
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe inversion of tetrahedral p-block element compounds: general trends and the relation to the second-order Jahn–Teller effect†
Lukas M.
Sigmund
 a,
Rouven
Maier
a and
Lutz
Greb
*ab
a,
Rouven
Maier
a and
Lutz
Greb
*ab
aAnorganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: greb@uni-heidelberg.de
bDepartment of Chemistry and Biochemistry – Inorganic Chemistry, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany
First published on 15th December 2021
Abstract
The tetrahedron is the primary structural motif among the p-block elements and determines the architecture of our bio- and geosphere. However, a broad understanding of the configurational inversion of tetrahedral compounds is missing. Here, we report over 250 energies (DLPNO-CCSD(T)) for square planar inversion of third- and fourth-period element species of groups 13, 14, and 15. Surprisingly low inversion barriers are identified for compounds of industrial relevance (e.g., ≈100 kJ mol−1 for Al(OH)4−). More fundamentally, the second-order Jahn–Teller theorem is disclosed as suitable to rationalize substituent and central element effects. Bond analysis tools give further insights into the preference of eight valence electron systems with four substituents to be tetrahedral. Hence, this study develops a model to understand, memorize, and predict the angular flexibility of tetrahedral species. Perceiving the tetrahedron not as forcingly rigid but as a dynamic structural entity might leverage new approaches and visions for adaptive matter.
Introduction
The tetrahedron dominates the structural chemistry of the p-block, e.g., that of carbon, aluminates, silanes, or ammonium ions. It thereby constitutes the fundamental building block for the molecular architecture of our bio- and geosphere (Fig. 1A). For species with eight valence electrons and four substituents (ER4), the tetrahedral preference is well-founded by molecular orbital theory or by the VSEPR model.1,2 But what do we know about the tetrahedron's structural deformation, its fluxionality, or its configurational inversion? Surprisingly little!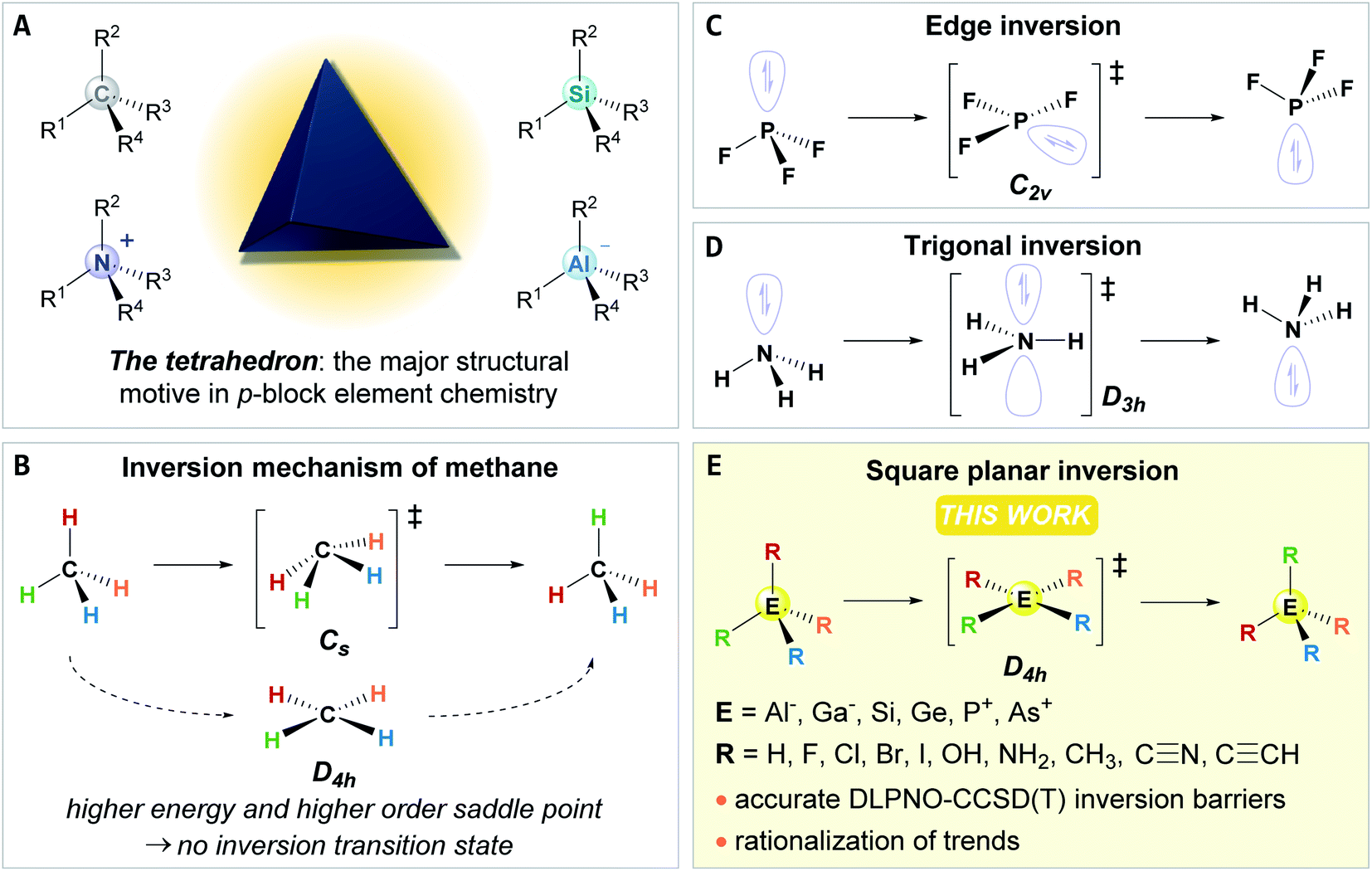 | ||
| Fig. 1 (A) Examples of eight valence electron p-block element species with a tetrahedral ground state. (B) The Cs symmetric inversion transition state for CH4 and its higher energy square planar state of D4h symmetry. (C) Edge inversion process for group 15 element fluorides. (D) Trigonal inversion of ammonia and other group 15 element compounds. (E) The inversion process of p-block tetrahedrons analyzed within this work. | ||
The potential energy surface for the structural deformation of the tetrahedron is by far the most developed for methane.3–5 The minimum energy inversion path was found to traverse a Cs symmetric transition state, whereas the square planar D4h symmetric state is significantly higher in energy with a higher-order saddle point on the potential energy surface (Fig. 1B).6–9 Accordingly, tremendous progress has been made in the field of planar tetra-, penta- and hexacoordinate carbon during the past decades.10–14 For the remaining p-block tetrahedrons, the inversion process is much less understood. For hydrides of less electronegative central elements (AlH4−, SiH4, PH4+), inversion transition states of D4h symmetry were suggested.15 For SiF4 and PF4+, analogies with the edge inversion of trigonal group 15 compounds with electronegative substituents were found (e.g., PF3, Fig. 1C).16,17 Strategies to stabilize square planar configurations for some other elements were probed computationally,18–24 and substantial progress was made for compounds featuring planar silicon.25–40 However, a systematic fundament of the inversion process in ER4 compounds or more general statements on their tetrahedral preference are elusive. This lack appears surprising compared with the extensive knowledge on the inversion process of trigonal group 15 compounds ER3 (Fig. 1D), which occupied generations of theoreticians and experimentalists.41–43 Different approaches to explain the inversion of ER3, like perturbational molecular orbital theory,1,44–46 the second-order Jahn–Teller effect (SOJTE),47–50 or recoupled pair bonding,51 afforded a profound base and thereby challenged our understanding of the chemical bonding.52,53 Likely, this diverging interest stems from the absent or potentially overlooked experimental observation of structural dynamics of the tetrahedron, which are classically considered configurationally stable.54 However, in recent years, several “anti-van't-Hoff–Le-Bel” species (i.e., stabilized inversion transition states), such as planar aluminates, silicon, or phosphonium ions, were isolated, offering unique reactivities.55–70 Hence, knowledge on the tetrahedral inversion transforms from fundamental to applied interest.
In the present study, we first recapture and develop a framework for the second-order Jahn–Teller effect (SOJTE) on square-planar inversion. Second, we provide a systematic discussion of inversion energies of p-block tetrahedrons of groups 13, 14, and 15 with homo- and heteroleptic substitution patterns (Fig. 1E). Surprisingly low barriers are identified, including compounds of industrial relevance, e.g. Al(OH)4−, but which have never been considered in light of configurational inversion. Finally, and most fundamentally, we prove that the SOJTE can rationalize and explain those trends. Additional bond analysis tools round off our understanding of the origin of the tetrahedral preference for ER4 compounds.
Qualitative remarks on the molecular orbitals during square planar inversion and the connection with the second-order Jahn–Teller effect (SOJTE).
The preference of ER4 molecules to adopt tetrahedral structures and the orbital variations upon distortion into the planar state are well-described by Walsh diagrams, as schematically shown for SiH4 and CH4 in Fig. 2A.1,54,71 In Td symmetry, four bonding (1a1 and 1t2) and four anti-bonding (2a1 and 2t2) molecular orbitals are built by combining the central elements' valence s- and p-orbitals with the symmetry-adapted linear combinations (SALCs) of the substituent fragment orbitals. The deformation into the planar D4h symmetric state occurs along the E harmonic vibration via intermediate D2d symmetry (Fig. 2B).16 Along this distortion coordinate, the 1a1 orbital experiences slight stabilization due to the increased overlap of the substituent-centered in-phase lobes. The 1t2 set of the Td state splits up into 1e and 1b2 molecular orbitals. The 1e set becomes slightly stabilized due to the increased overlap of substituent and central element orbitals. However, the 1b2 orbital turns from bonding to non-bonding and becomes significantly destabilized in the a2u molecular orbital of the final D4h symmetric state. The character of the highest occupied (HOMO) and lowest unoccupied molecular orbital (LUMO) in the square planar state depends on the electronegativity of the central element relative to the substituents.15 The a2u orbital is of π-character and is localized at the central atom, whereas the b1g orbital is of δ-type and resides at the substituents. The latter molecular orbital emanates from the 2t2 representation of the tetrahedral state and is energetically considerably lowered along the D2d symmetric distortion. Hence, for central elements which are less electronegative than hydrogen (e.g., SiH4, Fig. 2A), the substituent-centered b1g orbital is the HOMO, and the LUMO is of a2u symmetry (π-type orbital). For the inverse situation (e.g., CH4, Fig. 2A), the a2u orbital is the HOMO (primarily located at carbon), whereas the LUMO is the substituent-centered b1g molecular orbital. In total, this MO theoretical perspective assigns the primary cause for the inversion barrier to the significant energy increase of one of the orbitals of the occupied 1t2 set of molecular orbitals, which is not compensated by the stabilization of the other occupied orbitals.
