSMO)47,48 was used in all calculations to implement implicit solvent (water) effects. The first 8 excited states (Sn) were then calculated at the Franck–Condon geometry, using the same level of theory.
000 steps, NVT ensemble), density equilibration (50000 steps, NPT ensemble) and equilibration (50000 steps, NVT ensemble) procedures as detailed in the ESTEEM documentation, with T = 300 K applied by a Langevin thermostat with a collision frequency of 1 ps, resulting in boxes of sizes around 44 Å cubed. 200 snapshots each spaced by 2000 timesteps were then extracted, separated by 4 ps. Of these, 50 equally-spaced cluster snapshots were extracted from the trajectory. The extracted cluster incorporated the solute and solvent molecules, with any atom of the solvent within 6 Å of any atom of the solute (typically around 400 atoms). The explicit solvent is intended to be sufficient to well-converge the solvatochromic effect of the solvent. Calculations in Fig. S8† compare results for a subset of the results at 3 Å, 6 Å, and 9 Å cluster sizes, and indicate that this has been achieved by 6 Å. The average number of atoms in models of AQS and AH2QS in water for R = 6 is 385 and 395, respectively; for R = 9, for AQS it is 914, and for AH2QS it is 917.
From the MD trajectories, the ensemble of explicitly solvated structures is collected and Linear-Response TDDFT calculations are performed on extracted clusters, using the ONETEP (Order-N Electronic Total Energy Package) Linear-Scaling DFT package.61 Simulations are performed in a 26.5 Å simulation cell, in which the combined cluster of solute and solvent molecules is surrounded by implicit solvent to minimise any effects from spurious uncancelled dipole moments at the surface of the cluster.62 Optimisation of the support functions for the valence and conduction manifolds was performed.63 The density and transition kernels were left as dense matrices throughout as truncation was not necessary. Support functions were of radius 5.3 Å and represented with a cutoff energy of 800 eV, spanning to 6 eV above the LUMO. Finally, Linear-Response TDDFT calculations51 were performed for the first 12 excitations. The spectra resulting from these calculations was then subjected to a “spectral warp” procedure64 as motivated and discussed in ESI 6 to generate the final predicted spectra.
Results and discussion
Spectroelectrochemical instrumentation
Previous TA-SEC studies have employed a variety of electrodes and set-ups. These include: electrodes containing many holes (metal mesh or grid)18,65,66 or just a single hole,15,17 a flow cell arrangement where the electrode and spectroscopy components are spatially separated,67 or a thin layer spectroelectrochemical cell incorporating sufficiently thin planar electrodes.68 In the case of mesh/grid electrodes, especially metal electrodes, it is essential that the hole size is made large enough so there is no beam scattering, which can result in the production of charge carriers in the conduction band.69 Commercial Pt and Au mesh electrodes are available, however, there are limitations from the sizes of the mesh available, accuracy of the design, and, electrochemically, such electrodes have high background currents and are electrocatalytically efficient towards dissolved oxygen reduction and water electrolysis. This leads to a reduced solvent window in aqueous solution and a detectable dissolved oxygen signal, when scanning cathodically. With a flow cell arrangement,67 the range of systems available for study is limited due to the electrochemistry and spectroscopy components of the cell being spatially separated. Whilst sufficiently thin BDD films grown on quartz70–72 have been used for thin layer (steady-state) SEC experiments, the manufacturing process can be challenging. The arrangement in Fig. 2a, thus not only enables combined ultrafast TA-SEC measurements, but extends the range of electrodes/geometrical arrangements for conventional steady-state SEC measurements.16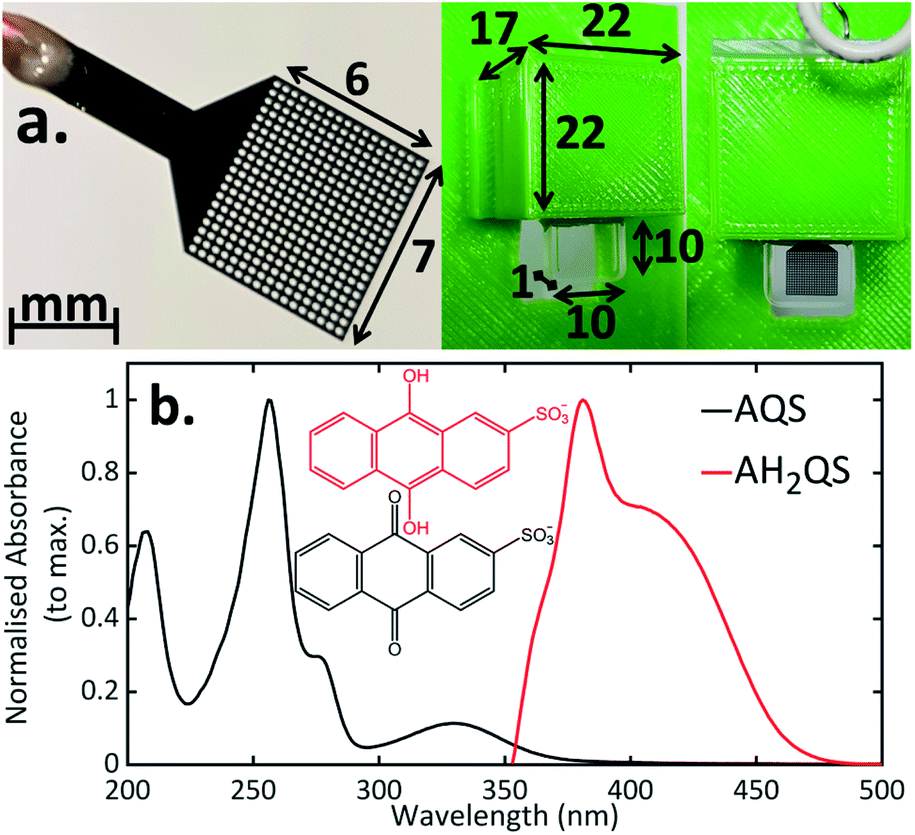 | ||
| Fig. 2 (a) Photograph of the BDD mesh working electrode and the 3D printed cell used in the three-electrode spectroelectrochemical set-up (measurements given in mm); (b) experimental UV/Vis spectra of AQS taken using a 0.2 mM AQS solution in 0.1 M acetate buffer pH 4.9, 0.2 M KCl. For the electrochemically reduced AH2QS, a 1 mM initial AQS solution was used (0.1 M acetate buffer pH 4.9, 0.2 M KCl) and a −0.65 V vs. SCE reduction potential was applied for 30 minutes before recording spectra, to ensure complete conversion. | ||
Reduction mechanism of AQS
