
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalysed C–H functionalisation of free carboxylic acids
Suparna
Dutta
a,
Trisha
Bhattacharya
a,
Finn J.
Geffers
b,
Marcel
Bürger
b,
Debabrata
Maiti
 *a and
Daniel B.
Werz
*b
*a and
Daniel B.
Werz
*b
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@iitb.ac.in; Web: https://www.dmaiti.com
bTechnische Universität Braunschweig, Institute of Organic Chemistry, Hagenring 30, 38106 Braunschweig, Germany. E-mail: d.werz@tu-braunschweig.de; Web: https://www.werzlab.de
First published on 10th January 2022
Abstract
Pd-catalysed C–H functionalisation of free carboxylic acids has drawn significant attention over the last few years due to the predominance of carboxylic acid moieties in pharmaceuticals and agrochemicals. But their coordinating ability was overlooked and masked by exogenous directing groups for a long time. Even other crucial roles of carboxylic acids as additives and steric inducers that directly influence the mode of a reaction have been widely neglected. This review aims to embrace all of the diverse aspects of carboxylic acids except additive and steric effects by concisely and systematically describing their versatile role in Pd-catalysed proximal and distal C–H activation reactions that could be implemented in the pharmaceutical and agrochemical industries. In addition, the mechanistic perspectives along with several recent strategies developed in the last few years discussed here will serve as educational resources for future research.
 Suparna Dutta | Suparna Dutta was born in 1997 in the state of West Bengal, India. She has done her BSc in Chemistry from Raja N. L. Khan Women's College affiliated to Vidyasagar University in 2018. After completing her MSc in Chemistry from the Indian Institute of Technology, Kharagpur, in 2020, she has joined the Maiti group at the Indian Institute of Technology, Bombay. Currently, she is pursuing her PhD in transition metal-catalyzed distal C–H bond activation. |
 Trisha Bhattacharya | Trisha Bhattacharya was born in 1993 in the state of West Bengal, India. She obtained her BSc degree in Chemistry from Asutosh College, University of Calcutta, in 2015. Then she completed her MSc in chemistry from the Indian Institute of Technology, Hyderabad, in 2017. After that, she joined the Maiti group at the Indian Institute of Technology, Bombay. Presently, she is pursuing her PhD in transition metal-catalyzed distal C–H bond activation. |
 Finn J. Geffers | Finn J. Geffers was born in 1991 in Lower Saxony (Germany) and studied chemistry at the TU Braunschweig where he received his BSc degree in 2016. In 2018 he received the MSc working in the lab of Prof. Werz. During his MSc studies, he spent 10 months abroad at the University of Utah in the lab of Prof. M. Sigman. Since 2018 he has been pursuing his PhD studies working on aminopalladation cascades across triple bonds in the Werz lab. |
 Marcel Bürger | Marcel Bürger was born in 1988 in Saxony, East Germany. He completed his BSc degree at the TU Bergakademie Freiberg (Germany) and did his MSc at the same place. Afterwards, he joined a spin-off led by Prof. H. Ehrlich and co-founded a biotech start-up. In 2016, he started his PhD studies in the group of Prof. Werz at the TU Braunschweig. His research is focused on the Pd-catalyzed activation of cyano-chalcogens and its implementation in cascade reactions for the synthesis of tetrasubstituted double bonds. |
 Debabrata Maiti | Debabrata Maiti received his PhD from Johns Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After postdoctoral studies at Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the Department of Chemistry at IIT Bombay in 2011. His research interests are focused on the development of new and sustainable synthetic and catalytic methods. |
 Daniel B. Werz | Daniel B. Werz received his diploma (2000) and PhD (2003, R. Gleiter) from the University of Heidelberg supported by a scholarship (Studienstiftung des deutschen Volkes). After a postdoctoral stay at the ETH Zurich (P. H. Seeberger), he began his independent research at the University of Göttingen in 2006 (mentor of the habilitation: L. F. Tietze). In 2013 he was appointed an associate professor of organic chemistry at TU Braunschweig where he was promoted to full professor in 2018. His research interests include donor–acceptor cyclopropanes, Pd catalysis, carbohydrates and organic dyes. |
1. Introduction
Carboxylic acids are one of the ubiquitous and most versatile functional motifs in the field of organic synthesis. The presence of carboxylate moieties in numerous pharmaceuticals, agrochemicals, natural products, and biologically active compounds makes the functionalisation of aromatic or aliphatic carboxylic acids extremely valuable for the synthesis of functional materials.1–4 In contrast to traditional synthetic routes, C–H activation has emerged as one of the most attractive strategies for the synthesis and derivatization of carboxylic acids over the past few years.5–11 Direct conversion of the substrate into a more complex molecule without any pre-functionalisation makes this strategy step economic. However, selective activation of specific C–H bonds present in a molecule is quite challenging. In this context, various directing groups, preinstalled with the carboxylic acid groups, can help the metal to come into the vicinity of a particular C–H bond through coordination. As a result, a wide array of functional groups can be incorporated within the molecule via directing group-assisted metal-catalyzed C–H bond activation reactions.12–14 However, such a three-step directing group-assisted protocol has some limitations: it contains two extra steps, (a) the incorporation of the directing group before functionalisation, and (b) the removal of that group after functionalisation. But, if the carboxylic acid group itself acts as a native directing group, functionalisation might occur in a single step (Fig. 1).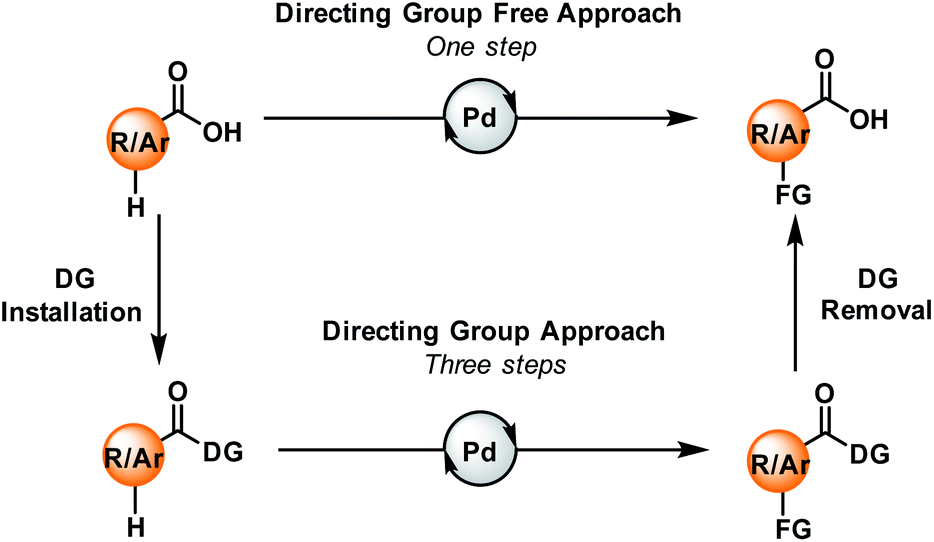 | ||
| Fig. 1 Carboxylic acids and their role in a directing group approach (bottom) and a directing group-free approach (top). DG = directing group. | ||
As a native directing group, it can activate both C(sp3)–H and C(sp2)–H bonds at proximal as well as distal positions of aliphatic and aromatic compounds through five- and six-membered metallacycle formation. Apart from that, it can also act as a traceless directing group,15 additive,16 and steric inducer17–20 that directly influence the mode of several C–H activation reactions.
While prior reviews have predominantly focused on the scope15,16,21–26 and mechanistic understanding27–34 of carboxylic acids, this review highlights the multifaceted roles of carboxylic acids as well as the developments in this area including recent studies and other important aspects. In this context, it is important to mention that free or mono-N-protected amino acids also utilize their carboxylic acid parts for coordination, but those substrates are not included here because they are a different class of substrates (or ligands). Since several reviews35–37 and mechanistic studies38–44 have already been dedicated to this topic, we decided not to include this compound class.
2. Coordination modes of carboxylates
Carboxylic acids (more precisely carboxylates in reaction media) have weaker coordinating ability6 to transition metals in comparison to many other functionalities or ligands. Therefore, the efficacy of the carboxylic acid moiety is reduced in the medium, which eventually affects the kinetics of the reaction. In the presence of a transition metal (e.g., Pd), it can adopt two types of coordination mode, κ1 and κ2. In the κ2 mode, the metal coordinates to two oxygen atoms of the carboxylate group in a bidentate fashion while in the κ1 mode, the metal remains attached to one oxygen atom of the carboxylate in a monodentate fashion. Both of them remain in equilibrium with a slight shift towards the κ2 mode that is not suitable for C–H bond activation.5,6,23,45,46 Several crystal structures of cyclic substrate–palladium complexes are available in the literature which represent the κ2 coordination mode of carboxylates shared by two Pd atoms.47–52It has been reported in many literature studies that hard Lewis acidic metal salts, such as alkali metal ions (Na+, K+, Cs+, etc.), make the κ1 mode preferred through coordination with a carboxylate in a bidentate fashion. Such binding allows Pd(II) to reorient towards the targeted C–H bond (Fig. 2). However, when such metals are absent the κ1 mode gets suppressed and the κ2 mode helps in C–H activation.53
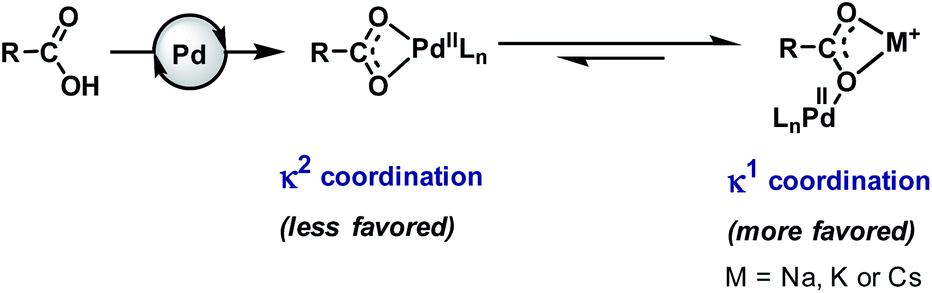 | ||
| Fig. 2 Coordination modes of carboxylates with palladium. | ||
3. Carboxylic acids as native directing groups
The major application of carboxylic acids can be witnessed as native directing groups in different C(sp3)–H as well as C(sp2)–H activation reactions. The carboxylate unit present in the substrate coordinates to the palladium and subsequently directs towards proximal as well as distal C–H bonds of that substrate via the formation of a five- or six-membered palladacycle. This part of the review discusses major advances in the functionalisation of free carboxylic acids using Pd catalysis.3.1. C(sp3)–H activation reactions of carboxylic acids
Aliphatic C–H bonds are rather stable and non-polar, preventing transition metals from being inserted into the C–H bond and thus making the C–H bond cleavage step difficult. However, a directing group that comes into close proximity of the aliphatic C–H bond facilitates the insertion step. Among all the directing groups, carboxylates have a weak coordination ability, which dictates the overall reaction efficiency. The weak coordinating nature can be improved by employing different ligands or electrophiles, which can help the carboxylate moiety to bind more strongly with the transition metal. To date, several research groups have reported their studies on Pd-catalyzed C(sp3)–H activation in proximal (α and β) as well as distal positions (γ and higher). Some asymmetric C(sp3)–H activation reactions were also reported on palladium catalysis. Most of the Pd-catalyzed α-C(sp3)–H activation reactions have been developed based on exogenous directing groups54 that masked the coordinating ability of the free carboxylates. But free carboxylic acids can also selectively activate β-, γ- and higher C(sp3)–H bonds with the help of suitable reagents and appropriate reaction conditions. Palladium/platinum-catalyzed C(sp3)–H activation was first reported by the Chang group in 2006 to prepare benzolactones from o-alkyl substituted aromatic carboxylic acid derivatives.55 Following this, in the same year, White and coworkers disclosed an interesting synthetic strategy for the Pd-catalyzed macrolactonization of ω-alkenoic acids using Pd(OAc)2 with a phenyl bissulfoxide catalyst (Scheme 1).56 Mechanistic studies revealed that the macrolactonization proceeded through a Pd-templated π-allyl carboxylate intermediate. In 2011, they extended this methodology to the total synthesis of an erythromycin precursor, 6-deoxyerythronolide B (6-dEB).57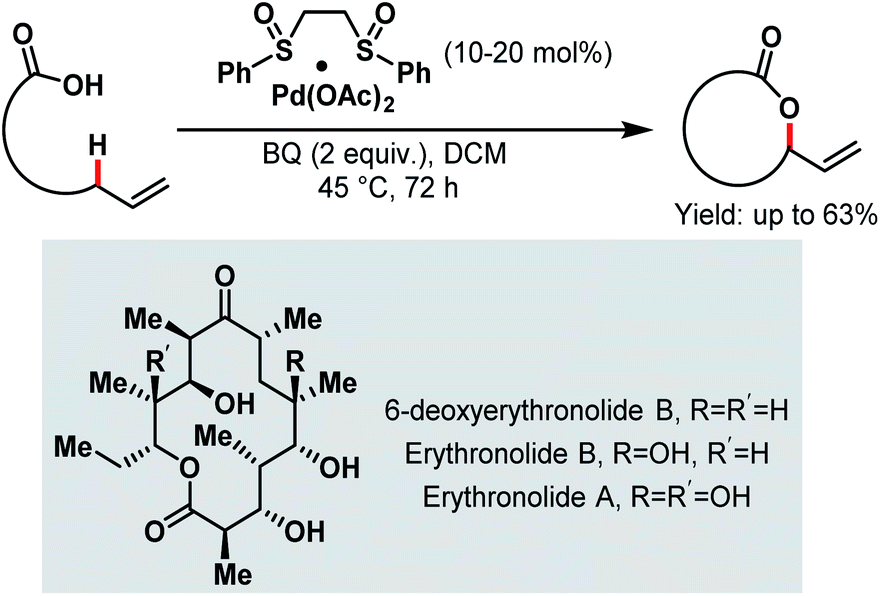 | ||
| Scheme 1 Pd bis-sulfoxide-catalyzed macrolactonization (White et al., 2006).56 | ||
3.1.1.1. β-C(sp3)–H arylation. In 2007, the Yu group first reported a Pd-catalyzed β-C(sp3)–H arylation of pivalic acid and similar aliphatic carboxylic acids containing quaternary centers at α-positions using two different arylating reagents, a phenylboronate ester and an aryl iodide (Scheme 2).58 In the former case, only the monoarylated product was formed, while in the latter one some amount of diarylated product was also produced along with the monoarylated product.
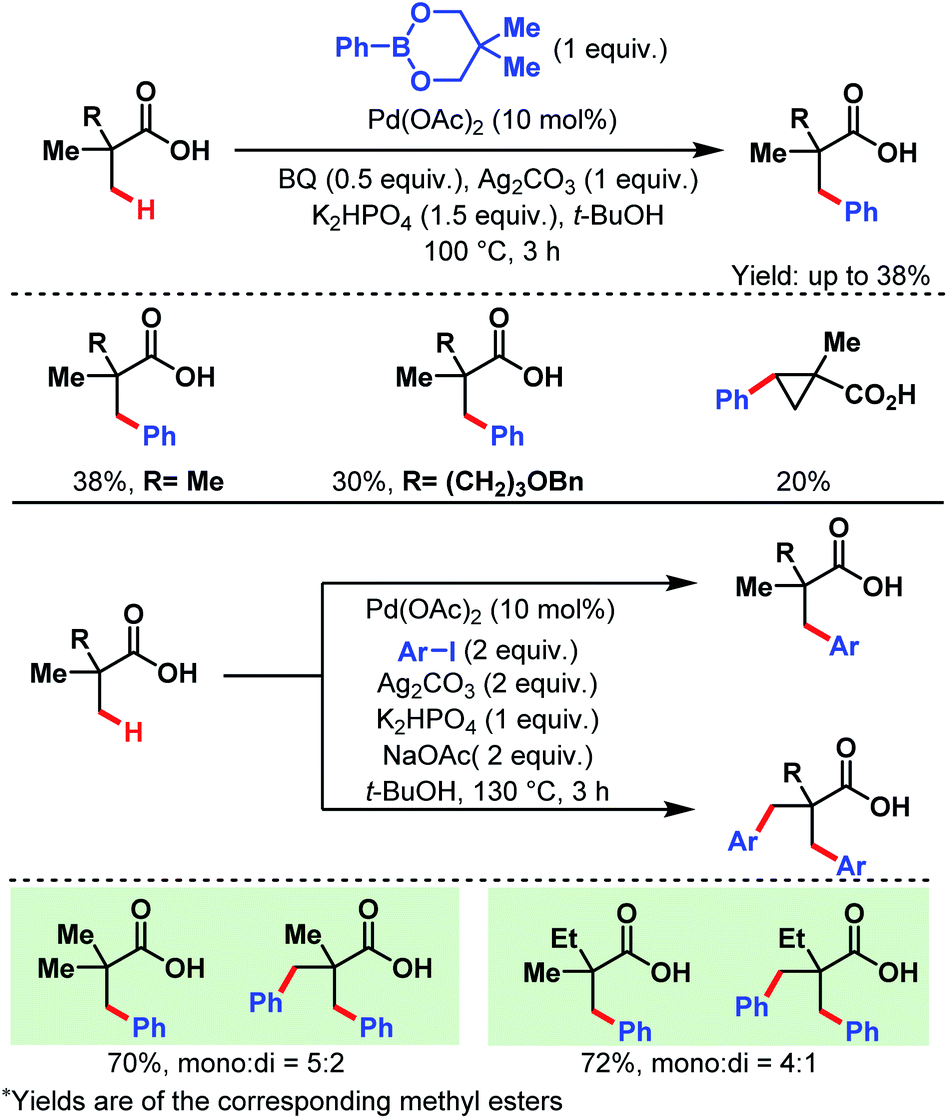 | ||
| Scheme 2 Pd-catalyzed β-C(sp3)–H arylation of α-quaternary carboxylic acids (Yu et al., 2007).57 | ||
Later, in 2017, the Zhao group came up with a new strategy to obtain arylated free carboxylic acids in good yields (up to 80%) using an amino acid as an exogenous ligand and aryl iodides as arylating reagents (Scheme 3).59 Here, they have used acetyl-protected glycine (L1) as a ligand. The scope of this work was also extended to β-C(sp3)–H arylation of N-protected alanine exploiting pyridine-based ligands which eventually led the way to unnatural amino acids.
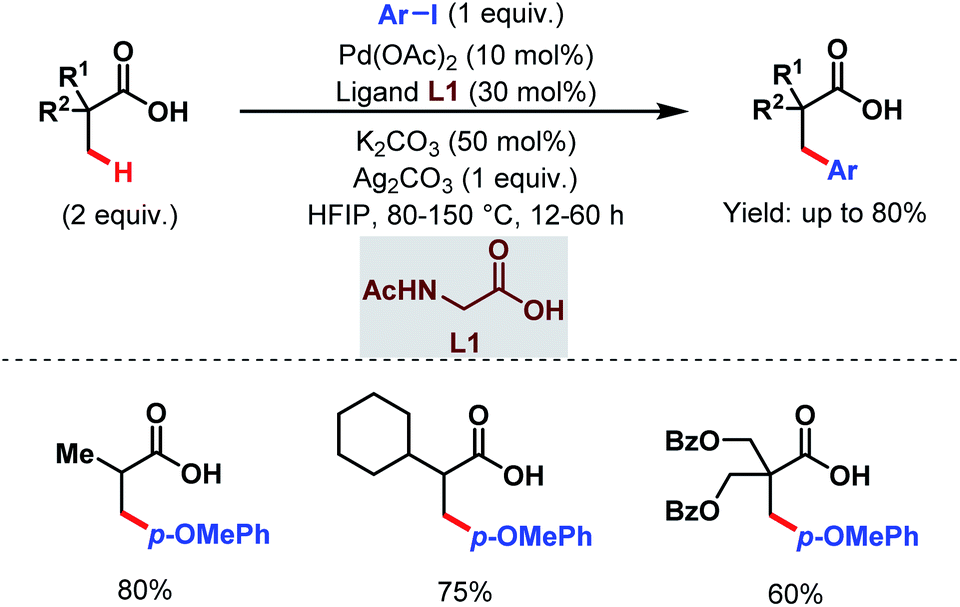 | ||
| Scheme 3 Pd-catalyzed β-C(sp3)–H arylation of aliphatic carboxylic acids (Zhao et al., 2017).59 | ||
Subsequently, the van Gemmeren group also reported a Pd-catalyzed β-C(sp3)–H arylation of free carboxylic acids using aryl iodides in excess relative to carboxylic acids and acetyl protected β-alanine (L2) as a ligand (Scheme 4).60
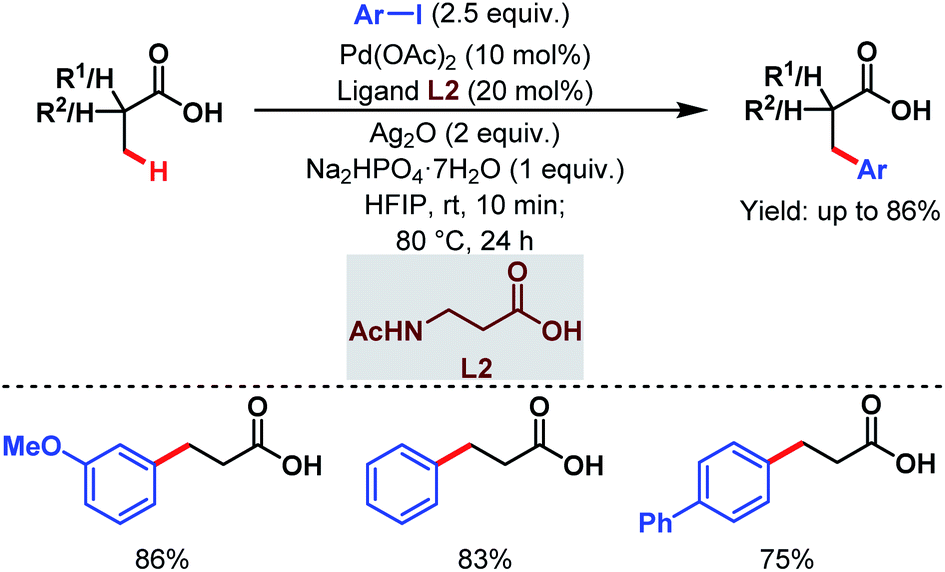 | ||
| Scheme 4 Pd-catalyzed β-C(sp3)–H arylation of aliphatic carboxylic acids (van Gemmeren et al., 2017).60 | ||
3.1.1.2. β-C(sp3)–H olefination. In 2018, the Yu group designed a new bidentate thioether-based ligand (L3) to promote β-C(sp3)–H olefination of a broad range of aliphatic carboxylic acids followed by subsequent lactonization of the olefinated product. This is the first example of a Pd-catalyzed β-C(sp3)–H olefination of free carboxylic acids without using any auxiliary. The methodology is supported by a large number of substrates being converted in good to excellent yields (up to 96%). The γ-lactone products can be further used for the production of various important aliphatic acids, e.g., β-vinylated acids and γ-hydroxylated acids (Scheme 5).61
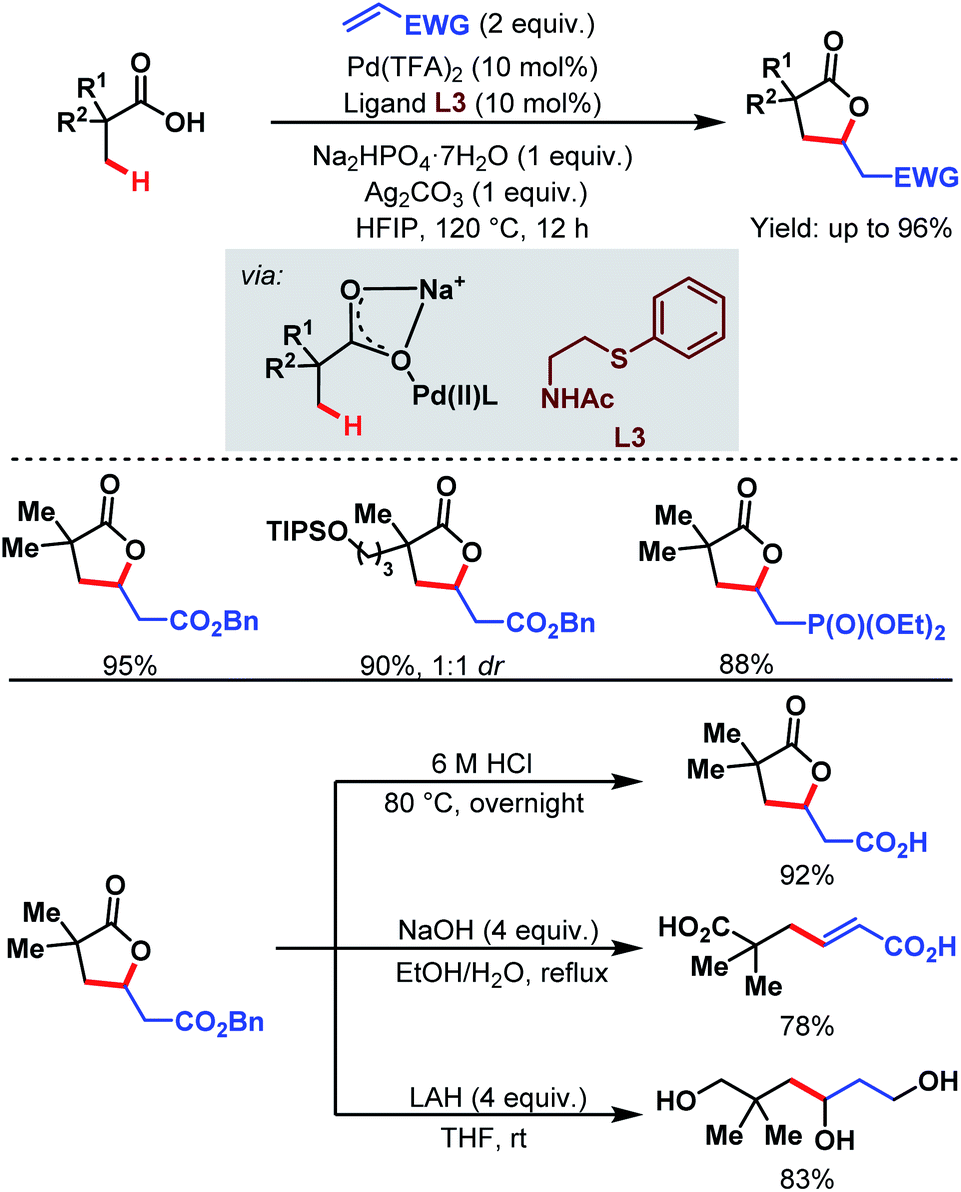 | ||
| Scheme 5 Pd-catalyzed β-C(sp3)–H olefination of aliphatic carboxylic acids (Yu et al., 2018).61 | ||
3.1.1.3. β-C(sp3)–H acetoxylation. Apart from arylation and olefination reactions, even a β-C(sp3)–H acetoxylation was reported by the van Gemmeren group in 2019 where phenyliodine(III) diacetate (PIDA) was used as an acetoxylating agent and the sodium salt of hexafluoroisopropanol (HFIP) as an additive. No external ligand was used here (Scheme 6).62
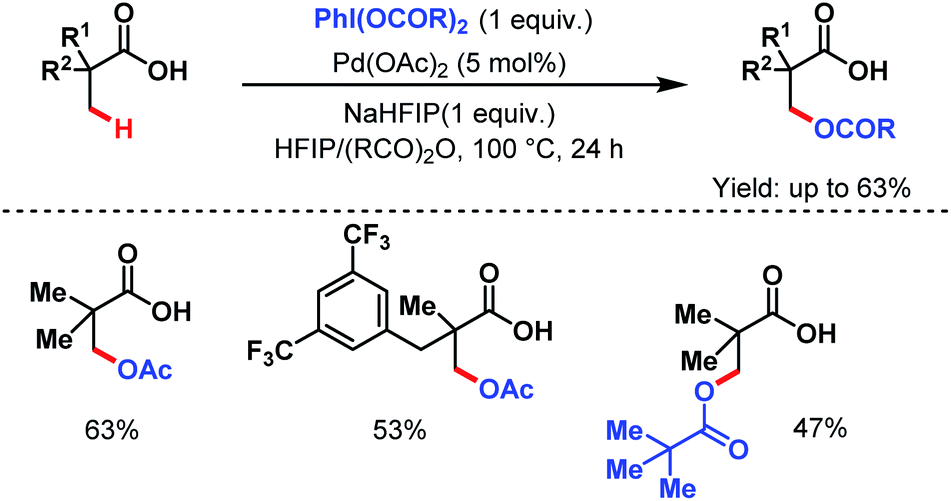 | ||
| Scheme 6 Pd-catalyzed direct β-C(sp3)–H acetoxylation of aliphatic carboxylic acids (van Gemmeren et al., 2019).62 | ||
However, this protocol demands an excess amount of acid substrate and is also restricted to tertiary carboxylic acids (mostly pivalic acid and its derivatives). This shortcoming was resolved by the Yu group who used acetic anhydride as the acetoxylating agent and an acetyl protected α-amino acid as a ligand (L4) in the presence of tert-butyl hydroperoxide (TBHP) as the sole oxidant (Scheme 7).63 An array of aliphatic carboxylic acids with or without α-quaternary centers was compatible under such reaction conditions.