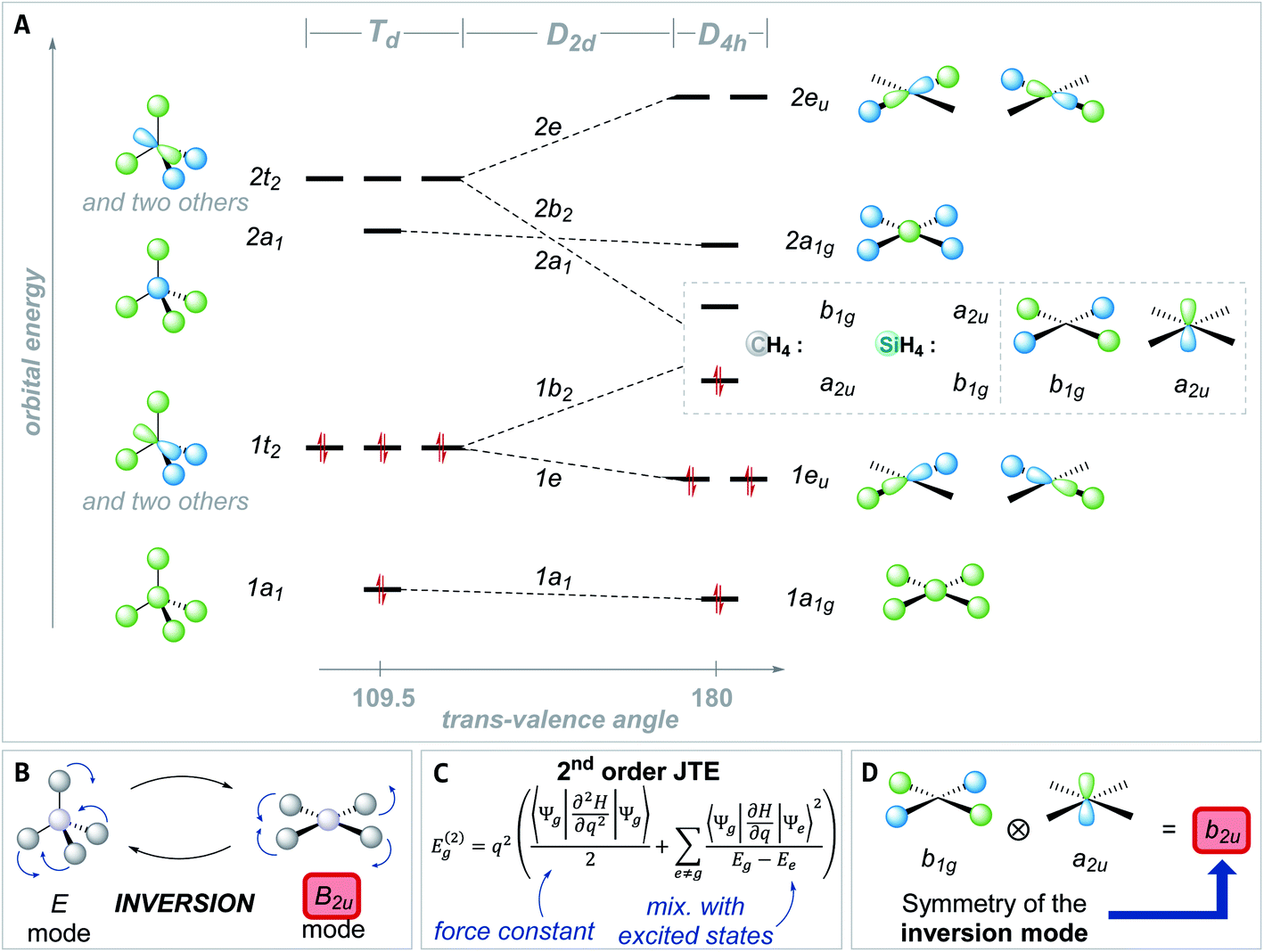 | ||
| Fig. 2 (A) Schematic Walsh diagram for the distortion from the tetrahedral to the square planar state of CH4 and SiH4. (B) Deformation vibrations that transform the tetrahedral ground state into the square planar inversion transition state and vice versa. (C) Second-order contributions for the deformation coordinate q to the energy of a given system. (D) Direct product of the b1g and a2uirrep within the D4h point group. It transforms under b2u, which is also the symmetry of the inversion vibration. | ||
Although the MO interpretation draws a qualitative picture, it cannot easily rationalize differences in inversion barriers for different molecules. Vibronic coupling theory provides an alternative approach through the second-order Jahn–Teller theorem (SOJTE).72–79 This model is more complex, but it allows to parametrize reaction pathways by considering the instability of transition states.80 Some crucial arguments on the SOJTE are revised first, in order to prepare for their application in a later section of this article.
The curvature of the adiabatic potential energy surface (PES) at a stationary state under consideration, such as a transition state, is inspected with respect to a structural deformation along the coordinate q. The second-order terms of the Jahn–Teller theorem relate the energy change for this structural deformation with the electronic ground state (Ψg) and contributions of higher excited states (Ψe) (Fig. 2C).74,81
The individual terms are interpreted as: (1) the force constant of the deformation (resistance toward deformation without electronic relaxation, first summand in Fig. 2C), (2) the symmetry and overlap of the electronic ground state coupling with excited states (“vibronic coupling constants”, the numerator of the second summand in Fig. 2C), and (3) their energy difference (denominator of the second term in Fig. 2C). Energy lowering upon distortion along q only happens, if the direct products of the irrep of the ground (Γ(Ψg)) and the excited state (Γ(Ψe)) contain the irrep under which the distortion coordinate q, i.e., its vibrational mode, transforms (Γ(Ψg) ⊗ Γ(Ψe) ∈ Γ(q)). The theory is only valid for small nuclear displacements from the reference structure, but the curvature may extrapolate the energy release upon structural relaxation.
For a quantitative analysis, all SOJTE-parameters of the equation given in Fig. 2C need to be evaluated along the entire adiabatic PES for all the states that fulfill the symmetry argument – a very tedious task.82 Several approximations offer simplification: (1) instead of using the full state wavefunctions, the coupling can be inspected at the level of one-electron molecular orbitals. The vibronic coupling constants become orbital vibronic coupling constants. (2) Instead of using the entire set of symmetry-allowed excited orbitals, only the two frontier molecular orbitals (FMO) can be considered. (3) If compounds with similar bonds are compared, it can be assumed that the vibronic coupling constants and the force constant, respectively, are not too different. Hence, the second-order contribution to the total energy gain (Eg(2), see Fig. 2C) turns out to be inversely proportional to the HOMO–LUMO gap in the initial structure.
In this FMO approximation, the SOJTE was used for several phenomena, advocated by Pearson and got popularized among chemists while sharing many characteristics with perturbational molecular orbital theory.79,83–86 Interpreted with precaution and bearing in mind its simplifications, it can be a valuable tool to obtain relations inaccessible by other approaches.87 For example, it has been used to rationalize the inversion barrier of group 15 and 16 elements or to discuss the structure of (SiO)4.1,44,45,47,88 In the context of tetra-coordinated species, the SOJTE has been mentioned, but was not further developed.89,90
Applied to the square planar inversion of ER4, the following relation can be formulated (Fig. 2D): In the D4h symmetric state, the HOMO and LUMO are of a2u and b1g symmetry, therefore orthogonal and cannot mix. The direct product of the symmetry species of both orbitals transforms under the b2u representation (a2u ⊗ b1g = b2u). Consequently, the vibronic interaction becomes allowed upon coupling with the b2u vibrational coordinate, which is just the deformation of the planar into the tetrahedral state (Fig. 2B). Thus, the qualitative hypothesis developed and to be probed in this manuscript reads as: the closer the FMOs are in the inversion transition state, the larger is the transition state instability, the larger is the inversion barrier. A small FMO gap in the inversion transition state results in a large contribution of the second summand in Fig. 2C's equation. Therefore, the relaxation into the tetrahedral structure promises the system significant energetic stabilization. Consequently, the energy span between ground and transition state must be large – a high inversion barrier is encountered. Complementarily, a large FMO gap diminishes the influence of the second summand (Fig. 2C), reduces the energetic stabilization due to structural deformation from the planar toward the tetrahedral state, and thus indicates a low inversion barrier.
