
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new route to polyoxometalates via mechanochemistry†
Manuel
Wilke
 * and
Nicola
Casati
* and
Nicola
Casati
Laboratory for Synchrotron Radiation – Condensed Matter, Paul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen PSI, Switzerland. E-mail: manuel.wilke@psi.ch
First published on 13th December 2021
Abstract
Mechanochemistry offers a new route to polyoxometalates (POMs) under mild conditions. The molybdenum isoPOM heptamolybdate and the molybdenum heteroPOMs of the Strandberg- and Keggin-type could be achieved from grinding together molybdenum oxide, potassium or ammonium carbonate and phosphate. The reactions were controlled by the stoichiometric ratio of the starting materials and the liquid used, with reaction times between 30 min and 3 h. In situ investigations of the syntheses reveal the formation of intermediates during the reactions. Their identification helps explaining the mechanism of formation of the intermediates as well as the final POMs.
Introduction
Polyoxometalates (POMs) are a material class of early transition metal oxygen cluster anions, with MoVI, WVI, and VV as the most described metals. The counter ions on the other hand range from hydrogen atoms over small inorganic cations to complex molecules.1,2 If the cluster includes heteroanions such as PO43− or SO43− they are called heteroPOMs, otherwise isoPOMs.3 The first reported POM was the nowadays-called Keggin-type [PM12O40]3−, which was also, in its form of phosphothungstic acid, the first one where the structure could be determined from powder X-ray diffraction data in 1933.4,5 In the last decades POMs were intensively investigated, showing a huge range of structural varieties and properties. The structures can not only differ in size of cluster, ranging from 6 to over 300 metal oxide building blocks, but also by the incorporation of organic molecules and the usage of POMs as building units for metal–organic frameworks.3,6–8 The resulting varying properties make them useful for a wide range of applications, e.g. in catalysis,3 energy storage3 and medicine7,9,10 or as ion-exchangers11 sensors,5 and electrochromic devices.12POMs are usually synthesized in aqueous solutions, containing the respective metal oxoanions. For the condensation reactions the pH of the solution plays a crucial role and has to be adjusted by the usage of acids. The syntheses are done at different conditions, ranging from ambient temperature over reflux to hydrothermal conditions.3 The use of alternative methods is scarce. Yan and co-workers used microwaves to synthesize POMs out of sodium molybdate.13,14 In a solid-state synthesis, the Keggin-type POM as K3PM12O40 (M = Mo, W) could be synthesized via heating a mixture of MO3, K2CO3 and K2HPO4 at 500 °C for 30 h.15
In the last decade mechanochemistry has gained a lot of interest as a synthetic method. The term is used for chemical reactions induced by mechanical energy and performed in hand mortars as well as ball, planetary and vibration mills.16 Mechanochemical reactions are increasingly used thanks to their easy handling, short reaction times, high conversion rates, a small production of waste, based on a high atom economy and the small amount of solvents used.17–20 Moreover, by using little quantities of liquids (liquid assisted grinding, LAG) the reaction outcome can be influenced regarding the yield, reaction times and the nature of the product. Mechanochemistry offers the possibility to create new compounds, not or only difficult achievable from solution.21–27 Thanks to the solid-state nature of the process, it is possible to follow in situ the reaction and several setups where developed, using X-ray diffraction, spectroscopy or temperature devices.28–34
Although POMs are used in mechanochemical synthesis, e.g. to exchange the cation35,36 or to encapsulate the POM inside of a metal–organic framework (MOF),37–40 there is, to the best of our knowledge, no description of a direct mechanochemical synthesis. Such an alternative route could make the use of strong acids obsolete as well as decrease reaction times and the energy consumption. Beside that, a different synthetic route could lead to new and unknown structures. Moreover, the relatively higher stability of solid products may be an advantage against a possible reorganization of POMs in solution. Therefore, the goal of this work was to produce POMs mechanochemically from the scratch and to investigate the reaction process by using in situ methods.