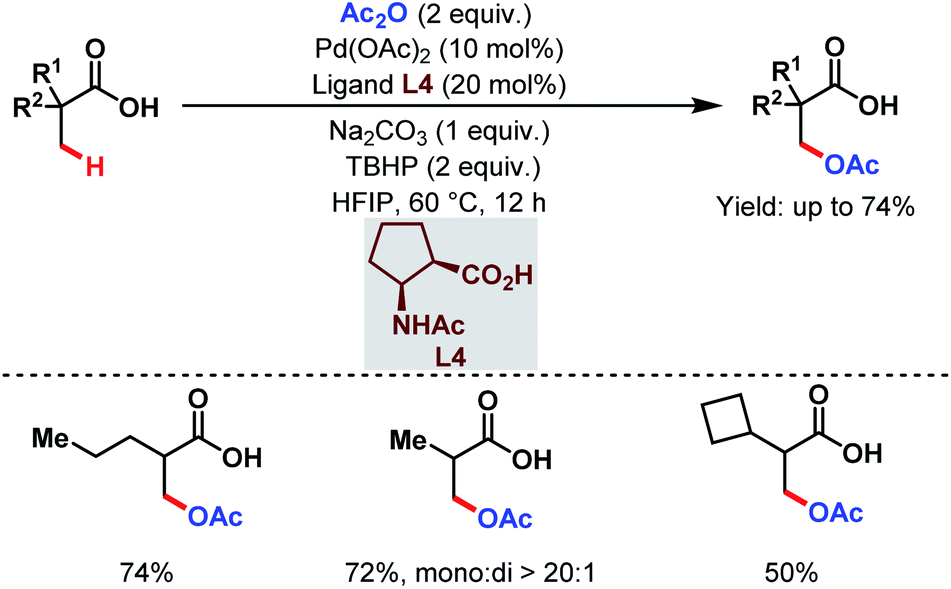 | ||
| Scheme 7 Pd-catalyzed ligand-enabled monoselective β-C(sp3)–H acetoxylation of free carboxylic acids using a practical oxidant (Yu et al., 2020).63 | ||
The transformation proceeds via a Pd(II)/Pd(IV) cycle, initiated with the ligand coordination to Pd(OAc)2 generating LPd(OAc). After that, Int-I gets generated via β-C(sp3)–H activation of sodium carboxylate. Subsequent oxidation of Int-I to Int-II by tert-butyl hydroperoxide (TBHP) and ligand exchange by acetic anhydride (Ac2O) produces Int-III. It is worth mentioning here that strained four-membered lactones can also be formed via selective reductive elimination from Int-II which upon attack by nucleophiles delivers a plethora of β-C(sp3)–H functionalised acids (discussed in detail in Scheme 8). On the other hand, reductive elimination of Int-III leads to the desired β-acetoxylated product and regenerates the active species LPd(OAc) (Scheme 8).
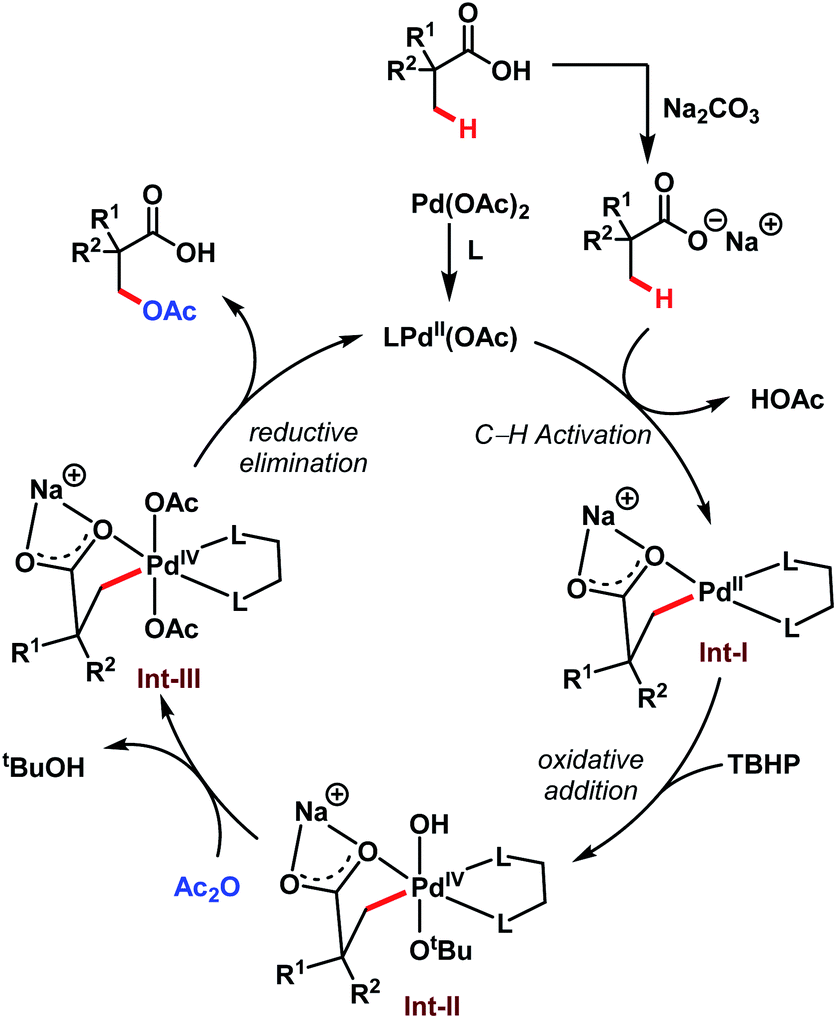 | ||
| Scheme 8 Proposed catalytic cycle for the Pd-catalyzed β-C(sp3)–H acetoxylation of free carboxylic acids (Yu et al., 2020).64 | ||
3.1.1.4. β-C(sp3)–H lactonization. Recently, a novel catalytic strategy for the synthesis of β-lactones directly from free carboxylic acids was achieved by the Yu group without using any olefin (Scheme 9).64 A variety of α-substituted aliphatic carboxylic acids were converted into the corresponding β-lactone derivatives in good to excellent yields using Pd(CH3CN)2Cl2 as a catalyst, a monoprotected amino acid (MPAA) as a ligand (L5), and tert-butyl hydroperoxide (TBHP) as an oxidant. TBHP helped to generate a Pd(IV) intermediate which upon reductive elimination generated a β-lactone. The stronger Pd–OtBu bond in the Pd(IV) intermediate mainly influenced the selective reductive elimination, i.e., β-lactone formation, preventing any other side product formation through other rapid reductive elimination processes. The steric bulk of the –OtBu group and the larger bite angle of the monoprotected amino acid (MPAA) ligand also played a crucial role in the reductive elimination step by enhancing the rate of lactone formation.
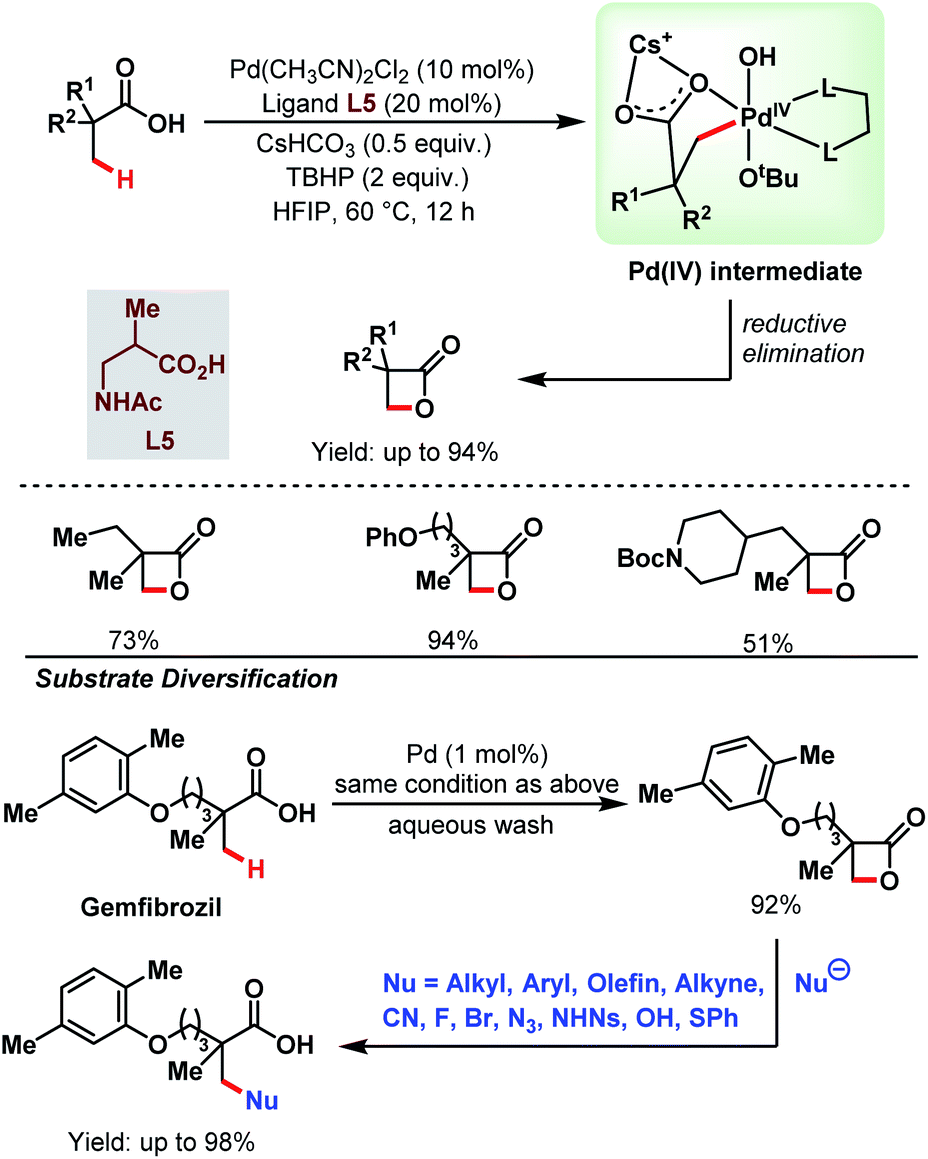 | ||
| Scheme 9 Pd-catalyzed β-C(sp3)–H lactonization of aliphatic carboxylic acids (Yu et al., 2020).64 | ||
This protocol was further extended to the late-stage modification of drug molecules. Gemfibrozil, a drug molecule used to regulate the lipid level in blood, was transformed into its β-lactone derivative with 92% yield. The β-lactone derivative formed was then functionalised with a variety of nucleophiles (e.g., alkyl, aryl, alkenyl, alkynyl, cyano, halogens, thiophenyl, etc.) to generate different types of bond, such as C–C, C–N, C–O, C–X (X = halogen), and C–S at the β-position of that drug molecule. Thus, the choice of an inexpensive oxidant TBHP and the strategy to form different bonds at the β-positions of the substrate acids makes this protocol highly versatile in the context of synthetic efficacy.
3.1.1.5. β-C(sp3)–H alkynylation. Last year, the van Gemmeren group identified a novel class of N-protected ethylene diamine-based ligands (L6 and L7) which enabled the direct β-C(sp3)–H alkynylation of free carboxylic acids (Scheme 10).65 Both α-quaternary and non-α-quaternary carboxylic acids performed well under such reaction conditions where triisopropylsilyl (TIPS) alkynyl bromide was used as an alkynylating agent.
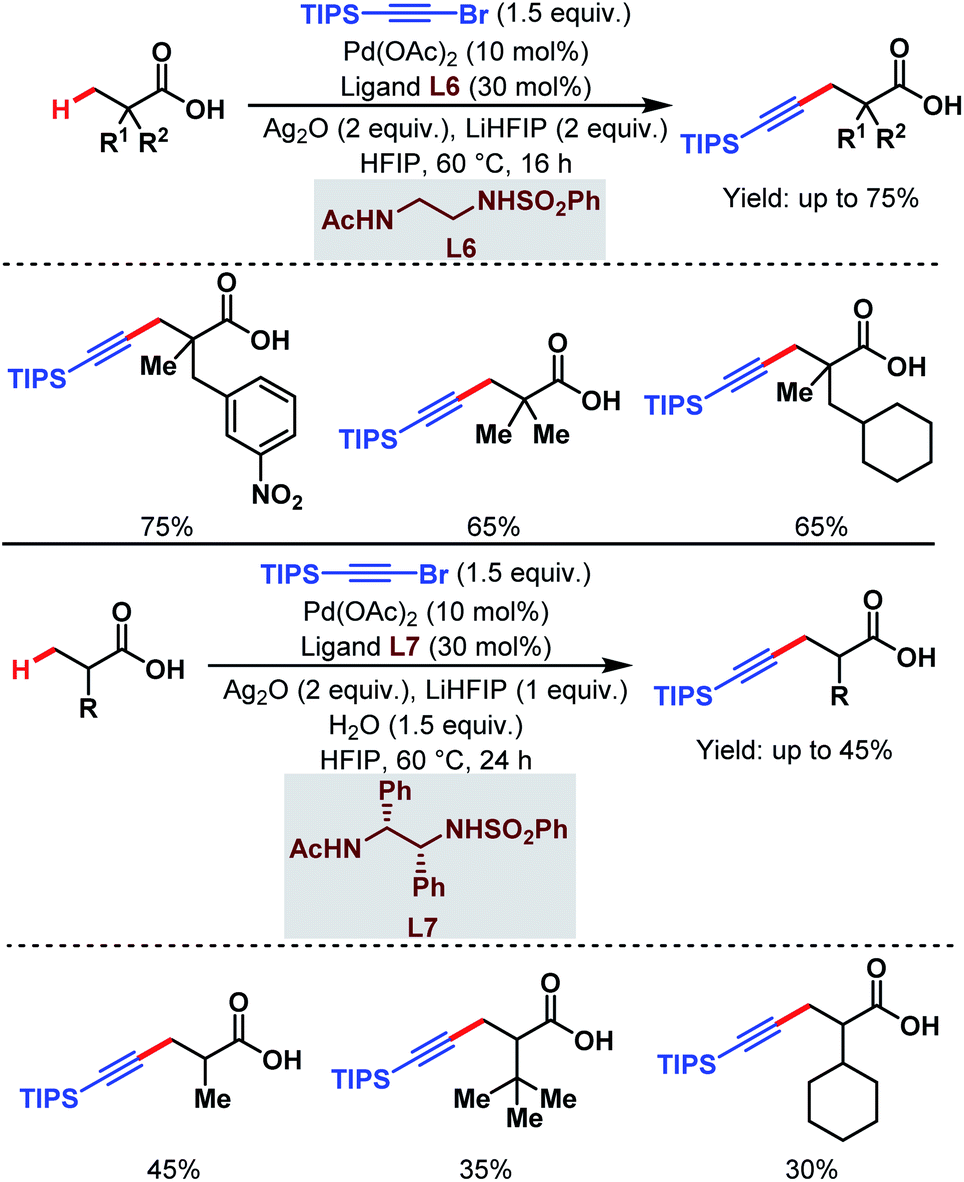 | ||
| Scheme 10 Pd-catalyzed direct β-C(sp3)–H alkynylation of free carboxylic acids (van Gemmeren et al., 2020).65 | ||
3.1.1.6. Late-stage β-C(sp3)–H deuteration. Very recently, the van Gemmeren group reported a regioselective late-stage β-C(sp3)–H deuteration of free carboxylic acids using ethylenediamine-based ligands having 2,6-disubstituted benzamide moieties (L8) and deuterated hexafluoroisopropanol (d1-HFIP) as a deuterating source (Scheme 11).66 This protocol was further extended to deuteration of a wide array of bioactive molecules having potential medicinal activities. Using this methodology, they were also able to achieve deuteration at nonactivated methylene β-C(sp3)–H bonds which had remained untouched so far.
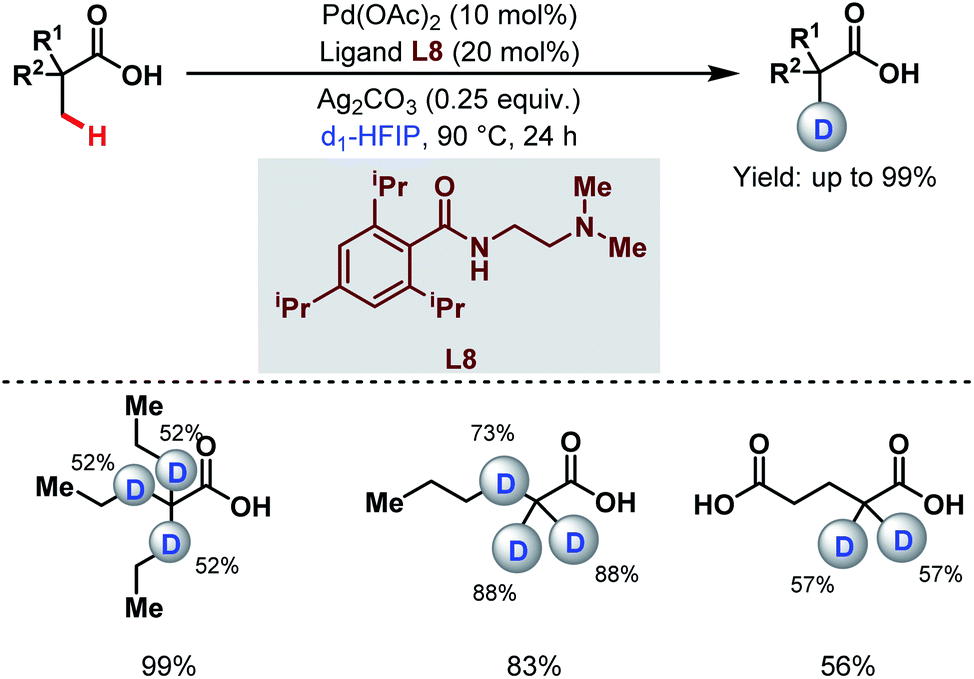 | ||
| Scheme 11 Pd-catalyzed late-stage β-C(sp3)–H deuteration of free carboxylic acids (van Gemmeren et al., 2021).66 | ||
3.1.2.1. γ-C(sp3)–H arylation. In 2019, one of our labs first reported the ligand-enabled Pd-catalyzed regioselective distal γ-C(sp3)–H arylation of aliphatic carboxylic acids without the help of any exogenous directing group. We used a monoprotected amino acid (L1) as a suitable ligand and base to switch the equilibrium from κ2 to κ1, thus allowing the palladium to activate the distal γ-C(sp3)–H bond and to access the γ-arylated aliphatic carboxylic acids in up to 88% yield. In contrast to the previous reports where the diarylated product was formed due to overreaction, it is important to mention that this protocol is compatible with iterative diarylation carried out by sequential incorporation of different aryl groups at other γ-positions. But the second arylation was performed at slightly higher temperature (110 °C). The methodology was further extended to synthesize substituted α-tetralone (Scheme 12).67 An optimization study revealed that the use of the Na+ salt was crucial for the reaction, which probably facilitated the κ1 coordination mode of the carboxylate with palladium, and kinetic isotope effect (KIE) experiments proved the C–H activation to be rate-limiting.
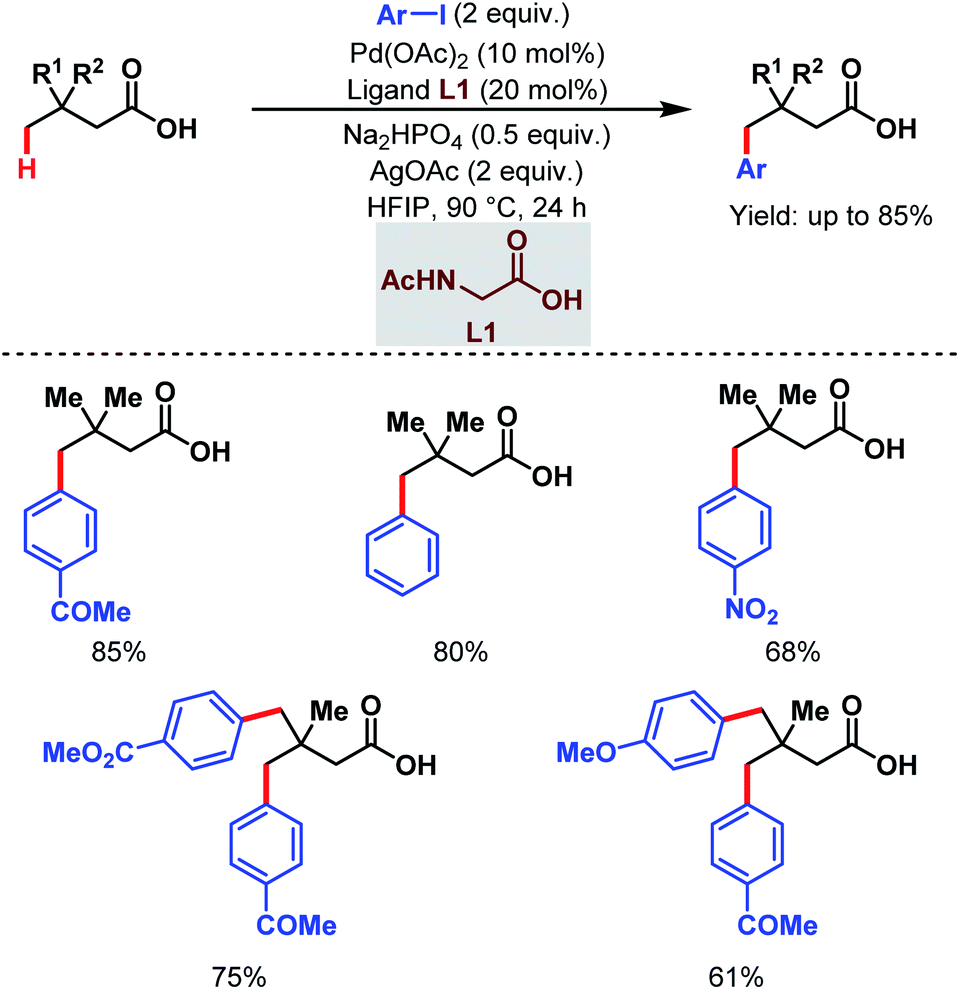 | ||
| Scheme 12 Ligand-enabled Pd-catalyzed direct γ-C(sp3)–H arylation of free aliphatic carboxylic acids (Maiti et al., 2019).67 | ||
Contemporarily, Shi and coworkers were also able to functionalise the distal γ-C(sp3)–H bond of amino acids and respective peptide derivatives without the help of any exogenous directing group using different electronically decorated aryl iodides as coupling partners and silver phosphate (Ag3PO4) as an additive which played a dual role of halide scavenger as well as basic species to facilitate the C–H activation step.68
3.1.2.2. γ-C(sp3)–H olefination. In 2020, the van Gemmeren group, the Yu group and one of our labs reported γ-C(sp3)–H olefination of aliphatic carboxylic acids. The van Gemmeren group designed a catalytic system to promote γ-C(sp3)–H olefination of aliphatic carboxylic acids followed by intramolecular Michael addition producing δ-lactones in moderate to good yields (Scheme 13).69N-Acetyl-β-alanine (L2) was found to be a suitable ligand. It is worth noting that this is the first report that supports a Pd(II)/Pd(0) catalytic cycle for distal γ-C(sp3)–H olefination.