Computational methods
The following set of molecules was included in the study: EH4−yRyn for En = Al−, Si, P+, and Ga−, Ge, As+ with R = F, Cl, Br, I, OH, NH2, CH3, CN, CCH and y = 0, 1, 2, 3, 4 (a total of 276 inversion barriers). The meta-GGA functional B97M, including Grimme's D3 correction, was combined with the cc-pVTZ basis set for structural optimization and frequency analysis.91 In six of the 276 cases, the optimization toward the desired transition structure remained unsuccessful (see the ESI† for details). In all other cases, proper transition structures for the inversion process were obtained by the applied DFT method as confirmed by numerical Hessian evaluations and IRC computations. For the final energies, the performance of several density functionals and approximated wave function methods were evaluated on a representative subset of molecules against CCSD(T)/CBS reference data. The DLPNO-CCSD(T)/cc-pVQZ method (tightPNO settings) showed a MAD of only 1.3 kJ mol−1 and was used to calculate all final single point energies. The influence of diffuse basis functions was also investigated with the aug-cc-pVQZ basis set but was found minor.The inversion transition states of D4h symmetry of PH4+ and AsH4+ have an open-shell singlet electronic configuration (1B2u state). This was found by ab initio calculations on the NEVPT2/cc-pVQZ//CASSCF(8,8)/cc-pVTZ theoretical level. The barriers amount to 500 (PH4+) and 498 kJ mol−1 (AsH4+). These numbers were used throughout the here presented work. The largest barriers (>480 kJ mol−1, group 15 cations) need to be taken with some caution due to a potential multiconfigurational character of the transition states (see the ESI† for further details). The possibility of a tunneling contribution during inversion, in particular for the hydrides, was not considered.9 For the SOJTE-analysis, the B97M-cc-pVTZ derived Kohn–Sham molecular orbital energies were used.92,93 For a selected number of compounds, the HOMO–LUMO gap of the inversion transition state was compared to the respective vertical excitation energy to the lowest energy excited singlet state obtained by TDDFT. Good correlations were obtained, and therefore the HOMO–LUMO gaps were used for the entire set of the investigated molecules. For any further details and specifics on natural bond orbital analysis (NBO), energy decomposition analysis (EDA), and the quantum theory of atoms in molecules (QTAIM) treatment, see the ESI.†
Results and discussion
Effects of substituent R and central element E in monosubstituted compounds EH3Rn
Comparing the inversion barriers for species EH3Rn with that of EH4n allowed to assess the influence of the respective substituent R (Fig. 3). For GaH3R− (R = Br, I, CN), the transition structure optimizations converged to a dissociated species and should not be considered (red stars in Fig. 3B). Stabilization energies range from ≈10 (CH3) to 180 kJ mol−1 (NH2) with respect to the barriers of the unsubstituted EH4n. For the group 13 halide anions EH3R− (R = F, Cl, Br, I), the transition state lowering effect increases for the heavier halides, whereas for the neutral group 14 compounds and the group 15 cations, fluoride, and iodide stabilize more efficiently than chloride and bromide. In groups 14 and 15, the stabilizing effect of the hydroxy group is larger than that of the halides. The amino group generally has the most powerful impact of all considered substituents (up to 180 kJ mol−1 in PH3(NH2)+versus PH4+). Substituents without the ability for π-donation (methyl, nitrile, and ethynyl) stabilize less effectively but still substantially (up to 40 kJ mol−1).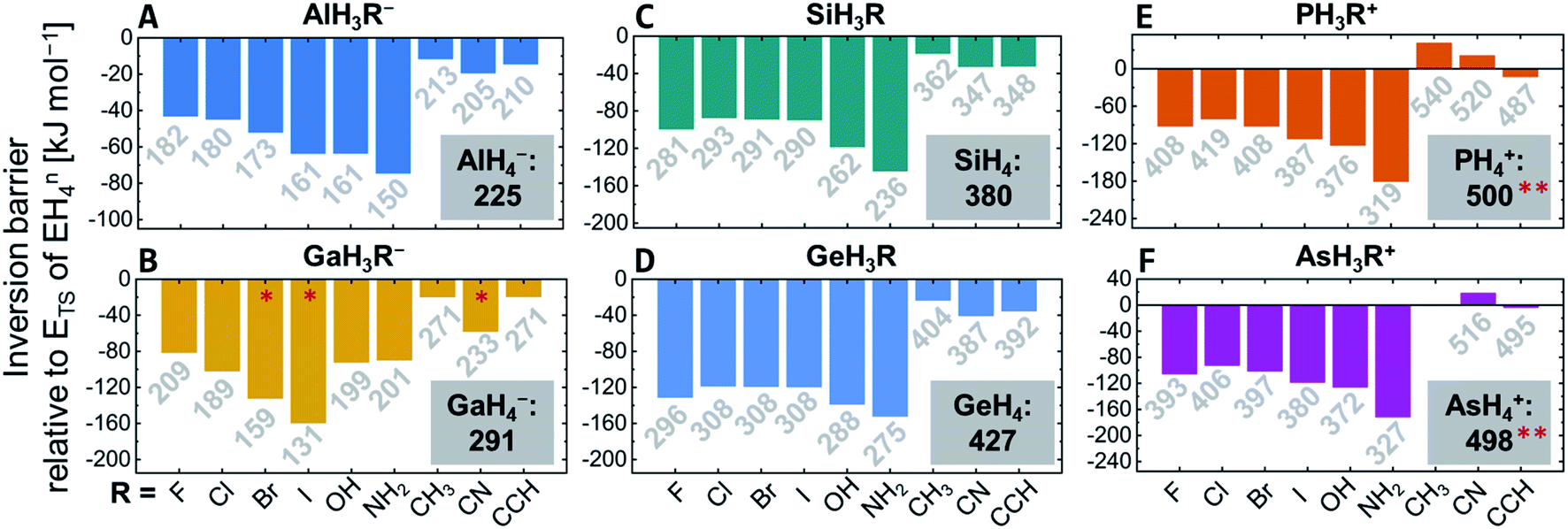 | ||
| Fig. 3 Relative inversion barriers of EH3Rn with respect to EH4n (grey panels), for E = Al (A), Ga (B), = Si (C), Ge (D), P (E), As (F). The label on each bar states the respective absolute inversion barrier of EH3Rn. No inversion transition state was found for AsH3(CH3)+.* The transition structure optimization converged to a dissociative structure (GaH3 and R−). ** The inversion barriers were obtained with NEVPT2/cc-pVQZ//CASSCF(8,8)/cc-pVTZ. | ||
The substituent effect is more pronounced for groups 14 and 15 versus group 13 and for the third period compared to the fourth period (Fig. 3). Concerning the central element, the inversion barriers increase significantly when going from left to right in the periodic table (Al < Si < P/Ga < Ge < As), with the group 15 element cations showing the largest inversion barriers (Fig. 3). Comparing the third- and fourth-period elements within one group, the trend of the inversion barrier is Al < Ga, Si < Ge but P > As. However, the difference between the periods is less pronounced than between the groups and is often outmatched by additive substitution effects.
Additive substitution effects along the series EH4−yRyn
Consecutive replacement of the hydrogen substituents against R groups revealed additive and counter-additive substituent effects on the inversion barrier heights.For the halido and amino series EH4−yRyn (R = F, Cl, Br, I, NH2), a maximum stabilization is reached with two substituents in trans-arrangement, i.e., the trans-EH2R2n transition states are lowest in energy as exemplarily shown for the silicon halides in Fig. 4A (for the plots of the other elements see the ESI†). The isomeric cis-EH2R2n transitions states are roughly 50 kJ mol−1 higher in energy than the trans-isomers and range in the region of monosubstituted derivatives. The trend along EH2R2n → EHR3n → ER4n depends on the central element. For the third-period elements (E = Al, Si, P), the barriers rise again, reaching energies like in the monosubstituted EH3Rn. Thus, counter-stabilizing effects occur upon substitution with more than two R groups. This trend is different for the fourth-period atoms: the barriers remain invariant along three and four substituents, i.e., the stabilizing effect and the counter-stabilizing effect cancel (Fig. 4B). Accordingly, for EH3Rn, the fourth-period species have the larger inversion barriers than the third-period compounds but lower inversion barriers for ER4.
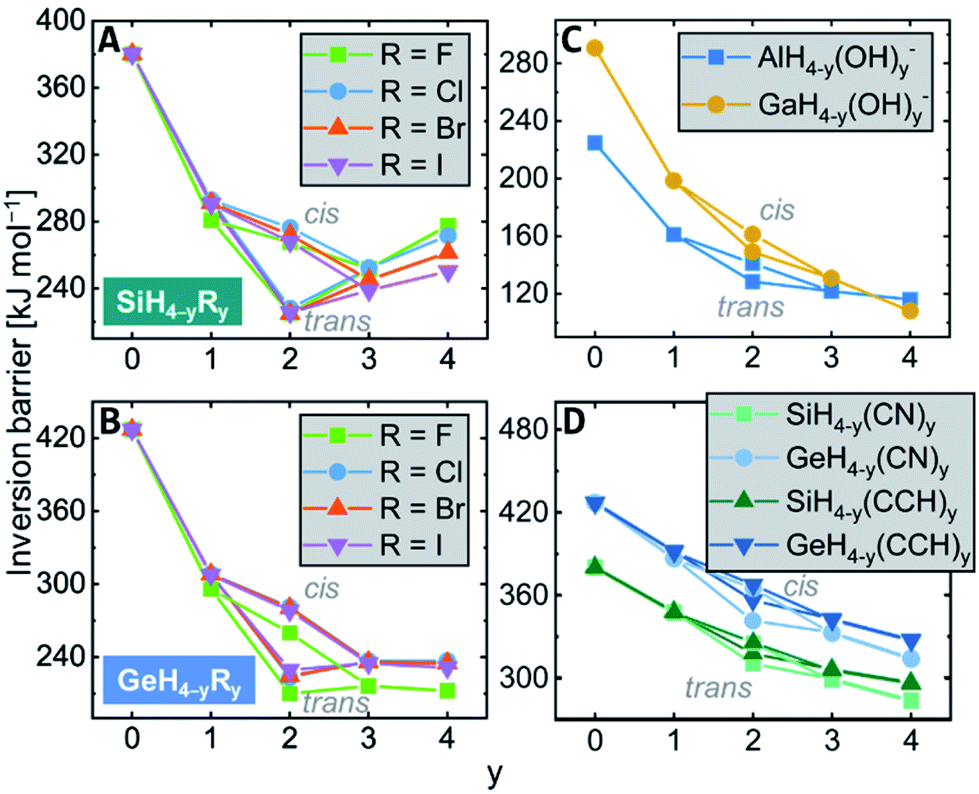 | ||
| Fig. 4 Additive substituent effects for the inversion barrier of (A) SiH4−yRy, (B) GeH4−yRy, with R = F, Cl, Br, I, of (C) AlH4−y(OH)y− and GaH4−y(OH)y−, and of (D) EH4−yRy, with E = Si, Ge and R = CN, CCH. | ||
Interestingly, the molecules with hydroxy groups show almost no saturation effect. In most cases, the four-fold substituted E(OH)4n have the lowest energy transition states (E = Al, Ga, Ge, As, Fig. 4C), and only for Si and P, a minimum for the trans-transition states of EH2(OH)2n is slightly developed. This deviation is explained by hydrogen bonding, which is more efficient in the transition than in the ground states (see Chapter S11 in the ESI† for a detailed discussion). A comparison of different transition state conformers of the EH(OH)3n class of compounds allowed to estimate the transition state stabilization for the individual elements to approximately 5 (E = Al), 10 (E = Ga), 12 (E = Si), 15 (E = Ge), and 21 (E = P, As) kJ mol−1 per hydrogen bond on top of the direct electronic effects. A similar trend is found for the nitrile and ethynyl substituents, which are σ-acceptors (Fig. 4D) – no saturation occurs. The inversion barriers steadily drop with each substitution, finding a minimum for the four-fold substituted derivatives ER4n (R = CN, CCH).