Results and discussion
Mechanochemical synthesis
Most syntheses from solution are starting from molybdate, MoO42−. To follow these routes in a solid-state synthesis would be difficult, since, depending on the desired POM, specific conditions regarding the solvent, pH, temperature and pressure are needed. Instead we wanted to use MoO3 and add O2− by using compounds who have good leaving group, e.g. carbonates with CO32−. Our reactions are done in a vibration ball mill, where the reaction mixture is weighted together with steel balls into a steel jar, which is closed and mounted into a vibrating holder. Any gas formation during reaction would therefore bee observed as a pressure release at the end of the reaction itself. The details of all lab syntheses can be found in the ESI.† The first idea was to create simple POMs with only one atom species. The isoPOM heptamolybdate [Mo7O24]6− is a well known member of the POM family. Other than most POMs it can be directly synthesized from solutions of MoO3 in ammonium hydroxide by evaporation.41 This simple approach and high tendency to self-assembly made it an ideal candidate to prove if our approach could work. The isoPOM heptamolybdate could be produced within the compounds (NH4)6/K6Mo7O24·4H2O (1a, 1b). They are formed already by neat grinding of MoO3 and NH4/KHCO3, but, as visible in the reaction equation in Fig. 1, for the full formation the addition of a small amount of water (30 μL to 500 mg) is needed. The respective Rietveld refinements can be found in the ESI.† Within 30 min a pure and highly crystalline compound could be produced.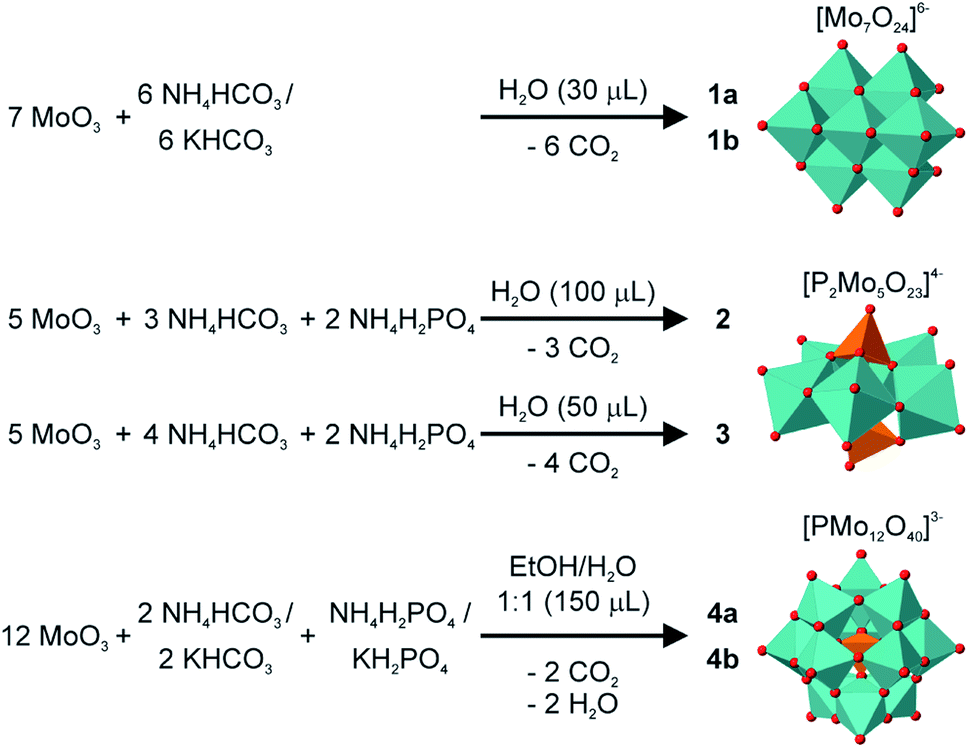 | ||
| Fig. 1 Reaction equations for the mechanochemically synthesized POMs in this work. For all syntheses a vibration ball mill Pulverisette 23 (Fritsch GmbH, Germany) and a total load of 500 mg was used. The reaction times varied between 30 min and 3 h. [Mo7O24]6− – heptamolybdate, [P2Mo5O23]4− – Strandberg-type, [PMo12O40]3− – Keggin-type; turquoise: molybdenum, orange: phosphorus, red: oxygen. | ||
In the next step we wanted to introduce heteroatoms in the POM structure. The Strandberg-type POM, [P2Mo5O23]4−, has a comparably high ratio of heteroatoms which is why we choose it as a first candidate. It could be realized within (NH4)5HP2Mo5O23·3H2O (2) and (NH4)6P2Mo5O23·5.5H2O (3), by grinding MoO3, NH4HCO3 and NH4H2PO4 in a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 and 5:4:2, respectively (Fig. 1). The addition of water (100 μL and 50 μL, respectively) was necessary for the formation of a pure and highly crystalline product. The reaction times varied from 30 to 60 min. For 2 a structure from literature was used for the Rietveld refinement while the structure of the new compound 3 could be solved from PXRD data (see ESI†).