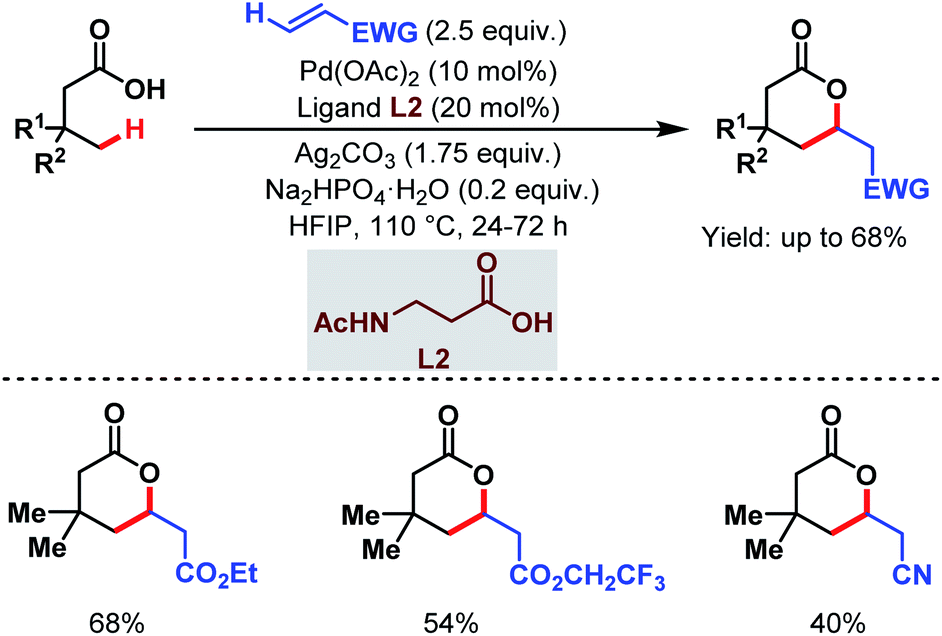 | ||
| Scheme 13 Ligand-enabled Pd-catalyzed direct γ-C(sp3)–H olefination of free carboxylic acids (van Gemmeren et al., 2020).69 | ||
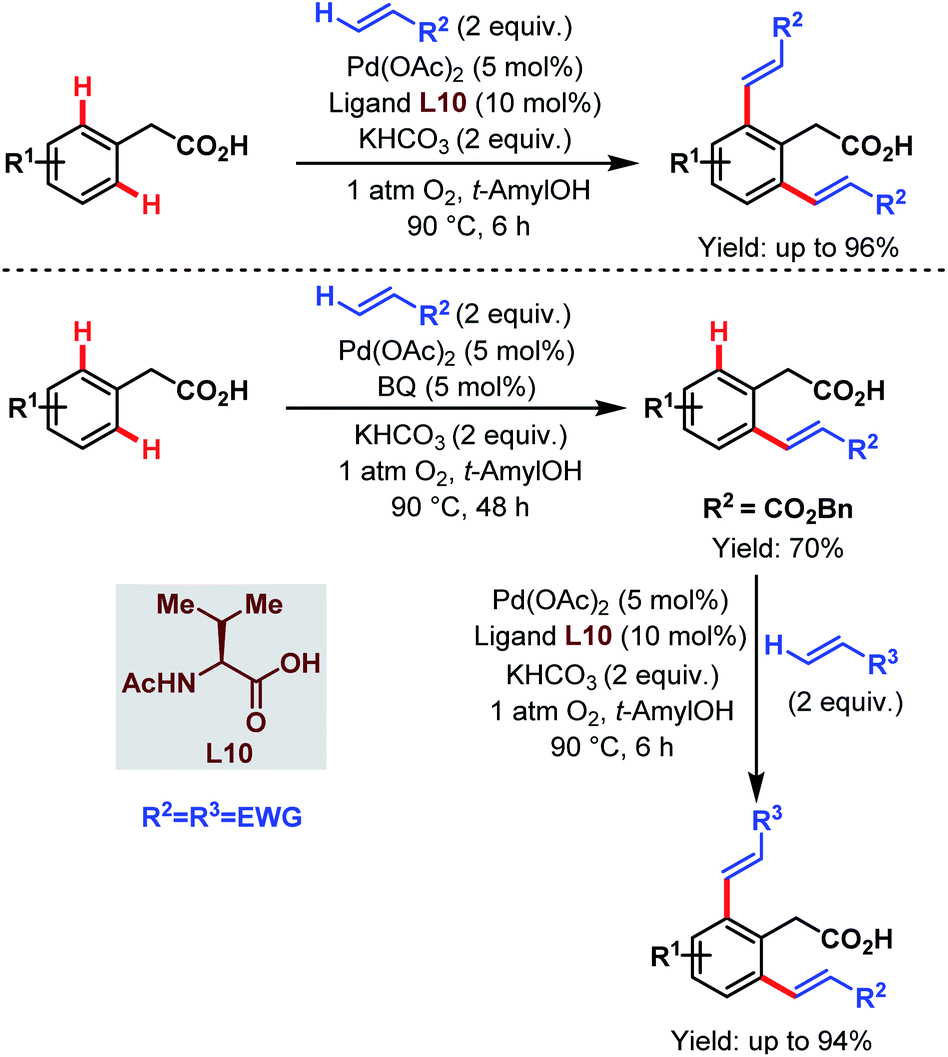
Contemporaneously, Yu and one of our groups reported a similar methodology to synthesize δ-lactones under modified conditions. In both cases, monoprotected α-amino acids (MPAAs) were crucial to achieve high yields. While in Yu's work, N-acetyl-α-phenylalanine (L9) (Scheme 14)70 turned out to be the best ligand for the scheme, we found N-acetyl-α-valine (L10) (Scheme 15)71 to be the most judicious choice of ligand. Using various activated olefins and maleimides as coupling partners we also achieved both δ-lactones and ε-lactones, respectively.
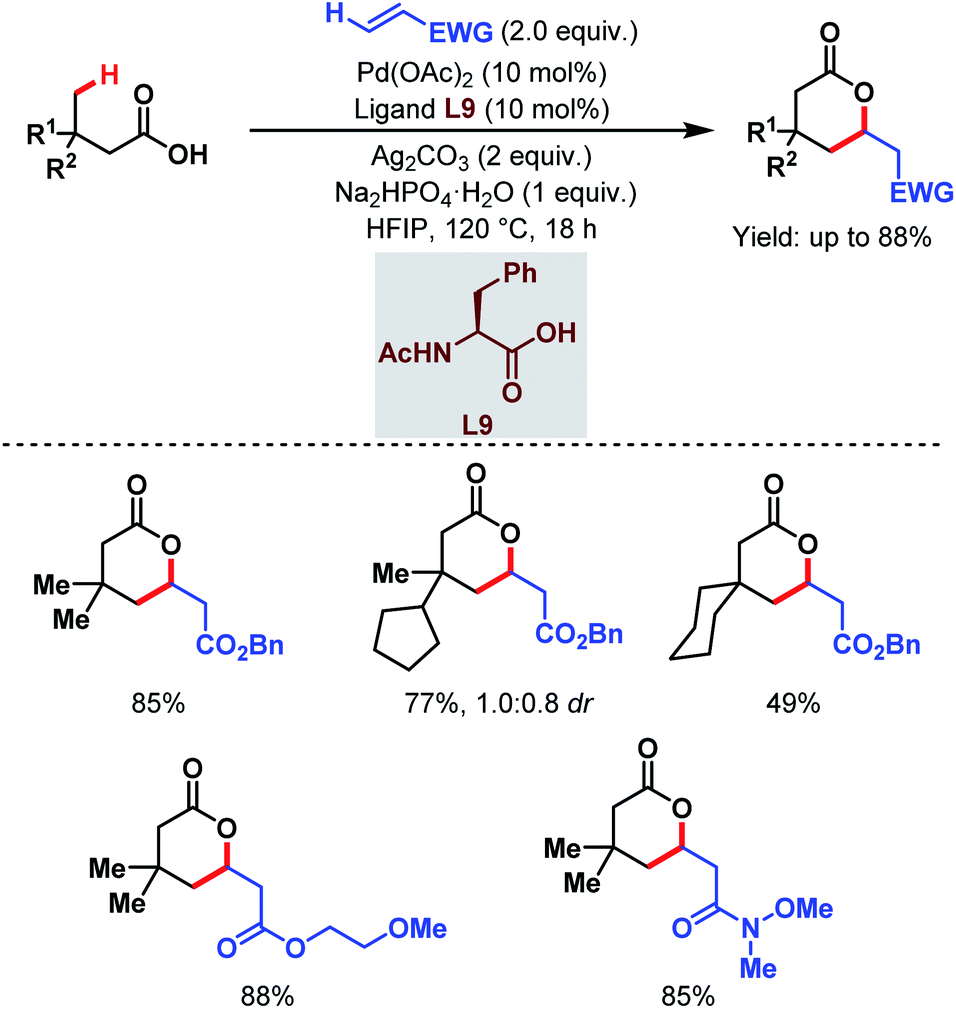 | ||
| Scheme 14 Pd-catalyzed distal γ-C(sp3)–H olefination of free carboxylic acids (Yu et al., 2020).70 | ||
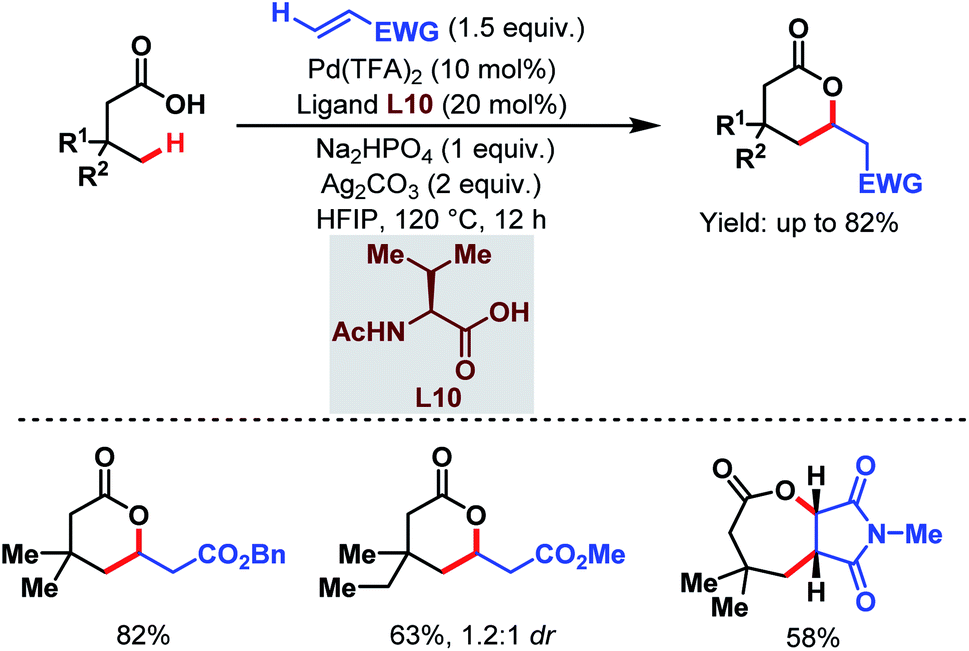 | ||
| Scheme 15 Lactone formation via Pd-catalyzed γ-C(sp3)–H activation of free aliphatic carboxylic acids (Maiti et al., 2020).71 | ||
The mechanism involves a Pd(II)/Pd(0) catalytic cycle in which a six-membered palladacycle gets formed because of the κ1 coordination mode of the carboxylate with Pd(II) in the presence of Na+ salt. Both six- and seven-membered lactones can be prepared using this protocol just by changing the olefinic coupling partner. A mechanistic study suggested C–H activation as the rate-determining step (Scheme 16).
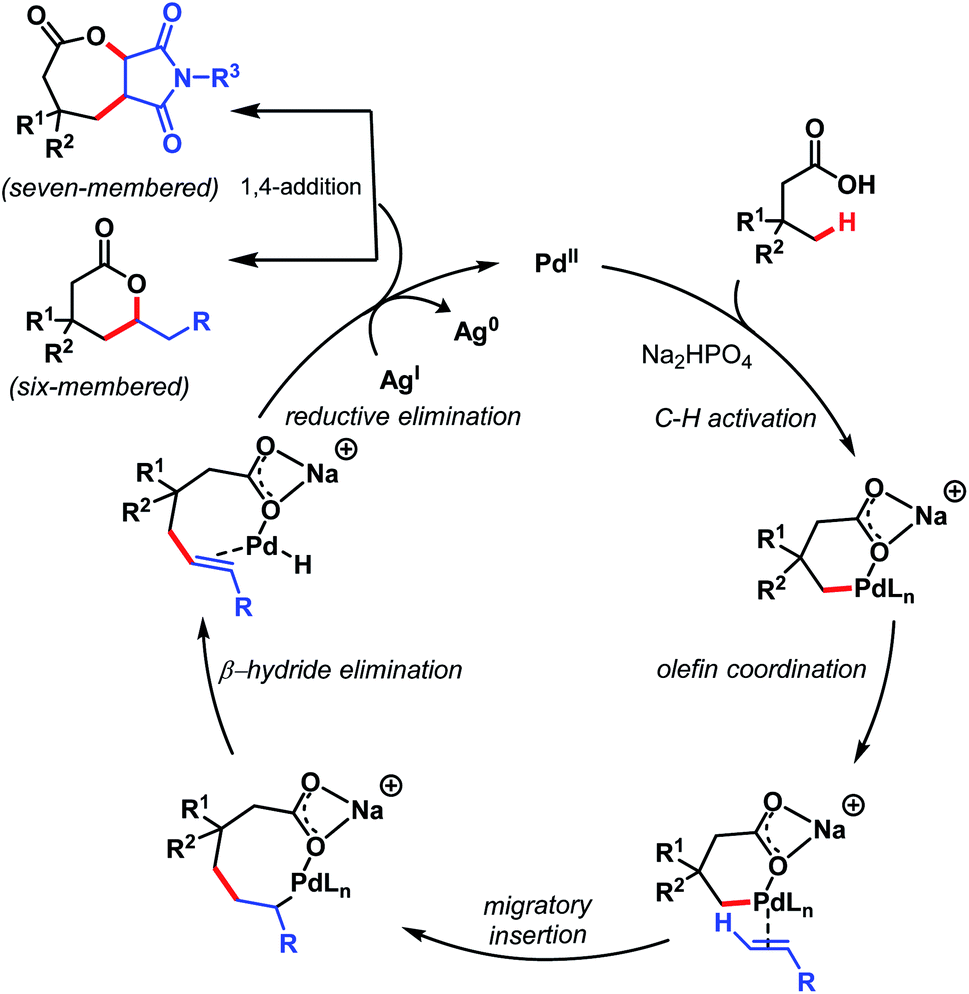 | ||
| Scheme 16 Proposed mechanistic cycle for γ-lactone formation (Maiti et al., 2020).71 | ||
3.1.2.3. γ-C(sp3)–H alkynylation. In 2020, the van Gemmeren group reported a γ-C(sp3)–H alkynylation of free aliphatic acids where a novel class of N-protected ethylene diamine-based ligand (L7) helps in the direct γ-C(sp3)–H activation (Scheme 17).65 Here, triisopropylsilyl (TIPS) alkynyl bromide was used as an alkynylating agent.
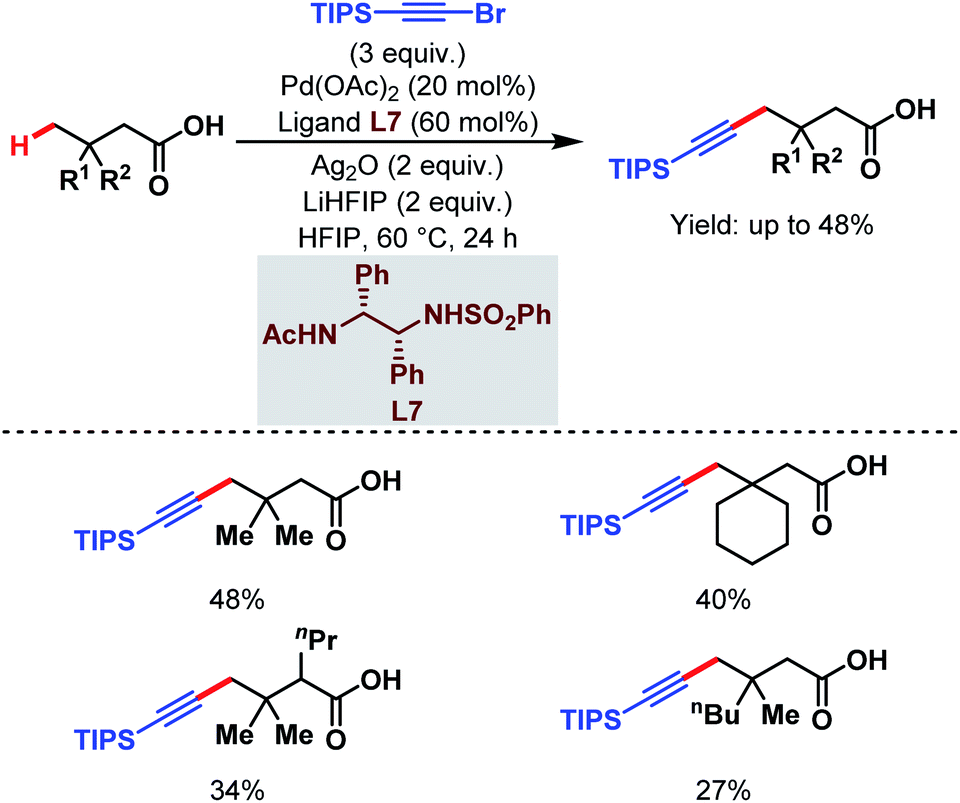 | ||
| Scheme 17 Pd-catalyzed direct γ-C(sp3)–H alkynylation of free carboxylic acids (van Gemmeren et al., 2020).65 | ||
 | ||
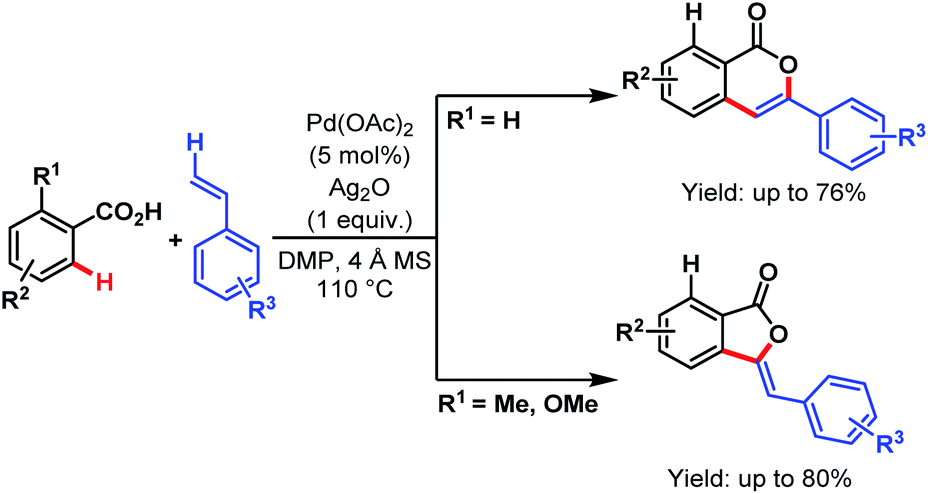
| Scheme 18 Synthesis of benzolactones via Pd-catalyzed direct benzylic C(sp3)–H activation of free carboxylic acids (Martin et al., 2011).72 | ||
A tentative mechanism proposed by the authors involves a Pd(II)/Pd(0) cycle, where Ag(I) played a dual role in regenerating the catalyst as well as participating in the early transmetalation step. Benzylic C(sp3)–H activation followed by C–O bond-forming reductive elimination leads to the formation of product (Scheme 19).