In all cases, the E–R bonds elongate upon going from the tetrahedral to square planar state (see Chapter S4 in the ESI†). Despite the larger s-orbital character at the central element in the transition states (Table S11 in the ESI†), this elongation agrees with the diminished formal bond order.
Excitingly, some compounds show inversion barriers that are easily surpassed at ambient conditions. For example, the di-substituted group 13 anions (EH2R2−) show barriers corresponding to configurational stability half-lives of several minutes at 25 °C. Most remarkably, Al(OH)4− has an inversion barrier of 116 kJ mol−1. This well-known anion occurs during the Bayer process in worldwide aluminum production.94 Process temperatures of 200 °C correspond to a tetrahedron inversion half-life for Al(OH)4− of ≈0.5 s. Thought-provokingly, a tetrahedral inversion process is, and was, happening for more than 130 years at a gigatonne scale but went unnoticed due to the degeneracy of products and educts.
Molecular orbitals during the deformation toward and in the transition state
For the rationalization of the inversion barrier energies, the discussion is narrowed to the homoleptic compounds, ER4n, as their inversion transition states are of D4h symmetry, facilitating MO theoretical considerations. In the following section, the intrinsic reaction coordinate (IRC) connecting the Td symmetric ground state with the inversion transition state is analyzed for SiH4, SiF4, and Si(CN)4. This is done with respect to the changes occurring in molecular orbital energies along the IRC (Walsh-type diagrams, Fig. 5). Comparisons among those three compounds illustrate the effects of π-donor or σ-acceptor substituents on the orbital energies. The generality of these findings was supported by sampling analysis throughout the data of other species.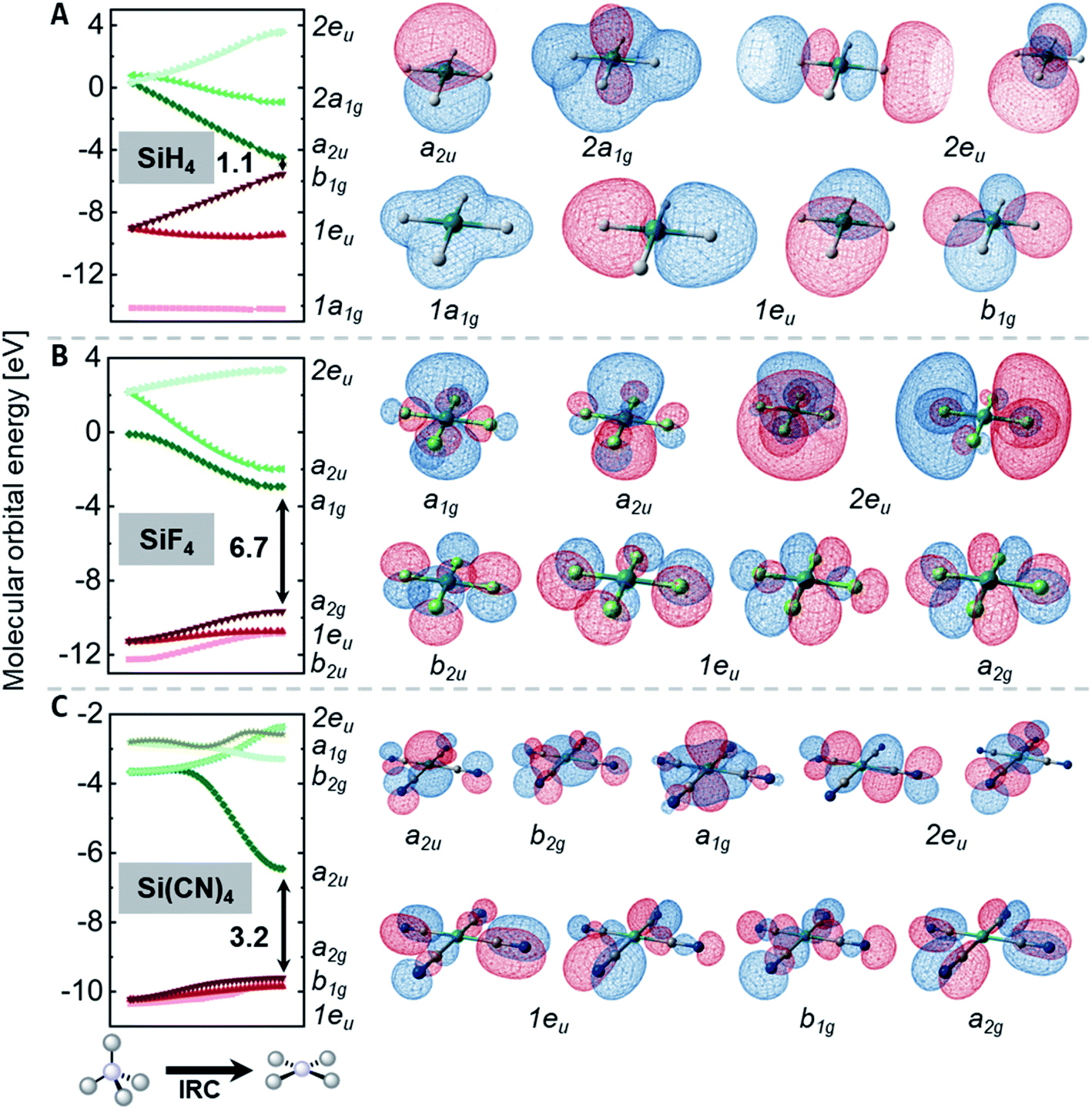 | ||
| Fig. 5 Kohn–Sham frontier molecular orbital energies along the inversion reaction coordinate and the corresponding molecular orbital isodensity plots (contour value 0.03 a.u.) of the D4h symmetric inversion transition state of (A) SiH4, (B) SiF4, and (C) Si(CN)4. | ||
For the D4h symmetric states of the element hydrides EH4n (E = Al−, Si, Ga−, Ge), the Kohn–Sham molecular orbitals are in line with the qualitative perturbational MO-picture given in Fig. 2A: a HOMO of b1g and LUMO a2u symmetry. This is quantitatively illustrated for SiH4 in Fig. 5A. SiH4 possesses a relatively small FMO gap of 1.1 eV in its inversion transition state. The LUMO+1 is the anti-bonding 2a1g molecular orbital which remains high in energy along the inversion reaction coordinate. In contrast, for systems with σ-acceptor/π-donor substituents, e.g. SiF4 (Fig. 5B), the energetic spacing between the FMOs (6.7 eV) is much wider in the D4h state than for the hydride. This difference is the consequence of the following:
(1) The a2u orbital in D4h symmetric SiH4 is non-bonding, whereas its counterpart (2t2) is fully anti-bonding in the tetrahedral ground state. Hence, it drops substantially by 5.3 eV upon planarization. For SiF4, this drop is mitigated to 4.1 eV, as the a2u orbital remains slightly anti-bonding in the D4h state due to the π-donor interaction with the fluorides (→ π-donor effect).
(2) All the highest occupied molecular orbitals of the square planar transition states are constituted from the substituents. Hence, they are substantially lower in energy for fluorides in SiF4 than for the hydrides in SiH4 (→ σ-acceptor effect).
These effects increase the FMO gap in the transition state and also influence the energetic ordering of the orbitals. The diminished energy drop of the empty a2u orbital in SiF4 causes the LUMO to be of a1g symmetry. The a2u molecular orbital turns out as LUMO+1 (Fig. 5B).
The MO-situation for Si(CN)4 settles between the hydrides and fluorides (Fig. 5C). As in the case of SiH4, the a2u orbital drops substantially, but the occupied orbitals remain little affected, resembling the situation of SiF4. This signature reflects the σ-acceptor character of the nitrile group but the inability for π-donation, which would prevent the a2u orbital from lowering in energy (as is the case for SiF4).