:3:2 and 5:4:2, respectively (Fig. 1). The addition of water (100 μL and 50 μL, respectively) was necessary for the formation of a pure and highly crystalline product. The reaction times varied from 30 to 60 min. For 2 a structure from literature was used for the Rietveld refinement while the structure of the new compound 3 could be solved from PXRD data (see ESI†).
With the proof that even heteroPOMs can be synthesized, the Keggin-type POM, as probably the best known representatives of the POM material class, was an attractive candidate to be synthesized by our method. Its synthesis requires strong acidic conditions, which can not be applied in an mechanochemical synthesis. Nevertheless, the Keggin-type POM containing compounds (NH4)3/K3PMo12O40 (4a, 4b) could both be synthesized by grinding MoO3, the respective bicarbonate and dihydrogen phosphate together (Fig. 1). It was necessary to add a mixture of water and ethanol to the reaction. With no solvent or ethanol alone almost no reaction could be observed, while with water as the only liquid the formation of a side product, with very broad peaks which could not be identified, was promoted. With 3 h the synthesis took much longer than the other ones presented in this paper. To prove the correct phase, we did Rietveld refinements (Fig. S3†). In both syntheses, a small amount of MoO3 can still be detected, 3 wt% and 2 wt%, respectively. Milling a longer time or with heavier balls did not help to fully complete the reaction, since it promoted the formation of the unidentified side product as well.
Our results show that the formation of POMs via mechanochemistry can be a valid alternative. The isoPOM heptamolybdate could be synthesized within 30 min by ball milling with a small amount of water. The products were highly crystalline powders. The Rietveld refinements prove the purity of the compounds and no further cleaning step was needed. For the heteroPOMs a small variation of the amounts of the starting materials is enough to get full control over the formed product. By using the respective stoichiometric ratio always the exact POM is formed. Using liquid assisted grinding the Strandberg-type POM was produced as a highly crystalline and pure product within maximum 60 min. The Rietveld refinements do not show any additional reflections. For the Keggin-type POM the synthesis was more complicated. The balance had to be found between a high conversion rate and the production of a second phase. By choosing the correct parameter it was possible to synthesize the Keggin-type POM wihtin 3 h with high purity.
In situ investigations
In our experiments we used MoO3 instead of MoO42−, which is mostly used in solution based syntheses. Also we worked under much milder conditions. In no synthesis strong acids were added to the mixture. To enable the full potential of the mechanochemical synthesis of POMs a better understanding of the mechanisms is needed. For this reason, we investigated in situ the formation of the three POMs which we synthesized.Both syntheses for the heptamolybdate in 1a and 1b were followed in situ and very similar pathways could be observed, (Fig. 2a and b, left). Among the reagents we could observe diffraction peaks of MoO3 from the very beginning, while no peak of the respective bicarbonates are visible at any time during the milling. In both cases an intermediate phase is formed immediately. During the synthesis the reflections of this phase vanish, while the ones of the product evolve. In the case of 1b, the peaks of MoO3 get very weak in the first minutes but can still be detected until the end. The lab synthesis instead produced a pure compound. We used the setup of the Materials Science beamline at the SLS, Paul Scherrer Institute in Switzerland.42 Due to the machinery of the in situ setup, where the powder is measured in a special probing area, there is the possibility of caking powder in this area, when additional liquid is used. This is possibly the reason for this observed slight difference. For both reactions we were able to identify the intermediates, which are novel molybdenum carbonates, namely and respectively (NH4)2MoO3CO3 (5a) and K2MoO3CO3·0.5H2O (5b). Both structures were solved from PXRD data. A detailed description, including crystallographic details and the Rietveld refinements can be found in the ESI.†
 | ||
| Fig. 2 2D-plot of the in situ investigation of the respective synthesis using PXRD (left), and comparison of the observed intermediate compound with a novel synthesized structure (right) for (a) (NH4)6Mo7O24·4H2O 1a and (b) K6Mo7O24·4H2O 1b. The molybdenum network for the intermediate is shown on the right for 5a and 5b, respectively. | ||
For 1b we were able to isolate the intermediate by reacting MoO3 with K2CO3 (1:1, LAG H2O). In the potassium molybdenum carbonate 5b, molybdenum is coordinated by three oxygen atoms and bidentate by a carbonate ligand, resulting in a 1D chain structure (Fig. 2b, right). A comparison with an in situ powder pattern after 2 min clearly confirms this as the found intermediate.