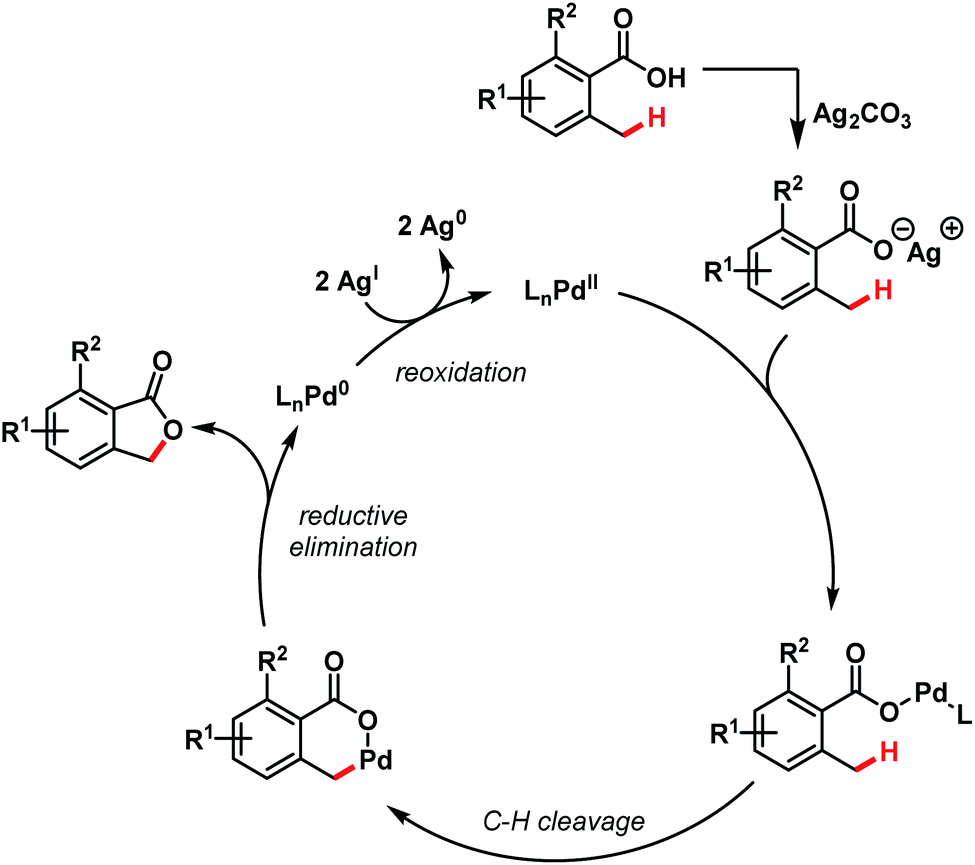 | ||
| Scheme 19 Mechanistic hypothesis for benzolactone synthesis (Martin et al., 2011).72 | ||
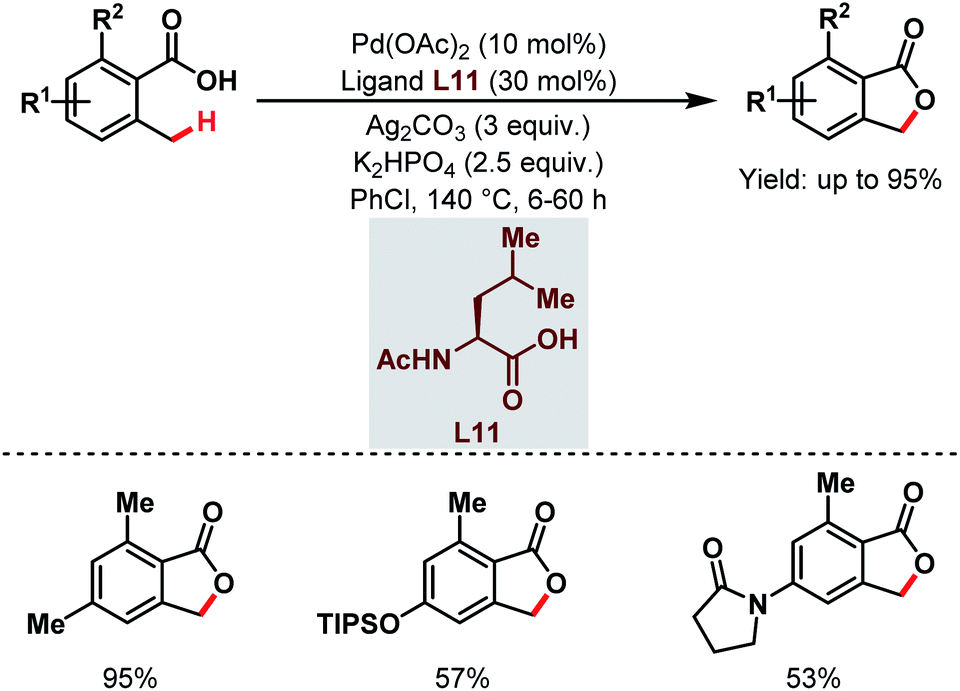
After that, a Pd-catalyzed benzylic C(sp3)–H lactonization of o-methyl benzoic acid substrates was reported by the Yu group in 2020 using a 3-trifluoromethyl-2-pyridone ligand (L12) and molecular oxygen as an oxidant to construct a broad range of important benzolactone molecules (Scheme 20).73
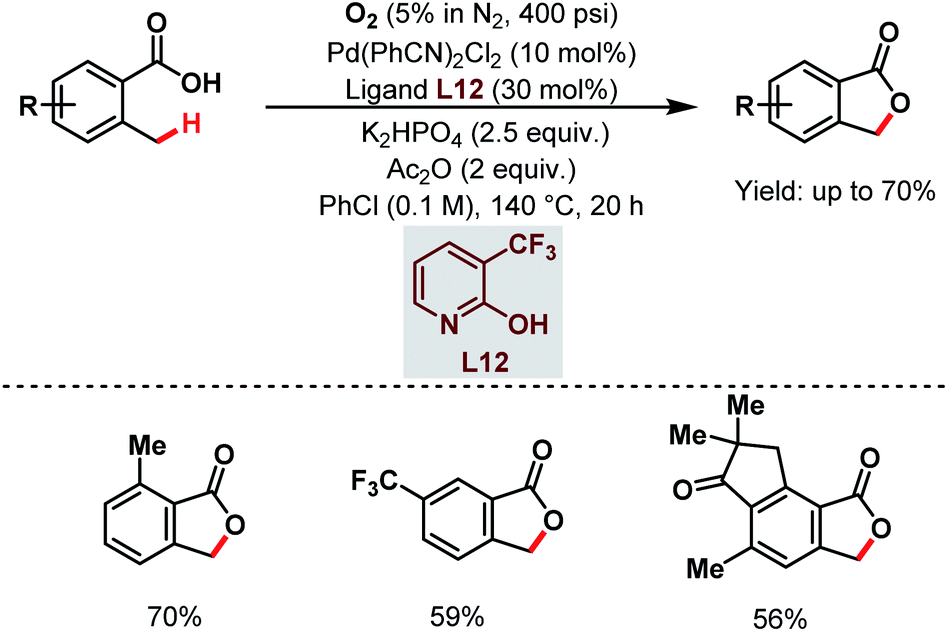 | ||
| Scheme 20 Ligand-enabled Pd-catalyzed C(sp3)–H lactonization using O2 as an oxidant (Yu et al., 2020).73 | ||
They also reported an olefination at the benzylic position of free carboxylic acids to synthesize δ-lactones under modified conditions. A monoprotected α-amino acid (L9) was used to achieve higher yields (Scheme 21).70
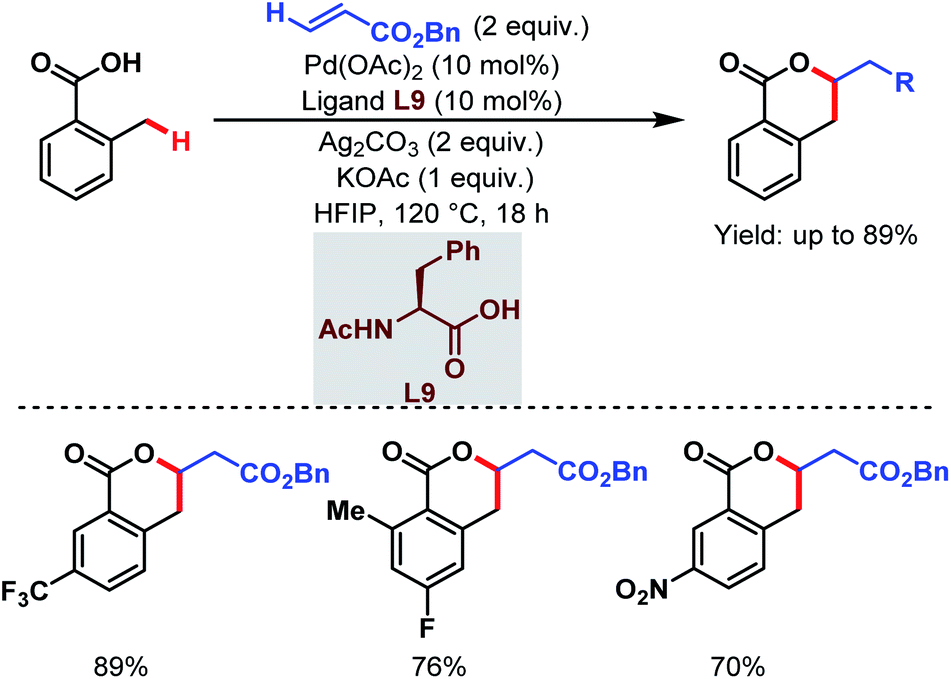 | ||
| Scheme 21 Pd-catalyzed distal γ-C(sp3)–H olefination of free carboxylic acids (Yu et al., 2020).70 | ||
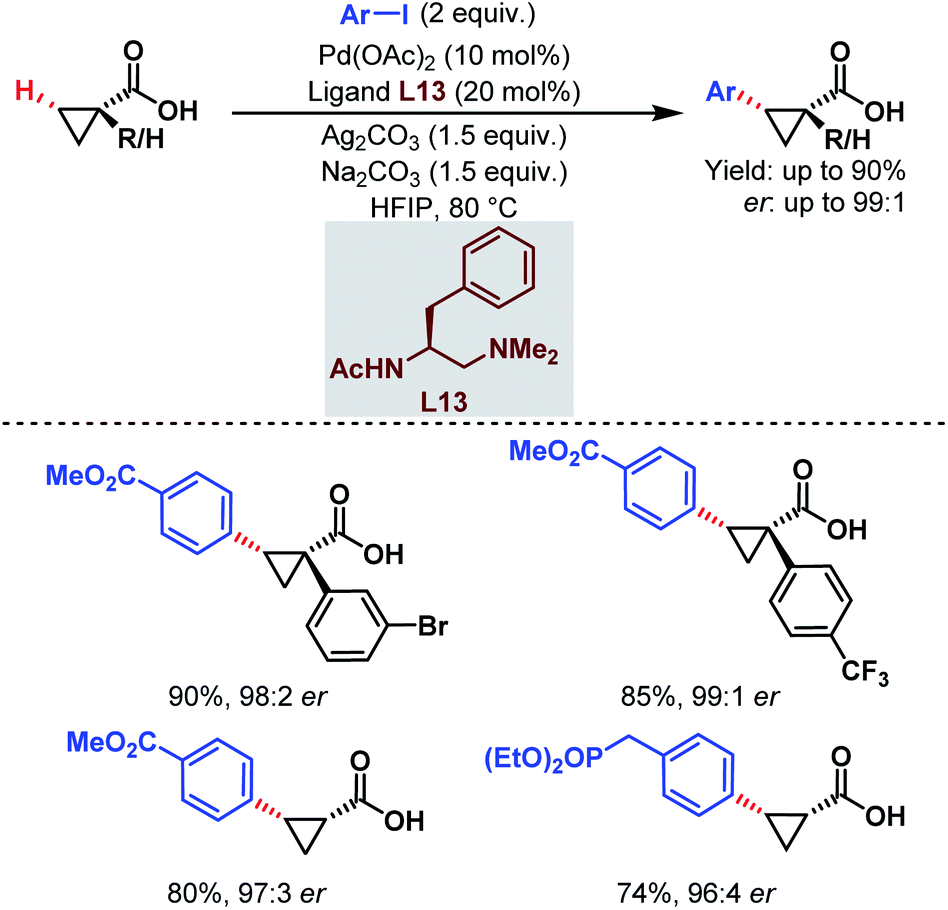 | ||
| Scheme 22 Pd-catalyzed enantioselective C(sp3)–H arylation of free carboxylic acids (Yu et al., 2018).74 | ||
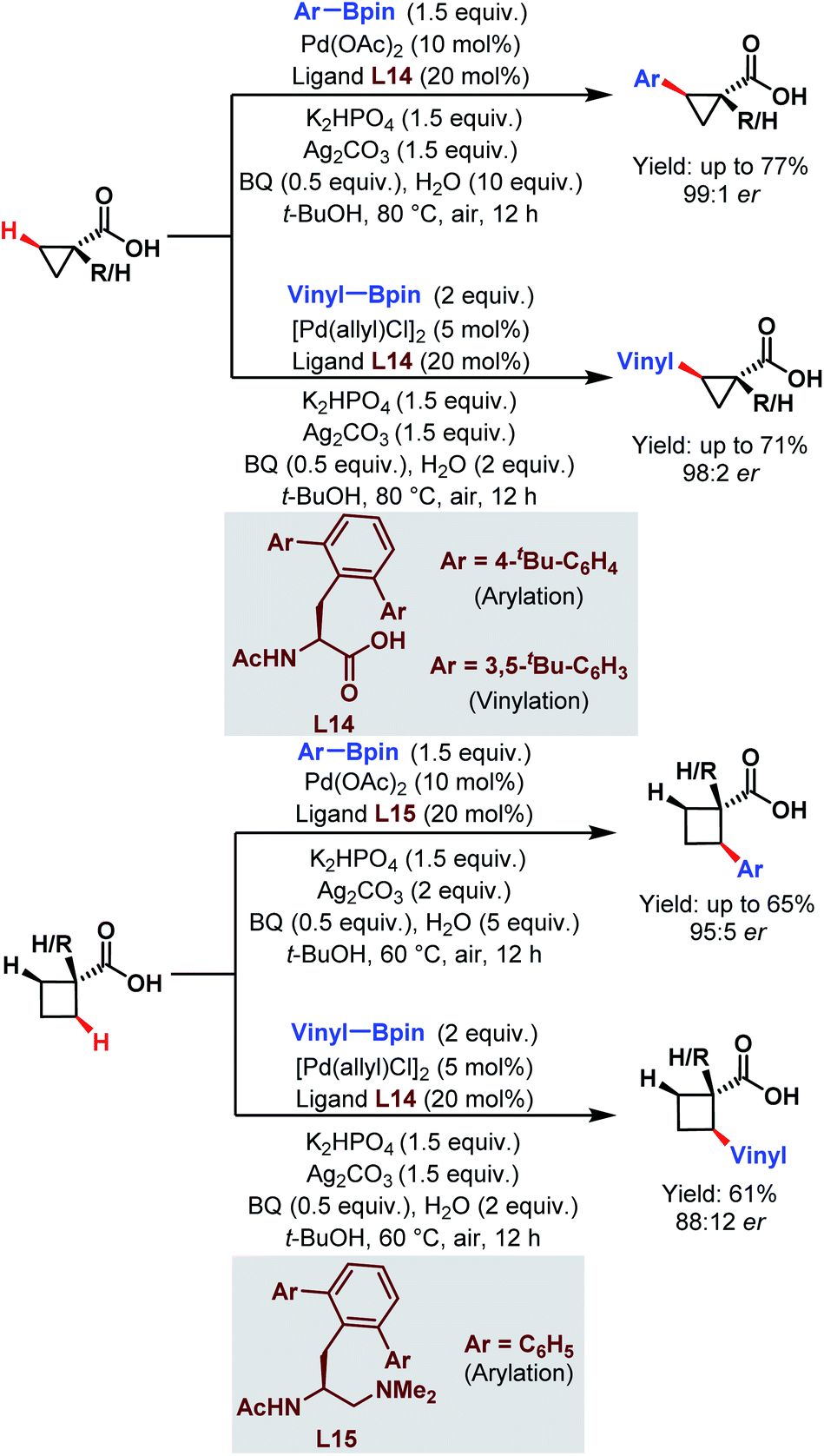 | ||
| Scheme 23 Enantioselective Pd-catalyzed C(sp3)–H arylation and vinylation of free carboxylic acids (Yu et al., 2019).75 | ||
3.2. C(sp2)–H activations of carboxylic acids
Though carboxylate-directed C(sp3)–H activation is still in its infancy, the use of carboxylic acids as native directing groups in C(sp2)–H activation is well established.3.2.1.1. ortho-C(sp2)–H activation.
3.2.1.1.1. ortho-C(sp2)–H arylation. In 2007, the Yu group first described an arylation of simple benzoic acids using phenylboronic acids as coupling partners to synthesize arylated benzoic acids in moderate to good yields (Scheme 24).58
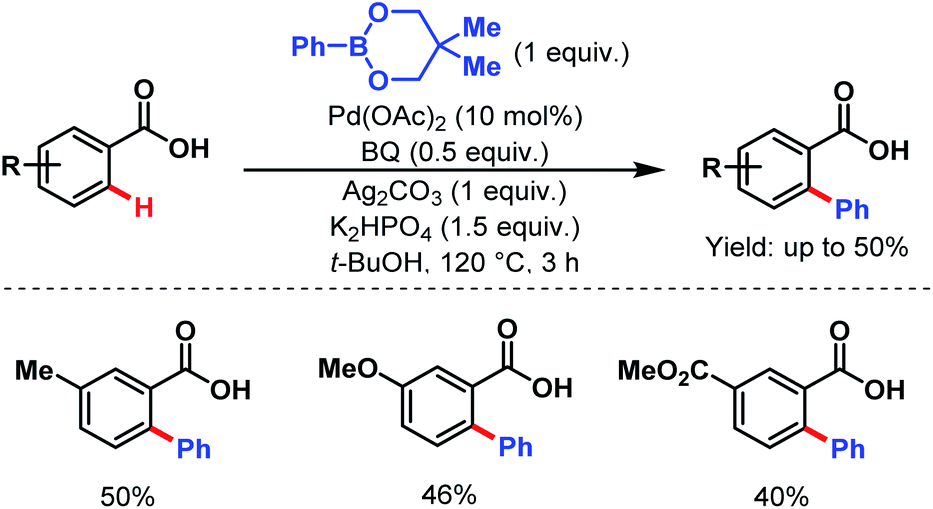 | ||
| Scheme 24 Pd-catalyzed ortho-C(sp2)–H arylation of benzoic acids (Yu et al., 2007).58 | ||
In the same year, the Daugulis group reported two catalytic methods for direct ortho-C(sp2)–H activation of benzoic acids. The first method is applicable to the arylation of electron-rich to moderately electron-deficient benzoic acids using an aryl iodide as a coupling partner in acetic acid as solvent and stoichiometric silver acetate for iodide removal. The second method employs an aryl chloride as a coupling partner, n-butyl-di-1-adamantylphosphine as a ligand, cesium carbonate as a base, and DMF as solvent. This procedure is suitable for both electron-rich and electron-deficient benzoic acids (Scheme 25).76
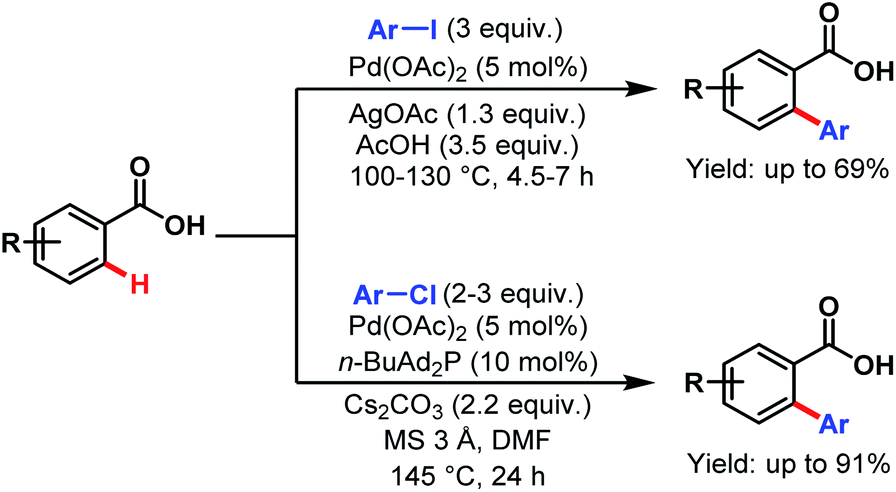 | ||
| Scheme 25 Pd-catalyzed direct ortho-C(sp2)–H arylation of benzoic acids (Daugulis et al., 2007).76 | ||
The following year, the Yu group disclosed another arylation strategy using aryl trifluoroborates as coupling partners in the absence of silver salt; instead, benzoquinone with oxygen (O2) was used as an oxidant. Both benzoic acids and phenylacetic acids gave the desired ortho-arylated products in good to excellent yields (Scheme 26).77
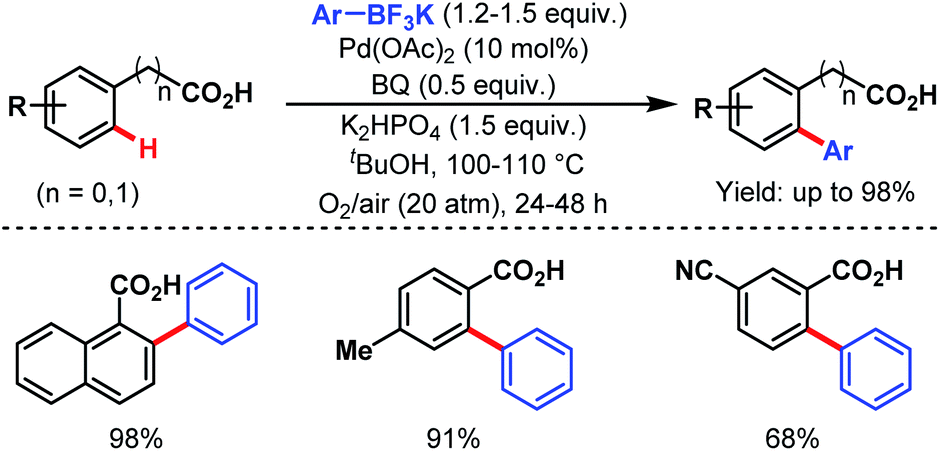 | ||
| Scheme 26 Pd-catalyzed ortho-C(sp2)–H arylation of benzoic acids and phenylacetic acids (Yu et al., 2008).77 | ||
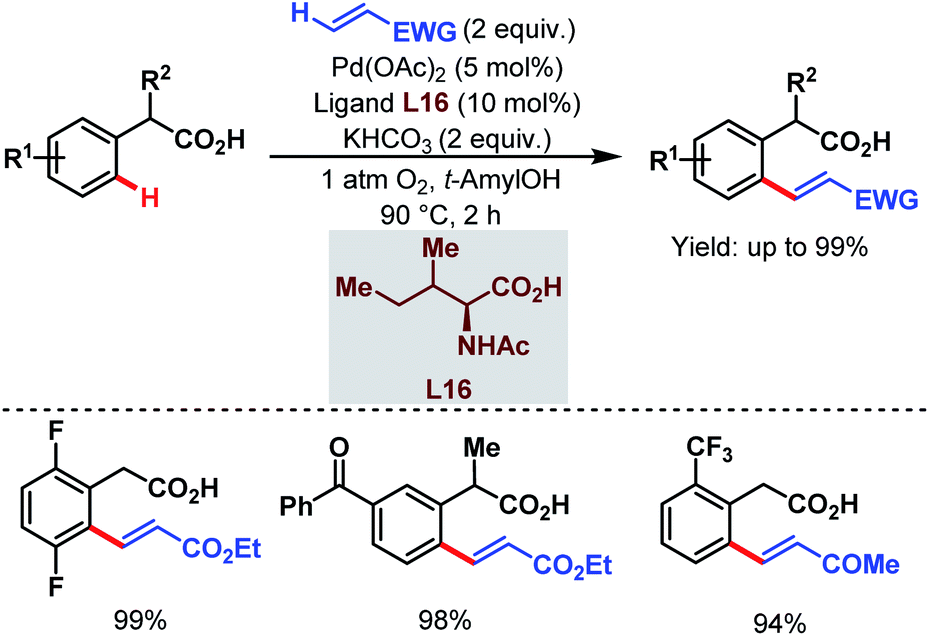
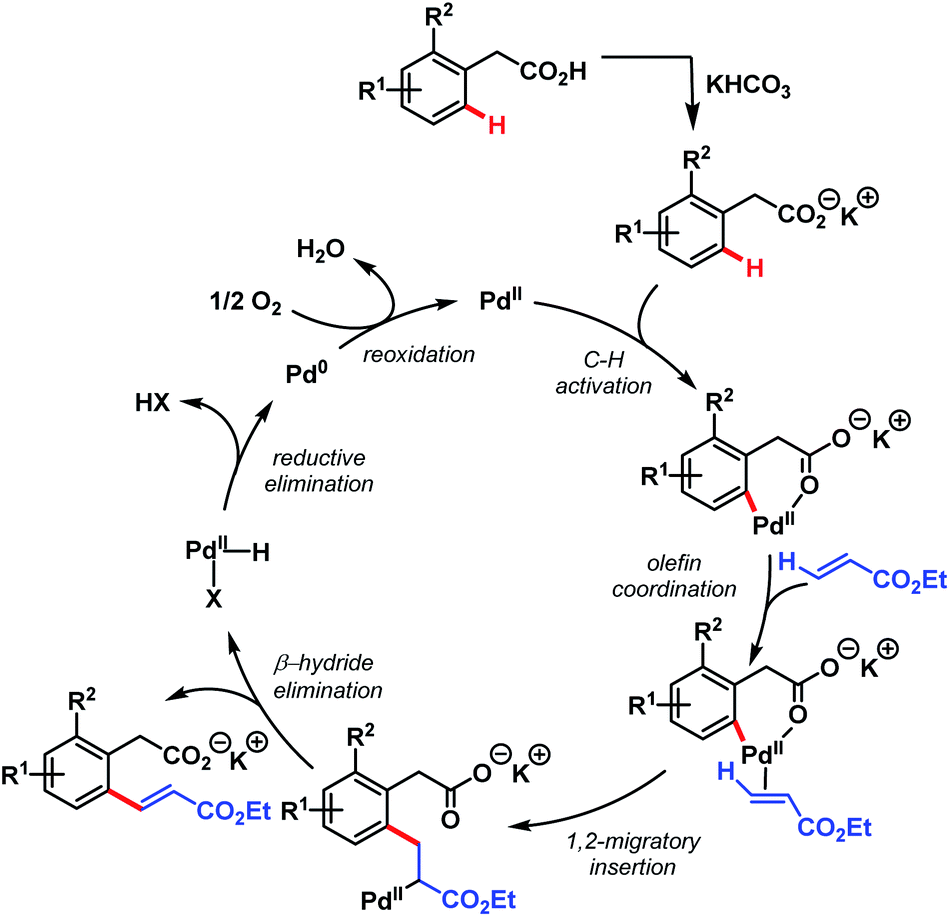
In 2011, they further extended the aforementioned arylation strategy using a mono-N-protected amino acid (L16) as a ligand, silver carbonate (Ag2CO3) as an oxidant, and the same aryltrifluoroborates as coupling partners. Following this protocol, they were able to synthesize a wide array of ortho-substituted phenylacetic acid derivatives in good yields (Scheme 27).78 The reaction underwent a Pd(II)/Pd(0) catalytic cycle initiated by substrate coordination followed by C–H bond cleavage. Subsequent transmetalation followed by reductive elimination gave the desired arylated product (Scheme 28).
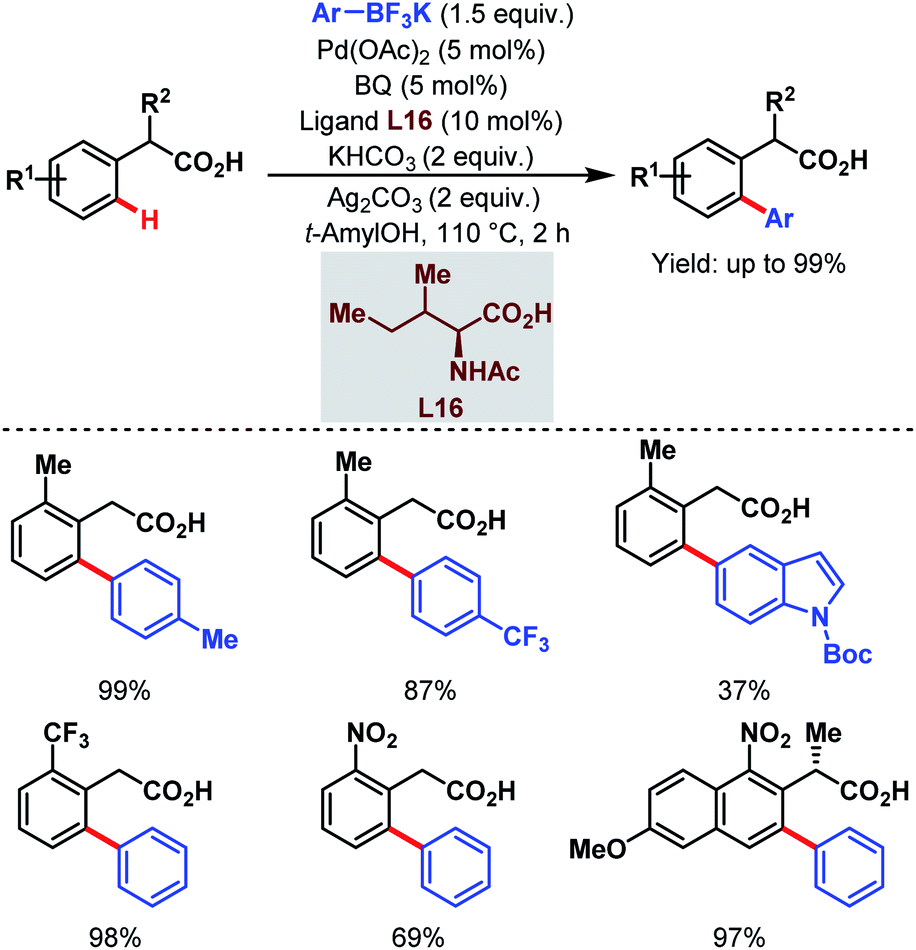 | ||
| Scheme 27 Pd-catalyzed ortho-C(sp2)–H arylation of phenylacetic acids (Yu et al., 2011).78 | ||
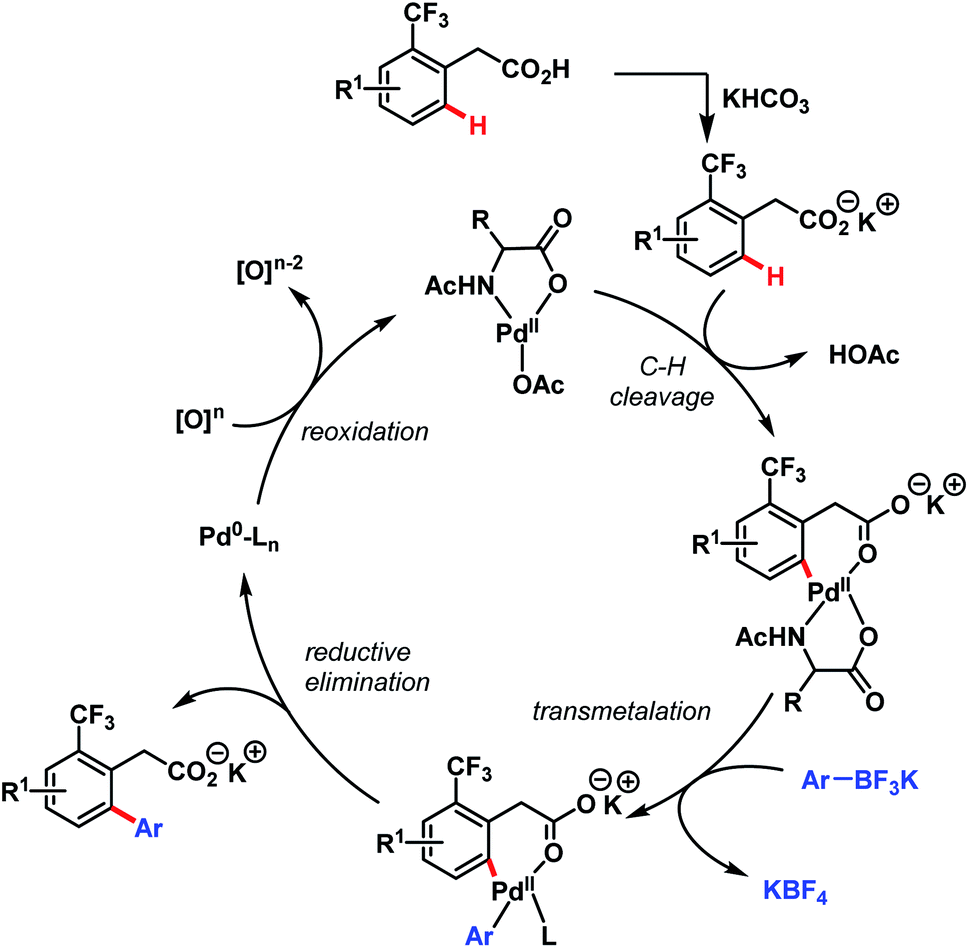 | ||
| Scheme 28 Proposed catalytic cycle for Pd-catalyzed ortho-C(sp2)–H arylation of phenylacetic acids (Yu et al., 2011).78 | ||
In 2013, Larrosa and coworkers devised a protocol for arylation of ortho-substituted benzoic acids.79 As ortho-disubstituted compounds readily undergo protodecarboxylation in the presence of a metal, to overcome such difficulty they used silver carbonate (Ag2CO3) as an oxidant and potassium carbonate (K2CO3) as an additive, and were able to synthesize several sterically hindered 2-aryl 6-substituted benzoic acids in good yields (Scheme 29). Later on, they replaced the Ag(I) salt with an inexpensive and organic analogue, tetramethylammonium acetate (NMe4OAc), used for in situ catalyst generation and developed a silver-free Pd-catalyzed ortho-C(sp2)–H arylation method with a variety of substrates, such as benzoic acids, benzamides, and different heterocyclic compounds.80
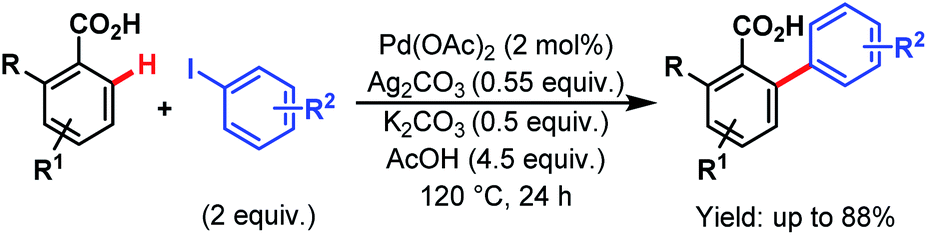 | ||
| Scheme 29 Pd-catalyzed ortho-C(sp2)–H arylation of ortho-substituted benzoic acids (Larrosa et al., 2013).79 | ||
After two years, the Su group developed a novel strategy for ortho-C(sp2)–H arylation of unactivated electron-deficient benzoic acids using aryl iodides as coupling partners and a monoprotected amino acid (L16) as a ligand (Scheme 30).81 Using this protocol, a wide range of ortho-arylated benzoic acids was synthesized in moderate to excellent yields at ambient temperature.