The inversion transition state and the associated barrier evaluated by the SOJTE–FMO approximation
The general SOJTE–FMO hypothesis developed in the introduction claims that the barrier should increase with a decreasing FMO gap in the inversion transition state. Indeed, for the element hydrides, EH4, a good correlation is obtained by plotting the FMO gap of the transition states against the inversion barrier heights (Fig. 6A). The smaller the FMO gap becomes, the larger is the inversion activation barrier. As shown in Fig. 6B, also σ-acceptor (nitrile and ethynyl) substituted systems show this correlation, indicating that these systems indeed obey the SOJTE-model sufficiently. It even holds reasonably for the entire set of EH4−yRyn (R = CN, CCH, see Fig. S26 in the ESI†). Plotting the hydride and the CN/CCH correlations in the same chart gives an offset between the two groups (see Fig. S27 in the ESI†). This is due to differences in vibrational coupling constants and the primary force constant, which are not included in the SOJTE–FMO model (cf.Fig. 2C). Interestingly, CCH and CN correlate rather well (Fig. S24 and S25 in the ESI†), as both groups bind through sp-hybridized carbon atoms.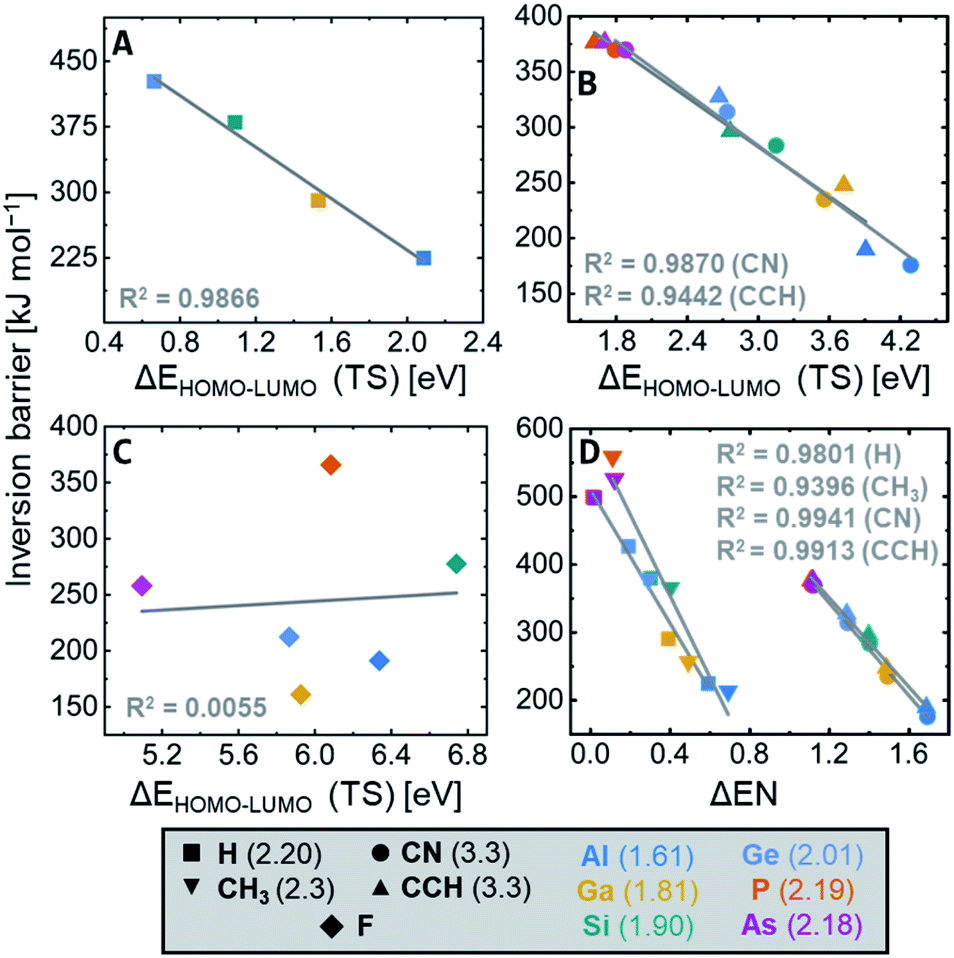 | ||
| Fig. 6 Correlation plots of the inversion barrier height versus the Kohn–Sham HOMO–LUMO gap in the inversion transition state for (A) EH4n, (B) E(CN)4n and E(CCH)4n, and (C) EF4n, for which the correlation vanishes. (D) Correlation plot of the inversion barrier height versus the difference in electronegativity between the central element and substituents for ER4n, R = H, CH3, CN, CCH. Pauling (group) electronegativities are given in parenthesis in the legend. | ||
Overall, these are unprecedented findings that allow explaining the influence of the central element and σ-acceptors on tetrahedral inversion barriers along the following arguments:
• The a2u LUMO is localized at the central element whose electronegativity (EN) determines its orbital energy.1 A larger EN causes a lower a2u energy which leads to a smaller FMO gap. Consequently, the inversion barrier increases with the EN of the central element.
• The b1g HOMO is located at the substituents. Thus, its energy is determined by the substituents' EN. An increasing EN lowers the HOMO energy and, therefore, the FMO gap. Consequently, the inversion barrier decreases with the EN of the substituents.
Indeed, the EN increases from left to right in the periodic table, as does the inversion barrier (Al < Si < P/Ga < Ge < As). The EN also increases from period three to period four (d-block effect), in line with the observed trend of inversion barriers between the periods (Al < Ga/Si < Ge/P ≈ As). If the FMO gap becomes too small, as for the most electronegative central elements P and As, multiconfigurational approaches are needed for an accurate description. Of course, one should consider that the absence of inner shell p-electrons leads to additional differences between the second and higher period elements, which are not considered here.95,96
Having found these correlations, it was tempting to probe the difference of electronegativities of central elements and substituents as a parameter for the inversion barrier. Indeed, for the non-π-donor substituents (H, CH3, CN, CCH), a proper correlation can be found (Fig. 6D). The observed offset again occurs due to differences in vibrational coupling constants and the primary force constant between E–H and E–C bonds. Interestingly, the group electronegativities of nitrile (3.3) and ethynyl (3.3) are identical.97 Accordingly, their effect on the inversion barriers is almost the same.
Coming back to the SOJTE-approximation, the correlation was probed for π-donor cases next. For the fluoride substituted derivatives, EF4n, the SOJTE-correlation breaks down (Fig. 6C). The same is observed for the hydroxy- and amido-substituted systems, which do not provide any apparent correlations between inversion barriers and FMO gaps in the D4h state. Although the model recaptures validity along Cl < Br < I with a steady increase in the correlation coefficient (Fig. S18–S20 in the ESI†), it is far from ideal. Hence, for π-donor substituted systems, additional factors cause deviations from the SOJTE–FMO model. Similar observations have been made for the trigonal inversion of group 15 halido pyramids.48 After all, one must not forget the participation of higher excited states that become particularly important for substituents with lone pairs but which are not considered in this approximation. However, the stabilizing effect of π-donor substituent can be understood qualitatively through the framework of the SOJTE.
• The lone pairs at the substituents interact with the a2u orbital at the central atom. Hereby, the a2u orbital becomes anti-bonding and increases in energy. The FMO gap in the transition state becomes larger for π-donors, and the inversion barrier decreases.
Summing up the ligand effects: if the electronegativity of the substituents increases, the b1g molecular orbital becomes stabilized, the FMO gap increases and the barrier becomes lower (σ-acceptor effect). For π-donor substituents, the donation leads to an FMO gap increase by raising the energy of the a2u molecular orbital. Gauged by the absolute values, π-donation appears more effective than σ-acceptance. For the hydride species and those substituted with less electronegative and non-π-donor groups, the SOJTE is a quantitatively fitting model. Here, the tetrahedral preference and the inversion barrier are dominated by orbital overlap effects. For π-donor substituents, the situation is more complex, and additional factors come into play that are elucidated next.
Analysis of differences in atom and bond properties by NBO, QTAIM, and EDA
To corroborate and extend the interpretations from the SOJTE analysis, the ground and inversion transition states for selected compounds were investigated by natural population analysis (see Chapter S14 of the ESI† for all data of the ER4n compounds).98 For SiH4 and Si(CN)4, the natural charge at Si gets more positive in the transition state. In contrast, for SiF4, the transition state has a less positively polarized silicon center (Table 1). These characteristics, namely, stronger positive polarization of the central element for hydride and σ-acceptor-substituted systems (H, CH3, CN, CCH) in the transition state and significantly less positive polarization for molecules with π-donor groups (F, OH, Cl, Br, NH2) are indicated throughout the entire set, substantiating a clear difference between both types.| Compound | NBO charges | QTAIM analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Si | R = H, F, CN | ρ BCP | ∇2ρBCP | K | DI | BE | ||
| SiH4 | GS | 0.560 | −0.140 | 0.122 | 0.173 | −0.0747 | 0.545 | 0.0008 |
| TS | 1.137 | −0.284 | 0.115 | 0.069 | −0.0742 | 0.503 | 0.0192 | |
| Diff. | 0.578 | −0.007 | −0.104 | 0.0005 | −0.042 | 0.0184 | ||
| SiF4 | GS | 2.531 | −0.633 | 0.154 | 1.135 | −0.0492 | 0.351 | 0.0004 |
| TS | 2.522 | −0.631 | 0.142 | 0.959 | −0.0495 | 0.359 | 0.0788 | |
| Diff. | −0.009 | −0.012 | −0.176 | −0.0003 | 0.008 | 0.0784 | ||
| Si(CN)4 | GS | 1.362 | −0.173 | 0.124 | 0.299 | −0.0696 | 0.408 | 0.0003 |
| TS | 1.580 | −0.219 | 0.119 | 0.266 | −0.0668 | 0.394 | 0.0596 | |
| Diff. | 0.218 | −0.006 | −0.033 | 0.0028 | −0.014 | 0.0593 | ||
Changes in bond characteristics that occur during the inversion process were further inspected by QTAIM.99 For molecules with hydride or σ-acceptor substituents, the electron density at the bond critical points (ρBCP) diminishes upon planarization (Table 1). The same trend is observed for the Laplacian of the electron density (∇2ρBCP), mirroring the generally diminished bonding strength. As an indicator of the amount of covalency, Kraka's energy density descriptor (K) and the delocalization index between Si and R (DI) were considered.100,101 These parameters are indicating a diminished amount of covalency in the transition state (DI ↓, K ↑), in line with molecular orbital theory. Strikingly, for the π-donor systems, the situation is different. In SiF4, the electron density and its Laplacian decrease, but the DI increases, and K decreases upon transformation to the transition state. These features indicate a diminished strength of bonding in the transition states with π-donors, but an increase in bond covalency! The bond ellipticity (BE) increases stronger for SiF4 as for SiH4 and Si(CN)4, supporting a π-back bonding effect already noticed by the analysis of natural atomic charges and assumed qualitatively by the perturbational MO/SOJTE-arguments. Again, those characteristics were supported by control sampling throughout the entire set of ER4n molecules.