The CV response for reduction of AQS is shown in ESI 1, Fig. S2,† recorded in a solution containing 1 mM AQS solution in 0.1 M acetate buffer pH 4.9, 0.2 M KCl, and at a scan rate of 0.1 V s−1. From the voltammetric response a potential and time were chosen, which were sufficient to drive the reduction of AQS at a rate appropriate for full conversion to reduced products in a reasonable timeframe (−0.65 V vs. SCE for 30 minutes). No evidence of AQS was observed in the UV/Vis spectrum (ESI 2, Fig. S3†), indicating complete electrochemical conversion on this timescale. Note, once the reducing potential was removed, or oxygen allowed back into the system, the characteristic absorption signatures of the electrochemically reduced species decreased with time (ESI 2, Fig. S3†).Fig. 2b shows the UV/Vis data for both AQS (solid black line) and electrochemically reduced AH2QS (solid red line). For AQS, in the region of interest, an absorption maximum at 330 nm (ref. 73 and 74) is present. For these studies, we focus on the low energy regions to avoid additional non radiative decay pathways from higher lying excited states when performing the subsequent ultrafast TAS (vide infra). The ground state electrochemical reduction mechanisms of quinones have been extensively studied in buffered aqueous media,33,74–77 with the many different pH dependent steps and intermediate species described by the ‘scheme of squares’ proposed by Jacq78 (see ESI 3, Fig. S4†). In an electrochemical proton-coupled electron transfer reaction, the reduction pathway can be tuned by varying the pH of the environment.32,33,78 For this redox couple, when pH < pKa,1 (= 7.6), i.e. in acidic solutions, the doubly protonated AH2QS species is formed (2e− + 2H+). As the pH is increased, such that pH > pKa,1 (= 7.6), AHQS− (negative charge denotes loss of a proton) forms, and the mechanism changes to 2e− + 1H+. For pH > pKa,2 (= 11), a 2e− only mechanism dominates, resulting in AQS2− formation.32,33 Thus, at pH 4.9, under conditions of exhaustive electrolysis where only the reduced form of the species is present in solution, the spectra observed (solid red line) corresponds to AH2QS alone. In particular, an absorption maximum at 382 nm is observed that tails off through the visible region of the electromagnetic spectrum.32
Theoretical analysis of predicted UV/Vis spectrum for AQS
TDDFT calculations using the NWChem Package (6-311++G** basis set; PBE0 functional; implicit solvent) were performed to identify the energy levels involved in the steady-state and ultrafast photoexcitation studies. The transitions observed for AQS are presented in ESI 4, Table S1,† with the peak around 330 nm (Fig. 2b) being assigned to a ππ* transition (S4 ← S0). The energy of the lowest lying triplet state (T1) was calculated using the Delta Self-Consistent Field (ΔSCF) method in the Franck–Condon region, which resulted in an energy of 418 nm. Triplet state vertical TDDFT calculations, although less reliable, indicate that there may be up to five triplet states with energies below the first excited singlet state (S1), with both nπ* and ππ* character (ESI 4, Table S1†). Intersystem crossing (ISC) is highly likely, with previous studies also suggesting the formation of a triplet state.73,79TAS of AQS
In our first step towards unravelling the influence of light on the model redox couple, we studied the dynamical processes following population of the excited state of the oxidised form of the redox couple, AQS. This was achieved through TAS36,37 upon photoexcitation at 330 nm. TA spectra were collated at a series of pump-probe time delays (Δt) out to the maximum time-window of our experiments (Δt = 2.5 ns). These are presented as a false colour map shown in Fig. 3a, with individual TA spectra at selected Δt shown in Fig. 3b. The TA spectra initially show an excited state absorption (ESA) feature centred at 395 nm. As this feature decays, two additional features centred at 460 nm and 590 nm are starting to form at Δt > 10 ps. A continuous blue shift of the 395 nm peak as it slowly decays, and a continuous red shift of the 460 nm peak can also be observed. These features are clearly resolved into two separate features, with peaks at 385 nm and 485 nm persisting out to our maximum available Δt of 2.5 ns. Linearity on the photon-induced dynamics was also determined through power dependence measurements (see ESI 5, Fig. S5a†).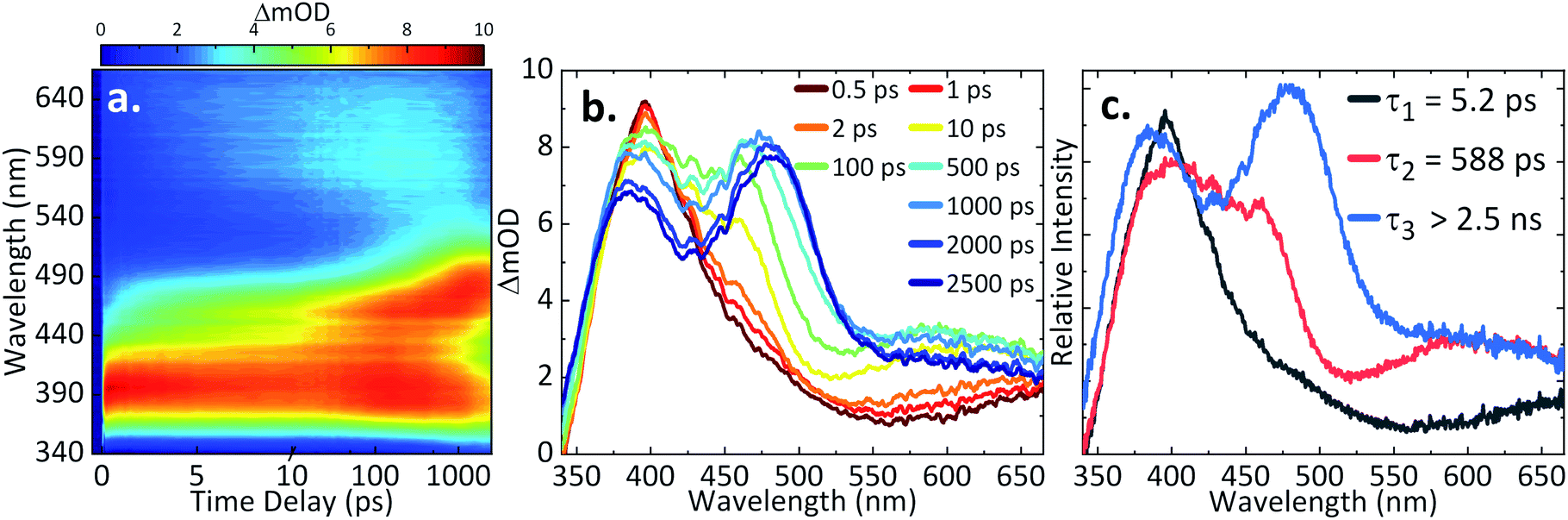 | ||
| Fig. 3 (a) TA spectra of 1.1 mM AQS in 0.1 M acetate buffer pH 4.9, 0.2 M KCl represented as a false colour map showing changes in optical density (ΔmOD) with photoexcitation at 330 nm; (b) different time delays (Δt) chosen for the TA spectra; (c) the corresponding evolution-associated difference spectra (EADS) from the global fit. | ||
To provide more quantitative insight into the dynamical processes involved, we employed a global sequential fit (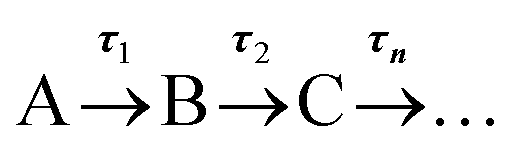 ) to extract the lifetimes contained in the TA spectra of AQS. This was achieved through the use of the Glotaran software package.38,39 Our sequential fit returned three time-constants with their associated errors, τ1 = 5.20 ± <0.05 ps, τ2 = 587.8 ± 9.8 ps, and τ3 > 2.5 ns, to model the dynamical processes. The evolution-associated difference spectra (EADS)80 linked to these time-constants are shown in Fig. 3c, aiding in the visualisation of the transient features within the TA spectra. The associated residuals (difference between raw and fitted data) shown in ESI 5, Fig. S6a,† also serve to indicate the good agreement between the experimental data and corresponding fit.
) to extract the lifetimes contained in the TA spectra of AQS. This was achieved through the use of the Glotaran software package.38,39 Our sequential fit returned three time-constants with their associated errors, τ1 = 5.20 ± <0.05 ps, τ2 = 587.8 ± 9.8 ps, and τ3 > 2.5 ns, to model the dynamical processes. The evolution-associated difference spectra (EADS)80 linked to these time-constants are shown in Fig. 3c, aiding in the visualisation of the transient features within the TA spectra. The associated residuals (difference between raw and fitted data) shown in ESI 5, Fig. S6a,† also serve to indicate the good agreement between the experimental data and corresponding fit.
In order to assign the three time-constants τ1 (5.20 ps), τ2 (587.8 ps), and τ3 (>2.5 ns) to dynamical processes, we draw on earlier photochemical and photophysical studies, and consolidate these with our TDDFT calculations. Previous studies have shown that AQS is promoted to its triplet state shortly after photoexcitation with a ns laser;73,74,81 the timescale for the formation of this process was previously unknown due to limitations in the experimental set-up. Close inspection of the absorption spectrum of the triplet state73,74,79,81 matches extremely well with the individual TA spectra shown in Fig. 3b for Δt > 10 ps, as well as the EADS for τ2 (which appears with τ1 and decays with τ2), strongly suggesting that the triplet state is formed with a time-constant of τ1. We add here, the 330 nm excitation peak can most likely be assigned to a ππ* transition involving S4 ← S0. The rate of ISC may be enhanced by higher lying triplet states (Tn) near resonant with the singlet states or internal conversion from the ππ* states to lower lying nπ* states which can subsequently lead to rapid ISC due to the change in orbital angular momentum.82,83 These, along with possible contributions of the heavy-atom (sulfur) in enhancing spin–orbit coupling (SOC)84 must contribute to the accelerated ISC we observe. Whether ISC occurs directly from the (likely) initially populated S4 → Tn or whether this is first preceded by internal conversion to the lowest-lying Sn followed by ISC, in agreement with Kasha's rule,85 is not known. From our calculations, we did not identify which of the excited states would exhibit the strongest SOC. Though, in accordance with El-Sayed's rule,86,87 an nπ* state is most likely being accessed. Subsequently, AQS remains in its triplet state.
From previous flash photolysis measurements,74,79 the photochemistry of AQS was shown to be affected by the environment used, with its excited triplet state forming what is known as transient species B and transient species C (both B and C unidentified); B has a strong, broad feature around 490 nm and C has a weaker feature around 590 nm. Thus, by comparing the time delays > 500 ps (Fig. 3b) with the flash photolysis measurements reported,74,79,81 it appears that the triplet state of AQS interacts with the aqueous buffer solution to form transient species B within τ2 = 587.8 ps, its lifetime persisting beyond the Δt = 2.5 ns time-window of our measurements, with τ3 > 2.5 ns. In addition, the smaller feature with a maximum peak at 590 nm could be species C, though spectral interferences from other transients may mask its formation. Moreover, given that the environment is slightly acidic, we do not expect to see it forming in high concentration; as previous work suggested, this characteristic feature was more evident in alkaline solutions.73,74,81 Importantly, however, it appears that on the timescales of the present measurements (fs–ns) not all of the triplet state reacts with the aqueous buffer environment.
TAS of AH2QS
For ultrafast TAS interrogation of the dynamics associated with the photoexcited, reduced form of AQS, as for the steady-state UV/Vis experiments, electrochemical reduction was initiated in situ by holding the BDD mesh at a potential of ca. −0.65 V vs. SCE for 30 minutes, to ensure complete conversion of AQS to AH2QS, in a buffered pH 4.9 solution. The BDD mesh electrode was coupled to the ultrafast TAS set-up (concept shown schematically in Fig. 4) and the photodynamics of the electrochemically reduced species, AH2QS, analysed. The TA spectra collected following photoexcitation of AH2QS at 382 nm are presented as a false colour map in Fig. 5a, with individual TA spectra at selected Δt shown in Fig. 5b. Initially (Δt of <1 ps), the TA spectra is comprised of two ESA features. The first feature is centred at 350 nm and the second broad feature, appears to have a main peak centred at 445 nm and a shoulder centred at 525 nm. The feature at 350 nm decays to baseline by Δt = 2.5 ns. In contrast, the features at 445 nm and 525 nm evolve to reveal further features at 400 nm and 475 nm (Δt of >300 ps) which persist beyond Δt = 2.5 ns. Power dependence measurements were also performed to ensure linearity on the photon-induced dynamics (see ESI 5, Fig. S5b†).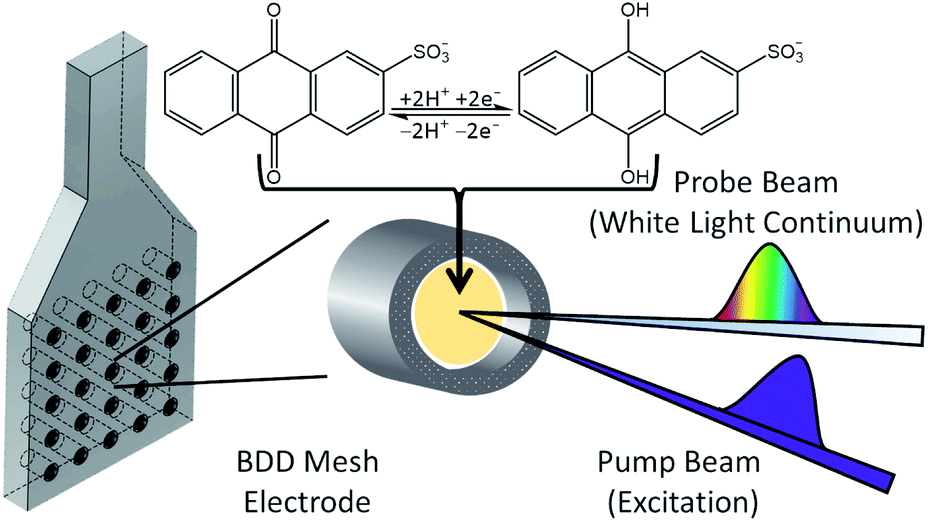 | ||
| Fig. 4 Schematic diagram of the spectroelectrochemical set-up highlighting its use on the TAS instrumentation. | ||
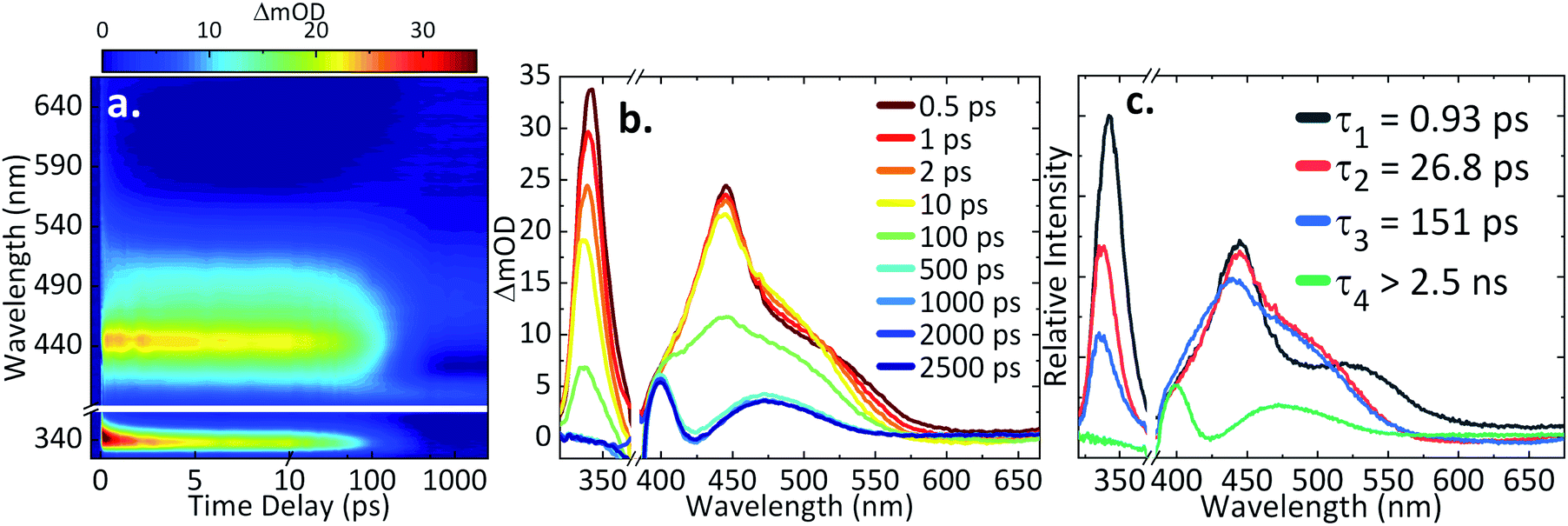 | ||
| Fig. 5 (a) TA spectra of the electrochemically reduced species (AH2QS; 1 mM AQS starting material, 0.1 M acetate buffer pH 4.9, 0.2 M KCl, −0.65 V vs. SCE), represented as a false colour map showing changes in optical density (ΔmOD) with photoexcitation at 382 nm; (b) different time delays (Δt) chosen for the TA spectra; (c) the corresponding evolution-associated difference spectra (EADS) from the global fit. Note: the AH2QS data was plotted by masking the spectral region of the pump leakage (370–390 nm). | ||
The TAS data was once again globally fit using a sequential model (vide supra for details) to extract the lifetimes contained in the TA spectra. The EADS linked to these time-constants are also shown in Fig. 5c, aiding in the visualisation of the transient features within the TA spectra. The associated residuals are shown in ESI 5, Fig. S6b.† In comparison to AQS, the data acquired for AH2QS is more complex and four time-constants (τ1–4) were now required to model the dynamical processes involved. A fifth time constant (≈60 fs) accounted for the instrument response (ESI 5, Fig. S7b†) due to contributions from the glass of the cuvette and the solvent itself, but was not taken into consideration when analysing the collected TA spectra. As seen in Fig. 5c, various features are formed in time, with their associated lifetimes and errors of τ1 = 0.93 ± <0.05 ps, τ2 = 26.8 ± 1.2 ps, τ3 = 151.2 ± 2.1 ps, and τ4 > 2.5 ns.
pH dependence measurements of the reduction process
In order to deconvolute the complex TA spectra and to understand the observed photochemical response, further steady-state UV/Vis measurements of AQS reduction were performed as a function of solution pH, in the wavelength range 350–600 nm. Given the reported pKa values of AQS,32Fig. 6a shows the structure of AH2QS (reductively formed under acidic conditions, < pH 7.6) and AHQS− (reductively formed under more basic conditions, > pH 7.6). The experiments were also complemented by excited-state calculations in explicit solvent; further information about the techniques used can be found in ESI 6.†Fig. 6b depicts the UV/Vis absorption spectra of the electrochemically reduced species at pH 4.9, pH 7.4, and pH 8.9. A clear difference can be observed between the pH 4.9 and pH 8.9 spectra, with the former associated with AH2QS and the latter with AHQS−, whilst the pH 7.4 spectrum shows features associated with both. At this juncture, it is worth pointing out the interesting similarity in the pH 8.9 UV/Vis absorption spectrum and the TA spectra for reduced AQS, in pH 4.9 buffer, at selected time delays ≥ 500 ps (Fig. 5b). We return to discuss the significance of this infra.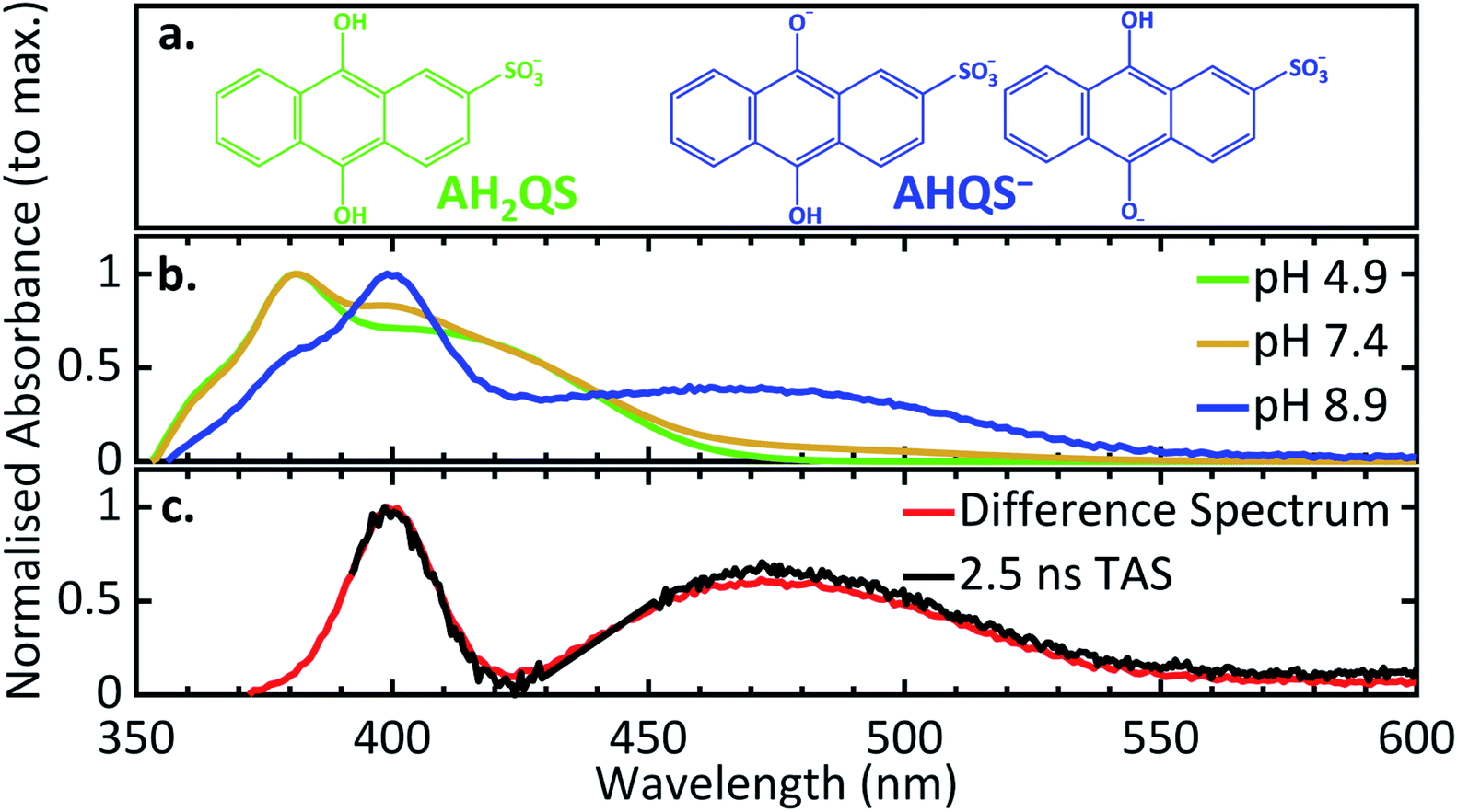 | ||
| Fig. 6 (a) The different protonated forms present at pH 4.9 and pH 8.9; (b) experimental UV/Vis spectra of the electrochemically reduced species at different pH (0.1 M acetate buffer pH 4.9, 0.1 M PBS buffer pH 7.4, 0.1 M carbonate–bicarbonate buffer pH 8.9; 0.2 M KCl added in all solutions; ca. −0.65 V vs. SCE applied potential for 30 minutes before recording spectra); (c) the UV/Vis difference spectrum between pH 8.9 and pH 4.9 in comparison to the 2.5 ns transient acquired from the TAS data of AH2QS at pH 4.9. | ||
Theoretical considerations of predicted UV/Vis spectra
The transitions observed from the implicit solvent singlet vertical excitations using the PBE0 functional for AH2QS and AHQS−, even though less accurate than for AQS, are presented in ESI 4, Table S2;† the lower energy peaks being assigned ππ* (S1 ← S0) transitions, and the higher energy peaks ππ* (S2 ← S0) transitions. The triplet state vertical excitation energies in the Franck–Condon region, albeit less reliable, are indicative of a triplet state Tn with energy below the first excited singlet state (S1) being accessed (ESI 4, Table S2†). To lend further weight to the identification of the species contributing to the UV/Vis spectra, ab initio explicit solvent theoretical calculations were performed on AQS, AHQS−, and AH2QS using ESTEEM51–53 (see Methods section and ESI 6† for detailed tasks performed). The different nature of the main excitations that contribute to the UV/Vis spectra in AQS and its derivatives, with differing charge-transfer contributions (see ESI 4, Table S1, 2†), mean that there is quite significant variation in peak energies between results for different functionals. We find that LC-ωPBE long-range corrected functional88 provided the most accurate results among all the functionals explored (ESI 6, Table S3†), correlating with the expectations that for large conjugated systems containing multiple excitations with different levels of charge transfer character, a range-separated hybrid is necessary for accurate theoretical spectroscopy. However, a fairly substantial blue shift in excitation energies was nevertheless observed,88,89 an effect that was also seen in range-separated functionals in previous work performed on a similar molecule, alizarin.52The theoretical UV/Vis spectrum from the explicit solvent calculations of AQS was 10 nm blue shifted for the lower-energy peak (320 nm) as seen in Fig. 7a and c, and 28 nm red shifted for the higher-energy peak (284 nm). Interestingly, for AH2QS and AHQS−, a blue shift was seen for both the higher and the lower-energy peaks; for AH2QS the lower-energy peak was shifted by 36 nm and the higher-energy peak by 48 nm (379 nm and 334 nm, respectively), whereas for AHQS−, both the lower and the higher-energy peaks (415 and 347 nm, respectively) were shifted ca. 60 nm (Fig. 7c). We note here that, due to the close similarity of the theoretical energies in explicit solvent for both structures studied for AHQS−, the results were merged into a combined spectrum for this molecule. A consistent blue shift of the theoretically predicted peak positions from the experimental data was observed throughout all the theoretical work in this paper when using range-separated functionals such as LC-ωPBE. This indicates that the different excitation characters contributing to the different peaks, make absolute peak positions more challenging to predict with TDDFT, no matter what functional is utilised. It should be noted, though, that the use of explicit solvent TDDFT has correctly predicted the state order of the transitions for the systems studied, their approximate peak shape and broadening, and the ratios of the heights of the peaks.
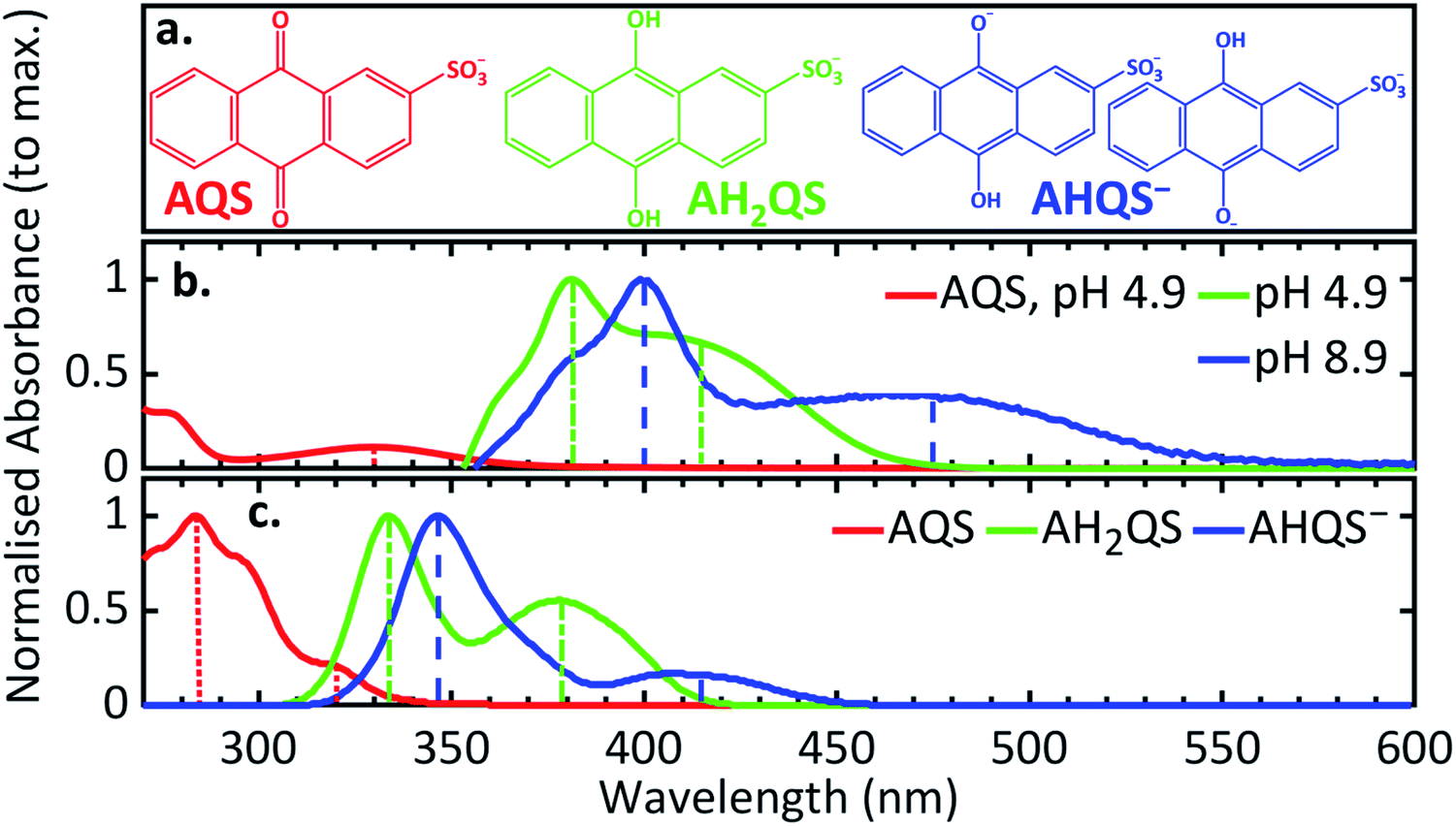 | ||
| Fig. 7 (a) The structures of AQS and the species present at pH 4.9 and pH 8.9; (b) experimental UV/Vis spectra of AQS (0.1 M acetate buffer pH 4.9, 0.2 M KCl) and the electrochemically reduced species at different pH (0.1 M acetate buffer pH 4.9, 0.1 M PBS buffer pH 7.4, 0.1 M carbonate–bicarbonate buffer pH 8.9; 0.2 M KCl added in all solutions; ca. −0.65 V vs. SCE for 30 minutes before recording spectra); (c) the predicted UV/Vis spectra from the LC-ωPBE explicit solvent TDDFT theoretical spectroscopy with spectral warp corrections applied (see ESI 6†). The vertical lines in (b, c) denote the experimental and theoretical peak positions corresponding to each feature. Note: the two structures studied for the species present at pH 8.9 showed very similar UV/Vis spectra. | ||
Further discussion of the AH2QS TAS data acquired
Taking into consideration the experimental data and the calculations above, we resume our discussion of the TAS data and associated lifetimes of AH2QS. At pH 4.9, photoexcitation (at 382 nm) is of the electrochemically reduced species AH2QS. Upon population of the excited state (Sn) of AH2QS, an evolution out of the FC window with concomitant solvent–molecule rearrangement/relaxation12 is described by τ1 = 0.93 ps in Fig. 5c, with EADS for τ1 appearing within the IRF and decaying with τ1. This, in turn, is followed by vibrational cooling from the excited state with a measured lifetime τ2 = 26.8 ps, with EADS for τ2 appearing with τ1 and decaying with τ2. We add here that the continuous spectral blue-shift in the maximum absorption at 345 nm (Fig. 5b) is also indicative of a vibrational relaxation along the excited state.90 These processes appear to take place within the same electronic state.More interestingly is the origin of τ3 and τ4. In Fig. 5c we notice the substantial difference between EADS for τ3 and EADS for τ4, which suggests either a new excited electronic state is being populated or the formation of a new species. We draw more insight into this when comparing the individual TA spectra (Fig. 5b), with the spectra in Fig. 6b. As the TA spectra represent difference spectra between the excited and ground states of AH2QS, for comparison, a ‘difference UV/Vis spectrum’ has been constructed in Fig. 6c (black line) by subtracting the UV/Vis spectrum at pH 4.9 (AH2QS) from pH 8.9 (AHQS−), Fig. 6b. The similarity between the 2.5 ns TA spectra (from Fig. 5b) and the “difference UV/Vis spectrum” is striking. The impressive agreement indicates that at this timescale,1 photoexcited AH2QS has most likely lost a proton and formed ground state AHQS−.
Taking into account the system used73,79 and the similarity between the TA spectra and the difference UV/Vis spectrum, we can deduce that the most likely cause of proton loss is a solute–solvent (or buffer) intermolecular single proton transfer between the excited state of AH2QS and the aqueous buffer solution, generating AHQS−; process occurring within τ3 = 151.2 ps. A caveat is in order here: proton transfer from AH2QS could be mediated by a triplet state, as discussed supra; we did not attempt to calculate the state arrangement at an ISC geometry in our present calculations but are planning to pursue this in the future to lend further insight. The formed AHQS− persists beyond the time-window of our measurements (Δt = 2.5 ns), its lifetime is assigned as τ4. These studies highlight the powerful insights that can be gained on early-stage mechanistic pathways from coupling ultrafast TAS with electrochemical methodology.