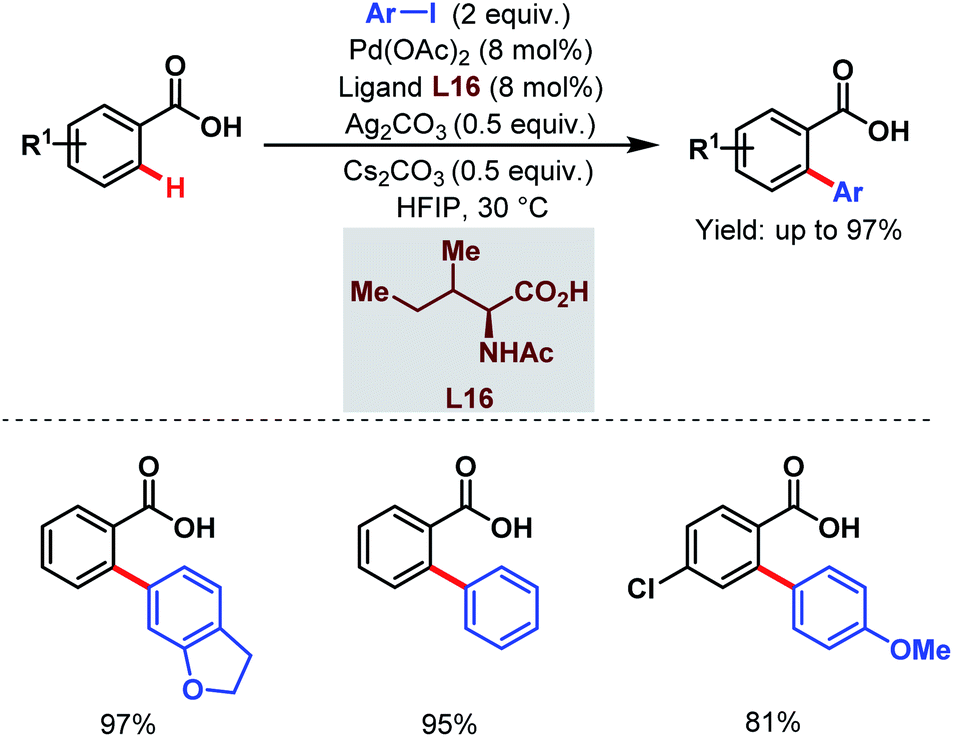 | ||
| Scheme 30 Pd-catalyzed ortho-C(sp2)–H arylation of benzoic acids at ambient temperature (Su et al., 2015).81 | ||
The choice of the solvent was crucial as hexafluoroisopropanol (HFIP) was responsible for such mild reaction conditions. HFIP, being a strong proton donor, helped the metal to come into close proximity of a particular C–H bond and stabilized the metallacycle formed.82 Furthermore, a kinetic study suggested the role of the ligand in accelerating the C–H activation step and also in improving the lifetime of the catalyst.
In the same year, Yu and coworkers reported a high yielding monoselective ortho-arylation of enantiopure pivaloyl- or acetyl-protected mandelic acid derivatives without using any external ligand (Scheme 31). Apart from arylation, they also developed different conditions for acetoxylation, iodination and olefination of mandelic acid and α-phenyl glycine derivatives. While other functionalisations do not require any external ligands, during olefination, monoselectivity was dependent on the choice of the monoprotected amino acid (MPAA ligand).83
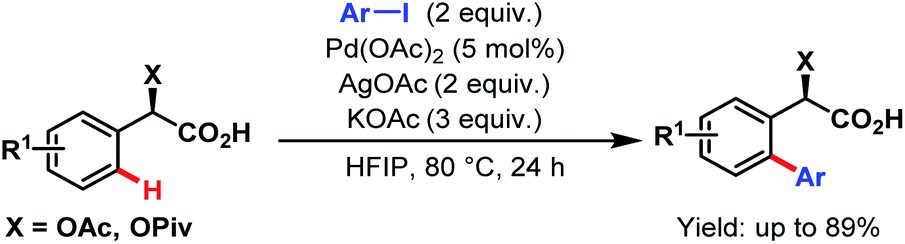 | ||
| Scheme 31 Pd-catalyzed ortho-C(sp2)–H arylation of mandelic acid and α-phenylglycine (Yu et al., 2015).83 | ||
3.2.1.1.2. ortho-C(sp2)–H alkylation. In 2007, the Yu group first reported the methylation of simple benzoic acids using methyl boronic acids as coupling partners to synthesize o-toluic acids in moderate to good yields (Scheme 32).58
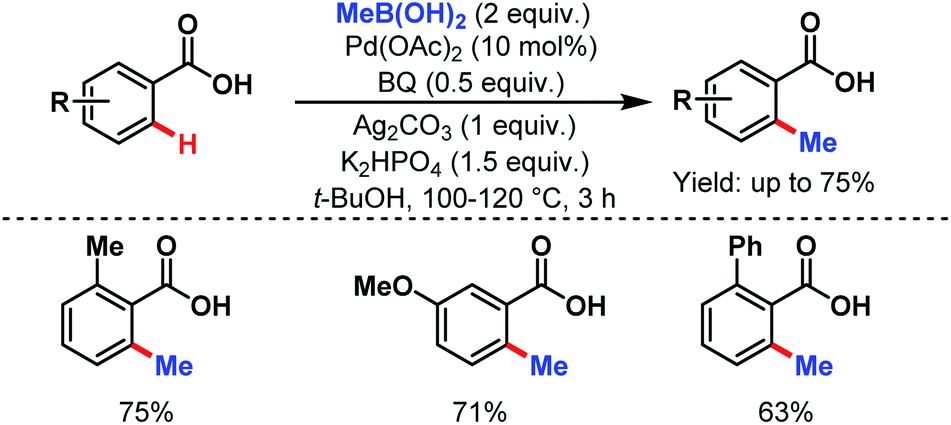 | ||
| Scheme 32 Pd-catalyzed ortho-C(sp2)–H methylation of benzoic acids (Yu et al., 2007).58 | ||
After that, in 2009 they developed another alkylation method using alkyl halides as coupling partners under silver-free conditions providing a new route to benzolactones (Scheme 33).84
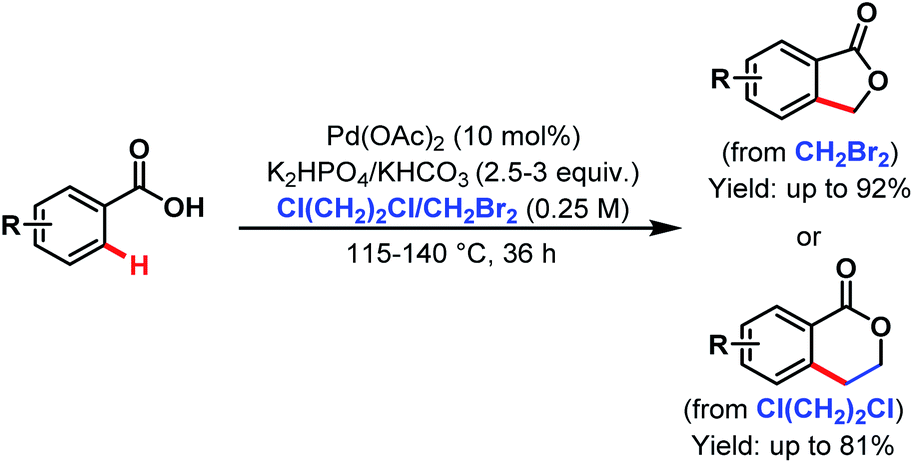 | ||
| Scheme 33 Pd-catalyzed ortho-C(sp2)–H alkylation of benzoic acids with alkyl halides (Yu et al., 2009).84 | ||
A few years later, they tried to disclose another ortho-alkylation method of aryl carboxylic acids using alkyl trifluoroborates as coupling partners and a monoprotected amino acid (L17) as a ligand which played a crucial role in C–H bond cleavage. They found that both electron-deficient and electron-rich substrates were compatible under the reaction conditions (Scheme 34).85
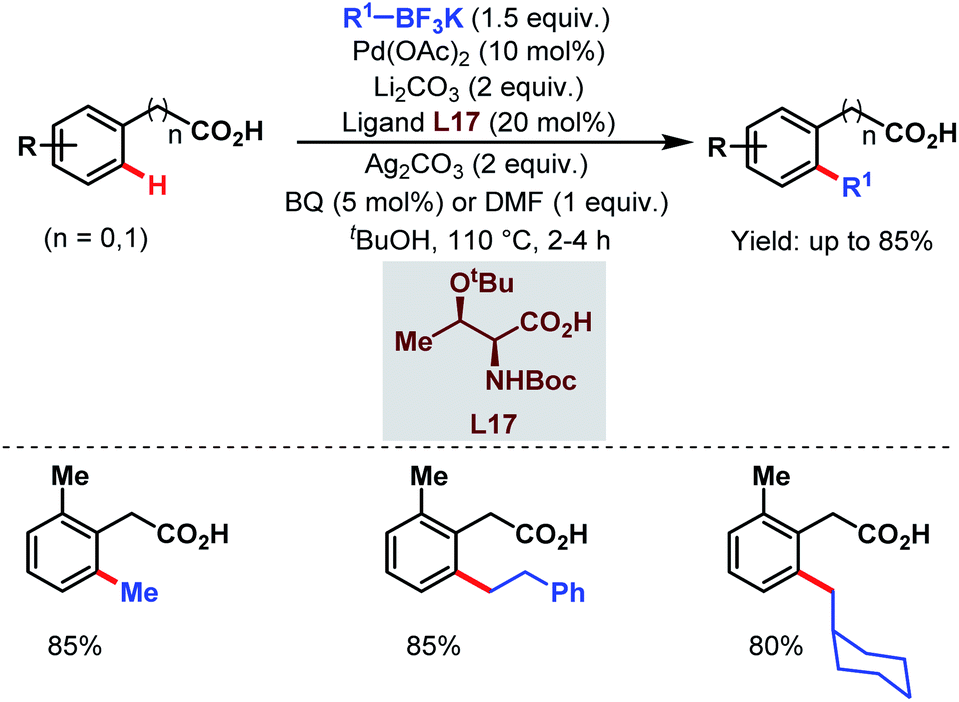 | ||
| Scheme 34 Ligand-accelerated Pd-catalyzed ortho-C(sp2)–H alkylation of aryl carboxylic acids using alkyl boron reagents (Yu et al., 2013).85 | ||
Despite the extensive applicability of such high yielding protocols in academia, Pd-catalyzed C–H activation reactions are often associated with high catalyst loading, harsh reaction conditions and the use of Ag salts as oxidants which result in some practical problems for industrial applications. To address such hurdles, in 2015 the Yu group came up with a relatively benign method using a new alkylating agent. An epoxide (in which the carbon atoms are in a higher oxidation state than in an alkene) was employed to synthesize 3,4-dihydroisocoumarin derivatives in one step. The reaction underwent ortho-C(sp2)–H alkylation followed by C–O cyclization. Both terminal and internal epoxides performed well (Scheme 35).86
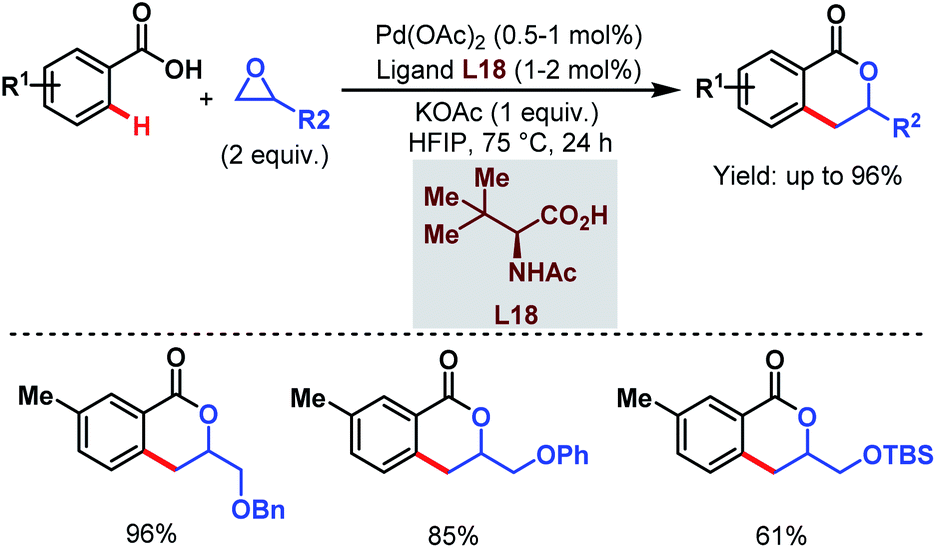 | ||
| Scheme 35 Synthesis of 3,4-dihydroisocoumarins via Pd-catalyzed ortho-C(sp2)–H alkylation using epoxides (Yu et al., 2015).86 | ||
3.2.1.1.3. ortho-C(sp2)–H olefination. Carboxylate directed C(sp2)–H olefination was first explored by the Larock group in 1984. The reaction proceeded via an electrophilic thallation, followed by a Pd-catalyzed olefination of the arylthallium intermediate with simple alkenes and subsequent cyclization to yield isocoumarin derivatives (Scheme 36).87
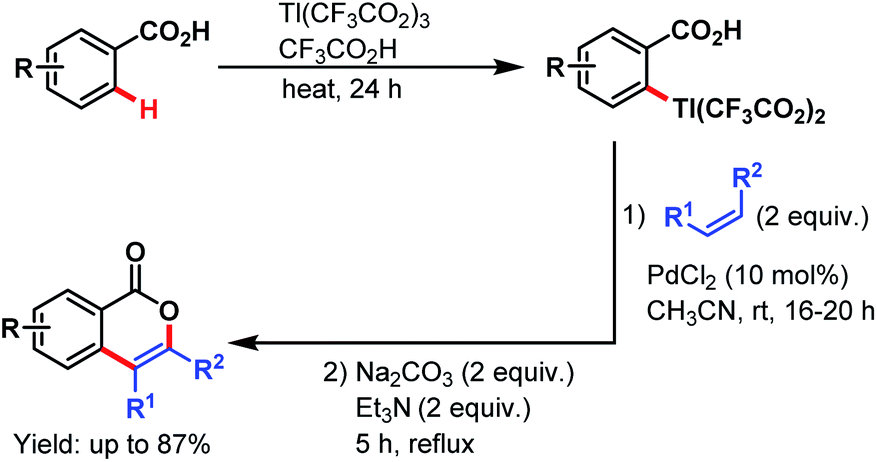 | ||
| Scheme 36 Synthesis of isocoumarins via Pd-catalyzed thallation–olefination of benzoic acids (Larock et al., 1984).87 | ||
In 1998, the Miura group came up with a Pd/Cu-co-catalyzed olefination strategy of several substituted benzoic acids with butyl acrylate and styrene to synthesize benzolactones under thallium-free conditions.88
After that, in 2010, Yu and coworkers designed a ligand-dependent catalytic system to perform C(sp2)–H olefination of a wide range of synthetically useful phenylacetic acids using molecular oxygen as the terminal oxidant (Schemes 37 (ref. 89) and 38 (ref. 90)). They clearly mentioned the role of a monoprotected amino acid as a ligand which was responsible for changing the mechanism of C–H bond cleavage from an electrophilic palladation to a concerted metalation–deprotonation (CMD) (Scheme 39).90 Using a similar concept, they further reported sequential ortho-C(sp2)–H olefination of phenylacetic acid derivatives (Scheme 40).91
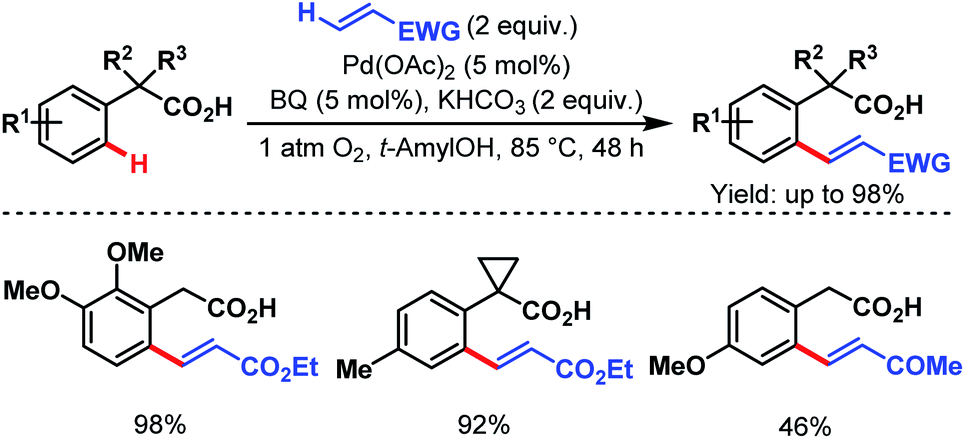 | ||
| Scheme 37 Pd-catalyzed ortho-C(sp2)–H olefination of arene carboxylic acids (Yu et al., 2010).89 | ||
 | ||
| Scheme 38 Ligand-accelerated Pd-catalyzed ortho-C(sp2)–H olefination of arene carboxylic acids (Yu et al., 2010).90 | ||
 | ||
| Scheme 39 Proposed catalytic cycle for ligand-accelerated Pd-catalyzed ortho-C(sp2)–H olefination of arene carboxylic acids (Yu et al., 2010).90 | ||
 | ||
| Scheme 40 Sequential Pd-catalyzed ortho-C(sp2)–H olefination of arene carboxylic acids (Yu et al., 2010).91 | ||
After that, in 2013, the Lee group developed a straightforward protocol for the synthesis of isocoumarins and 3-benzylidenephthalides via Pd-catalyzed C–H olefination followed by oxidative coupling of benzoic acids and vinyl arenes. The directing ability of various substituents helps in the formation of both lactones in pure form (Scheme 41).92
 | ||
| Scheme 41 Synthesis of isocoumarins and 3-benzylidenephthalides via ligand-free Pd-catalyzed oxidative coupling of benzoic acids and vinyl arenes (Lee et al., 2013).92 | ||
In 2019, the Zeng group designed a carboxylate-directed domino C(sp2)–H activation strategy to synthesize pyrano[4,3-b]indole-1[5H]-one motifs from readily available indole-3-carboxylic acids and olefins via carboxylate-directed sequential ortho-C(sp2)–H alkenylation and remote olefinic C(sp2)–H activation (or lactonization). This strategy proved to be superior with respect to the selectivity being obtained in the reaction of the carboxylic acids with acrylates through a conventional Michael addition (Scheme 42).93
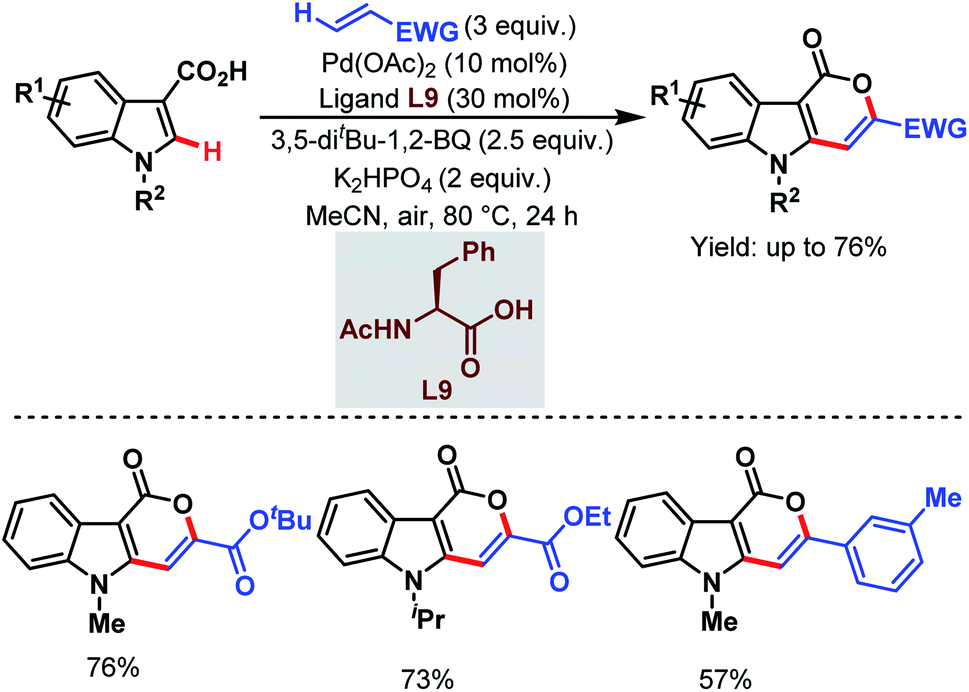 | ||
| Scheme 42 Pd-catalyzed carboxylate-directed domino C(sp2)–H activation strategy (Zeng et al., 2019).93 | ||
3.2.1.1.4. ortho- and other proximal C(sp2)–H carboxylations. In 2008, Yu and coworkers designed a simple protocol to synthesize dicarboxylic acids, which was the first Pd-catalyzed reaction for direct carboxylation of benzoic acids and phenylacetic acid derivatives. The first palladacycle generated from a carboxylic acid was characterized by X-ray crystallography. They further extended this protocol for carboxylation of vinylic C–H bonds (Scheme 43).45
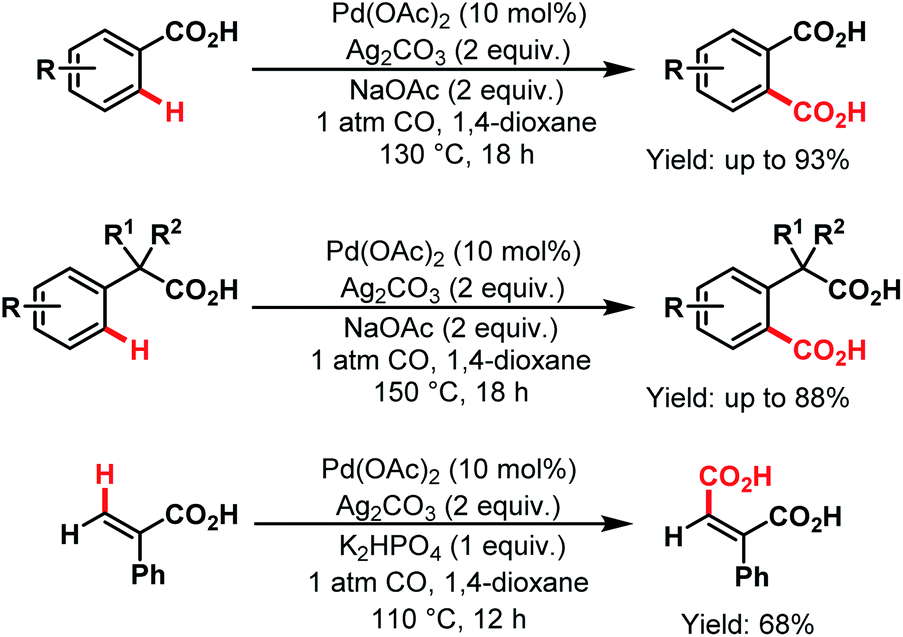 | ||
| Scheme 43 Pd-catalyzed carboxylation of aryl and vinyl C(sp2)–H bonds (Yu et al., 2008).45 | ||
3.2.1.1.5. ortho-C(sp2)–H halogenation. In 2008, the Yu group first reported ortho-C(sp2)–H halogenations of a wide range of arene carboxylic acids using iodoacetic acid (IOAc) and N-bromosuccinimide (NBS) as the halogenating source (Scheme 44).94