To verify and extend these hypotheses, the interaction between substituents and central elements was inspected by Ziegler and Rauk's version of the energy decomposition analysis (EDA).102–105 Specifically, it was considered how Pauli repulsion (ΔEPauli), electrostatic (ΔECoul), and orbital interactions (ΔEOrb) between substituent fragments and the central element change during planarization. Moreover, the process was dissected in a radial (change of bond lengths) and angular deformation (change in bond angle, Table 2), as recently demonstrated elsewhere.106,107 Heterolytic fragmentation was chosen due to, on average, less sizeable orbital relaxation energy, as frequently suggested (Table 2).108–110 Further, it was verified that the general conclusions are neither depending on the type (heterolytic vs. homolytic) or the number (four bonds vs. one bond) of fragmentation(s), nor on the order of deformation (i.e. radial/angular yields practically identical numbers as angular/radial, see Chapter S15 in the ESI†). The most significant points are illustrated here for SiH4 and SiF4.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| GS | TS′ | TS | Angular contribution (GS → TS′, ΔΔEA) | Radial contribution (TS' → TS, ΔΔER) | Total (GS → TS, ΔΔE) | ||
| SiH4 | ΔEPauli | 692.0 | 709.5 | 582.4 | 17.5 | −127.1 | −109.6 |
| ΔECoul | −7847.3 | −7807.0 | −7795.3 | 40.3 | 11.7 | 52.0 [12%] | |
| ΔEOrb | −6241.3 | −6018.8 | −5872.7 | 222.6 | 146.0 | 368.6 [88%] | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| ΔΔEInt | 280.4 | 30.6 | 311.0 (85%) | ||||
| ΔΔEPrep | 97.4 | −43.3 | 54.1 (15%) | ||||
| Δ(ΔEInt + ΔEPrep) | 377.8 | −12.6 | 365.2 | ||||
| SiF4 | ΔEPauli | 957.8 | 961.9 | 831.5 | 4.1 | −130.4 | −126.3 |
| ΔECoul | −10558.3 | −10406.3 | −10370.2 | 152.0 | 36.1 | 188.2 [66%] | |
| ΔEOrb | −4277.9 | −4317.1 | −4182.5 | −39.2 | 134.7 | 95.5 [34%] | |
|
|||||||
| ΔΔEInt | 117.0 | 40.4 | 157.3 (62%) | ||||
| ΔΔEPrep | 154.8 | −59.5 | 95.3 (38%) | ||||
| Δ(ΔEInt + ΔEPrep) | 271.7 | −19.1 | 252.6 | ||||
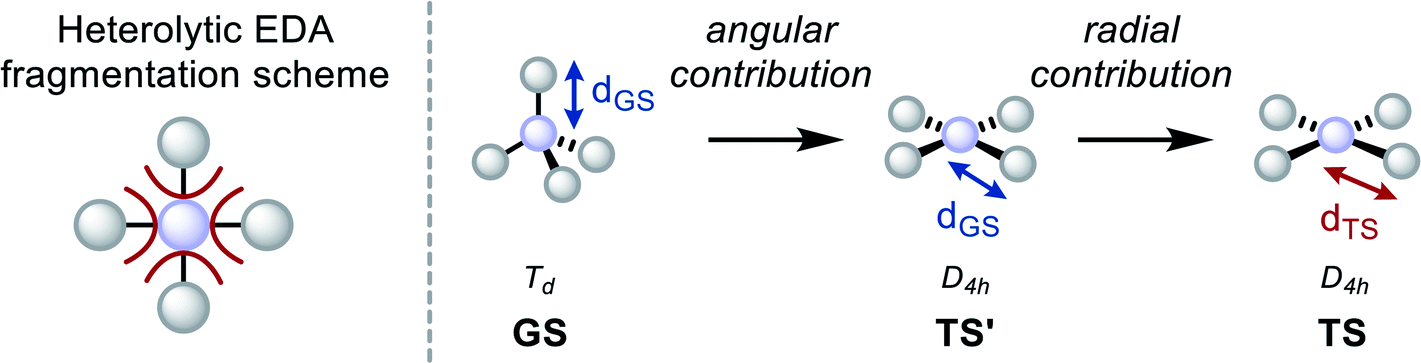
The energy differences upon planarization for SiH4 reveal that the barrier is dominated by a loss of ΔEOrb, whereas ΔΔECoul is less decisive (Table 2). The relief of ΔEPauli is a consequence of the bond elongation in the square planar state. This becomes evident by inspecting the results of angular and radial deformation contributions. Upon distorting the ground state to square planarity, while conserving the bond lengths (“angular contribution”), the major portion of ΔEOrb is lost, but ΔEPauli increases. Subsequent bond length relaxation (“radial contribution”) leads to a substantial reduction in ΔEPauli and a further reduction of ΔEOrb. Thus, the bond elongation in the TS is a result of ΔEPauli-lowering, similar to the recently described effects for CH-bond lengths in the series of spn (n = 1–3) hybridized carbon atoms.111 The differences in preparation energy (ΔΔEPrep) need to be added to reproduce the final inversion barrier energies. ΔΔEPrep can be understood as the part of the inversion barrier caused by substituent–substituent repulsion. For SiH4, ΔΔEPrep is relatively small.
The situation is different for SiF4. Here, the primary cause for the inversion barrier is the loss in ΔECoul (Table 2) and a larger relative amount of ΔΔEPrep. As can be seen from the angular/radial analysis, both energies are mainly the consequence of the bending deformation. Remarkably, ΔEOrb even increases when going from the ground state to the D4h state with GS-bond lengths. This means that the covalent bonding of the fluorides to silicon is more effective in a compressed D4h state. As for SiH4, relaxation of the bond lengths to the optimized distance leads to an overall diminished ΔEOrb and ΔEPauli. The larger value of ΔΔEPrep can be understood as a consequence of electrostatic repulsion of the more negatively polarized fluorides and actual intersubstituent steric Pauli repulsion.
Comparing the contributions in bond energy changes and the percental contribution of the preparation energy on the overall barrier (values in parentheses in the Table 2) emphasizes the statements. The inversion barrier in SiH4 is dominated by an energy loss in ΔEOrb, but the barrier in SiF4 is due to a loss in ΔECoul and a larger substituent–substituent repulsion (ΔΔEPrep). Indeed, the loss of ΔEOrb in the square planar state is not decisive at all. This picture is in line with the interpretation from real-space bonding analyses, which do not rely on artificially chosen reference states (cf. increased covalent bonding in the D4h state of SiF4, see Table 1).
Conclusions
The trigonal inversion of ER3 species is well understood and this knowledge has been used in various contexts (e.g., phosphines can serve as chiral ligands whereas amines cannot). In contrast, the inversion of ER4 species and their configurational stability has been virtually unexplored. The present study gives accurate barrier heights for p-block tetrahedron inversions through planar transition states. Barriers increase from group 13 to group 15 and decrease with π-donor/σ-acceptor substituents. Fundamentally, the second-order Jahn–Teller effect (SOJTE) is identified as a solid model to explain these general trends qualitatively. The core argument is: the closer the energies of the HOMO (b1g) and the LUMO (a2u) are in the D4h symmetric inversion transition state, the stronger they mix, and the more energy is released upon relaxation into the tetrahedral ground state (see step-by-step explanation in the introduction). In turn, a larger energy release for D4h → Td corresponds to a larger inversion barrier Td → D4h → Td (Fig. 7). The inverse correlation of HOMO–LUMO gap and energy also agrees with Pearson's principle of maximum hardness – the larger the gap, the more stable (the lower) the inversion transition state.112 Electronegative central elements lower the LUMO (a2u) energy; the inversion barrier increases. With more electronegative substituents, the HOMO (b1g) energy is lowered; the inversion barrier decreases (σ-acceptor effect). π-Donor substituents increase the LUMO energy; the inversion barrier decreases (π-donor effect). Interestingly, these substituent effects are inverse to that found for carbon-based tetrahedra, owing to the inverted FMO symmetries (cf.Fig. 2A).4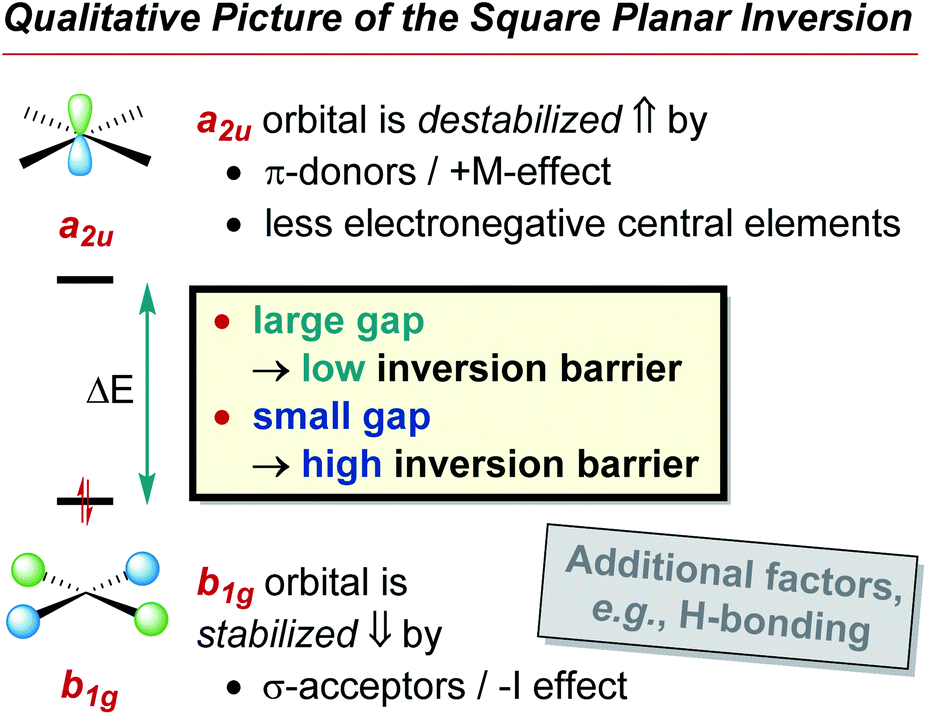 | ||
| Fig. 7 Simplified presentation of the factors that determine the height of the planar inversion barrier of tetrahedral p-block element-based species, for which the substituents are more electronegative than the central elements. | ||
Correlations between FMO gap and inversion barrier are obtained for hydrides, sustain for sole σ-acceptor substituents, but almost vanish for π-donor groups. NBO, EDA, and QTAIM calculations shed light on these deviations. For less electronegative substituents, the tetrahedral preference is determined by optimal orbital overlap, thus conforming to the SOJTE-model. For σ-acceptor/π-donor substituents, the tetrahedral preference is dominated by favorable electrostatics and diminished ligand–ligand repulsion. Strikingly, the covalency in Td and D4h configurations is almost equal in compounds such as SiF4 due to the favorable π-back bonding in the planar state.
There might be more than only fundamental interest in “molecular fluctionality”.113 One-third of the compounds studied in this contribution show transition state energies corresponding to configurational instability at ambient to elevated temperatures. One prominent example is Al(OH)4−, which is right now exhibiting tetrahedral inversion at tonne scale (Baeyer process) without being recognized.
What are the consequences of low inversion barrier heights in our geosphere (aluminates) or materials properties (silicones)? Are there features that can be achieved by exploiting tetrahedral “hinges” for three-dimensional connectivity with shape “adaptive” behavior?114 Another aspect is the connection of molecular flexibility with the phenomenon of Lewis acidity. It has been noticed that the energy required to deform a Lewis acid into the structure of the Lewis adduct effectively quenches Lewis acidity.115,116 In other words, compounds with a low deformation energy yield potentially strong Lewis acids. Of course, the deformation energy of tetrahedral compounds is entangled with the inversion barrier. Hence, the knowledge of tetrahedral inversion offers strategies to design Lewis acids that garner strengths not only from electron withdrawal but also from a low structural deformation penalty. Indeed, this has been proven most recently.117,118
The herein-developed perspective of the process of tetrahedral inversion might be in parts too simplified (neglected effects of vibronic coupling constants and higher excited states). Still, it bolsters our fundamental understanding of molecular flexibility and might propel further realizations of anti-van't-Hoff–Le-Bel configurations by guided choice.
Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
All authors produced and interpreted the data. L. M. S. and L. G. wrote the manuscript. L. G. supervised the project.Conflicts of interest
No conflict of interest is declared by the authors.Acknowledgements
Prof. H.-J. Himmel is thanked for his constant support. Financial support was provided by the DFG (GR5007/2-1) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 948708). L. M. S. is grateful to the Studienstiftung des deutschen Volkes for a scholarship. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster).Notes and references
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley, Hoboken, NJ, 2013 Search PubMed.
- R. J. Gillespie, Coord. Chem. Rev., 2008, 252, 1315–1327 CrossRef CAS.
- H. J. Monkhorst, Chem. Commun., 1968, 1111–1112, 10.1039/C19680001111.
- R. Hoffmann, R. W. Alder and C. F. Wilcox, J. Am. Chem. Soc., 1970, 92, 4992–4993 CrossRef CAS.
- V. I. Minkin, R. M. Minyaev and R. Hoffmann, Russ. Chem. Rev., 2002, 71, 869–892 CrossRef CAS.
- S. Durmaz, J. N. Murrell and J. B. Pedley, J. Chem. Soc., Chem. Commun., 1972, 933–934, 10.1039/C39720000933.
- D. C. Crans and J. P. Snyder, J. Am. Chem. Soc., 1980, 102, 7152–7154 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, J. Am. Chem. Soc., 1993, 115, 7486–7492 CrossRef CAS.
- M. J. M. Pepper, I. Shavitt, P. V. R. Schleyer, M. N. Glukhovtsev, R. Janoschek and M. Quack, J. Comput. Chem., 1995, 16, 207–225 CrossRef CAS.
- D. Röttger and G. Erker, Angew. Chem., Int. Ed., 1997, 36, 812–827 CrossRef.
- R. Keese, Chem. Rev., 2006, 106, 4787–4808 CrossRef CAS PubMed.
- G. Merino, M. A. Méndez-Rojas, A. Vela and T. Heine, J. Comput. Chem., 2007, 28, 362–372 CrossRef CAS PubMed.
- V. Vassilev-Galindo, S. Pan, K. J. Donald and G. Merino, Nat. Rev. Chem., 2018, 2, 0114 CrossRef CAS.
- L. Leyva-Parra, L. Diego, O. Yañez, D. Inostroza, J. Barroso, A. Vásquez-Espinal, G. Merino and W. Tiznado, Angew. Chem., Int. Ed., 2021, 60, 8700–8704 CrossRef CAS PubMed.
- M. B. Krogh-Jespersen, J. Chandrasekhar, E. U. Wuerthwein, J. B. Collins and P. von Ragué Schleyer, J. Am. Chem. Soc., 1980, 102, 2263–2268 CrossRef CAS.
- D. A. Dixon and A. J. Arduengo, J. Phys. Chem., 1987, 91, 3195–3200 CrossRef CAS.
- D. A. Dixon and A. J. Arduengo, J. Chem. Soc., Chem. Commun., 1987, 498–500, 10.1039/C39870000498.
- E.-U. Würthwein and P. von Ragué Schleyer, Angew. Chem., Int. Ed., 1979, 18, 553–554 CrossRef.
- A. I. Boldyrev, P. von Ragué Schleyer and R. Keese, Mendeleev Commun., 1992, 2, 93–95 CrossRef.
- P. Belanzoni, G. Giorgi, G. F. Cerofolini and A. Sgamellotti, J. Phys. Chem. A, 2006, 110, 4582–4591 CrossRef CAS PubMed.
- P. Belanzoni, G. Giorgi, G. F. Cerofolini and A. Sgamellotti, Theor. Chem. Acc., 2006, 115, 448–459 Search PubMed.
- L. Fang, C. Zhang, X. Cao and Z. Cao, J. Phys. Chem. C, 2020, 124, 18660–18669 CrossRef CAS.
- Y. Zhang, C. Zhang, Y. Mo and Z. Cao, Chem.–Eur. J., 2021, 27, 1402–1409 CrossRef CAS PubMed.
- D. Szieberth, M. Takahashi and Y. Kawazoe, J. Phys. Chem. A, 2009, 113, 707–712 CrossRef CAS PubMed.
- P. v. R. Schleyer and A. E. Reed, J. Am. Chem. Soc., 1988, 110, 4453–4454 CrossRef CAS.
- S.-D. Li, C.-Q. Miao, J.-C. Guo and G.-M. Ren, J. Am. Chem. Soc., 2004, 126, 16227–16231 CrossRef CAS PubMed.
- S.-D. Li, G.-M. Ren and C.-Q. Miao, Inorg. Chem., 2004, 43, 6331–6333 CrossRef CAS PubMed.
- T. N. Gribanova, R. M. Minyaev and V. I. Minkin, Russ. J. Gen. Chem., 2005, 75, 1651–1658 CrossRef CAS.
- S.-D. Li, J.-C. Guo, C.-Q. Miao and G.-M. Ren, J. Phys. Chem. A, 2005, 109, 4133–4136 CrossRef CAS PubMed.
- S.-D. Li and C.-Q. Miao, J. Phys. Chem. A, 2005, 109, 7594–7597 CrossRef CAS PubMed.
- J.-C. Guo and S.-D. Li, J. Mol. Struct.: THEOCHEM, 2007, 816, 59–65 CrossRef CAS.
- W. Tiznado, N. Perez-Peralta, R. Islas, A. Toro-Labbe, J. M. Ugalde and G. Merino, J. Am. Chem. Soc., 2009, 131, 9426–9431 CrossRef CAS PubMed.
- Y. Li, F. Li, Z. Zhou and Z. Chen, J. Am. Chem. Soc., 2011, 133, 900–908 CrossRef CAS PubMed.
- J.-C. Guo, C.-Q. Miao and G.-M. Ren, Comput. Theor. Chem., 2014, 1032, 7–11 CrossRef CAS.
- J. Xu and Y.-h. Ding, J. Comput. Chem., 2015, 36, 355–360 CrossRef CAS PubMed.
- M.-J. Sun, X. Cao and Z. Cao, Nanoscale, 2018, 10, 10450–10458 RSC.
- V. S. Thimmakondu and K. Thirumoorthy, Comput. Theor. Chem., 2019, 1157, 40–46 CrossRef CAS.
- M.-h. Wang, X. Dong, Z.-h. Cui, M. Orozco-Ic, Y.-h. Ding, J. Barroso and G. Merino, Chem. Commun., 2020, 56, 13772–13775 RSC.
- Y. Wang, Y. Li and Z. Chen, Acc. Chem. Res., 2020, 53, 887–895 CrossRef CAS PubMed.