For 1a the ammonium molybdenum carbonate 5a could only be synthesized within a mixture with 5a as the main compound with 66 wt% (see ESI†). The motif is similar to 5b (Fig. 2a, right), except that no water of crystallization is present. The comparison between a calculated powder pattern of 5a and the powder pattern after 80 s of the in situ investigation of 1a is proving that it is the quickly formed intermediate. In both cases the formation of the heptamolybdate happened via the formation of a molybdenum carbonate. The carbonate coordinates to the molybdenum, activating the MoO3 and then CO2 is evolved.
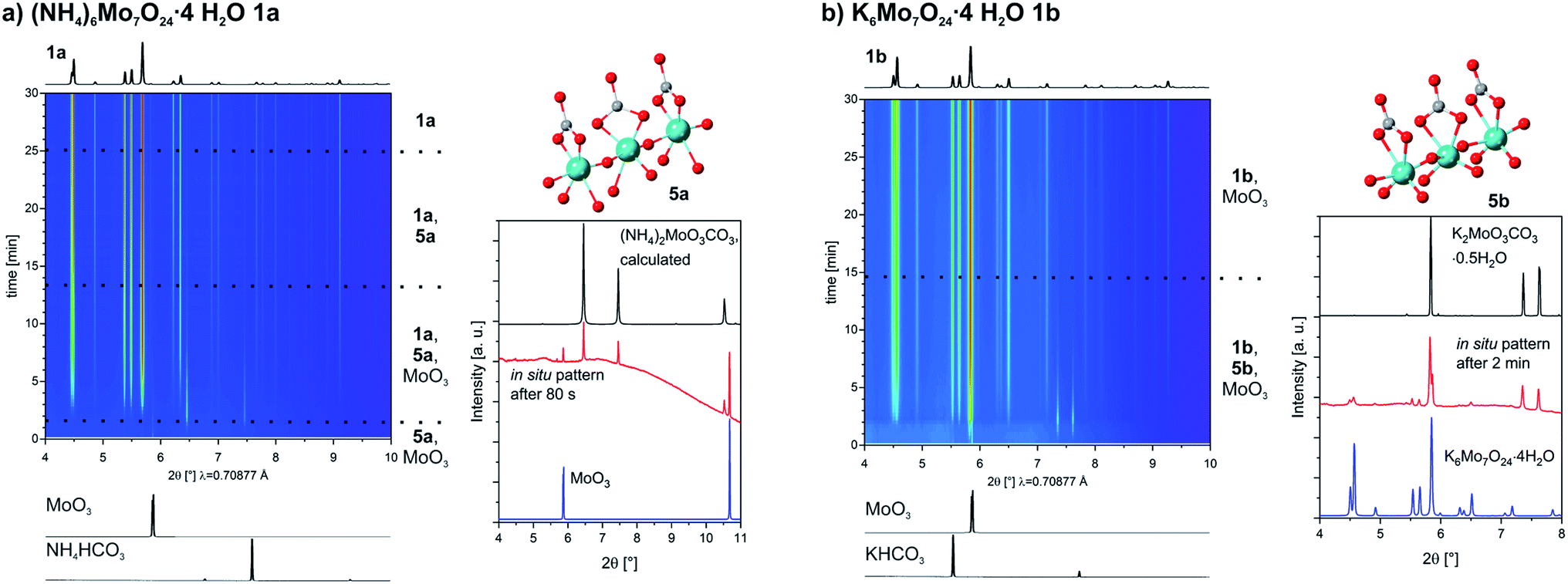
For the Strandberg-type POM we followed in situ the formation of 2. To prevent caking we halved the amount of water used. However, at the end the powder was wet and caked in the probing area of the in situ jar. Nevertheless, the reaction is proceeding and our gained data can still shed light on the reaction pathway. The 2D plot of the time-resolved powder patterns is shown in Fig. 3. As observed for the heptamolybdates, MoO3 can be detected from the start. The additional reflections can not be assigned to the other starting materials, but to (NH4)2HPO4. Its reflections can be detected until 9 min of milling, showing that there is also an interaction between the non-metallic starting materials. After 6 min another intermediate is fast forming. We identified it as a new ammonium phosphomolybdate containing the Strandberg-type POM ((NH4)6−xHxP2Mo5O23·yH2O 6). After 36 min also 2 can be detected and its peaks get more intense with time while at the same time the ones of 6 start to vanish. After 80 min, all peaks of the intermediate and the ones of MoO3 vanish, and the reaction is completed.
 | ||
| Fig. 3 2D-plot of the in situ investigation for the synthesis of (NH4)5HP2Mo5O23·3H2O 2 using PXRD (left, * = position of the peaks of (NH4)2HPO4(N2HP)), and comparison of the observed intermediate compound with the pattern of the novel synthesized structure (NH4)6−xHxP2Mo5O23·yH2O 6 and of the calculated pattern of this compound using x = 0.5, y = 2.5 (right). The pattern from the lab synthesis does also contain peaks for NH4H2PO4 and (NH4)2HPO4. | ||
For a certain period, the unknown intermediate and MoO3 are the only two phases visible. We were not able to isolate the intermediate, but were able to identify it as another ammonium phosphomolybdate containing the Strandberg-type POM. The formula is (NH4)6−xHxP2Mo5O23·yH2O (6, x = 2–0, y = 2–4). The competition with the very similar compounds 2 and 3 prevented a pure synthesis. The details are described in the ESI.† In Fig. 3, on the right a comparison between an in situ pattern after 10 min and patterns from 6 (from the lab synthesis and calculated with x = 0.5, y = 2.5) is showing that 6 is the observed intermediate.
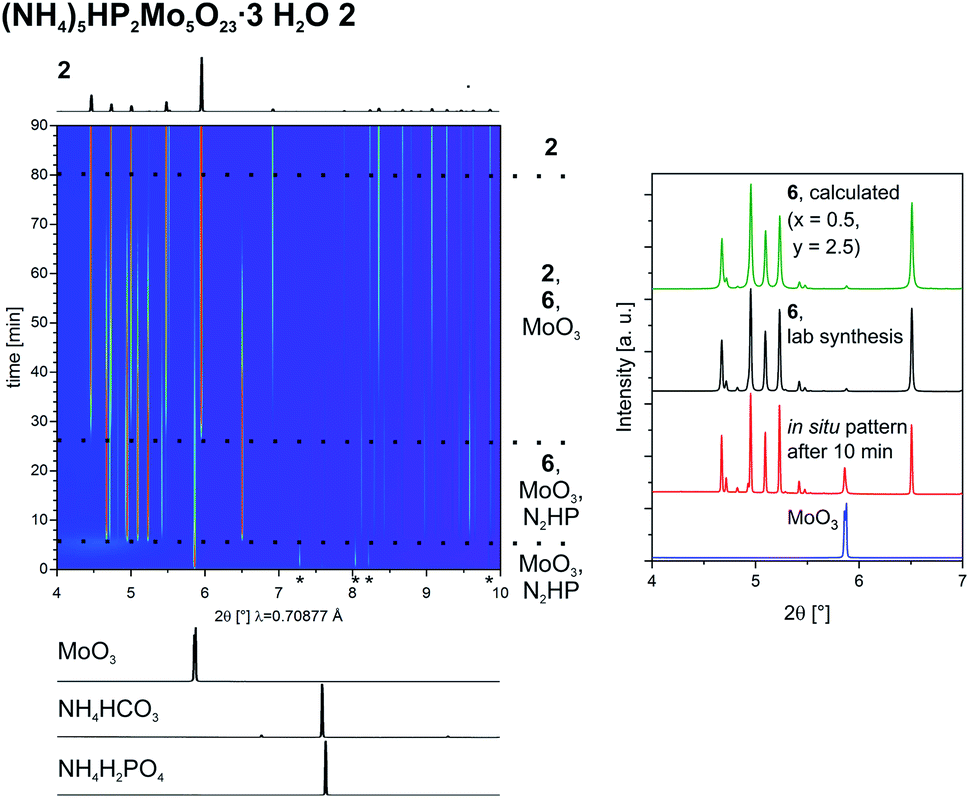
To investigate the formation of the Keggin-type POM we followed the synthesis of 4ain situ (Fig. 4, on the left). To lower the amount of caking we reduced the amount of liquid to 50 μL. Nevertheless, at the end the powder was caked and after 4 h the synthesis did not proceed anymore but was also not completed. As seen before, also this reaction proceeds with the formation of intermediates. At the beginning only peaks of MoO3 can be detected. No other starting materials could be observed. After 1.5 min 6 is formed as an intermediate, as in the synthesis of 2, followed by the formation of 2 after 3.5 min. Shortly after the peaks of 6 are vanished. The peaks of 2 get less intense with time as well and are very weak after 4 h. The product 4a can be detected for the first time after 18 min. In the following hours it is formed more and its crystallinity improves, as visible from the more intense and sharper diffraction peaks. After 4 h, no changes can be observed. The reaction is not complete, very likely because of the observed caking.
 | ||
| Fig. 4 2D-plot of the in situ investigation of the formation of (NH4)3PMo12O404a using PXRD (left), and comparison of the observed intermediate compounds with the calculated pattern of the novel structure 6 (with x = 0.5, y = 2.5), after 160 s (middle) and with 2, after 9 min (right). | ||
In all conducted syntheses we could observe intermediates, formed before the final product evolves. In the syntheses of heptamolybdate, molybdenum carbonates are formed intermediately. These compounds where not described before, probably based on the reactivity of the carbonate ion and, for 5a, the short life time. During the synthesis of the Keggin-type POM the Strandberg-type POM is formed as an intermediate. The intermediates differ for the isoPOM and the heteroPOM reactions, but they have in common that they include a bigger amount of the basic anion, either carbonate or phosphate (Fig. 5). In all reactions a pressure release could be observed when the jar was opened, showing the evolution of CO2. Therefore, the formation of molybdenum carbonates seems to be possible also in the heteroPOM reactions. Nevertheless, the respective compounds could not be observed during the ball milling. Based on the small amount of carbonate during the reactions, it might be possible that the formed amount is too small or the reaction is too fast. Grinding only MoO3 and NH4H2PO4, neat or as LAG with water, did not lead to a reaction. (NH4)2HPO4 instead reacts with MoO3 even without the addition of ammonium carbonate, indicating that also the ammonium-ion and the acidity play an important role. Based on these results we can deduce that MoO3 is activated by a nucleophilic attack. The activated species react with each other or with more starting material to form the final POM.
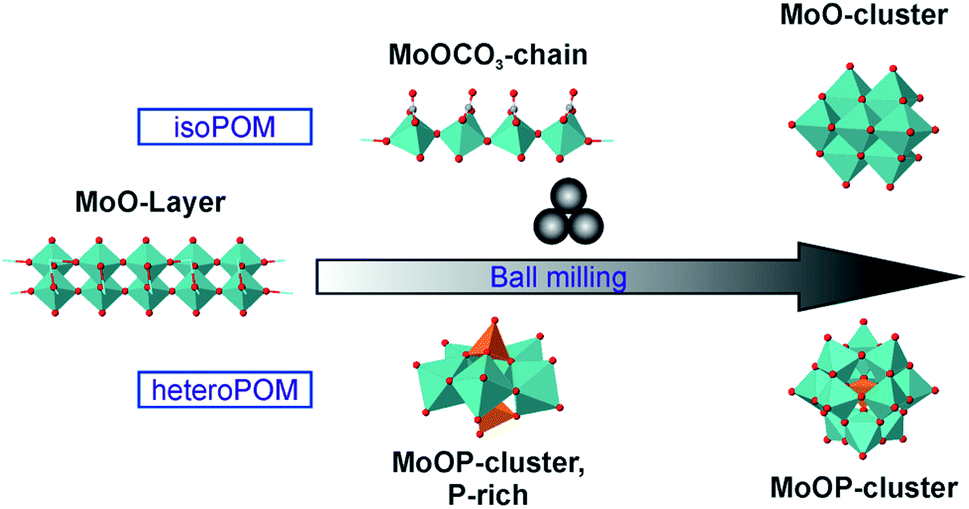 | ||
| Fig. 5 Formation scheme for the mechanochemical synthesis of POMs. | ||
A comparison to the classical, solution based synthesis of POMs is difficult, since there mostly the more aggressive molybdate MoO42− is used. The formation process is supposed to happen via condensation and addition reactions.43 In the in situ investigations of our mechanochemical reactions we could not observe any MoO42− species or its condensation products, except for heptamolybdate, which is the final product of the synthesis of 1a and 1b. Additionally, NH4MoO4 was formed as a side product in the synthesis of 5a. Nevertheless, our results suggest that different mechanisms lead to the POM formation in the mechanochemical reaction than in the solution based methods.
With the possibility, to synthesize POMs via mechanochemistry, come several opportunities. The short reaction times lower the energy consumption of the process. The absence of strong acids, the low amount of liquids and the high atomic economy are a huge asset for less waste production and go towards green chemistry.44 As typical in mechanochemical reactions, the yield only depends on the possibility to remove the powder from the jar since the conversion is almost complete. Moreover, new compounds can also be stabilized in these conditions, as demonstrated by the synthesis of two new POMs containing the Strandberg-type POM and especially with the two new ammonium molybdenum carbonates. Host-guest compounds instead, where POMs are encapsulated into MOFs are already done mechanochemically. Now it might be possible, to create the POM on the side, while the MOF and the host-guest structure is formed. This might not only safe one production step, it also might allow the encapsulation of different POMs and create different structures. Also the new field of POMs as secondary building-units in MOFs is open for mechanochemical tries.
Conclusion
Our results show that POMs can be formed mechanochemically from simple building blocks. Under mild conditions and in a fast and easy manner we could synthesize iso- and heteroPOMs with varying counter ions. In situ investigations revealed that the formation goes via metal oxygen clusters, incorporating the basic anions of the starting materials. This first mechanistic insight shows that the molybdenum oxide is activated by a nucleophilic attack before the final product is formed. This new route to polyoxometalates provides a big range of possibilities, from synthesizing POMs in short times, to finding new structures or to find new ways to encapsulate them into porous frameworks.Author contributions
Manuel Wilke planed and conducted the research and wrote the manuscript. Nicola Casati participated in the data analysis and supported the writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the funding received from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No. 701647.Notes and references
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef
.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS PubMed
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed
- J. F. Keggin, Nature, 1933, 131, 908–909 CrossRef CAS
- D. Wang, L. L. Liu, J. Jiang, L. J. Chen and J. W. Zhao, Nanoscale, 2020, 12, 5705–5718 RSC
- D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed
- W. Xu, X. Pei, C. S. Diercks, H. Lyu, Z. Ji and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 17522–17526 CrossRef CAS PubMed
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed
- J. Zhao, K. Li, K. Wan, T. Sun, N. Zheng, F. Zhu, J. Ma, J. Jiao, T. Li, J. Ni, X. Shi, H. Wang, Q. Peng, J. Ai, W. Xu and S. Liu, Angew. Chem., Int. Ed., 2019, 58, 18032–18039 CrossRef CAS PubMed
- S. Uchida, Chem. Sci., 2019, 10, 7670–7679 RSC
- H. Gu, C. Guo, S. Zhang, L. Bi, T. Li, T. Sun and S. Liu, ACS Nano, 2018, 12, 559–567 CrossRef CAS PubMed
- X.-x. Jin, W.-d. Yu, Y.-M. Nie, M.-S. Liu and J. Yan, Dalton Trans., 2016, 45, 3268–3271 RSC
- Y.-M. Nie, S. Liang, W.-D. Yu, H. Yuan and J. Yan, Chem.–Asian J., 2018, 13, 1199–1205 CrossRef CAS PubMed
- F. Goubin, L. Guénée, P. Deniard, H. J. Koo, M. H. Whangbo, Y. Montardi and S. Jobic, J. Solid State Chem., 2004, 177, 4528–4534 CrossRef CAS
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC
- A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Commun., 2015, 6, 6662 CrossRef CAS PubMed
- M. Wilke, L. Batzdorf, F. Fischer, K. Rademann and F. Emmerling, RSC Adv., 2016, 6, 36011–36019 RSC
- M. Wilke, A. G. Buzanich, U. Reinholz, K. Rademann and F. Emmerling, Dalton Trans., 2016, 45, 9460–9467 RSC
- C. Mottillo and T. Friščić, Molecules, 2017, 22, 144 CrossRef PubMed
- A. E. M. Beedle, M. Mora, C. T. Davis, A. P. Snijders, G. Stirnemann and S. Garcia-Manyes, Nat. Commun., 2018, 9, 3155 CrossRef PubMed
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimaki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed
- X. H. Ma, W. B. Yuan, S. E. J. Bell and S. L. James, Chem. Commun., 2014, 50, 1585–1587 RSC
- L. Batzdorf, F. Fischer, M. Wilke, K. J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS PubMed
- H. Kulla, M. Wilke, F. Fischer, M. Rollig, C. Maierhofer and F. Emmerling, Chem. Commun., 2017, 53, 1664–1667 RSC
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930–5933 CrossRef CAS PubMed
- K. Užarević, N. Ferdelji, T. Mrla, P. A. Julien, B. Halasz, T. Friščić and I. Halasz, Chem. Sci., 2018, 9, 2525–2532 RSC
- P. F. M. de Oliveira, A. A. L. Michalchuk, A. G. Buzanich, R. Bienert, R. M. Torresi, P. H. C. Camargo and F. Emmerling, Chem. Commun., 2020, 56, 10329–10332 RSC
- S. Hu, C. Ma, F. Zhan, Y. Cao, P. Hu and Q. Zhen, Chem. Pap., 2017, 71, 1323–1329 CrossRef CAS
- F. M. Santos, H. I. S. Nogueira, A. M. V. Cavaleiro, E. D. Gomes and M. S. Belsley, Inorg. Chim. Acta, 2017, 455, 600–606 CrossRef CAS
- R. Mao, F. Zhan, N. Bu, Y. Cao, P. Hu, G. Gong and Q. Zhen, Mater. Lett., 2016, 173, 111–114 CrossRef CAS
- M. Wilke, M. Klimakow, K. Rademann and F. Emmerling, CrystEngComm, 2016, 18, 1096–1100 RSC
- X. Zhao, Y. Duan, F. Yang, W. Wei, Y. Xu and C. Hu, Inorg. Chem., 2017, 56, 14506–14512 CrossRef CAS PubMed
- X. Zhao, L. Gong, C. Wang, C. Wang, K. Yu and B. Zhou, Chem.–Eur. J., 2020, 26, 4613–4619 CrossRef CAS PubMed
- H. T. Evans, B. M. Gatehouse and P. Leverett, J. Chem. Soc., Dalton Trans., 1975, 505–514 RSC
- V. Ban, Y. Sadikin, M. Lange, N. Tumanov, Y. Filinchuk, R. Černý and N. Casati, Anal. Chem., 2017, 89, 13176–13181 CrossRef CAS PubMed
- E. Petrus, M. Segado and C. Bo, Chem. Sci., 2020, 11, 8448–8456 RSC
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, including a chemical list, synthesis protocols, and the structure solution and refinement description with plots of the Rietveld refinements followed by a discussion of the observed results. CCDC 2067564–2067567. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05111c |
| This journal is © The Royal Society of Chemistry 2022 |