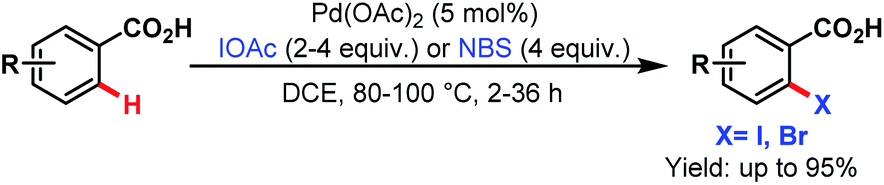 | ||
| Scheme 44 Pd-catalyzed monoselective ortho-halogenation of C(sp2)–H bonds of arene carboxylic acids (Yu et al., 2008).94 | ||
Two years later, they devised another novel strategy for monoselective ortho-iodination of phenylacetic acids using Pd(OAc)2 as a catalyst. Late-stage functionalisation and concise synthesis of drug molecules make this protocol versatile in the context of synthetic utility (Scheme 45).95
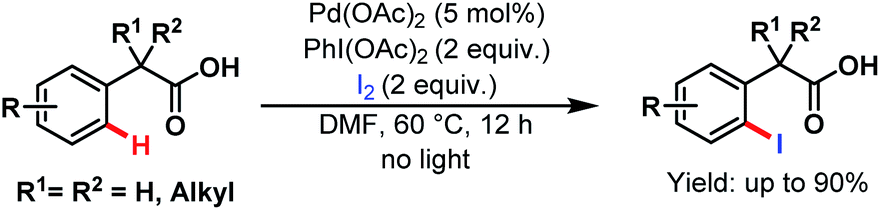 | ||
| Scheme 45 Pd-catalyzed monoselective ortho-iodination of phenylacetic acids (Yu et al., 2010).95 | ||
Very recently, the Li group reported ortho-selective iodination of hydrocinnamic acids utilizing an aryl iodide as the unique source of iodine employing the very carboxylate based directing group (Scheme 46).96
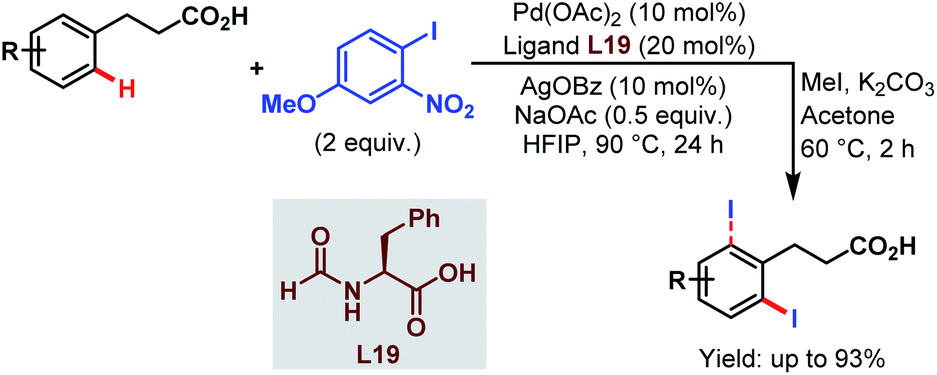 | ||
| Scheme 46 Pd-catalyzed carboxylate-assisted ortho-C(sp2)–H iodination of arenes (Li et al., 2021).96 | ||
3.2.1.1.6. ortho-C(sp2)–H amidation. Pd-catalyzed C(sp2)–H amidation was first reported by the Yu group in 2012. They synthesized anthranilic acids from lithium carboxylates with high regioselectivity using ethyl mesitylsulfonyloxycarbamates as amidating reagents (Scheme 47).97
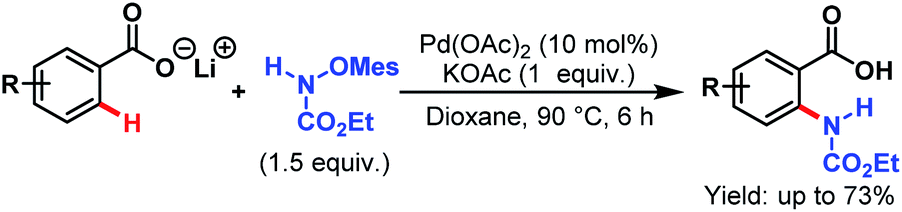 | ||
| Scheme 47 Pd-catalyzed ortho-amidation of arene carboxylic acids (Yu et al., 2012).97 | ||
3.2.1.1.7. ortho-C(sp2)–H acylation. Pd-catalyzed ortho-acylation of benzoic acids was first explored by Ge and coworkers by employing α-oxocarboxylic acids as acylating reagents. An array of benzoic acids and α-oxocarboxylic acids performed well under such reaction conditions and gave the desired acylated products in good to excellent yields (Scheme 48).98
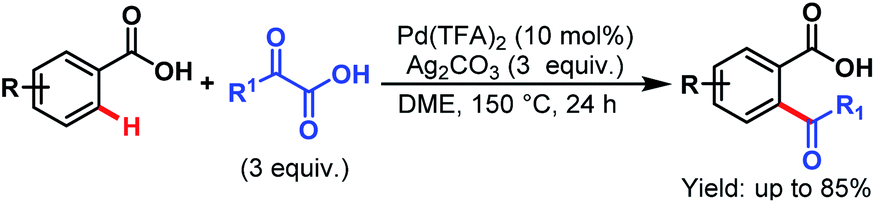 | ||
| Scheme 48 Pd-catalyzed ortho-acylation using α-oxocarboxylic acids (Ge et al., 2013).98 | ||
3.2.1.1.8. ortho-C(sp2)–H deuteration. In 2014, the Yu group reported a Pd-catalyzed ortho-deuteration of several arene carboxylic acids using [D4]acetic acid as a deuterium source. A number of substituted phenylacetic acids and benzoic acids were deuterated at ortho-positions in good to excellent yields (Scheme 49).99
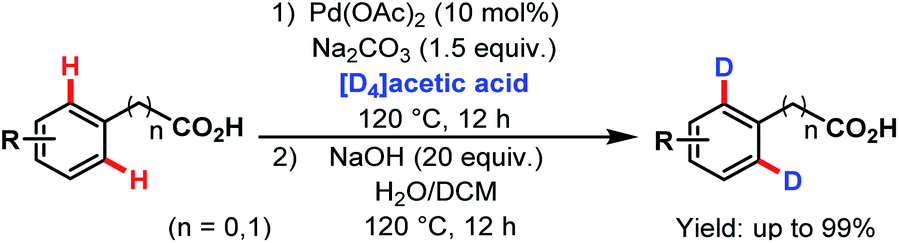 | ||
| Scheme 49 Pd-catalyzed ortho-deuteration of arene carboxylic acids (Yu et al., 2014).99 | ||
3.2.1.1.9. ortho-C(sp2)–H hydroxylation. ortho-C(sp2)–H hydroxylation was first explored by the group of Yu in 2009. An 18O labeling study supported the role of oxygen (O2) as the hydroxylation source (Scheme 50).100
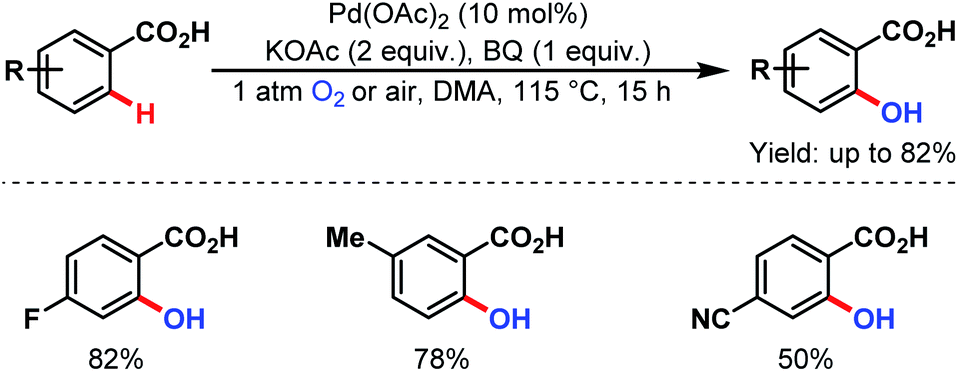 | ||
| Scheme 50 Pd-catalyzed ortho-hydroxylation of C(sp2)–H bonds of arene carboxylic acids (Yu et al., 2009).100 | ||
Very recently, they designed a bifunctional tautomeric ligand (L20) containing pyridone and pyridine moieties to hydroxylate the ortho-C(sp2)–H bonds of a wide array of arene carboxylic acids including the heterocyclic compounds which were previously inaccessible. O2 was used as a hydroxylating source and Pd(OAc)2 as a catalyst. The pyridone motif of the ligand accelerates the C–H bond cleavage step and the pyridine motif helps in O2 activation (Scheme 51).101
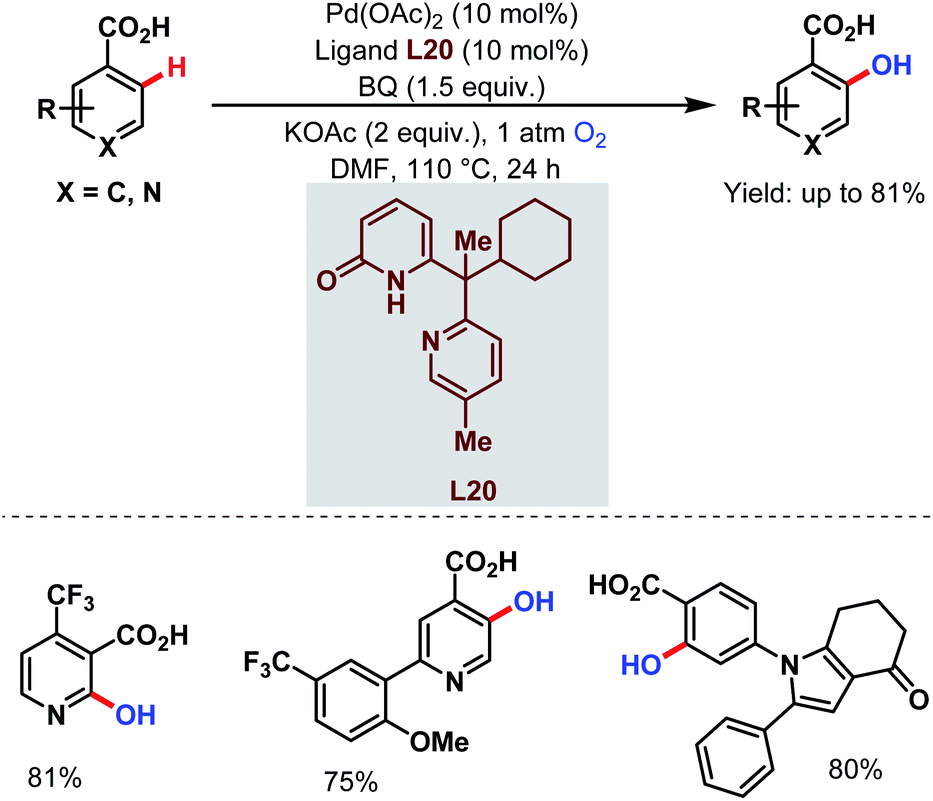 | ||
| Scheme 51 Tautomeric ligand-enabled Pd-catalyzed ortho-C(sp2)–H hydroxylation of arene carboxylic acids (Yu et al., 2021).101 | ||
3.2.1.2. Distal C(sp2)–H activation.
3.2.1.2.1. meta-C(sp2)–H arylation. Distal C(sp2)–H activation is mainly dependent on the template (or directing group) based strategy which proceeds via a high energy demanding macrocyclic transition state to reach a particular distal C(sp2)–H bond. The main drawback of this strategy is that installation and removal of the template (or directing group) require extra steps. In this context, the Catellani reaction offers a good alternative to this previously well-explored strategy.102 It was introduced by Prof. Marta Catellani103 and later brilliantly applied in meta-C(sp2)–H alkylation by the Yu group104 employing norbornene as a transient mediator. This strategy involves a Pd-relay chemistry which helps in switching the ortho-selectivity to meta-selectivity in C(sp2)–H activation of the same substrate bearing a simple ortho-directing group by catalyst control.
In 2017, the Yu group applied this Pd-relay chemistry in ligand-controlled auxiliary-free meta-C(sp2)–H arylation of phenylacetic acids using various aryl iodides as coupling partners, 2-carbomethoxynorbornene (NBE-CO2Me) as a transient mediator, and monoprotected 3-amino-2-hydroxypyridine (L21) as a ligand. They further extended this strategy for the free meta-arylation of mandelic acid and phenylglycine (Scheme 52).105
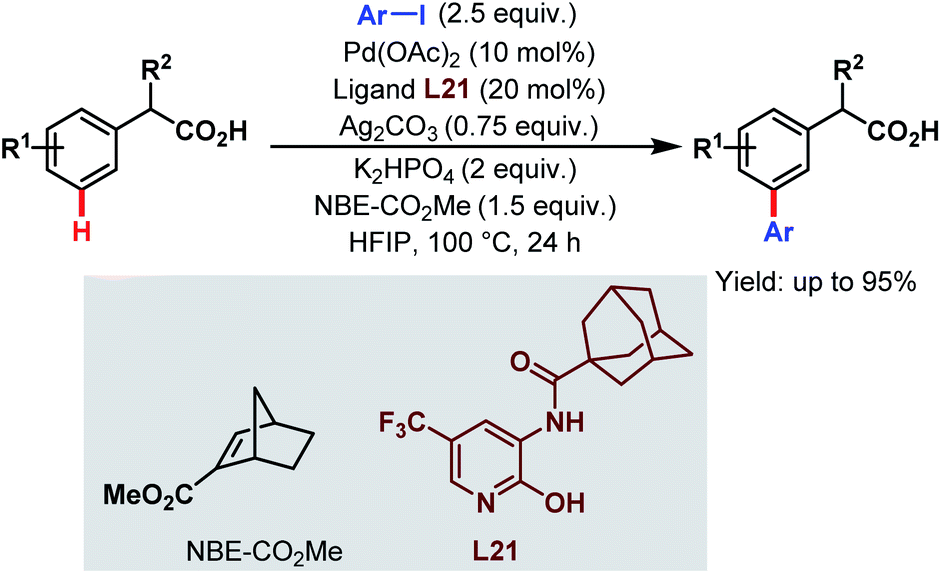 | ||
| Scheme 52 Pd-catalyzed auxiliary-free meta-C(sp2)–H arylation of phenylacetic acids (Yu et al., 2017).105 | ||
While previously the κ1 coordination mode of carboxylates was utilized extensively for ortho-functionalisations of benzoic acids, in 2019, Li and co-workers reported for the first time a remote meta-selective directing group exploiting the κ2 coordination of the carboxylates. It was hypothesized that the absence of metal counterions made the PdII carboxylate complex adopt κ2 coordination over κ1. This strategy was found to be well compatible with both Pd/Pd0 and PdII/PdIV (or PdII/PdIII) catalytic cycles featuring meta-arylation of hydrocinnamic acid derivatives (Schemes 53).106
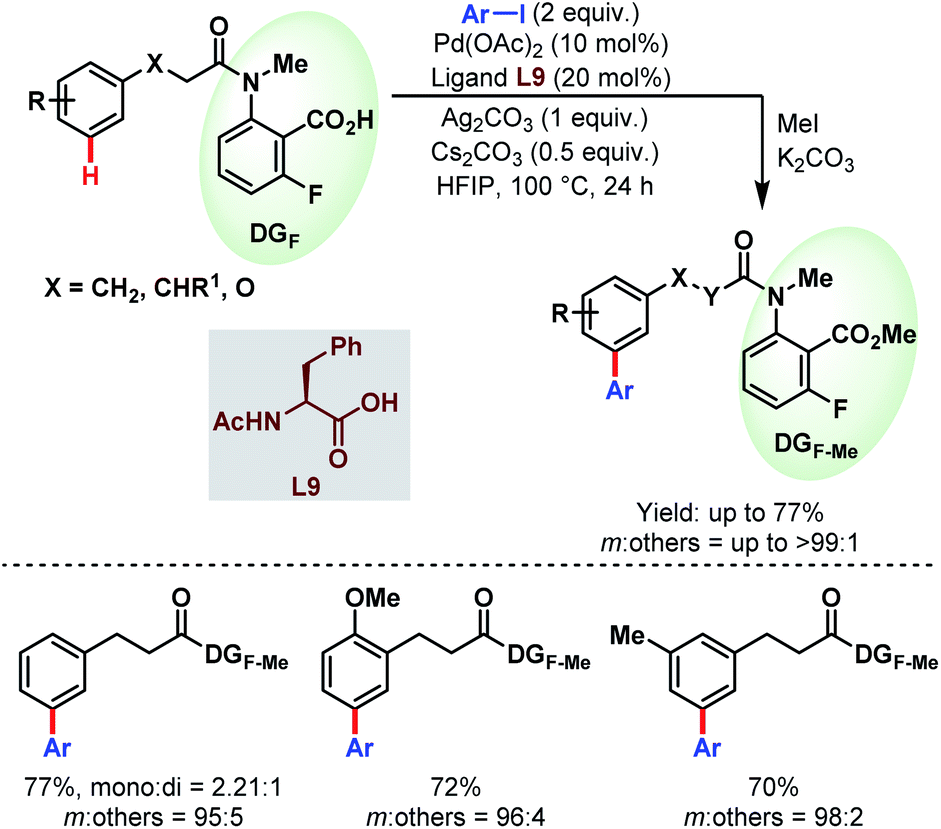 | ||
| Scheme 53 Pd-catalyzed carboxylate-assisted meta-C(sp2)–H arylation of hydrocinnamic acid derivatives (Li et al., 2019).106 | ||
This work was further extended to a series of benzylsulfonamide derivatives by the same group in 2020 (Schemes 54).53
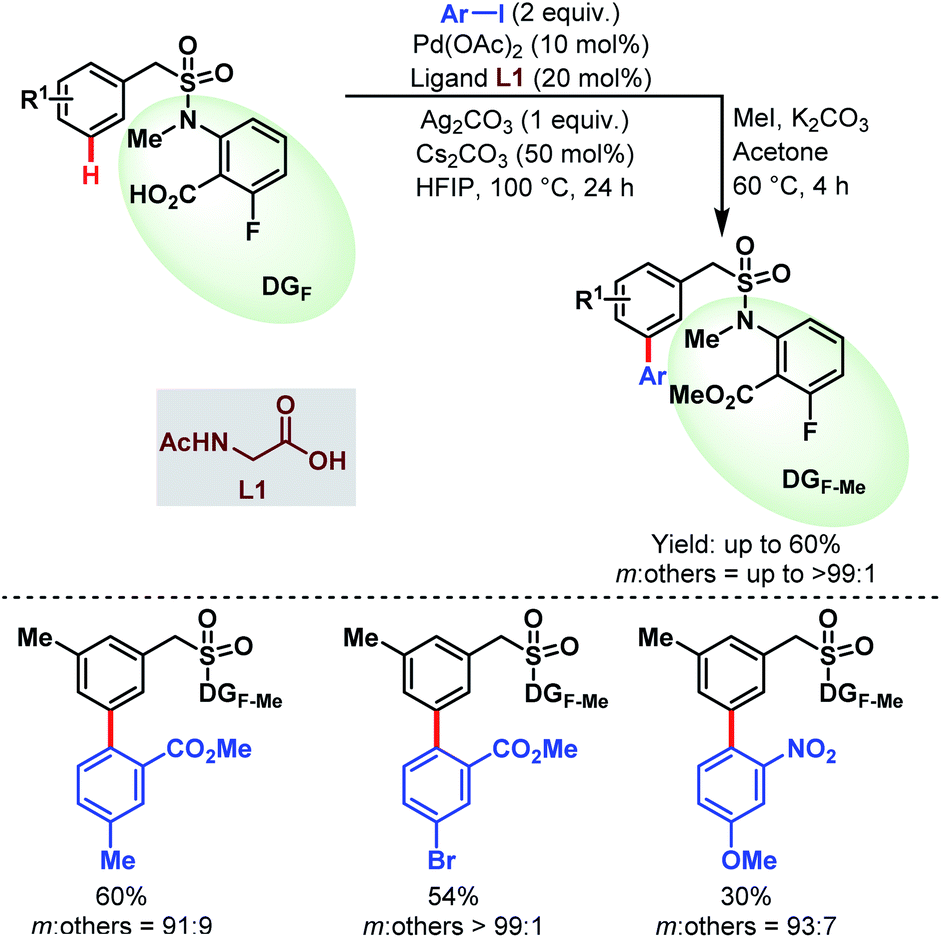 | ||
| Scheme 54 Pd-catalyzed carboxylate-assisted meta-C(sp2)–H arylation of benzylsulfonamides (Li et al., 2020).53 | ||
3.2.1.2.2. meta-C(sp2)–H olefination. In 2019, the Li group reported the first carboxylate-assisted remote meta-selective olefination of hydrocinnamic acid derivatives. Here, in the absence of metal counterions κ2 coordination of the carboxylates became more facile than κ1 coordination and the protocol was compatible with both Pd/Pd0 and PdII/PdIV (or PdII/PdIII) catalytic cycles (Schemes 55).106 The following year, they applied the same strategy to a series of benzylsulfonamide derivatives (Schemes 56).53
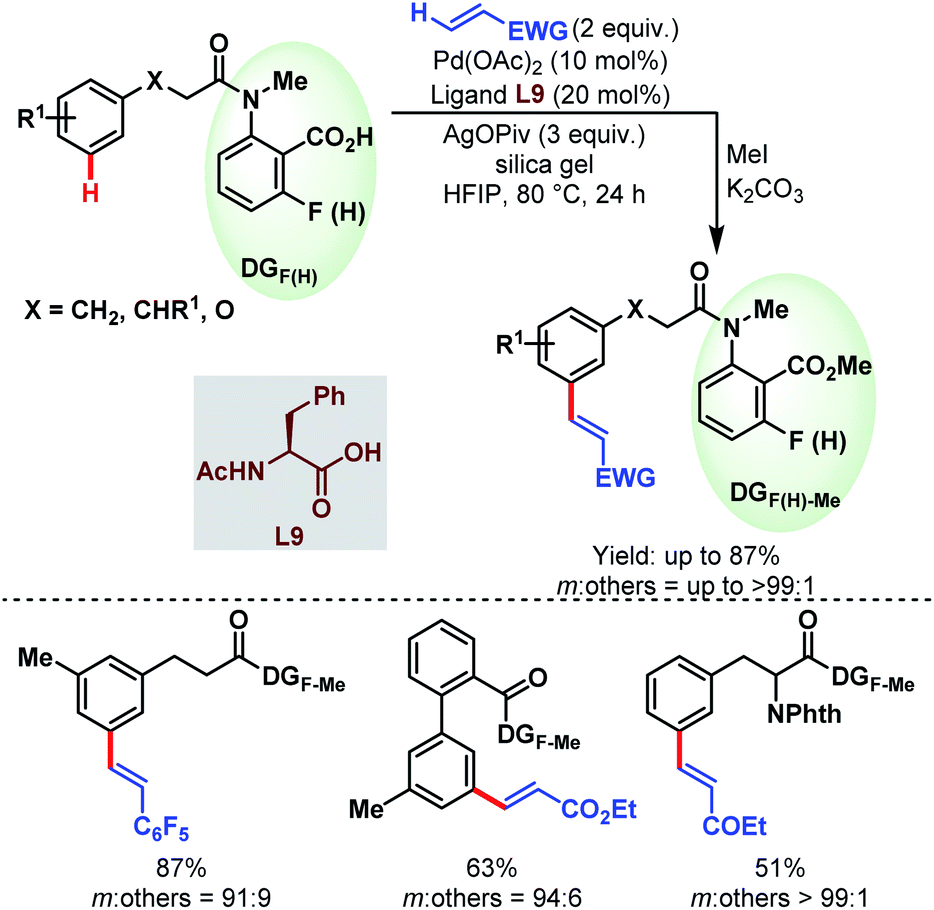 | ||
| Scheme 55 Pd-catalyzed carboxylate-assisted meta-C(sp2)–H olefination of hydrocinnamic acid derivatives (Li et al., 2019).106 | ||
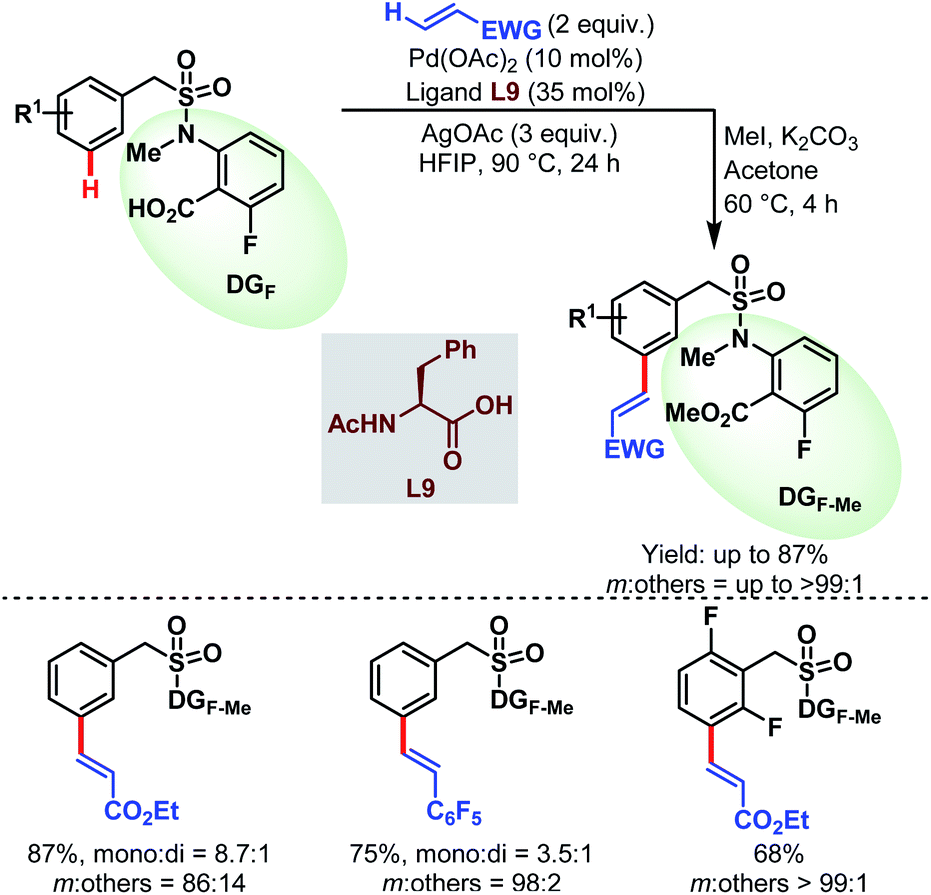 | ||
| Scheme 56 Pd-catalyzed carboxylate-assisted meta-C(sp2)–H olefination of benzylsulfonamides (Li et al., 2020).53 | ||
3.2.1.2.3. meta-C(sp2)–H iodination. Very recently, the Li group devised a meta-selective iodination strategy of hydrocinnamic amides utilizing an aryl iodide as the iodinating source employing the very carboxylate based directing group (Scheme 57).96
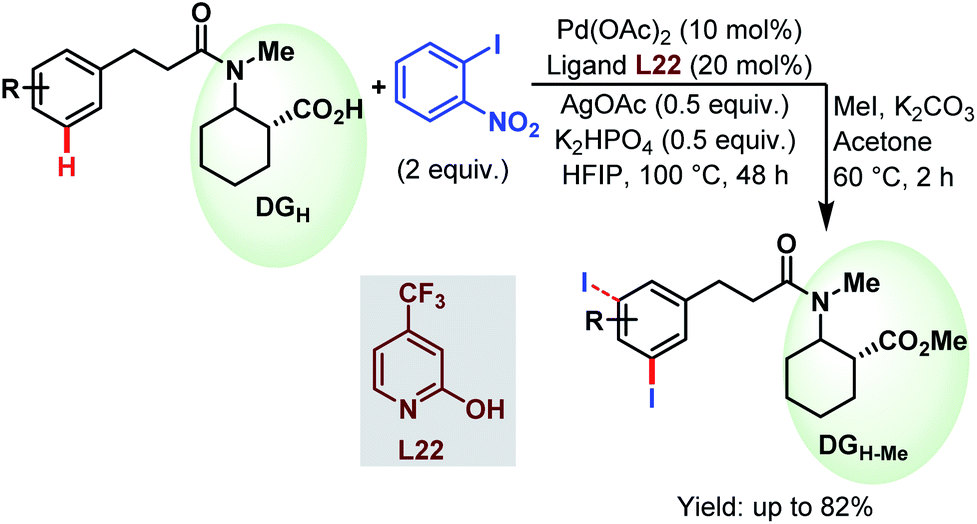 | ||
| Scheme 57 Pd-catalyzed carboxylate-assisted meta-C(sp2)–H iodination of arenes (Li et al., 2021).96 | ||