- C. Zhang, Z. Tian and W. Jia, J. Phys. Chem. A, 2021, 125, 843–847 CrossRef CAS PubMed.
- J. M. Lehn, in Top. Curr. Chem., Springer Berlin Heidelberg, 1970, vol. 15/3, ch. 2, pp. 311–377 Search PubMed.
- A. Rauk, L. C. Allen and K. Mislow, Angew. Chem., Int. Ed., 1970, 9, 400–414 CrossRef CAS.
- A. I. Boldyrev and O. P. Charkin, J. Struct. Chem., 1985, 26, 451–475 CrossRef.
- C. C. Levin, J. Am. Chem. Soc., 1975, 97, 5649–5655 CrossRef CAS.
- W. Cherry and N. Epiotis, J. Am. Chem. Soc., 1976, 98, 1135–1140 CrossRef CAS.
- R. F. See, A. D. Dutoi, K. W. McConnell and R. M. Naylor, J. Am. Chem. Soc., 2001, 123, 2839–2848 CrossRef CAS PubMed.
- P. Schwerdtfeger, L. J. Laakkonen and P. Pyykkö, J. Chem. Phys., 1992, 96, 6807–6819 CrossRef CAS.
- P. Schwerdtfeger, P. D. W. Boyd, T. Fischer, P. Hunt and M. Liddell, J. Am. Chem. Soc., 1994, 116, 9620–9633 CrossRef CAS.
- M. Atanasov and D. Reinen, J. Phys. Chem. A, 2001, 105, 5450–5467 CrossRef CAS.
- M. Atanasov and D. Reinen, J. Am. Chem. Soc., 2002, 124, 6693–6705 CrossRef CAS PubMed.
- L. T. Xu, T. Y. Takeshita and T. H. Dunning, Theor. Chem. Acc., 2014, 133, 1493 Search PubMed.
- S. Pelzer, K. Wichmann, R. Wesendrup and P. Schwerdtfeger, J. Phys. Chem. A, 2002, 106, 6387–6394 CrossRef CAS.
- Z. Varga, P. Verma and D. G. Truhlar, J. Phys. Chem. A, 2019, 123, 301–312 CrossRef CAS PubMed.
- B. M. Gimarc, Acc. Chem. Res., 1974, 7, 384–392 CrossRef CAS.
- A. I. Boldyrev and J. Simons, J. Am. Chem. Soc., 1998, 120, 7967–7972 CrossRef CAS.
- M. Driess, J. Aust, K. Merz and C. van Wullen, Angew. Chem., Int. Ed., 1999, 38, 3677–3680 CrossRef CAS PubMed.
- X. Li, L.-S. Wang, A. I. Boldyrev and J. Simons, J. Am. Chem. Soc., 1999, 121, 6033–6038 CrossRef CAS.
- L.-S. Wang, A. I. Boldyrev, X. Li and J. Simons, J. Am. Chem. Soc., 2000, 122, 7681–7687 CrossRef CAS.
- D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 501–502 CrossRef CAS PubMed.
- A. I. Boldyrev, X. Li and L.-S. Wang, Angew. Chem., Int. Ed., 2000, 39, 3307–3310 CrossRef CAS PubMed.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed.
- E. J. Thompson, T. W. Myers and L. A. Berben, Angew. Chem., Int. Ed., 2014, 53, 14132–14134 CrossRef CAS PubMed.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed. Engl., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- K. Lee, A. V. Blake, A. Tanushi, S. M. McCarthy, D. Kim, S. M. Loria, C. M. Donahue, K. D. Spielvogel, J. M. Keith, S. R. Daly and A. T. Radosevich, Angew. Chem., Int. Ed., 2019, 58, 6993–6998 CrossRef CAS PubMed.
- F. Ebner, H. Wadepohl and L. Greb, J. Am. Chem. Soc., 2019, 141, 18009–18012 CrossRef CAS PubMed.
- T. M. Bass, C. R. Carr, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2020, 59, 13517–13523 CrossRef CAS PubMed.
- F. Ebner, L. M. Sigmund and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 17118–17124 CrossRef CAS PubMed.
- L. M. Sigmund and L. Greb, Chem. Sci., 2020, 11, 9611–9616 RSC.
- P. Ghana, J. Rump, G. Schnakenburg, M. I. Arz and A. C. Filippou, J. Am. Chem. Soc., 2021, 143, 420–432 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- K. Yoshizawa and A. Suzuki, Chem. Phys., 2001, 271, 41–54 CrossRef CAS.
- H. C. Longuet-Higgins and C. A. Coulson, Proc. R. Soc. London, Ser. A, 1956, 235, 537–543 CAS.
- U. Öpik and M. H. L. Pryce, Proc. R. Soc. London, Ser. A, 1957, 238, 425–447 Search PubMed.
- R. F. W. Bader, Can. J. Chem., 1962, 40, 1164–1175 CrossRef CAS.
- I. B. Bersuker, Chem. Rev., 2001, 101, 1067–1114 CrossRef CAS PubMed.
- I. B. Bersuker, Chem. Rev., 2013, 113, 1351–1390 CrossRef CAS PubMed.
- I. B. Bersuker, Chem. Rev., 2021, 121, 1463–1512 CrossRef CAS PubMed.
- R. G. Pearson, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2104–2106 CrossRef CAS PubMed.
- R. G. Pearson, J. Mol. Struct.: THEOCHEM, 1983, 103, 25–34 CrossRef.
- R. G. Pearson, Symmetry Rules for Chemical Reactions: Orbital Topology and Elementary Processes, J. Wiley & Sons, New York, 1976 Search PubMed.
- H. A. Jahn and E. Teller, Proc. R. Soc. London, Ser. A, 1937, 161, 220–235 CAS.
- W. Hermoso, A. R. Ilkhani and I. B. Bersuker, Comput. Theor. Chem., 2014, 1049, 109–114 CrossRef CAS.
- L. S. Bartell, J. Chem. Educ., 1968, 45, 754 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1969, 91, 4947–4955 CrossRef CAS.
- W. Cherry, N. Epiotis and W. T. Borden, Acc. Chem. Res., 1977, 10, 167–173 CrossRef CAS.
- J. K. Burdett, Chem. Soc. Rev., 1978, 7, 507–526 RSC.
- F. Cimpoesu and K. Hirao, in Adv. Quantum Chem., Academic Press, 2003, vol. 44, pp. 369–387 Search PubMed.
- S. Kozuch, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2013, 135, 19282–19291 CrossRef CAS PubMed.
- I. B. Bersuker, N. N. Gorinchoi and V. Z. Polinger, J. Mol. Struct., 1992, 270, 369–380 CrossRef CAS.
- A. F. Saturno, Theor. Chim. Acta, 1967, 8, 273–276 CrossRef CAS.
- A. Najibi and L. Goerigk, J. Comput. Chem., 2020, 41, 2562–2572 CrossRef CAS PubMed.
- E. J. Baerends, O. V. Gritsenko and R. van Meer, Phys. Chem. Chem. Phys., 2013, 15, 16408–16425 RSC.
- R. van Meer, O. V. Gritsenko and E. J. Baerends, J. Chem. Theory Comput., 2014, 10, 4432–4441 CrossRef CAS PubMed.
- J. C. Southern, in Materials & Equipment/Whitewares: Ceramic Engineering and Science Proceedings, ed. W. M. Carty, The American Ceramic Society, Westerville, OH, 2nd edn, 2001, vol. 22, ch. 12, p. 59 Search PubMed.
- W. Kutzelnigg, Angew. Chem., Int. Ed., 1984, 23, 272–295 CrossRef.
- Z.-L. Wang, H.-S. Hu, L. von Szentpály, H. Stoll, S. Fritzsche, P. Pyykkö, W. H. E. Schwarz and J. Li, Chem.–Eur. J., 2020, 26, 15558–15564 CrossRef CAS PubMed.
- P. R. Wells, in Progress in Physical Organic Chemistry, ed. A. J. Streitwieser and R. W. Taft, John Wiley and Sons, Inc., New York, 1968, vol. 6, ch. 3, pp. 111–145 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- C. Outeiral, M. A. Vincent, Á. M. Pendás and P. L. A. Popelier, Chem. Sci., 2018, 9, 5517–5529 RSC.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed.
- M. v. Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 Search PubMed.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- M. v. Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 Search PubMed.
- D. Rodrigues Silva, L. de Azevedo Santos, M. P. Freitas, C. F. Guerra and T. A. Hamlin, Chem.–Asian J., 2020, 15, 4043–4054 CrossRef CAS PubMed.
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed.
- A. Krapp, K. K. Pandey and G. Frenking, J. Am. Chem. Soc., 2007, 129, 7596–7610 CrossRef CAS PubMed.
- R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3260–3272 CrossRef CAS PubMed.
- C. Mohapatra, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 10429–10432 CrossRef CAS PubMed.
- P. Vermeeren, W.-J. van Zeist, T. A. Hamlin, C. Fonseca Guerra and F. M. Bickelhaupt, Chem.–Eur. J., 2021, 27, 7074–7079 CrossRef CAS PubMed.
- R. G. Pearson, Acc. Chem. Res., 1993, 26, 250–255 CrossRef CAS.
- M. L. McKee, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 943–951 CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- R. M. Guidry and R. S. Drago, J. Am. Chem. Soc., 1973, 95, 759–763 CrossRef CAS.
- H. Fleischer, Eur. J. Inorg. Chem., 2001, 2001, 393–404 CrossRef.
- D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845–15851 CrossRef CAS PubMed.
- A. Ben Saida, A. Chardon, A. Osi, N. Tumanov, J. Wouters, A. I. Adjieufack, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2019, 58, 16889–16893 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational details, all calculated data, additional plots, further discussions. All xyz coordinates and molecular energies are given in a separate file. See DOI: 10.1039/d1sc05395g |
| This journal is © The Royal Society of Chemistry 2022 |