Conclusions
A compact spectroelectrochemical methodology based on BDD mesh electrodes, that can be used in a wide range of spectroscopic techniques, from primary (fs–ns) photodynamics all the way through to steady-state measurements, has been successfully developed to assess the time-resolved photochemical and photophysical properties of both the oxidised and reduced forms of a quinone (AQS/AH2QS). Whilst in many systems, the reduced form of the electron donating quinone is formed partway through a reaction pathway, we show how electrochemical methods can be used to directly isolate the reduced quinone for photodynamic interrogation. A bespoke freestanding BDD mesh electrode was developed to facilitate these experiments, with the hole size in the electrode, tuned to ensure no light scattering from the electrode itself. Such electrodes also offered low background currents, an extended solvent window in aqueous solution, were easy to fabricate, show reduced electrode fouling, and were also suitable for steady-state UV/Vis spectroelectrochemical measurements.Ultrafast TAS (UV/Vis) measurements on AQS in pH 4.9 buffer solution highlighted the formation of a triplet state within τ1 = 5.20 ps, with a proportion of the excited AQS molecules interacting with the aqueous buffer to form different transient species within τ2 = 587.8 ps. Both pathways had lifetimes persisting beyond Δt = 2.5 ns. Formation of the reduced species, AH2QS, was achieved via electrochemical reduction of AQS at the BDD mesh electrode. Upon photoexcitation to the lowest energy absorption peak maximum of AH2QS (382 nm), excited state relaxation dynamics occurred via solute–solvent (or buffer) intermolecular proton transfer within 151.2 ps, between AH2QS and the aqueous buffer, which persisted beyond Δt = 2.5 ns. This resulted in the formation of ground state AHQS−, at a pH not expected based on pKa considerations alone. The TAS data was supported by steady state UV/Vis measurements at different pH values. Explicit solvent TDDFT theoretical calculations were also performed on the proposed structures using the ESTEEM package for the spectroscopic and excited-state characterisation of molecules in a solvent environment. A consistent blue shift of the predicted energies was observed for all molecules, except for the higher-energy peak of AQS which was red shifted. This effect was observed across all functionals studied, LC-ωPBE being the most suitable for this system as the shape, broadening, and height ratios of peaks, along with the state order of transitions were correctly determined.
Whilst the present study has focused on one system, the data showcases the potential for providing routes into a mechanistic understanding of processes involving a change in oxidation state and formation of a photoactivated reaction intermediate. The approach also adds an ultrafast dynamic time dimension to the broad field of spectroelectrochemistry, adding to the suite of techniques available to tackle the many outstanding challenges associated with photoactivated processes.
Data availability
All the relevant data to this study is presented in the main text and the ESI.† The underlying raw data used in this publication can be accessed via the internet through the Zenodo repository at DOI: 10.5281/zenodo.5778925.Author contributions
S. G., B. R. R., A. B., and V. G. S. conceived the presented idea. S. G. developed the methodology, built the system, and carried out the experiments. A. J. B., S. J. C., J. T., and J. V. M. contributed in building the spectroelectrochemical cell and the BDD electrode, and in conducting the electrochemical experiments. J. M. W., M. D. H., and M. S. contributed in conducting TEAS measurements. N. D. M. H., M. A. P. T., and S. G. performed the computational calculations. S. G., M. A. P. T., J. M. W., M. S., N. D. M. H., S. J. C., J. V. M., and V. G. S. analysed the results. S. G. and M. A. P. T. designed the figures. S. G. wrote the manuscript with input from all authors. V. G. S., J. V. M., B. R. R., and A. B. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Warwick Centre for Ultrafast Spectroscopy (https://warwick.ac.uk/fac/sci/wcus) for the use of the Cary 60 UV/Vis Spectrometer and the TAS set-up. Computing facilities were provided by the Scientific Computing Research Technology Platform of the University of Warwick through the use of the High Performance Computing (HPC) cluster Tinis, and the use of Athena and Sulis Tier 2 platforms at HPC Midlands+ funded by the Engineering and Physical Sciences Research Council (EPSRC), grant numbers EP/P020232/1 and EP/T022108/1. N. D. M. H. and M. A. P. T. acknowledge the support of EPSRC grant number EP/P02209X/1, whilst J. V. M. acknowledges EP/V056778/1. S. G. would like to thank Prof. Scott Habershon for assistance with the theory work and Dr Dugan Witherick for installation of required modules on the HPC platforms. S. G. would also like to thank Syngenta and EPSRC for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1. J. M. W. is grateful to EPSRC and Newport Spectra-Physics Ltd for a joint studentship. M. D. H. thanks the Leverhulme Trust for postdoctoral funding. A. J. B. and S. J. C. thank the Centre for Doctoral Training in Diamond Science and Technology (EP/L015315/1). J. J. T. acknowledges The Royal Society for financial support under the Industry Fellows PhD studentship scheme (INF/PHD/180016). V. G. S. would like to thank the Royal Society for a Royal Society Industry Fellowship.References
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef CAS.
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef CAS.
- T. T. Eisenhart and J. L. Dempsey, J. Am. Chem. Soc., 2014, 136, 12221–12224 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 954–959 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 1–4 Search PubMed.
- C. L. Pitman and A. J. M. Miller, ACS Catal., 2014, 4, 2727–2733 CrossRef CAS.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS PubMed.
- A. J. Orr-Ewing, Struct. Dyn., 2019, 6, 010901 CrossRef PubMed.
- J. C. Owrutsky, D. Raftery and R. M. Hochstrasser, Annu. Rev. Phys. Chem., 1994, 45, 519–555 CrossRef CAS PubMed.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- S. Bold, L. Zedler, Y. Zhang, J. Massin, V. Artero, M. Chavarot-Kerlidou and B. Dietzek, Chem. Commun., 2018, 54, 10594–10597 RSC.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
- Y. Zhang, L. Zedler, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2019, 21, 10716–10725 RSC.
- L. M. Kiefer, L. B. Michocki and K. J. Kubarych, J. Phys. Chem. Lett., 2021, 12, 3712–3717 CrossRef CAS PubMed.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- S. J. Cobb, Z. J. Ayres and J. V. Macpherson, Annu. Rev. Anal. Chem., 2018, 11, 463–484 CrossRef CAS PubMed.
- J. V. Macpherson, Phys. Chem. Chem. Phys., 2015, 17, 2935–2949 RSC.
- N. Yang, S. Yu, J. V Macpherson, Y. Einaga, H. Zhao, G. Zhao, G. M. Swain and X. Jiang, Chem. Soc. Rev., 2019, 48, 157–204 RSC.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed.
- G. Mitchell, E. D. Clarke, S. M. Ridley, D. T. Greenhow, K. J. Gillen, S. K. Vohra and P. Wardman, Pestic. Sci., 1995, 44, 49–58 CrossRef CAS.
- F. Müh, C. Glöckner, J. Hellmich and A. Zouni, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 44–65 CrossRef PubMed.
- W. Zhang, J. Gacs, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2017, 9, 3821–3826 CrossRef CAS PubMed.
- P. Reddy Maddigapu, C. Minero, V. Maurino, D. Vione, M. Brigante and G. Mailhot, Chemosphere, 2010, 81, 1401–1406 CrossRef PubMed.
- D. Vione, P. R. Maddigapu, E. De Laurentiis, M. Minella, M. Pazzi, V. Maurino, C. Minero, S. Kouras and C. Richard, Water Res., 2011, 45, 6725–6736 CrossRef CAS PubMed.
- J. Sun and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 10904–10908 CrossRef CAS PubMed.
- R. S. K. A. Gamage, A. J. McQuillan and B. M. Peake, J. Chem. Soc., Faraday Trans., 1991, 87, 3653–3660 RSC.
- C. Batchelor-McAuley, Q. Li, S. M. Dapin and R. G. Compton, J. Phys. Chem. B, 2010, 114, 4094–4100 CrossRef CAS PubMed.
- L. A. Hutton, J. G. Iacobini, E. Bitziou, R. B. Channon, M. E. Newton and J. V. Macpherson, Anal. Chem., 2013, 85, 7230–7240 CrossRef CAS PubMed.
- S. J. Cobb, F. H. J. Laidlaw, G. West, G. Wood, M. E. Newton, R. Beanland and J. V. Macpherson, Carbon, 2020, 167, 1–10 CrossRef CAS.
- R. Berera, R. van Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2007, 18, 1–46 CrossRef.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 85, 064104 CrossRef PubMed.
- S. A. Kovalenko, A. L. Dobryakov, J. Ruthmann and N. P. Ernsting, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 2369–2384 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. De Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- E. Aprà, E. J. Bylaska and W. A. De Jong, et al. , J. Chem. Phys., 2020, 152, 184102 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. M. York and M. Karplus, J. Phys. Chem. A, 1999, 103, 11060–11079 CrossRef CAS.
- E. Clementi, C. C. J. Roothaan and M. Yoshimine, Phys. Rev., 1962, 127, 1618–1620 CrossRef CAS.
- R. O. Jones and O. Gunnarsson, Rev. Mod. Phys., 1989, 61, 689–746 CrossRef CAS.
- T. J. Zuehlsdorff, P. D. Haynes, M. C. Payne and N. D. M. Hine, J. Chem. Phys., 2017, 146, 124504 CrossRef CAS PubMed.
- M. A. P. Turner, M. D. Horbury, V. G. Stavros and N. D. M. Hine, J. Phys. Chem. A, 2019, 123, 873–880 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen and J. Blomqvist, et al. , J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- M. Kraus, W. Wintz, U. Seifert and R. Lipowsky, Phys. Rev. Lett., 1996, 77, 3685–3688 CrossRef CAS PubMed.
- E. R. Davidson, Chem. Phys. Lett., 1996, 260, 514–518 CrossRef CAS.
- E. Gross and E. Runge, Phys. Rev. Lett., 1984, 52, 997 CrossRef.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- J. C. A. Prentice, J. Aarons and J. C. Womack, et al. , J. Chem. Phys., 2020, 152, 174111 CrossRef CAS PubMed.
- G. Lever, D. J. Cole, N. D. M. Hine, P. D. Haynes and M. C. Payne, J. Phys.: Condens. Matter, 2013, 25, 152101 CrossRef PubMed.
- L. E. Ratcliff, N. D. M. Hine and P. D. Haynes, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 165131 CrossRef.
- X. Ge, I. Timrov, S. Binnie, A. Biancardi, A. Calzolari and S. Baroni, J. Phys. Chem. A, 2015, 119, 3816–3822 CrossRef CAS PubMed.
- R. W. Murray, W. R. Heineman and G. W. O’Dom, Anal. Chem., 1967, 39, 1666–1668 CrossRef CAS.
- T. P. DeAngelis and W. R. Heineman, J. Chem. Educ., 1976, 53, 594–597 CrossRef CAS.
- J. Heitmüller, K. Eckstein, R. Renner, M. Stolte, T. Hertel, F. Würthner and T. Brixner, Spectrochim. Acta, Part A, 2021, 253, 119567 CrossRef PubMed.
- Y. El Khoury, L. J. G. W. Van Wilderen, T. Vogt, E. Winter and J. Bredenbeck, Rev. Sci. Instrum., 2015, 86, 083102 CrossRef PubMed.
- A. D. Modestov, J. Gun and O. Lev, Surf. Sci., 1998, 417, 311–322 CrossRef CAS.
- J. Stotter, J. Zak, Z. Behler, Y. Show and G. M. Swain, Anal. Chem., 2002, 74, 5924–5930 CrossRef CAS PubMed.
- J. Stotter, S. Haymond, J. K. Zak, Y. Show, Z. Cvackova and G. M. Swain, Electrochem. Soc. Interface, 2003, 12, 33–38 CrossRef.
- J. K. Zak, J. E. Butler and G. M. Swain, Anal. Chem., 2001, 73, 908–914 CrossRef CAS PubMed.
- A. Bedini, E. De Laurentiis, B. Sur, V. Maurino, C. Minero, M. Brigante, G. Mailhot and D. Vione, Photochem. Photobiol. Sci., 2012, 11, 1445–1453 CrossRef CAS PubMed.
- P. R. Maddigapu, A. Bedini, C. Minero, V. Maurino, D. Vione, M. Brigante, G. Mailhot and M. Sarakha, Photochem. Photobiol. Sci., 2010, 9, 323–330 CrossRef CAS PubMed.
- Y. B. Shim and S. M. Park, J. Electroanal. Chem., 1997, 425, 201–207 CrossRef CAS.
- P. S. Guin, S. Das and P. C. Mandal, Int. J. Electrochem., 2011, 1–22 Search PubMed.
- Q. Li, C. Batchelor-Mcauley, N. S. Lawrence, R. S. Hartshorne and R. G. Compton, Chem. Commun., 2011, 47, 11426–11428 RSC.
- J. Jacq, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29, 149–180 CrossRef CAS.
- G. L. Zhu, L. W. Zhang, Y. C. Liu, Z. F. Cui, X. S. Xu and G. Z. Wu, Chin. J. Chem. Phys., 2016, 29, 140–146 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, 2004, 1657, 82–104 CrossRef CAS PubMed.
- I. Loeff, A. Treinin and H. Linschitz, J. Phys. Chem., 1983, 87, 2536–2544 CrossRef CAS.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- A. Saha, M. Chen, M. Lederer, A. Kahnt, X. Lu and D. M. Guldi, Chem. Sci., 2017, 8, 1360–1368 RSC.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS PubMed.
- M. A. Rohrdanz and J. M. Herbert, J. Chem. Phys., 2008, 129, 034107 CrossRef PubMed.
- J. A. Mondal, V. Samant, M. Varne, A. K. Singh, T. K. Ghanty, H. N. Ghosh and D. K. Palit, ChemPhysChem, 2009, 10, 2995–3012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectroelectrochemical set-up used and CV measurements, UV/Vis spectra monitoring the redox cycle, ‘scheme of squares’ for the AQS/AH2QS redox couple, implicit solvent TDDFT results, power measurements, residual data, and solvent scans for the TAS data, explicit solvent TDDFT results. See DOI: 10.1039/d1sc04993c |
| This journal is © The Royal Society of Chemistry 2022 |