In 2010, the Yu group demonstrated the Pd-catalyzed enantioselective C(sp2)–H olefination of diphenylacetic acids using a monoprotected α-amino acid (L23) as a chiral ligand (Scheme 58).107
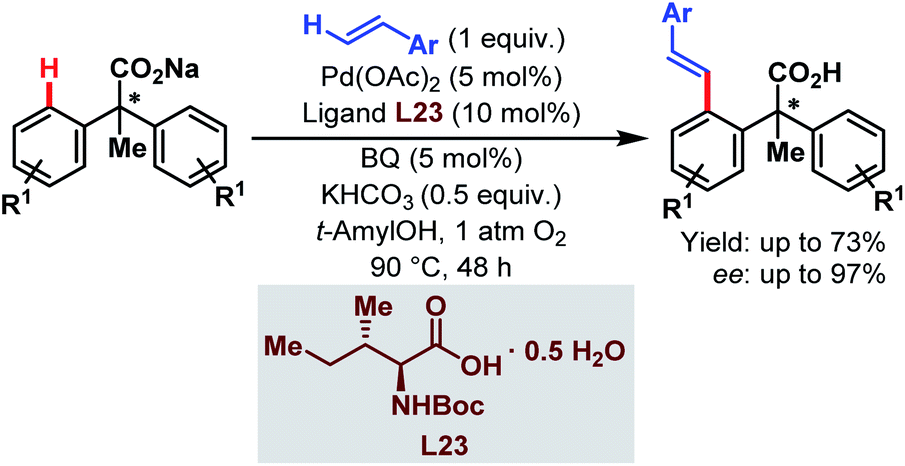 | ||
| Scheme 58 Pd-catalyzed enantioselective C(sp2)–H olefination of diphenylacetic acids (Yu et al., 2010).107 | ||
Three years later, they reported the synthesis of chiral benzofuranone motifs via Pd-catalyzed C(sp2)–H activation followed by intramolecular C–O bond formation. This was the first example of a Pd-catalyzed enantioselective C(sp2)–H activation through a rare Pd(II)/Pd(IV) catalytic cycle (Scheme 59).108
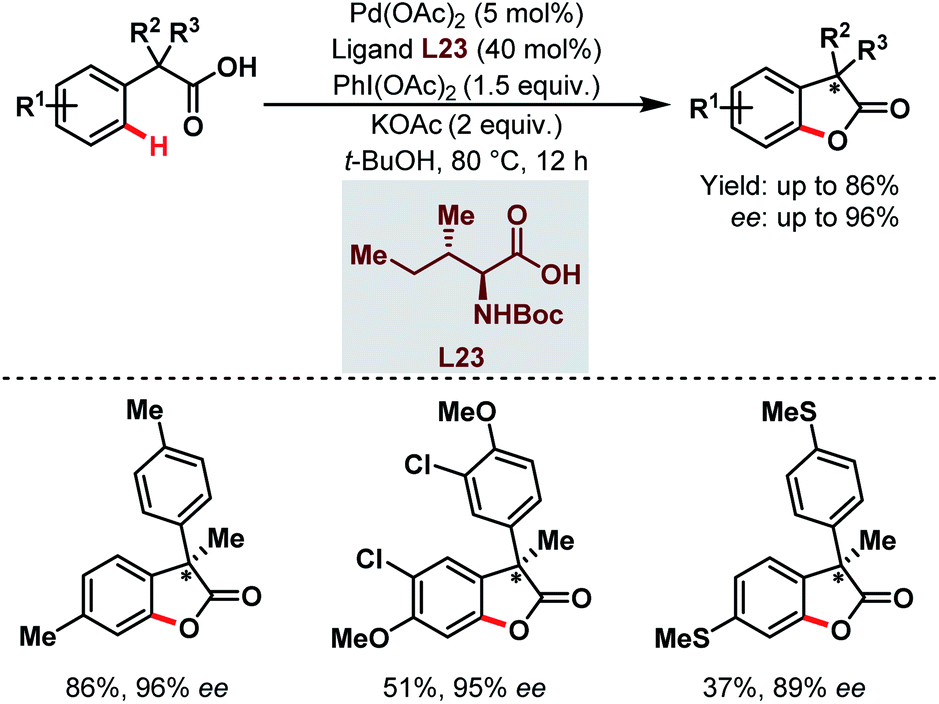 | ||
| Scheme 59 Pd-catalyzed enantioselective C(sp2)–H activation/C–O bond formation (Yu et al., 2013).108 | ||
4. Carboxylic acids as traceless directing groups
The utilization of carboxylic acids is not limited to native directing groups; they can also be used as removable directing groups for C–H activation of arenes. In such a case, they at first direct to their proximal C–H bond for the metalation and eventually dissociate via a decarboxylation process upon completion of the directed C–H functionalisation. Several reports109–115 are already available in the literature. All of them have been systematically compiled by Larrosa and coworkers in a review,15 and some of the examples are discussed here.Kozlowski and coworkers first developed Pd-catalyzed decarboxylation of aromatic carboxylic acids in 2007. They showed that electron-rich bis-ortho-substituted aromatic acids underwent decarboxylation under mild conditions. This reaction worked especially well for sterically hindered carboxylic acids (Scheme 60).109
 | ||
| Scheme 60 Pd-catalyzed decarboxylation of aromatic carboxylic acids (Kozlowski et al., 2007).109 | ||
Later, in 2011, the Larrosa group came up with a new protocol of formal meta-selective direct C–H arylation using iodoarenes as coupling partners and carboxylic acids as traceless directing groups (Scheme 61).105 This method was compatible with a wide variety of electron-donating and electron-withdrawing groups at the meta-position through bypassing the electronic preferences of substituents in arene rings. This strategy provides an efficient alternative to expensive Suzuki couplings to access meta-substituted biaryl compounds.
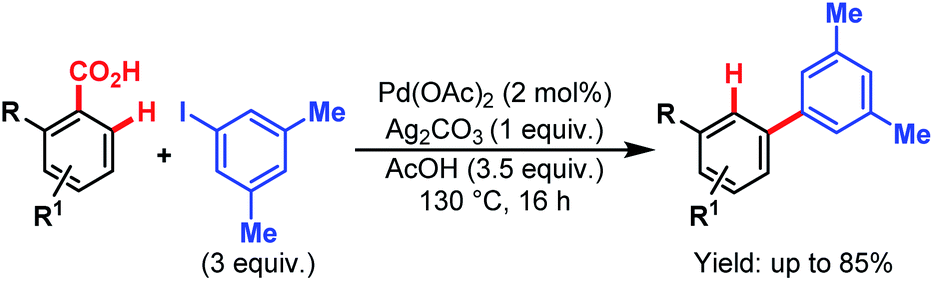 | ||
| Scheme 61 Pd-catalyzed meta-selective direct C–H arylation (Larrosa et al., 2011).110 | ||
Then, in 2015, they utilized a similar concept to investigate the Pd-catalyzed tandem arylation/protodecarboxylation of readily available salicylic acids (Scheme 62).106 Here, [1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene](3-chloropyridyl)palladium(II) dichloride (PEPPSI™-IPr) was found to be the best catalyst, leading to a wide array of meta-arylated phenols with good regioselectivity. These meta-arylated phenols can be easily transformed into a variety of meta-substituted biaryls to highlight the versatility of this approach.
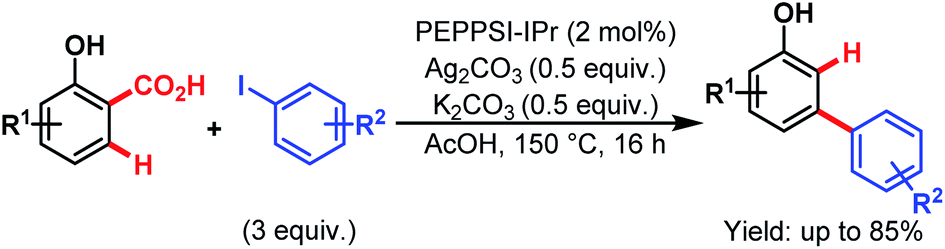 | ||
| Scheme 62 Pd-catalyzed C–H arylation/decarboxylation of salicylic acids (Larrosa et al., 2015).111 | ||
The following year, Hong and coworkers devised a Pd-catalyzed cascade reaction via a traceless directing group approach starting from di(hetero)aryl acetic acids and acrylates (Scheme 63).112
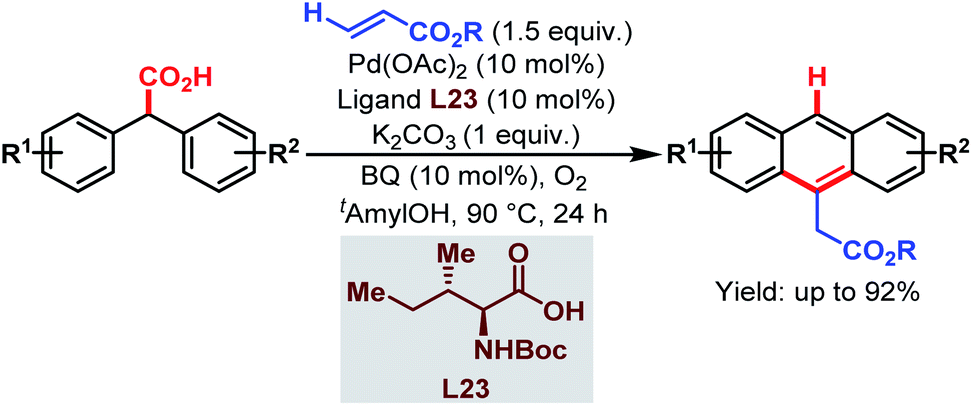 | ||
| Scheme 63 Pd-catalyzed cascade synthesis of acenes (Hong et al., 2016).112 | ||
The overall strategy involved carboxyl-directed C–H alkenylation/carboxyl-directed secondary C–H activation and rollover/intramolecular C–C bond formation/decarboxylative aromatization as key steps. This method was compatible with a wide range of substituents, leading to several synthetically useful anthracene derivatives. It also provides a convenient route to access various useful structural motifs, polyacenes, heteroacenes, etc. The mechanism proposed by the author is shown in Scheme 64.
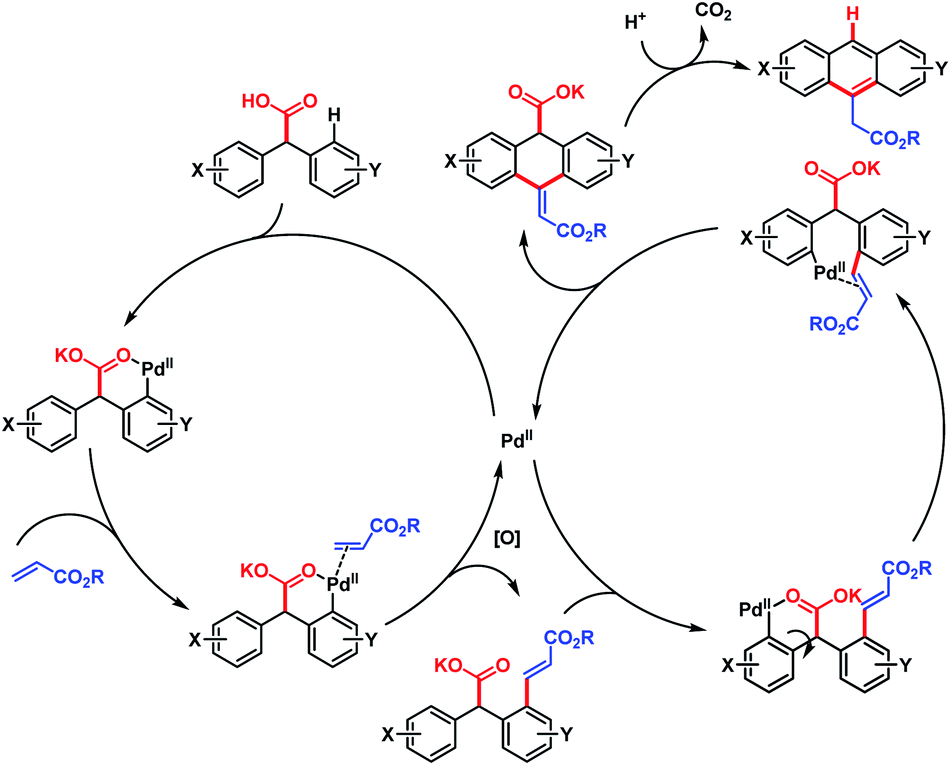 | ||
| Scheme 64 Proposed mechanism for Pd-catalyzed cascade synthesis of acenes (Hong et al., 2016).112 | ||
5. Synthetic applications
As shown in the previous chapters, the Pd-catalyzed carboxylate-assisted C–H activation has been established as a powerful tool in synthetic chemistry. Along with the development of the methodologies, the range of synthetic applications was also investigated because carboxylic acids not only enable easier access to valuable products via synthetic precursors or key intermediates but are also important functionalities in numerous drug molecules, agrochemicals and natural products.The synthesis of 2-tetralone and naphthoic acid natural product cores, and the diversification of commercial drug scaffolds were demonstrated by the Yu group in 2009 via an aryl C–H olefination approach (Scheme 65).89 Through this, a simple one-step elaboration of nonsteroidal anti-inflammatory drugs (NSAIDs, e.g. ibuprofen and naproxen) was shown. Furthermore, 2,3-dimethoxyphenylacetic acid was used as a simple feedstock to obtain an olefinated intermediate (Scheme 65, third row) that subsequently gave, in a four-step straightforward sequence, a 2-tetralone derivative. Additionally, structural components of the natural products of neocarzinostatin and kedarcidin were obtained with the aid of either protecting group- or ligand-controlled position selectivity. In the first case, a triisopropylsilyl group affords the selective synthesis of the olefin which is then further transformed by procedures reported in the literature (Scheme 65, PG-controlled olefination). Notably, a regioselective olefination can be achieved by the introduction of amino acid derivatives as ligands. An interesting example is given by the synthesis of the naphthoic acid core of kedarcidin via an olefinated intermediate (Scheme 65, ligand-controlled olefination).
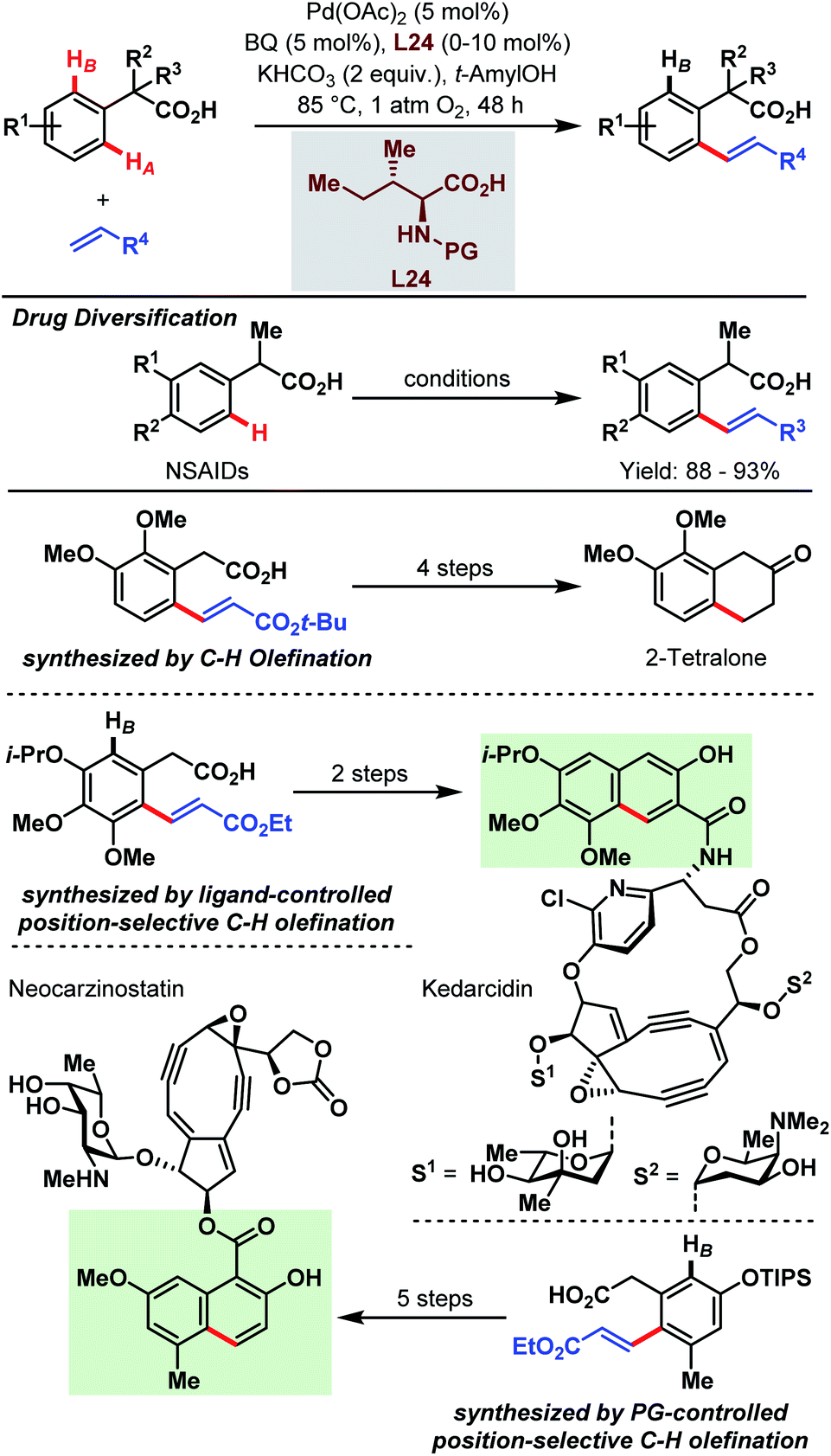 | ||
| Scheme 65 Site-selective Pd-catalyzed aryl C–H olefination (Yu et al., 2009).89 | ||
In 2010, Yu and co-workers again amended the synthetic toolbox of carboxylate-assisted C–H activation by an ortho-iodination protocol (Scheme 66).95 Besides the general utility of this methodology, they present another possibility for an expedient alteration of several nonsteroidal anti-inflammatory drugs (NSAIDs). Furthermore, a short two-step procedure for both lumiracoxib and diclofenac was developed.
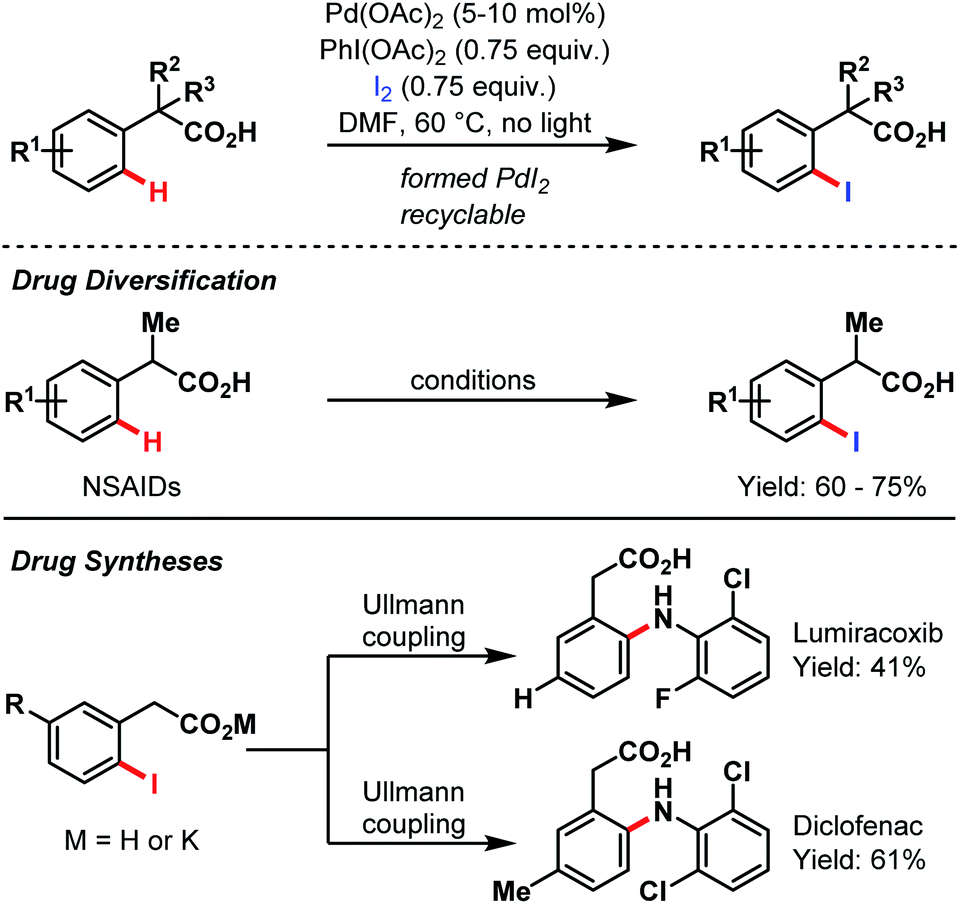 | ||
| Scheme 66 Pd-catalyzed drug synthesis and diversification (Yu et al., 2010).95 | ||
As mentioned at the beginning of this chapter, carboxylic acids are ubiquitous motifs in nature. An example is (+)-lithospermic acid that bears even two –COOH groups. Concerning the C–H olefination approach mentioned above, this has to be considered as the key step in the total synthesis of natural products. Indeed, in 2011, a highly convergent and simplified route was published to obtain an antiviral agent by a late-stage coupling of the olefin unit and the dihydrobenzofuran core (Scheme 67).116 Despite uncertainties like side reactions on more electron-rich arene moieties, and the possibility of a favorable or unfavorable dihedral angle, the anticipated transformation succeeded with a yield of 93%. This result emphasizes not only the directing power of the carboxylate but also the importance of the amino acid ligand (cf. table in Scheme 67).
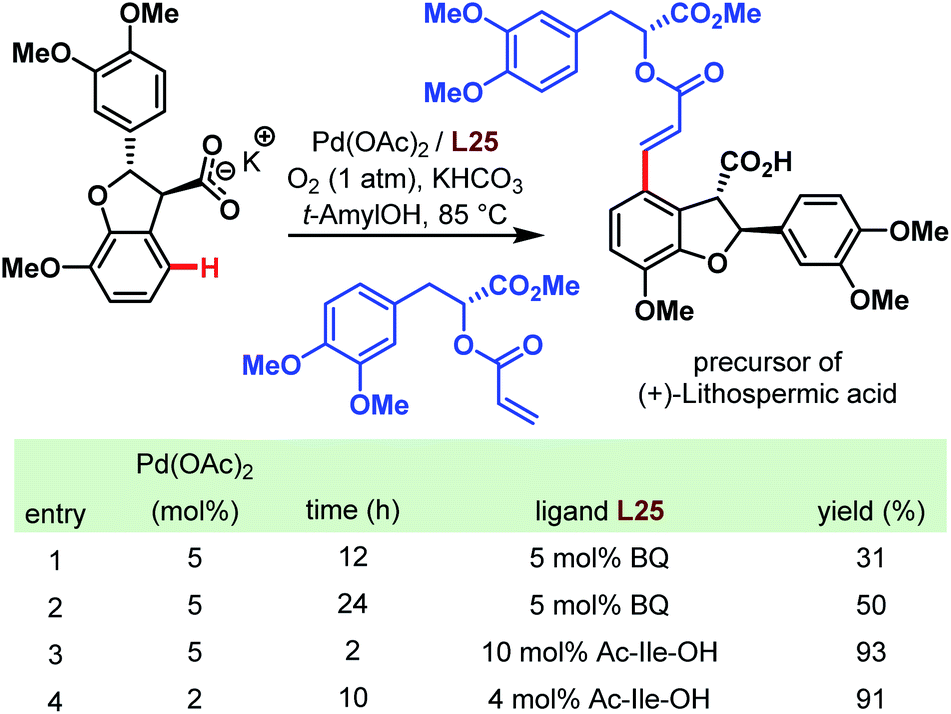 | ||
| Scheme 67 Total synthesis of (+)-lithospermic acid via late-stage Pd-catalyzed C–H olefination (Yu et al., 2011).116 | ||
Two years later, the groups of Baran and Yu collaborated for a ligand-accelerated C–H alkylation. The site-selective reaction was integrated into the synthesis of the natural product (+)-hongoquercin and thereby served as another example for the feasibility of the Pd-catalyzed C–H bond activation directed by a carboxylic acid (Scheme 68).117 After optimization of the reaction using m-toluic acid as a model substrate, potassium methyltrifluoroborate (MeBF3K) emerged as the best alkyl source and Boc-protected phenylalanine as a ligand of choice. The site-selective methylation afforded the (+)-hongoquercin A precursor in a reasonable yield of 45% along with 15% of doubly methylated side product and 32% of recovered starting material. However, the final and subsequent Pd-catalyzed C–H oxidation step resulted in a diminished yield of only 15%.
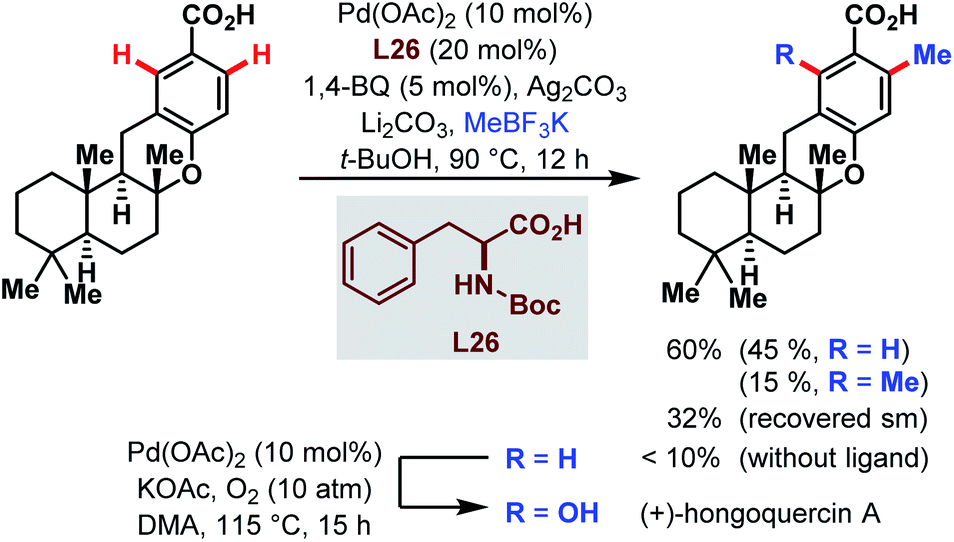 | ||
| Scheme 68 Pd-catalyzed C–H alkylation in the synthesis of (+)-hongoquercin A (Yu and Baran et al., 2013).117 | ||
Later, in 2015, the Lei group used the power of the Pd-catalyzed C–H activation in a challenging total synthesis of the monoamine oxidase inhibitor (−)-incarviatone A (Scheme 69).118 Starting with inexpensive phenylacetic acid they performed a C–H alkylation as an initial step for the introduction of a propyl substituent on a gram scale (A). After further steps that built up the indane backbone, the benzylic carboxylic acid paved the way for iodination in the distal position (B).
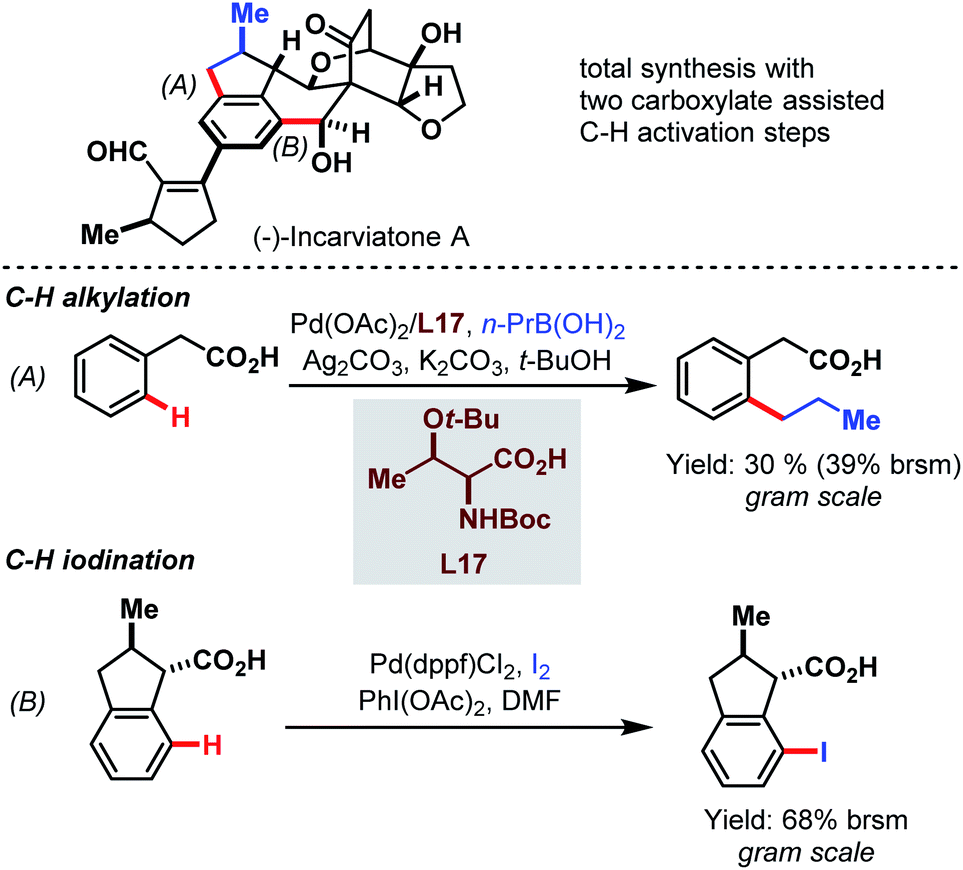 | ||
| Scheme 69 Pd-catalyzed gram-scale procedures for the total synthesis of (−)-incarviatone A (Lei et al., 2015).118 | ||
The van Gemmeren lab provided a method for the β-C(sp3)–H activation of propionic acid and other aliphatic acids in 2017 (Scheme 70).60 Also, this transformation was accelerated by an amino acid derivative and gave reasonable yields even without the supporting Thorpe–Ingold effect. In addition to a broad substrate scope, the authors disclosed another possibility to obtain the bait marker iophenoxic acid in three steps starting from 2-methylbutanoic acid.
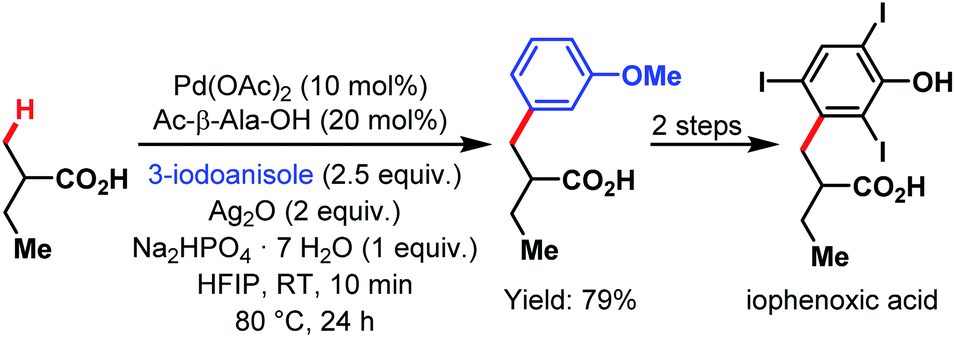 | ||
| Scheme 70 Synthesis of iophenoxic acid via β-C(sp3)–H arylation (van Gemmeren et al., 2017).60 | ||
Recently, the group of Yu published another study that highlights the synthetic utility of the carboxylate-assisted C–H activation catalyzed by palladium and thereby provides a fast access to tetralin, chromane and indane core structures. Finally, they applied their approach in a four-step synthesis of (±)-russujaponol F (Scheme 71).119 In the first coupling, pivalic acid was attached to the phenylacetic acid derivative using Pd(OAc)2 and an acetylated aminoethyl phenyl sulfide as a ligand. Subsequently, this product was subjected to optimized conditions with an F+-source as an oxidant to afford the indane motif by cyclizing C(sp3)–H/C(sp2)–H coupling.
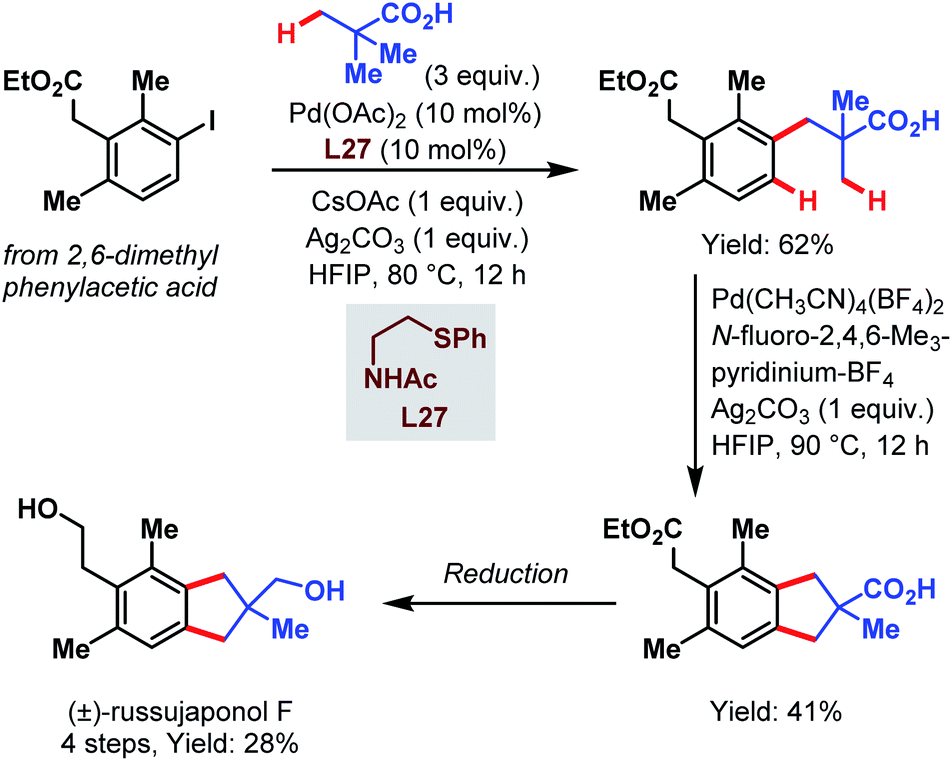 | ||
| Scheme 71 Total synthesis of (±)-russujaponol F (Yu et al., 2021).119 | ||
6. Conclusion and outlook
Pd-catalyzed C–H activation of free carboxylic acids has evolved as an interesting and widely recognized topic in the domain of C–H functionalisation. Extensive research has been carried out around the globe on this topic by a number of renowned research groups showcasing the multifaceted role of carboxylic acids in C–H activation. The major advantage of carboxylic acids is that it is a potential directing group for C(sp3)–H and C(sp2)–H bond functionalisations. Being a native directing group, it excludes extra effort to attach and detach an external unit as a directing entity. The extent of this directing ability has reached distal γ-positions of free aliphatic carboxylic acids, while in case of arene systems carboxylate motifs have been used for meta-C–H olefination. The current state-of-the-art demands propagation towards even farther positions of aromatic and aliphatic systems. In this aspect, exposure to base metals and rare transition metals other than palladium is scarce. A systematic development to close this loophole is of utmost importance apart from their application as chelating groups, carboxylic acids are well-established additives. However, a thorough mechanistic study to shine light on their role as additives is yet to be performed. Therefore, a number of aspects in this emerging topic are expected to reveal a lot more, which will eventually add significant value to the work of chemists in academia and industry.Author contributions
S. D., T. B., F. J. G., and M. B. prepared the original draft. D. M. and D. B. W. supervised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by SERB India (CRG/2018/003951) and research fellowships from PMRF-India (S. D.) and UGC-India (T. B.). D.M. and D. B. W. are grateful to the Alexander von Humboldt Foundation (Humboldt Research Fellowship for Experienced Researchers to D. M.). D. B. W. thanks the German Science Foundation (We2932/7-2).References
- C. Lamberth and J. Dinges, Bioact. Carboxylic Compd. Classes Pharm. Agrochem., 2016, vol. 1 Search PubMed.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed.
- G. D. Hartman, M. S. Egbertson, W. Halczenko, W. L. Laswell, M. E. Duggan, R. L. Smith, A. M. Naylor, P. D. Manno, R. J. Lynch, G. Zhang, C. T. Chang and R. J. Gould, J. Med. Chem., 1992, 35, 4640 CrossRef CAS PubMed.
- V. Vranova, K. Rejsek and P. Formanek, Sci. J., 2013, 2013, 524239 Search PubMed.
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571 CrossRef CAS PubMed.
- U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, 701 CrossRef PubMed.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820 CrossRef CAS PubMed.
- M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584 RSC.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569 CrossRef CAS.
- T. Fujihara, A. Yoshida, M. Satou, Y. Tanji, J. Terao and Y. Tsuji, Catal. Commun., 2016, 84, 71 CrossRef CAS.
- Y. Tanji, N. Mitsutake, T. Fujihara and Y. Tsuji, Angew. Chem., Int. Ed., 2018, 57, 10314 CrossRef CAS PubMed.
- Y. Tanji, R. Hamaguchi, Y. Tsuji and T. Fujihara, Chem. Commun., 2020, 56, 3843 RSC.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654 CrossRef CAS.
- A. Uttry and M. van Gemmeren, Synlett, 2018, 29, 1937 CrossRef CAS.
- A. Uttry and M. van Gemmeren, Synthesis, 2020, 52, 479 CrossRef CAS.
- J. Das, D. K. Mal, S. Maji and D. Maiti, ACS Catal., 2021, 11, 4205 CrossRef CAS.
- A. Das and B. Maji, Chem.–Asian J., 2021, 16, 397 CrossRef CAS PubMed.
- A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618 CrossRef CAS PubMed.
- A. J. Canty, A. Ariafard, B. F. Yates and M. S. Sanford, Organometallics, 2015, 34, 1085 CrossRef CAS.
- A. Gray, A. Tsybizova and J. Roithova, Chem. Sci., 2015, 6, 5544 RSC.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed.
- C. Athira and R. B. Sunoj, Org. Biomol. Chem., 2017, 15, 246 RSC.
- Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820 RSC.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- B. P. Carrow, J. Sampson and L. Wang, Isr. J. Chem., 2020, 60, 230 CrossRef CAS PubMed.
- D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43, 5009 RSC.
- K. M. Engle, Pure Appl. Chem., 2016, 88, 119 CAS.
- S. Shabani, Y. Wu, H. G. Ryan and C. A. Hutton, Chem. Soc. Rev., 2021, 50, 9278 RSC.
- B. E. Haines and D. G. Musaev, ACS Catal., 2015, 5, 830 CrossRef CAS.
- G. J. Cheng, Y. F. Yang, P. Liu, P. Chen, T. Y. Sun, G. Li, X. Zhang, K. N. Houk, J. Q. Yu and Y. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed.
- G. J. Cheng, P. Chen, T. Y. Sun, X. Zhang, J. Q. Yu and Y. D. Wu, Chem.–Eur. J., 2015, 21, 11180 CrossRef CAS.
- D. E. Hill, K. L. Bay, Y. F. Yang, R. E. Plata, R. Takise, K. N. Houk, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 18500 CrossRef CAS PubMed.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746 RSC.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, ACS Catal., 2019, 9, 11386 CrossRef CAS.
- C. A. Salazar, J. J. Gair, K. N. Flesch, I. A. Guzei, J. C. Lewis and S. S. Stahl, Angew. Chem., Int. Ed., 2020, 59, 10873 CrossRef CAS PubMed.
- R. Giri and J. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed.
- G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419 CrossRef CAS.
- R. Pratap, D. Parrish, P. Gunda, D. Venkataraman and M. K. Lakshman, J. Am. Chem. Soc., 2009, 131, 12240 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308 CrossRef CAS PubMed.
- K. F. Peng and C. T. Chen, Eur. J. Inorg. Chem., 2011, 5182 CrossRef CAS.
- D. Ghorai, V. Müller, H. Keil, D. Stalke, G. Zanoni, B. A. Tkachenko, P. R. Schreiner and L. Ackermann, Adv. Synth. Catal., 2017, 359, 3137 CrossRef CAS.
- P. Saxena and R. Murugavel, J. Organomet. Chem., 2018, 868, 76 CrossRef CAS.
- A. Vasseur, R. Membrat, D. Palpacelli, M. Giorgi, D. Nuel, L. Giordano and A. Martinez, Chem. Commun., 2018, 54, 10132 RSC.
- L. Cai, S. Li, C. Zhou and G. Li, Org. Lett., 2020, 22, 7791 CrossRef CAS PubMed.
- J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887 RSC.
- J. M. Lee and S. Chang, Tetrahedron Lett., 2006, 47, 1375 CrossRef CAS.
- K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032 CrossRef CAS PubMed.
- E. M. Stang and M. C. White, Angew. Chem., Int. Ed., 2011, 50, 2094 CrossRef CAS PubMed.
- R. Giri, N. Maugel, J. J. Li, D. H. Wang, S. P. Breazzano, L. B. Saunders and J. Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed.
- Y. Zhu, X. Chen, C. Yuan, G. Li, J. Zhang and Y. Zhao, Nat. Commun., 2017, 8, 14904 CrossRef CAS PubMed.
- K. K. Ghosh and M. van Gemmeren, Chem.–Eur. J., 2017, 23, 17697 CrossRef CAS PubMed.
- Z. Zhuang, C. B. Yu, G. Chen, Q. F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J. Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363 CrossRef CAS PubMed.
- K. K. Ghosh, A. Uttry, A. Koldemir, M. Ong and M. van Gemmeren, Org. Lett., 2019, 21, 7154 CrossRef CAS PubMed.
- Z. Zhuang, A. N. Herron, Z. Fan and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 6769 CrossRef CAS PubMed.
- Z. Zhuang and J. Q. Yu, Nature, 2020, 577, 656 CrossRef CAS PubMed.
- F. Ghiringhelli, A. Uttry, K. K. Ghosh and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 23127 CrossRef CAS PubMed.
- A. Uttry, S. Mal and M. van Gemmeren, J. Am. Chem. Soc., 2021, 143, 10895 CrossRef CAS PubMed.
- P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773 CrossRef CAS PubMed.
- L. Liu, Y. H. Liu and B. F. Shi, Chem. Sci., 2020, 11, 290 RSC.
- K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848 CrossRef CAS PubMed.
- H. S. Park, Z. Fan, R. Y. Zhu and J. Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853 CrossRef CAS PubMed.
- J. Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakash and D. Maiti, Chem. Sci., 2020, 11, 9697 RSC.
- P. Novák, A. Correa, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef PubMed.
- S. Qian, Z. Q. Li, M. Li, S. R. Wisniewski, J. X. Qiao, J. M. Richter, W. R. Ewing, M. D. Eastgate, J. S. Chen and J. Q. Yu, Org. Lett., 2020, 22, 3960 CrossRef CAS PubMed.
- P. X. Shen, L. Hu, Q. Shao, K. Hong and J. Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545 CrossRef CAS PubMed.
- L. Hu, P. X. Shen, Q. Shao, K. Hong, J. X. Qiao and J. Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134 CrossRef CAS PubMed.
- H. A. Chiong, Q. N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS PubMed.
- D. H. Wang, T. S. Mei and J. Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676 CrossRef CAS PubMed.
- K. M. Engle, P. S. Thuy-Boun, M. Dang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183 CrossRef CAS PubMed.
- C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910 CrossRef CAS PubMed.
- C. Arroniz, J. G. Denis, A. Ironmonger, G. Rassias and I. Larrosa, Chem. Sci., 2014, 5, 3509 RSC.
- C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418 CrossRef CAS PubMed.
- T. Bhattacharya, A. Ghosh and D. Maiti, Chem. Sci., 2021, 12, 3857 RSC.
- N. Dastbaravardeh, T. Toba, M. E. Farmer and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877 CrossRef CAS PubMed.
- Y. H. Zhang, B. F. Shi and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097 CrossRef CAS PubMed.
- P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508 CrossRef CAS PubMed.
- G. Cheng, T. J. Li and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950 CrossRef CAS PubMed.
- R. C. Larock, S. Varaprath, H. H. Lau and C. A. Fellows, J. Am. Chem. Soc., 1984, 106, 5274 CrossRef CAS.
- M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211 CrossRef CAS.
- D. H. Wang, K. M. Engle, B. F. Shi and J. Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed.
- K. M. Engle, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed.
- K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169 CrossRef CAS PubMed.
- D. Nandi, D. Ghosh, S. J. Chen, B. C. Kuo, N. M. Wang and H. M. Lee, J. Org. Chem., 2013, 78, 3445 CrossRef CAS PubMed.
- H. Wang, Z. X. Zhou, M. Kurmoo, Y. J. Liu and M. H. Zeng, Org. Lett., 2019, 21, 2847 CrossRef CAS PubMed.
- T. S. Mei, R. Giri, N. Maugel and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS PubMed.
- T. S. Mei, D. H. Wang and J. Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef CAS PubMed.
- S. Li, C. Zhang, L. Fu, H. Wang, L. Cai, X. Chen, X. Wang and G. Li, CCS Chem., 2021, 3, 2360–2371 CrossRef.
- K. H. Ng, F. N. Ng and W. Y. Yu, Chem. Commun., 2012, 48, 11680 RSC.
- J. Miao and H. Ge, Org. Lett., 2013, 15, 2930 CrossRef CAS PubMed.
- S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J. Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734 CrossRef CAS PubMed.
- Y. H. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed.
- Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K. S. Yeung, W. R. Ewing and J. Q. Yu, Science, 2021, 372, 1452 CrossRef CAS PubMed.
- J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478 CrossRef CAS PubMed.
- M. Catellani, E. Motti and N. Della Ca, Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS PubMed.
- X. C. Wang, W. Gong, L. Z. Fang, R. Y. Zhu, S. Li, K. M. Engle and J. Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed.
- G. C. Li, P. Wang, M. E. Farmer and J. Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 6874 CrossRef CAS PubMed.
- S. Li, H. Wang, Y. Weng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 18502 CrossRef CAS PubMed.
- B. F. Shi, Y. H. Zhang, J. K. Lam, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 460 CrossRef CAS PubMed.
- X. F. Cheng, Y. Li, Y. M. Su, F. Yin, J. Y. Wang, J. Sheng, H. U. Vora, X. S. Wang and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed.
- J. S. Dickstein, C. A. Mulrooney, E. M. O'Brien, B. J. Morgan and M. C. Kozlowski, Org. Lett., 2007, 9, 2441 CrossRef CAS PubMed.
- J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 123, 9601 CrossRef.
- J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127 RSC.
- K. Kim, D. Vasu, H. Im and S. Hong, Angew. Chem., Int. Ed., 2016, 55, 8652 CrossRef CAS PubMed.
- M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818 CrossRef CAS PubMed.
- M. Font, A. R. A. Spencer and I. Larrosa, Chem. Sci., 2018, 9, 7133 RSC.
- M. Kapoor, P. Chand-Thakuri and M. C. Young, J. Am. Chem. Soc., 2019, 141, 7980 CrossRef CAS PubMed.
- D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767 CrossRef CAS PubMed.
- B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J. Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317 CrossRef CAS PubMed.
- B. Hong, C. Li, Z. Wang, J. Chen, H. Li and X. Lei, J. Am. Chem. Soc., 2015, 137, 11946 CrossRef CAS PubMed.
- Z. Zhuang, A. N. Herron, S. Liu and J. Q. Yu, J. Am. Chem. Soc., 2021, 143, 687 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |