
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal substrate catalysis in the confined space for platinum drug delivery†
Susana
Velasco-Lozano‡
 a,
Silvia Alonso-de
Castro‡
b,
Carlos
Sanchez-Cano
abc,
Ana I.
Benítez-Mateos
a,
Fernando
López-Gallego
*ac and
Luca
Salassa
*bcd
a,
Silvia Alonso-de
Castro‡
b,
Carlos
Sanchez-Cano
abc,
Ana I.
Benítez-Mateos
a,
Fernando
López-Gallego
*ac and
Luca
Salassa
*bcd
aCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramon 182, San Sebastián, 20014, Spain. E-mail: flopez@cicbiomagune.es
bDonostia International Physics Center, Paseo Manuel de Lardizabal 4, Donostia, 20018, Spain. E-mail: lsalassa@dipc.org
cIkerbasque, Basque Foundation for Science, Bilbao, 48011, Spain
dPolimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, Paseo Manuel de Lardizabal 3, Donostia, 20018, Spain
First published on 29th November 2021
Abstract
Catalysis-based approaches for the activation of anticancer agents hold considerable promise. These principally rely on the use of metal catalysts capable of deprotecting inactive precursors of organic drugs or transforming key biomolecules available in the cellular environment. Nevertheless, the efficiency of most of the schemes described so far is rather low, limiting the benefits of catalytic amplification as strategy for controlling the therapeutic effects of anticancer compounds. In the work presented here, we show that flavin reactivity within a hydrogel matrix provides a viable solution for the efficient catalytic activation and delivery of cisplatin, a worldwide clinically-approved inorganic chemotherapy agent. This is achieved by ionically adsorbing a flavin catalyst and a Pt(IV) prodrug as substrate into porous amino-functionalized agarose beads. The hydrogel chassis supplies high local concentrations of electron donating groups/molecules in the surrounding of the catalyst, ultimately boosting substrate conversion rates (TOF >200 min−1) and enabling controlled liberation of the drug by light or chemical stimuli. Overall, this approach can afford platforms for the efficient delivery of platinum drugs as demonstrated herein by using a transdermal diffusion model simulating the human skin.
Introduction
Recent studies demonstrate how metal complexes,1–4 artificial metalloenzymes5,6 and nanomaterials7–10 can catalyze abiotic reactions in biological environments, including in vitro and in vivo. In the context of chemotherapy, metal complexes have been designed to function as catalysts for the deprotection of organic anticancer agents11 and for the oxidation/reduction of biomolecules that are key for the cell homeostasis.2,12 Likewise, metal-loaded catalytic nanocarriers have been engineered for delivery to tumors, or to be placed in their proximity by surgery where they can trigger the conversion of prodrugs into their biologically active counterparts.9,11,13,14Despite proving the potential of catalysis-based approaches in medicine, the great majority of the schemes proposed so far display rather modest catalytic efficiencies, even in buffer solutions, as evidenced by low turnover numbers (TONs) and slow reaction kinetics.15 The latter aspect is especially overlooked by the scientific community working in this field, yet of key importance in the development of drug activation strategies. Apart from few exceptions,2,12,16,17 catalysts currently reported in the literature typically achieve substrate conversion rates in solution that are in the order of 0.1–10 h−1 (10−3 to 10−1 min−1). This undoes the benefits of catalytic amplification since catalysts with low turnover frequency (TOF) require long exposure periods and/or high loadings for transforming enough substrate (i.e. prodrug) to induce the desired therapeutic effects. In such a scenario, the catalyst intrinsic toxicity is critical15 and the choice of drugs with high-potency (sub-μM) becomes often mandatory, overall limiting the therapeutic scope of several catalysis-based strategies for prodrug activation.
Thus, catalysts with high TOFs are preferable for rapidly generating lethal doses of drug before both the prodrug and catalyst are metabolized and cleared by the treated tissue or organism. In that regard, our groups recently reported that flavins and selected flavoproteins photocatalytically convert Pt(IV) prodrug precursors into clinically-approved cisplatin and carboplatin with unanticipated efficiency, exhibiting TOFs as high as 25 min−1 and TONs up to 500.18,19 Reaction mechanism studies indicate that the active catalyst is the flavin hydroquinone species (doubly reduced flavin) whose generation is dramatically accelerated by photoirradiation in the presence of selected electron donors.18 Notably, these inorganic transformations catalyzed by innocuous biomolecules unconventionally use metal complexes as substrates20 and display bioorthogonal selectivity,19,21,22i.e. they occur in cell culture media avoiding interference from other chemical and biochemical entities.
Considering the exceptional anticancer activity of Pt chemotherapeutics and the tremendous research efforts devoted to design delivery platforms capable of reducing their off-target effects,23 we speculated that we could capitalize on the catalytic efficiency of these flavin-mediated reactions and devise convenient solutions for the administration of cisplatin and its derivatives. To such aim, we sought inspiration in Nature which confines biocatalysts (i.e. enzymes) and substrates (i.e. metabolites) inside organelles to control biosynthetic pathways and boost catalytic performance.24 Catalysts confined into supramolecular structures have proved very effective in favoring chemical reactions for different technological purposes.25 In biomedicine, enzymes and chemical catalysts entrapped into artificial envelops have afforded therapeutic weapons to target cancer.26,27 However, none of the state-of-the-art systems involve confinement of the catalyst and its corresponding prodrug substrate into an artificial chassis that can operate as drug depot. Besides, catalysis-based systems for prodrug activation have been so far designed to function under the very diluted conditions that they encounter during in vitro and in vivo studies. In such a scenario, catalysts hardly find substrate molecules to react with, ultimately displaying low catalytic efficiencies. In confined spaces this is not the case since high concentrations of substrate are available in the proximity of the catalyst, therefore maximizing reaction kinetics when rate laws are equal or higher than 1.
Hydrogels have been widely used as solid supports to immobilize different types of catalysts,28 including for therapy purposes.29 Furthermore, they are appealing materials for delivery, because of their excellent biocompatibility and high water content.30 These soft materials can be applied in surgical implants, locally injected or administered systematically via intravenous infusion,31 and used to fabricate therapeutic patches for intra- and transdermal release of pharmaceuticals.32 Binding of drugs to hydrogel polymer networks can be extremely diverse, ranging from covalent conjugation, electrostatic and hydrophobic interactions, as well as physical trapping. The nature of the interaction governs drug release properties.31
Stimuli responsive hydrogels are particularly promising because they introduce an additional level of control on the drug action. In the specific case of platinum-based anticancer agents, hydrogel polymers have generally been employed to release their Pt(II) and Pt(IV) cargos upon swelling and degradation of the gel.33,34 In such systems, Pt complexes are loaded by host guest chemistry or by conjugating prodrugs with polymer monomer units. Recent studies showed that release of active platinum species from hydrogels can also be triggered by short-wavelength light.35
In this contribution, we demonstrate how loading of a Pt(IV) prodrug complex and a flavin catalyst onto agarose porous microbeads enables performing in situ the confined catalytically-driven generation and subsequent release of cisplatin in a stimuli responsive manner, by light and chemical activation. Catalysis has so far been employed in drug release to accelerate the degradation of hydrogels and thus control the delivery of active agents,36 rather than activating a confined prodrug. Our approach is different and may provide a general method to produce high local concentrations of Pt drugs in short bursts upon remote control. In the long run, this can afford viable strategies to develop topical medicaments and implantable devices37 that could overcome some of the issues associated with the systemic administration of Pt chemotherapeutic agents.
Results and discussion
Confinement of flavin catalyst and Pt(IV) prodrug into agarose beads
We selected agarose porous microbeads activated with diethylaminoethyl groups (AGM), the complex cis,cis,trans-[Pt(NH3)2(Cl2)(O2CCH2CH2CO2H)2] (1) and riboflavin-5′-phosphate (FMN) as components to assemble a hydrogel capable of functioning simultaneously as catalytic reactor and drug release system for cisplatin. According to previous work, 1 is a suitable prodrug candidate to treat cancer cells since it is substantially unreactive under blue light irradiation (460 nm) and displays no dark toxicity in a number of cancer cell lines due to its robustness against biological reducing agents.22 Besides, we recently proved that FMN is an efficient photocatalyst for the conversion of 1 into cisplatin and that such flavin can also perform the same activation reaction in the dark and in the absence of O2 when high concentrations of NADH are present.18 Finally, porous agarose microbeads emerge as ideal biocompatible microreactors to both confined catalytic reactions and deliver substances, as documented for a wide range of applications such as biosensing and biocatalysis.38–40 Furthermore, agarose macro-sized gels containing stem cells have been tested in clinical trials as implants for the treatment of resistant metastatic cancers.41Thus, we loaded the Pt(IV) substrate and the flavin catalyst onto transparent AGM functionalized with diethylaminoethyl groups (pKa ≈ 9.5), exploiting non-covalent interactions between FMN, 1, and the microbead network (Fig. 1).
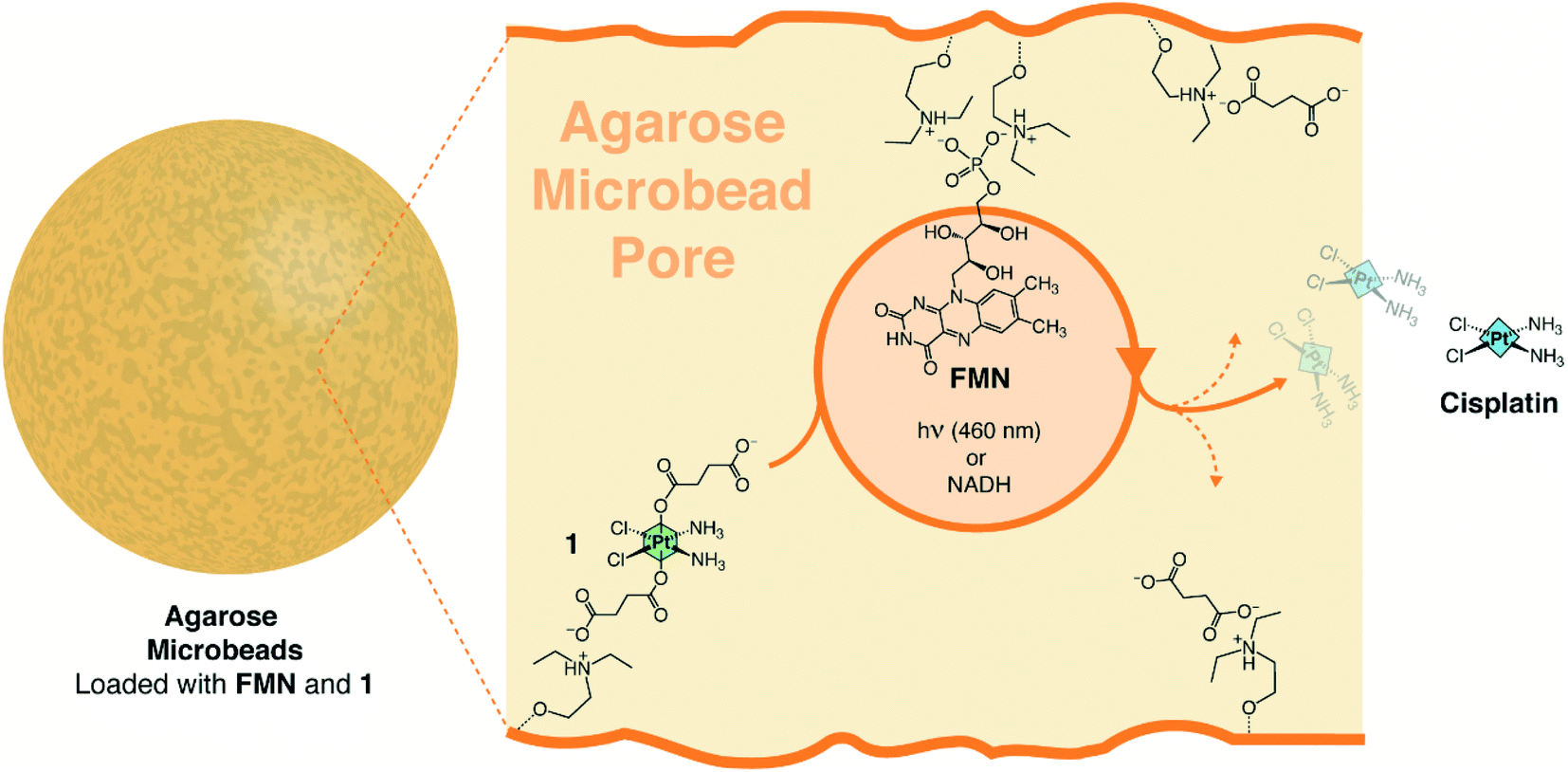 | ||
| Fig. 1 Flavin-catalyzed generation of cisplatin from a Pt(IV) prodrug precursor inside diethylaminoethyl agarose microbeads (AGM). | ||
The immobilization is based on the electrostatic interactions established by the phosphate and succinates of FMN and 1, respectively, and the positively charged tertiary amino groups of AGM. The association/dissociation equilibrium that rules these ionic interactions also enables the flavin to diffuse within the bead pores and act over the electrostatically anchored Pt(IV) complexes.42
Confinement of the photocatalyst and the metal substrate was achieved by sequential incubation of 1 mM 1 and 75 μM FMN solutions with AGM, under low ionic strength and neutral pH conditions (10 mM Tris–HCl at pH 7.6). After buffer washing, the resulting 1-FMN@AGMs could be loaded with concentrations up to 0.165 ± 0.06 and 4.70 ± 0.02 μmol g−1 of flavin and 1, respectively, as determined by UV Vis and UPLC-MS. When 1-FMN@AGMs were incubated with 1 M NaCl, 1 and FMN were majorly released from the matrix, demonstrating that both metal substrate and catalyst were ionically absorbed to the AGMs.
Flavin-mediated photocatalytic delivery of cisplatin
Next, we irradiated 1-FMN@AGM in correspondence of the lowest-energy absorption band of FMN (λexc = 460 nm) and investigated the substrate conversion and release of cisplatin from the polymer matrix by UPLC-MS and 1H NMR (Fig. 2 and S1–S6†). UPLC-MS enables monitoring of the flavin-catalyzed reaction by the direct quantification of cisplatin. Instead, 1H NMR can determine reaction progression by means of the diagnostic signals relative to the axial succinato ligands of 1 (Fig. S1†). Once generated by the on-bead photoreductive elimination reaction of 1, cisplatin is either in its neutral form or positively-charged upon hydrolysis of chlorido ligands.43 Therefore, it cannot longer interact with the carrier positively-charged diethylaminoethyl groups and is then released. Conversely, high salt concentrations must be added to the reaction medium after activation, for the negatively-charged free succinate and residual 1 to escape the AGM porous network and allow 1H NMR and UPLC-MS detection.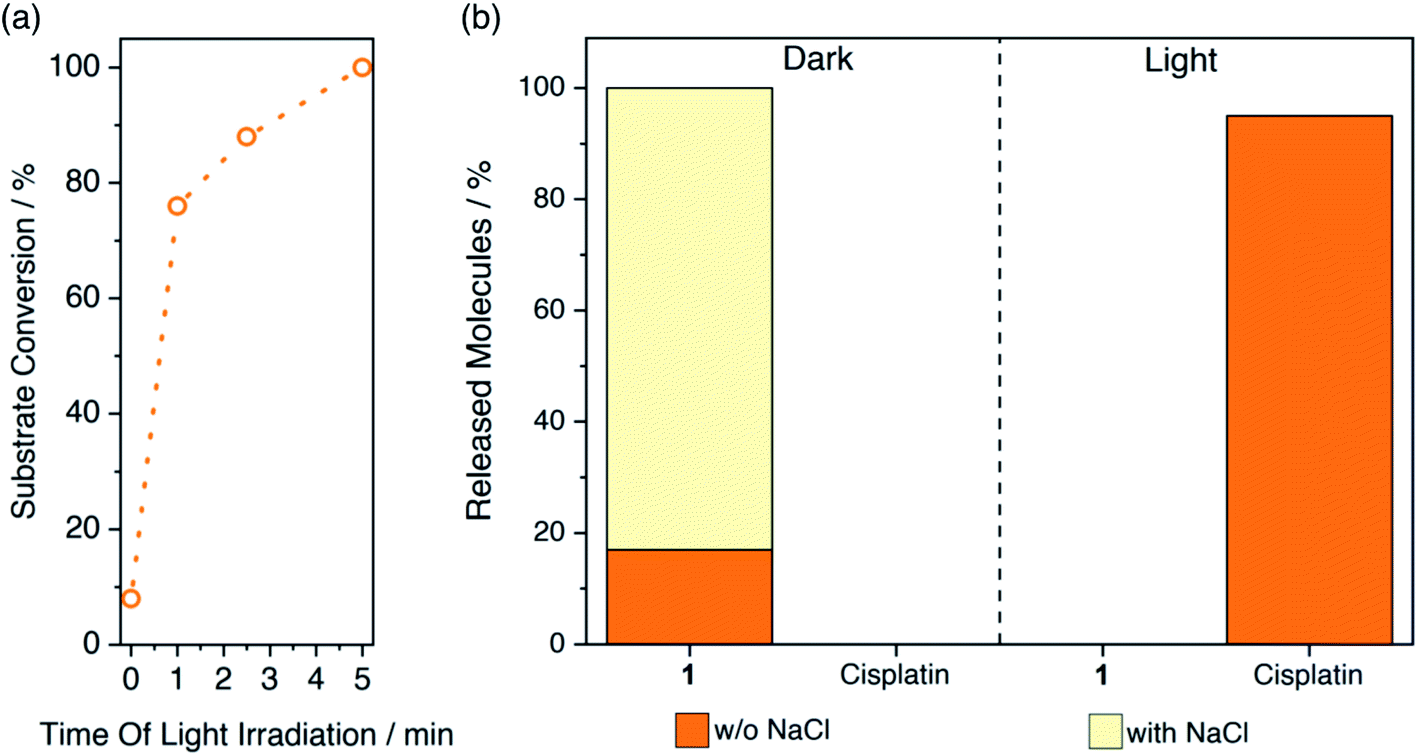 | ||
| Fig. 2 Flavin-catalyzed photoactivation of 1-FMN@AGM and subsequent cisplatin release monitored by (a) 1H NMR and (b) UPLC-MS. Supernatant and solutions eluted with (pale yellow) and without (orange) 1 M NaCl were employed for UPLC-MS analysis to quantify the amount of cisplatin liberated by light-activated 1-FMN@AGM (5 min, 6 mW cm−2) or residual 1 trapped in the beads, respectively (ESI†). | ||
With that in mind, we observed by 1H NMR that 5 min of low-power light irradiation (6 mW cm−2) were indeed sufficient to catalytically convert 100% of immobilized 1 into its photoproducts and release free succinate upon addition of 1 M NaCl (Fig. 2a). UPLC-MS (Fig. 2b and S2†) confirmed the simultaneous generation and release of cisplatin (yet without the need of adding NaCl), demonstrating that the process took place entirely, since no presence of 1 could be detected in the supernatant after light-irradiation and upon incubation with salt (Fig. 2b). After 1 min of exposure to light, 1-FMN@AGM achieved a TOF of 20.1 ± 0.6 min−1, in agreement with results previously reported for reactions in diluted solution.18 Remarkably, we measured a 10-fold increase in TOF (244 ± 10 min−1) by reducing the catalyst loading from 0.165 to 0.008 μmol g−1 (Table S1†) which indicated that confined spaces are indeed advantageous for improving the efficiency of these catalytic transformations.
Control experiments showed that cisplatin release was not taking place in the dark or in the absence of FMN under light irradiation (Fig. 2b and S3–S6†). Moreover, when kept in the dark and washed with 1 M NaCl, 1-FMN@AGM liberated approximately 80% of 1 (Fig. 2b), confirming that light was required for the flavin to trigger the catalysis.
In order to demonstrate that 1-FMN@AGM was capable of delivering therapeutic doses of cisplatin in a short period, we photoirradiated for 5 min a solution containing the microbeads (≈3 μmol g−11) and supplied 5 and 10 μL aliquots of supernatant to in vitro cultures of MCF-7 cells (breast cancer). Under the tested conditions, the cisplatin delivered from irradiated 1-FMN@AGM induced over 80% reduction in cell viability, corresponding to full conversion of the non-toxic Pt(IV) precursor and administration of 25–50 μM cisplatin, depending on the supplied volume. On the contrary, light-irradiated 1@AGM and dark controls did not have any effect on cell proliferation, in line with their inability to generate and release the Pt(II) drug (Fig. S7†).
Role of the agarose matrix in the flavin-mediated photocatalytic reduction of the Pt(IV) prodrug
Zwitterionic buffers such as MES and HEPES have been previously employed as electron donors in the photocatalytic conversion of Pt(IV) substrates, as well as in other flavin-mediated catalytic reactions.22,44 On the contrary, we found that the Tris buffer used in this study did not play the same role when a solution of 1 was irradiated with 460 nm light in the presence of catalytic amounts of FMN (Fig. S8†). Surface morphology analysis of light-irradiated AGM by SEM (Scanning Electron Microscopy) suggested that the agarose microstructure likely participated in the catalytic cycle as electron donor, besides functioning as chassis to confine the reaction. In fact, loading of 1 and FMN onto the beads kept in the dark did not cause any significant visual change in AGM, while we observed a notably rougher surface for 1-FMN@AGM after 1 min of light irradiation (Fig. 3a and S9–S11†).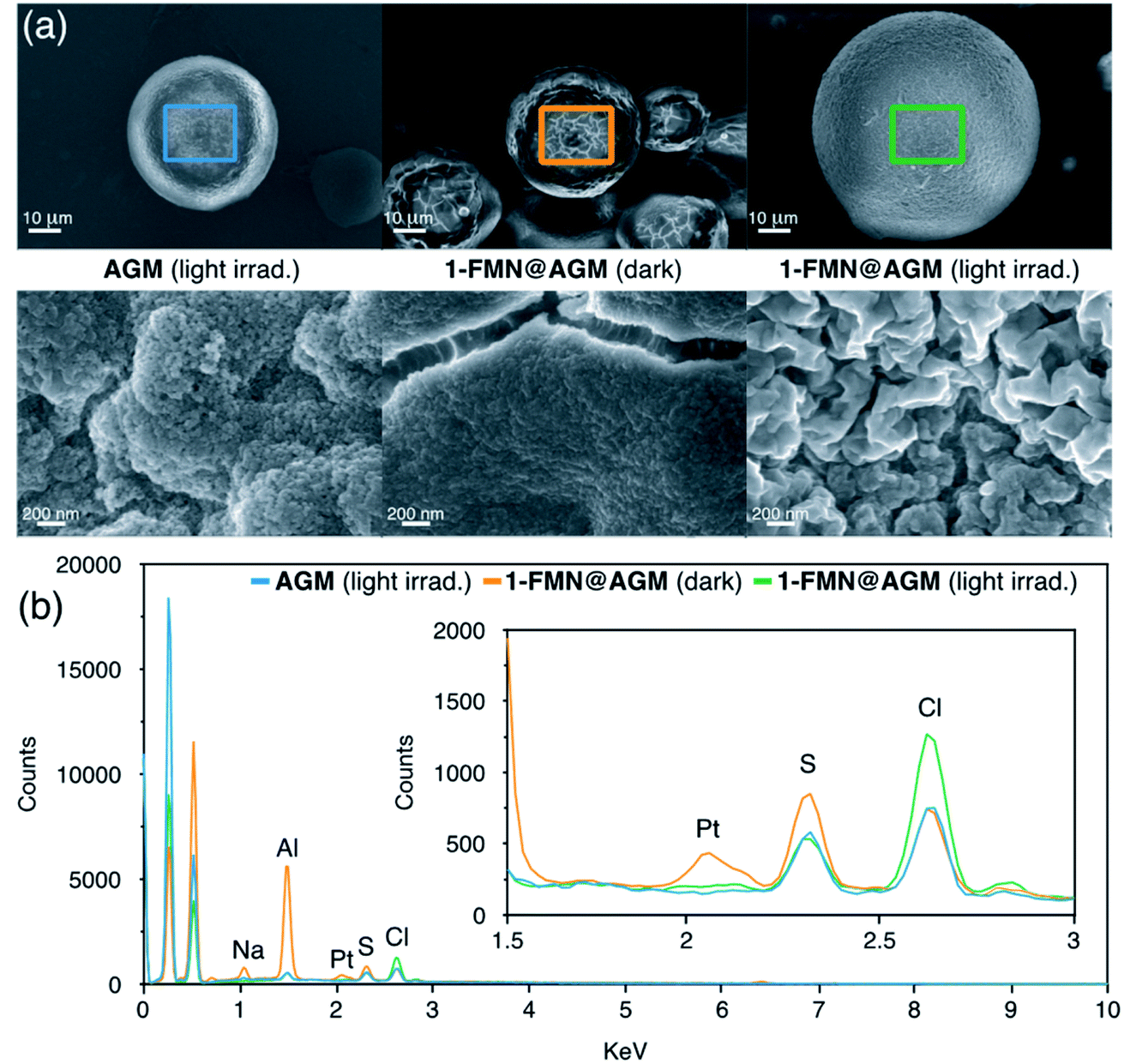 | ||
| Fig. 3 (a) Low (top) and high (bottom) magnification SEM micrographs of AGM, 1-FMN@AGM in the dark and 1-FMN@AGM under light irradiation (1 min, 460 nm, 6 mW cm−2); (b) EDX spectra of AGM, 1-FMN@AGM in the dark and 1-FMN@AGM under light irradiation. EDX data were collected from the sample region indicated by the colored squares in the lower magnification SEM micrographs. | ||
EDX (Energy-Dispersive X-ray) spectroscopy further confirmed the capacity of 1-FMN@AGM of photoreleasing cisplatin (Fig. 3b and S12†). EDX spectra collected for 1-FMN@AGM and controls clearly showed that a signal corresponding to Pt was present on the material surface after loading 1 in the dark but disappeared upon light irradiation.
To gain further insights, we performed catalysis experiments in solution using galactose, diethylaminoethanol and agarose as electron donors to identify which component of AGM was directly involved in the photoreaction. Among these co-reactants, only diethylaminoethanol was capable of promoting the catalytic conversion of the Pt(IV) substrate, albeit at lower rate than the confined system (Fig. S13†). The higher catalytic efficiency observed for 1-FMN@AGM with respect to free diethylaminoethanol further confirmed the key role played by the microbead chassis in providing a high local concentration of electron donating groups and substrate molecules in the surrounding of the entrapped FMN. Others already demonstrated that confinement improves the outcome of catalytic reaction giving access to pathways that are forbidden or kinetically unfavored in diluted solutions.45 In photocatalysis, confined spaces enhance photochemical processes by altering several key steps, such as light absorption, lifetime of excited species and formation of key redox intermediates.46
Cisplatin photocatalytic delivery under anaerobic conditions
To expand the versatility of this system, we assessed the performance of light-irradiated 1-FMN@AGM under anaerobic conditions motivated by the importance of O2 as electron acceptor in flavin catalysis47 and by the relevance of hypoxic environments in cancer.48 To this purpose, we co-immobilized glucose oxidase (GOX, 1 mg mL−1) on AGM to afford 1-FMN-GOX@AGM. This enzyme selectively oxidizes glucose using O2 as electron acceptor producing gluconic acid and hydrogen peroxide. In such a way, GOX effectively depletes O2 from aqueous solutions in the presence of an excess of glucose.49 Our previous work also demonstrated that GOX, despite being a flavoenzyme, was catalytically inactive towards 1 under catalysis conditions similar to the ones employed here.191H NMR results revealed that the anaerobic environment produced by GOX inside AGM prompted a 4-fold acceleration in the generation of cisplatin compared to aerobic conditions, upon the same light irradiation time (Fig. S14†). O2 competes with 1 as terminal electron acceptor in the oxidation of the catalytically-active FMN hydroquinone (FMNH−).18,19 Therefore, depletion of O2 inside irradiated 1-FMN-GOX@AGM resulted in a faster production of the Pt(II) drug. These insights suggest that the GOX capability to starve cells50 and concurrently deplete O2 can be of value for increasing cisplatin photogeneration and delivery to the glucose-rich extracellular environments of tumors.
Cisplatin catalytic delivery triggered by NADH as electron donor
In a recent work, we demonstrated that flavoenzymes such as NADH oxidase catalytically activated Pt(IV) substrates also in the dark when NADH was used as electron donor.19 In the enzyme binding site, both flavin and NADH redox cofactors are confined, enabling the efficient formation of the catalytically active species FMNH− (or FADH−) which triggers the Pt reduction reaction. Inspired by this example, and in general by the ability of flavoenzymes to extract electrons from NAD(P)H cofactors,51 we hypothesized that entrapment of both FMN and NADH within the porous environment of AGM could result in the activation of 1 also under dark conditions. Therefore, we incubated 1-FMN@AGM with 0.5 mM NADH and examined by 1H NMR the accumulation of free succinate ligand into the reaction bulk after addition of 1 M NaCl. Data demonstrated that NADH was able to reduce the immobilized FMN and concurrently transform 1 into cisplatin in the dark (Fig. 4a and S15†). However, incubation of 1-FMN@AGM with NADH in the dark during 5 min reduced 60% of the immobilized 1 (Fig. S16 and S17†) compared to the substrate quantitative reduction observed under light irradiation in absence of NADH (Fig. 2a). In the case of NADH-activated 1-FMN@AGM, EDX experiments also revealed disappearance of the Pt signal after addition of the electron donor (Fig. S18†). Comparative amounts of NADH, FMN and 1 in solution showed no conversion of the metal substrates.18Therefore, confinement of the reaction components enabled a proficient electron transport from NADH to the Pt(IV) substrate, which conversely is precluded in diluted solutions without the use light. A similar behavior was observed by Chen et al.52 using a zeolitic imidazolate framework where both FMN and NADH were co-immobilized. As in our design, such nanozyme architecture was able to convert ferric cytochrome C to the ferrous form, supporting that confinement of the catalyst and electron donor is crucial to transfer electrons to metal centers in an efficient manner under dark conditions.
In order to better understand the dynamics of the catalytic reaction inside the beads, we employed fluorescence microscopy to monitor at a single-particle level how NADH and FMN fluorescence signals of AGM evolved in the presence of 1 (Fig. 4b and c). Upon excitation at 365 nm, NADH is fluorescent (λem = 402–448 nm) only in its reduced form, whereas FMN (λexc = 470 nm) emits at 520 nm, becoming non-emissive once reduced to FMNH−.53 After NADH addition, we monitored the fluorescence within each bead during the first 10 min, observing a progressive increase of NADH emission. This process corresponded to the immobilization of NADH on 1-FMN@AGM beads. Simultaneously, FMN fluorescence decreased due to the generation of the catalytically active FMNH−. After this onset period, however, NADH-associated emission diminished rapidly because of its FMN-promoted efficient conversion to NAD+. Alongside, FMN fluorescence was restored consistently by the catalytic Pt(IV)-to-Pt(II) reduction triggered by FMNH− and the regeneration of the emissive FMN catalyst in its oxidized form. Control experiments (Fig. S19†) confirmed this scenario, demonstrating that AGM containing NADH only displayed slower fluorescence decay than in presence of the FMN-1 pair, likely due to simple photobleaching of the electron donor. Furthermore, FMN-loaded beads showed a constant emission profile with limited photodecomposition in the absence of NADH.
 | ||
| Fig. 4 (a) Time-course of the flavin-catalyzed activation of 1-FMN@AGM and subsequent cisplatin release in the presence of 0.5 mM NADH, under dark conditions. Beads were washed at different time-points with high ionic strength solutions (1 M NaCl) and the supernatants were analyzed by 1H NMR; (b and c) Single-particle fluorescence of 1-FMN@AGM activated via NADH-triggered reduction. Plots in (c) were recorded from time-lapse fluorescence microscopy experiments (average of 5 particles of 59 μm diameter) monitoring the fluorescence of NADH (dark cyan, λexc = 365 nm, λem = 402–448 nm) and FMN (orange, λexc = 470 nm, λem = 500–557 nm) in the presence of 1. Shadowed region depicts the time NADH takes to be immobilized on AGM. | ||
Transdermal photocatalytic delivery of cisplatin through synthetic human skin models
Encouraged by the catalytic performance of these hydrogels, we explored the capacity of 1-FMN@AGM to function as delivery platform for cisplatin. As a proof of concept, we tested how light-activated 1-FMN@AGM delivered the Pt(II) drug through the STRAT-M® membrane,54 a transdermal diffusion model predictive of the human skin that is employed to avoid the use of animal tissues. We chose skin as target tissues for our studies motivated by the relevance of photodynamic therapy in the treatment of pre-cancerous and cancerous skin lesions.55,56 Furthermore, we envision that 1-FMN@AGM can be tailored and its features optimized for application to other types of tissues, in which release devices are of interest for the controlled administration of anticancer drugs.In our experiments (Fig. 5 and S20†), 1-FMN@AGM was formulated with different concentrations of polypropylene glycol (PPG, 50–100%), since previous work demonstrated that this macromolecule aided the percutaneous penetration of drugs through the skin of mammals.57 The obtained viscous solutions were first applied to the STRAT-M® membrane and then light-irradiated for 5 min to fully convert 1 into cisplatin. Immediately afterwards, we incubated the whole system at 298 K in Tris buffer for 18 h in the dark, allowing cisplatin to permeate through the STRAT-M® and reach an agarose film placed beneath it. Quantification of the cisplatin that diffused through the membrane and accumulated in the collecting compartment was achieved by ICP-MS. Under these experimental conditions, we observed that light-activated 1-FMN@AGM in PPG did show significantly higher levels of membrane permeation than controls in the dark or in the absence of FMN. According to ICP-MS data, photoirradiated 1-FMN@AGM delivered approximately 36 ± 17 nM cisplatin (0.5% of loaded 1), whereas the concentration of cisplatin was either non-detected or <10 nM in the case of 1@AGM under the same conditions. After lyophilization, the agarose film collecting the permeated cisplatin was administered to MCF-7 cell cultures, observing a small (10%) but significant cell viability reduction for light-activated 1-FMN@AGM only, in agreement with the dose-toxicity profile of cisplatin (Fig. S21†).
 | ||
| Fig. 5 (a) Scheme for the flavin-catalyzed photoactivation of 1-FMN@AGM and transdermal delivery of cisplatin through the synthetic STRAT-M® membrane. 1-FMN@AGM was formulated using different concentrations of polypropylene glycol (PPG, 50–100% v/v). The resulting creamy formulation was spread over the synthetic membrane placed at the top of an agarose film. Components are not drawn to scale and are exaggerated in size for illustration purposes. (b) Accumulation of Pt in the agarose collecting compartment measured by ICP-MS. Results are presented as a mean ± SD (n = 2). *p <0.05; n.s.: non significative. | ||
Conclusions
In summary, 1-FMN@AGM is a versatile and efficient delivery platform that exploits the flavin-catalyzed transformation of a Pt(IV) precursor into the clinically-approved anticancer drug cisplatin. In 1-FMN@AGM, the generation and release of high concentrations of cisplatin can be controlled either by light employing very low light doses or by a chemical stimulus (e.g. NADH) in the dark. In both cases, the microenvironment of the AGM chassis and its functionalization with diethylaminoethyl groups is key for the efficacy of the system. In fact, the agarose confined space ensures high local concentration of reactants and intraparticle diffusion of catalyst and substrate molecules, ultimately boosting the activation rate of the Pt(IV) prodrug to levels unmatched by previous catalytic systems developed for the activation of therapeutic drugs (TOF >200 min−1).Our study shows at a proof-of-concept level that a device based on flavin catalysis towards Pt substrates has potential applications for the controlled transdermal delivery of cisplatin. Careful refinement of this strategy in terms of formulation can afford creams and ointments for the topic treatment of specific cancerous lesions of the skins. Nevertheless, the versality of these catalytic reactions enables extending our approach towards other applications, for example by employing diverse polymeric architectures and fine-tuning their functional chemical groups for anchoring different flavins (or flavoproteins) and Pt substrates. Overall, this can afford real catalytic therapies for the cure of several types of cancers with new modes of action and fewer side effects.
Data availability
All experimental data and procedures are provided in the ESI.†Authors contributions
S. V. L., S. A. dC., C. S. C., A. I. B. M. F. L. G. and L. S. contributed to the design of the study. S. V. L., S. A. dC., C. S. C., A. I. B. M. conducted the experiments and data analysis. F. L. G. and L. S. provided technical advice and result interpretation. F. L. G. and L. S. wrote the manuscript and ESI.† All the authors commented on and amended both documents. All the authors discussed and contributed to the work.Conflicts of interest
The authors declare that 1-FMN@AGM is protected under the patent application EP20383067.4.Acknowledgements
We acknowledge financial support from the Spanish State Research Agency (grants CTQ2016-80844-R, PID2019-109111RB-I00, RTI2018-094398-B-I00, BIO2014-61838-EXP) and the Basque Government (Eusko Jaurlaritza, grant PIBA_2021_1_0034). S. V. L. thanks the Mexican Council of Science and Technology (CONACyT) for the postdoctoral fellowship she received (ref. CVU-267390). C. S. C. thanks Gipuzkoa Foru Aldundia (Gipuzkoa Fellows program; grant number 2019-FELL-000018-01/62/2019) for financial support. L. S. thanks the Spanish Multi-MetDrugs network (RED2018-102471-T) for fruitful discussion. FLG thanks the Spanish Biocatalysis network (RED2018-102403-T) and the European Research Council (ERC-Co-2018 818089). This work was performed under the Maria de Maeztu and Severo Ochoa Centres of Excellence Programme run by the Spanish State Research Agency, Grant No. MDM-2017-0720 (CIC biomaGUNE) and CEX2018-000867-S (DIPC).References
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef.
- J. P. C. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- S. Infante-Tadeo, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Chem. Sci., 2021, 12, 9287–9297 RSC.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1–9 CAS.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780–792 CrossRef CAS.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS.
- R. M. Yusop, A. Unciti-Broceta, E. M. V Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS.
- R. Martínez, C. Carrillo-Carrión, P. Destito, A. Alvarez, M. Tomás-Gamasa, B. Pelaz, F. Lopez, J. L. Mascareñas and P. del Pino, Cell Rep. Phys. Sci., 2020, 1, 100076 CrossRef.
- T. L. Bray, M. Salji, A. Brombin, A. M. Pérez-López, B. Rubio-Ruiz, L. C. A. Galbraith, E. E. Patton, H. Y. Leung and A. Unciti-Broceta, Chem. Sci., 2018, 9, 7354–7361 RSC.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef.
- Z. Chen, H. Li, Y. Bian, Z. Wang, G. Chen, X. Zhang, Y. Miao, D. Wen, J. Wang, G. Wan, Y. Zeng, P. Abdou, J. Fang, S. Li, C.-J. Sun and Z. Gu, Nat. Nanotechnol., 2021, 16, 933–941 CrossRef CAS.
- F. Mancuso, M. Rahm, R. Dzijak, H. Mertlíková-Kaiserová and M. Vrabel, ChemPlusChem, 2020, 85, 1669–1675 CrossRef CAS PubMed.
- S. Ji, B. Jiang, H. Hao, Y. Chen, J. Dong, Y. Mao, Z. Zhang, R. Gao, W. Chen, R. Zhang, Q. Liang, H. Li, S. Liu, Y. Wang, Q. Zhang, L. Gu, D. Duan, M. Liang, D. Wang, X. Yan and Y. Li, Nat. Catal., 2021, 4, 407–417 CrossRef CAS.
- R. Huang, C. H. Li, R. Cao-Milán, L. D. He, J. M. Makabenta, X. Zhang, E. Yu and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 10723–10729 CrossRef CAS.
- J. Gurruchaga-Pereda, V. Martínez-Martínez, E. Rezabal, X. Lopez, C. Garino, F. Mancin, A. L. Cortajarena and L. Salassa, ACS Catal., 2020, 10, 187–196 CrossRef CAS.
- S. Alonso-de Castro, A. L. Cortajarena, F. López-Gallego and L. Salassa, Angew. Chem., Int. Ed., 2018, 57, 3143–3147 CrossRef CAS PubMed.
- G. Salassa and L. Salassa, ACS Omega, 2021, 6, 7240–7247 CrossRef CAS PubMed.
- S. Alonso-de Castro, A. Terenzi, S. Hager, B. Englinger, A. Faraone, J. C. Martínez, M. Galanski, B. K. Keppler, W. Berger and L. Salassa, Sci. Rep., 2018, 8, 17198 CrossRef.
- S. Alonso-de Castro, E. Ruggiero, A. Ruiz-de-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. López-Gallego and L. Salassa, Chem. Sci., 2017, 8, 4619–4625 RSC.
- R. J. Browning, P. J. T. Reardon, M. Parhizkar, R. B. Pedley, M. Edirisinghe, J. C. Knowles and E. Stride, ACS Nano, 2017, 11, 8560–8578 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed.
- M. Vázquez-González, C. Wang and I. Willner, Nat. Catal., 2020, 3, 256–273 CrossRef.
- Q. Wei, S. Jiang, R. Zhu, X. Wang, S. Wang and Q. Wang, iScience, 2019, 14, 27–35 CrossRef CAS.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS.
- P. Zucca, R. Fernandez-Lafuente and E. Sanjust, Molecules, 2016, 21, 1–25 Search PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, T. Valero, R. Contreras-Montoya, L. Álvarez De Cienfuegos, V. Sebastián, J. Santamaría and A. Unciti-Broceta, J. Med. Chem., 2020, 63, 9650–9659 CrossRef PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- M. N. Pastore, Y. N. Kalia, M. Horstmann and M. S. Roberts, Br. J. Pharmacol., 2015, 172, 2179–2209 CrossRef CAS.
- W. Shen, J. Luan, L. Cao, J. Sun, L. Yu and J. Ding, Biomacromolecules, 2015, 16, 105–115 CrossRef CAS PubMed.
- W. Shen, X. Chen, J. Luan, D. Wang, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 40031–40046 CrossRef CAS.
- V. Venkatesh, N. K. Mishra, I. Romero-Canelón, R. R. Vernooij, H. Shi, J. P. C. Coverdale, A. Habtemariam, S. Verma and P. J. Sadler, J. Am. Chem. Soc., 2017, 139, 5656–5659 CrossRef CAS PubMed.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933 RSC.
- S. A. Chew and S. Danti, Adv. Healthcare Mater., 2017, 6, 1600766 CrossRef.
- M. Rinaudo, Polym. Int., 2008, 57, 397–430 CrossRef CAS.
- J. V Jokerst, J. Chou, J. P. Camp, J. Wong, A. Lennart, A. A. Pollard, P. N. Floriano, N. Christodoulides, G. W. Simmons, Y. Zhou, M. F. Ali and J. T. McDevitt, Small, 2011, 7, 613–624 CrossRef.
- J. Rocha-Martín, B. de las Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- B. H. Smith, T. Parikh, Z. P. Andrada, T. J. Fahey, N. Berman, M. Wiles, A. Nazarian, J. Thomas, A. Arreglado, E. Akahoho, D. J. Wolf, D. M. Levine, T. S. Parker, L. S. Gazda and A. J. Ocean, Cancer Growth Metastasis, 2016, 9, CGM.S39442 CrossRef PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- L. C. P. Goncąlves, H. R. Mansouri, S. Pourmehdi, M. Abdellah, B. S. Fadiga, E. L. Bastos, J. Sá, M. D. Mihovilovic and F. Rudroff, Catal. Sci. Technol., 2019, 9, 2682–2688 RSC.
- C. Gaeta, P. La Manna, M. De Rosa, A. Soriente, C. Talotta and P. Neri, ChemCatChem, 2021, 13, 1638–1658 CrossRef CAS.
- M. Häring, A. Abramov, K. Okumura, I. Ghosh, B. König, N. Yanai, N. Kimizuka and D. Díaz Díaz, J. Org. Chem., 2018, 83, 7928–7938 CrossRef.
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed.
- R. G. Bristow and R. P. Hill, Nat. Rev. Cancer, 2008, 8, 180–192 CrossRef CAS PubMed.
- A. S. Jeevarathinam, F. Guo, T. Williams, J. A. Smolen, J. A. Hyde, M. J. McShane, P. de Figueiredo and D. L. Alge, Mater. Today Bio, 2021, 9, 100092 CrossRef CAS PubMed.
- L. H. Fu, C. Qi, J. Lin and P. Huang, Chem. Soc. Rev., 2018, 47, 6454–6472 RSC.
- S. Weber and J. M. Walker, Flavins and Flavoproteins, Springer New York, New York, NY, 2014, vol. 1146 Search PubMed.
- J. Chen, Q. Ma, M. Li, W. Wu, L. Huang, L. Liu, Y. Fang and S. Dong, Nanoscale, 2020, 12, 23578–23585 RSC.
- Y.-T. Kao, C. Saxena, T.-F. He, L. Guo, L. Wang, A. Sancar and D. Zhong, J. Am. Chem. Soc., 2008, 130, 13132–13139 CrossRef CAS PubMed.
- T. Uchida, W. R. Kadhum, S. Kanai, H. Todo, T. Oshizaka and K. Sugibayashi, Eur. J. Pharm. Sci., 2015, 67, 113–118 CrossRef CAS PubMed.
- K. Yamagishi, I. Kirino, I. Takahashi, H. Amano, S. Takeoka, Y. Morimoto and T. Fujie, Nat. Biomed. Eng., 2019, 3, 27–36 CrossRef CAS.
- Z. Yu, P. Zhou, W. Pan, N. Li and B. Tang, Nat. Commun., 2018, 9, 1–9 CrossRef.
- V. Carrer, C. Alonso, M. Pont, M. Zanuy, M. Córdoba, S. Espinosa, C. Barba, M. A. Oliver, M. Martí and L. Coderch, Arch. Dermatol. Res., 2020, 312, 337–352 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05151b |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |