
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiving in a transient world: ICP-MS reinvented via time-resolved analysis for monitoring single events
M.
Resano
 *a,
M.
Aramendía
ab,
E.
García-Ruiz
a,
A.
Bazo
a,
E.
Bolea-Fernandez
c and
F.
Vanhaecke
*c
*a,
M.
Aramendía
ab,
E.
García-Ruiz
a,
A.
Bazo
a,
E.
Bolea-Fernandez
c and
F.
Vanhaecke
*c
aDepartment of Analytical Chemistry, Aragón Institute of Engineering Research (I3A), University of Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: mresano@unizar.es
bCentro Universitario de la Defensa de Zaragoza, Carretera de Huesca s/n, 50090 Zaragoza, Spain
cGhent University, Department of Chemistry, Atomic & Mass Spectrometry – A&MS Research Unit, Campus Sterre, Krijgslaan 281-S12, 9000 Ghent, Belgium. E-mail: frank.vanhaecke@Ugent.be
First published on 14th March 2022
Abstract
After 40 years of development, inductively coupled plasma-mass spectrometry (ICP-MS) can hardly be considered as a novel technique anymore. ICP-MS has become the reference when it comes to multi-element bulk analysis at (ultra)trace levels, as well as to isotope ratio determination for metal(loid)s. However, over the last decade, this technique has managed to uncover an entirely new application field, providing information in a variety of contexts related to the individual analysis of single entities (e.g., nanoparticles, cells, or micro/nanoplastics), thus addressing new societal challenges. And this profound expansion of its application range becomes even more remarkable when considering that it has been made possible in an a priori simple way: by providing faster data acquisition and developing the corresponding theoretical substrate to relate the time-resolved signals thus obtained with the elemental composition of the target entities. This review presents the underlying concepts behind single event-ICP-MS, which are needed to fully understand its potential, highlighting key areas of application (e.g., single particle-ICP-MS or single cell-ICP-MS) as well as of future development (e.g., micro/nanoplastics).
1. Introduction: adapting to an ephemeral world
For a long time, Analytical Chemistry has been coping with the challenge of providing as much information as possible from ever smaller sample amounts. As an example, bioanalysis is driven towards the development of methods that can provide more relevant information in a faster and less invasive way for the benefit of the patient. The concept of personalized medicine is affecting the way health control is addressed, and strong efforts are made to develop methods capable of assisting in the regular/continuous automated monitoring of patients.The current Covid-19 pandemic situation has only accelerated this trend, further showing the need to develop methods that require a few droplets of blood or other biofluids only (e.g., dried blood spots1,2). This type of analytical methodology enables patients to collect their own samples at home, in a simple and painless way, and send them to the laboratory by postal mail. It is a field that is very likely to grow, because it improves the quality of life of patients needing frequent controls, particularly when they live in remote areas with no hospitals nearby or when their mobility is compromised.
The development of such methods only brings advantages to the patients but poses some challenges for the professionals who need to carry out the corresponding analyses. Among other issues, such as potential contamination or inadequate sampling, the sample volume is limited as, instead of obtaining as much sample as desired (typically 5–10 mL), volumes in the range of 10–100 μL only will become standard. Therefore, there is a need to develop novel analytical methods that minimize sample consumption.3
Self-evidently, in the context of this review paper, we are referring to analysis with instruments that can hardly be miniaturized, such that this strategy (sending the sample to the specialized lab) makes sense, while for the determination of some clinical parameters the use of sensors enables development of direct approaches relying on remote in situ monitoring. However, when multi-element analysis at trace levels is required, the use of inductively coupled plasma-mass spectrometry (ICP-MS) provides an unparalleled performance in terms of sample throughput and detection power, but this type of instrumentation should be operated in a lab, under sufficiently “clean” conditions, by an experienced analyst.
When analyzing such “micro-samples” with ICP-MS, the traditional sample introduction system, consuming sample solution at flows of about 1 mL min−1, needs to be replaced with an alternative device limiting sample uptake rates, such as a miniaturized nebulizer/spray chamber combination with or without a desolvation unit,4 a flow injection (FI) device,5 a laser ablation (LA) unit6 or an electrothermal vaporization (ETV) set-up.7 Some of these sample introduction strategies give rise to fairly short transient signals, while the instrumentation still is expected to acquire trace multi-element information from them. New sample introduction devices (e.g., based on microfluidics8) and faster detectors are therefore needed, and this is an area where substantial improvements have been made in recent years.
The expectations that the detection systems have to match become even much more stringent if the transient nature of the signals is not caused by the limited amount of sample available and thus, the adapted way of sample introduction, but by the very nature of the sample itself, i.e., if ICP-MS is no longer used for “bulk analysis” of homogeneous/homogenized solutions, but for suspensions containing individual entities for which elemental information is desired. This type of application changes the way in which ICP-MS instrumentation needs to be operated and the corresponding data are handled.
In this sense, for many years, atomic spectrometric techniques in general, and ICP-MS in particular, have been deployed with the aim of achieving average elemental concentrations. This strategy assumes that the target sample presents a high degree of homogeneity, and, thus, this average value is representative and allows one to adequately assess the situation. Of course, in practice, an uncertainty budget always accompanies such value. Many contributions related to the sample itself and all of the individual steps the measurement protocol consists of affect the overall uncertainty. Only in particular cases (e.g., direct analysis of solid microsamples) this uncertainty is dominated by the contribution stemming from the sample heterogeneity.
Many examples of a situation in which average results suffice can be given, such as cases in which a patient has his/her blood analyzed to check if the levels of some elements are abnormally high or low. The relevant aspect here is to compare the average value based on a few replicate measurements with a threshold, to assess the occurrence of a potential problem. A high concentration of elements such as Co, Cr, Ni, or Ti can, e.g., indicate prosthesis malfunction;9,10 a high content level of Hg may be the result of excessive consumption of food from marine origin,11 such that a dietary change may be considered; verifying that the concentration of Li is within the therapeutic levels is required when a patient is treated with this metal for a major depressive disorder, as too high levels may be toxic, while too low levels may not suffice in solving the disorder.12 These are just a few cases related with biomonitoring, but analogous situations are encountered in environmental and food analysis too, as well as in many other fields. Bulk analysis, therefore, has been and still is the norm in most cases.
However, new applications have shifted this paradigm. One important case is cell analysis. Cells are complex structures and, unfortunately, cell cultures are not necessarily homogenous. In fact, both in vivo and in vitro, a collection of cells typically contains cells of different ages and in different stages. Therefore, when aiming at establishing the cellular uptake of a particular element in a cell population, the traditional approach for achieving bulk (averaged) information is of limited value only. The quantitative study of single cells, which consists of counting and sorting of cells, is called flow cytometry and plays an essential role in today's medical sciences and diagnostics.13,14 This is easy to understand if we think of a simple example. In the case of chemotherapeutic treatment with a metallodrug (e.g., cisplatin), the success depends on ensuring that all cancer cells take up a sufficient dose, while ideally healthy cells do not take up too much, such that we can selectively kill tumoral cells without (or with minimal) side effects. Bulk analysis is characterized by just providing average values, but this approach may be misleading in this kind of situation in which knowing the inter-cell variation may be critical. As a result, methods for (high-throughput) single cell analysis capable of providing information on inter-cell variability are highly demanded in cell biology.15,16
Another important and novel application concerns the characterization of metallic engineered nanoparticles (ENPs). While ENPs are already used in many biomedical applications, their increasing presence in even more types of consumer products is inevitably accompanied by their release into the environment and their incorporation into the human body. As a result, there is a growing demand for the development of methodologies capable of detecting and fully characterizing ENPs,17 particularly in all types of biomedical and environmental samples. However, a full characterization does not only mean mass concentration (mass of the element concerned per unit of volume) and average size, but also information on their size distribution and particle number concentration (PNC, i.e., number of particles per unit of volume) is of crucial importance. Furthermore, these methods should be capable of differentiating such particles from other chemical forms in which the same metal may be present in the samples of interest (e.g., ionic species). In other words, it is necessary to obtain signals corresponding to individual NPs, even when they are present in complex media. The methodologies proposed should be selective and require none or minimal sample preparation only, to avoid species interconversion.
Finally, also the occurrence of micro and nanoplastics is another emerging threat to the environment and to human health18 and their proper characterization shares many of the characteristics described before. A population of particulate plastics typically resulting from degradation of plastic debris cannot be expected to be homogeneous in nature (neither in size, nor in polymer composition) and, therefore, information from individual plastic particles needs to be obtained.19
It is actually a bit surprising that the use of ICP-MS can help in solving all the problems mentioned before. Such applications would probably have been considered as out of scope just a decade ago, at least without the hyphenation of a separation technique to ICP-MS, while now, they can be carried through in a simple (at least conceptually) way: i.e., by modifying the frequency of data acquisition, thus effectively uncovering information that was hidden by the averaging of signals.
The purpose of this review is therefore to explain how ICP-MS operating in time-resolved analysis (TRA) mode, also known as single event mode (e.g., single particle (SP) or single cell (SC), depending on the target entities), has effectively expanded its field of application beyond expectations, meeting scientific and societal needs. The fundamentals of such working strategy will be presented, and selected applications will be discussed.
It is important to indicate that other strategies that require the coupling of an alternative sample introduction system (e.g., LA20) or separation technique (e.g., chromatography or flow field fractionation –FFF-) will not be covered in this review, but will only be mentioned when appropriate, for the sake of focus and simplicity. Furthermore, this review discusses the basic fundamental concepts and will refer to essential literature for specific topics, such as a detailed discussion of the equations supporting the calculations in single event mode.
2. ICP-MS and the need for speed
As discussed in the previous section, there are basically two ways in which an ICP-MS can be operated. The first is the traditional one, and still is perfectly valid for all those situations in which the purpose of the analysis is to achieve averaged or “bulk” information. Such approach is represented in Fig. 1a. In short, a sample is pretreated (if needed, which is often the case) and then, after digestion, extraction and/or dilution, a portion of the resulting homogeneous liquid is pumped towards the sample introduction system. The liquid will then undergo nebulization, using an argon (Ar) gas to produce a fine aerosol that is further processed in a spray chamber, such that it can be adequately handled by the Ar plasma, as described below. A large amount of sample (90–98%, depending on the nebulizer/spray chamber combination) will not be transformed into sufficiently small droplets (of a size of a few microns or less) during nebulization, will hence fall out due to gravity or will impact the spray chamber wall due to inertia and will finally end up in the waste solution. The fine aerosol leaving the spray chamber will then be transported to the plasma by this Ar gas flow, and the high plasma temperature (ionization temperature Tion ≈ 7500 K) will lead to desolvation, vaporization, atomization, and ionization, thus rendering most of the atoms in ionic form, such that they can be subsequently separated from one another on the basis of their mass (more accurately: mass-to-charge – m/z – ratio) and quantified by the mass spectrometer.21 | ||
| Fig. 1 (a) Scheme highlighting the main steps of an ICP-MS measurement in conventional bulk analysis mode; (b) scheme highlighting the main steps of an ICP-MS measurement in single event mode for analysis of discrete entities. | ||
The type of signal that is generated by this system is quasi stable. A typical sample flow varies between 200 and 1000 μL min−1, meaning that 1 mL of sample is aspirated in 60–300 s. The scanning speed of all types of current mass spectrometers is sufficient to provide enough data points for each of the target nuclides (ICP-MS does not measure elements, but their nuclides) in this period, such that data acquisition speed is not very critical. A typical value for acquisition duration would be of the order of 50 ms, thus resulting in 1200 data points (assuming that no detector settling time is needed, as it is often the case nowadays for single m/z monitoring) during 60 s of measurement. Since variations in this type of signal are not expected to be very significant, as the ions will arrive at the detector at a fairly constant flow, that is more than enough to obtain a representative average value.
And such is also the case when not only one nuclide but tens of them are measured, thus spreading the total acquisition time over all m/z ratios of interest. This is relevant because the majority of ICP-MS devices, such as the traditional quadrupole-based ICP-MS (q-ICP-MS) and the more sensitive high-resolution sector field ICP-MS (SF-ICP-MS), do not measure all the signals in a truly simultaneous fashion, but in fast sequential (e.g., some ms per isotope) mode, thus producing less measurement values for each nuclide as the total number of nuclides monitored increases while keeping the total acquisition time constant. The only exception in practice (multicollector ICP-MS devices are simultaneous but are typically dedicated to high-precision isotopic analysis, not to trace element determination) to this rule is time-of-flight ICP-MS (ICP-TOF-MS), which measures all the nuclides sampled from the plasma at the same moment in time.
When the nature of the signal changes and short transient events are to be measured (TRA mode), the situation becomes more challenging. It is necessary then to carefully optimize the acquisition parameters to monitor all the nuclides of interest in a much shorter period. The strategy for dealing with transient signals has already been abundantly discussed in the literature for years for a variety of sampling approaches (e.g., LA, ETV, FI, etc.) but, in such cases, the signals still lasted for a few seconds to hundreds of milliseconds.
A much more demanding case is depicted in Fig. 1b. This new “extreme” TRA mode tries to detect single events resulting from the introduction of discrete entities. In other words, the idea is to measure a single nano/microstructure on a one-by-one basis, whether it is a colloid, a nanoparticle (NP), a cell, a micro/nanoplastic particle, etc. The duration of such individual event is much shorter, below 1 ms, and each event will be different and can appear randomly within the total acquisition time. In order to cope with this situation, ultrafast signal acquisition is required and recording a sufficiently high number of events (for proper statistics) is mandatory.
The basic principle of this mode is based upon working with highly diluted suspensions containing the entities in question. In such way, for a considerable fraction of the time, only solvent will be aspirated and, unless the target analyte is also present in dissolved form, no signal will be generated, except for the background (BG). Then, at a given moment in time, when one of these small entities is aspirated and introduced into the ICP (it needs to be stressed that the transport efficiency is never 100%, and often is far below this value, such that not every individual entity will actually reach the ICP), a burst of ions will be generated in the plasma. This bunch of ions will travel towards the MS and the ions of a given m/z value present will be detected during a very short period of time. In other words, unlike what occurs in Fig. 1a, in which a constant flow of ions is reaching the detector all the time, almost no signal is detected in single event mode, until suddenly a pack of ions arrives (all ions almost together), thus generating an intense signal pulse above the BG (see Fig. 1b). Instead of a quasi-stable signal, a number of very short discrete pulses are detected.
The signal intensity of such pulse is directly proportional to the number of ions that contribute to it, that is, to the mass of the analyte present in the entity of interest, while the number of events detected in a defined period of time is proportional to the number concentration of entities in the suspension. Therefore, provided suitable calibration of the instrument sensitivity, sample uptake rate, and transport efficiency (TE), information on both the number of entities and the mass of the analyte present in them is simultaneously acquired.
Thus, by increasing the speed of data acquisition, new information is unraveled. There are several interesting aspects associated with this single event methodology that differ substantially from traditional approaches in analytical chemistry. One of these is the effect of sample dilution. Under normal conditions, diluting a sample may be beneficial (e.g., to mitigate matrix effects), but if the sample is diluted too much, the signal may become indistinguishable from the BG. In single event mode, this is not the case. Each entity will still give rise to a signal of the same magnitude, only the frequency with which such transient signals occur will become lower upon dilution. Hence, the sample needs to be diluted sufficiently to minimize the risk of aspiring two entities at the same time (double event), but not to the extent that the low frequency of events detected leads to excessively long measurement times, taking into account that a minimum number of such events should be recorded to obtain representative data.
Quite often the number of events recorded is about 2000. If the analyst wants to devote about 60 s to each sample and the TE of the sample introduction system under the conditions used is 5%, it will be thus necessary to aspirate 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entities in these 60 s. If the sample flow rate is 1 mL min−1, then the suspension must contain 40000000 entities per L.22 If the sample is diluted more, then a longer acquisition time will be required to record that number of events, but the risk of suffering from double events will further decrease.
000 entities in these 60 s. If the sample flow rate is 1 mL min−1, then the suspension must contain 40000000 entities per L.22 If the sample is diluted more, then a longer acquisition time will be required to record that number of events, but the risk of suffering from double events will further decrease.
The probability of occurrence of such double events can be calculated based on Poisson statistics. Still, the signal generated by each of the entities remains the same and is not affected at all by the dilution factor. In fact, further dilution may even help to improve the signal-to-background ratio (i.e., the ratio between the signal intensity generated by the entity and that of the BG). This is very unique in analytical chemistry. In fact, the concept of sensitivity changes, as it is no longer related to the concentration of the element in the solution, but with the absolute amount of it in the particular target entity. In an extreme scenario, if only a few particles are present in a suspension as large as the ocean that is aspirated during a long enough time, many of them will end up in the waste, but every particle reaching the detector will still produce a transient signal with an integrated signal intensity proportional to the absolute amount of analyte element in that particle. It is a case of 0 or 1, where the dilution factor plays no role.22 Only if the mass of the analyte present in the structure is too low, the corresponding signal will not be distinguishable from the BG signal. Thus, metallic ENPs of a larger size are easier to detect than those of a smaller size with the same chemical composition, while evidently, pure metallic NPs give rise to higher integrated signal intensities compared to, for instance, multi-metal oxide NPs of a similar size. For a specific cell, a certain number of atoms of analyte need to be present to allow quantitative determination of the absolute amount (mass) of that analyte per cell. And for a polymeric particle, the amount of C atoms needs to be above a threshold. That is where the sensitivity of the technique applies. Below a certain number of ions, no signal significantly different from the BG will be obtained.
The detection efficiency of an ICP-MS unit depends on the type of detector and a few other factors (e.g., the ionization energy of an element governs the ionization efficiency, while lighter ions are transported less effectively to the detector than heavier ones), but typical values range between 10−4 and 10−6. This entails that a minimum number of ions between 10000 and 1000000 are needed to produce a detectable signal. This is one limitation of the technique. The other major limitation is the potential occurrence of a significant BG signal. In single event mode, a pulse needs to be detected above the BG signal. Obviously, the lower the BG, the easier the event can be distinguished. However, this BG signal is sometimes not close to zero. This is often related to analyte ions in the dissolved phase of the suspension, i.e., outside the target nano/microstructures. Also, the occurrence of spectral interference, a common issue in ICP-MS, may enhance the BG signal, potentially hampering the detection of individual events.
In short, ICP-MS operated in signal event mode allows differentiation between the elements present in the target entities and those found in the dissolved phase of the suspension containing them, as the latter will produce a constant BG signal instead of a pulse, but only to some extent. If the BG signal becomes too high, then the signal from the single events will not be properly discerned.
These are general concepts that should be kept in mind for the different applications of single event-ICP-MS that will be discussed in the following sections, where also some related concepts (e.g., influence of the acquisition time on the signal-to-background ratio and means to overcome spectral overlap) will be discussed in detail.
3. Colloids, nanoparticles, and the big bang of single particle-ICP-MS
The use of ICP-MS in TRA mode is not completely novel. As discussed before, the use of some sampling introduction devices changes the nature of the signal obtained from quasi stable to transient.23 The attempt to go beyond that and be able to detect single events is more novel, but again not as much as one could anticipate. Prior to its application in ICP-MS, attempts to monitor discrete entities were made using atomic absorption spectrometry and ICP-optical emission spectrometry (ICP-OES) back in the 1960s to 80s, as described in detail by Montaño et al.24 Later, the group of Kawaguchi demonstrated the potential of ICP-MS to detect individual airborne particles in a series of works from the 1990s and early 2000s.25–27 Despite the novelty of these works, their immediate impact in terms of citations was moderate, as most of such citations originate from 2010 onwards only.In 2003, Degueldre and Favarger published their seminal paper,28 in which they demonstrated the use of single event-ICP-MS for the characterization of colloids of different compositions (alumina, clay, goethite and rutile) and already established the fundamental concepts described in the previous section. In that paper, these authors stated: “A feasibility study of the single particle analysis of water bearing colloid suspensions by ICP-MS has been conducted. The transient signal induced by the flash of ions due to the ionisation of a colloidal particle in the plasma torch can be detected and measured by the mass spectrometer. The intensity of the signal is determined by the size of the particles for the matrix elements and the frequency of the flashes is directly proportional to the concentration of particles in the initial colloidal suspension. After developing the theory of ion flash intensities, composition and detection, tests were performed on model colloids and on natural clay colloids…”. Degueldre et al. published a few more papers on this topic in the following years,29–32 devoted to different types of colloids.
Again, despite the promising results, the initial impact of these works was very moderate. A search in Scopus (02/02/2022) provides 242 citations for the first of these papers, but only 8 of those citations originate from papers published before 2010 (and four of those come from follow-up works of the authors of this initial work themselves).
It could be said that analytical chemistry already had a solution for a problem that did not exist yet at that time. However, this situation changed in the last decade with the increasing importance of nanotechnology and the need to characterize ENPs. Fig. 2a shows the number of publications reporting on single particle (SP)-ICP-MS and the trend upwards during the last decade can be clearly appreciated.
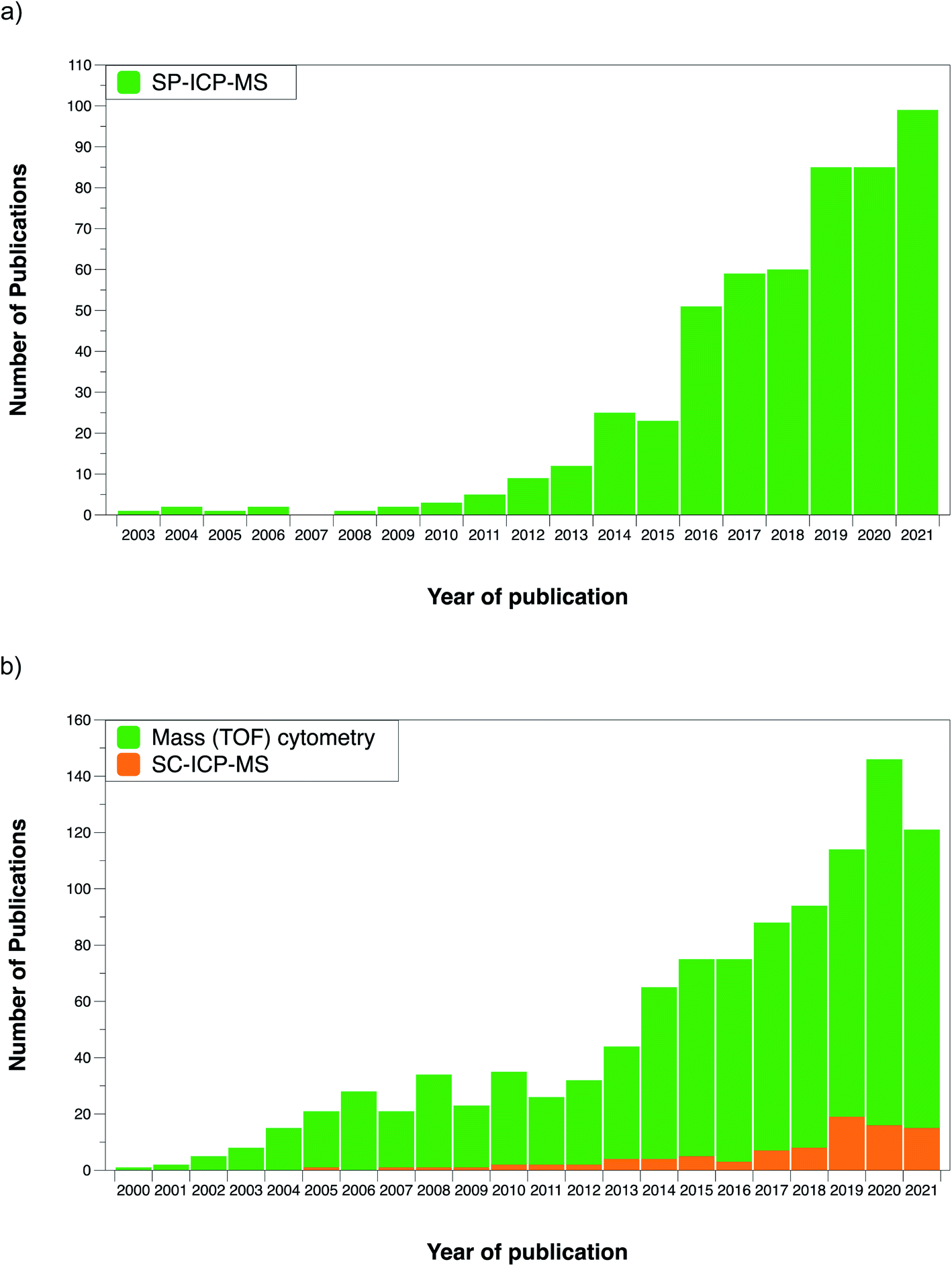 | ||
| Fig. 2 (a) Evolution of the number of publications devoted to single particle-ICP-MS over the last two decades; (b) evolution of the number of publications devoted to single cell-ICP-MS and to mass cytometry (using the CyTOF) over the last two decades. Source Scopus, January 2022. | ||
It can also be highlighted that several relevant articles, further cementing the methodology, were published at the beginning of the 2010–2020 decade, favoring the ulterior development of many real-world applications. But before discussing these works, we would like to point out to a few interesting articles published before that decade.
In 2005, Yau and Chan published a paper on “a novel detection scheme of trace elements using ICP-MS”.33 Basically, the approach consisted of “preconcentrating” the elements of interest onto suspended Fe(OH3) particles. The suspension could be then analyzed using ICP-MS in TRA mode, thus improving the limits of detection by a factor of 20. This paper reinforces the basic concept behind single event detection and one of its main advantages: concentrating the analyte onto these nanostructures improves the detection power because a very short but high pulse is easier to detect (providing the detector is fast enough) than a longer lasting but much less intense one. The signal-to-noise ratio drastically improves and the influence of spectral interference decreases, unless the interference occurs due to the presence of a parent element in the target nanostructures.
In 2009, Hu et al. published a work34 in which AuNPs were used as antibody tags to determine α-fetoprotein in serum by monitoring Au in TRA mode. This work links in fact two of the main application fields covered in this review, as tagging is a strategy widely used for analysis of cell compounds. In this regard, Lores-Padín et al. describe the advantages of using NPs for signal amplification in the context of biomolecule determination in a recent review paper.35 The work by Hu et al. was followed by another article by some of the same authors comparing figures of merit for the determination of Au-labeled IgG using ICP-MS in both conventional and single particle mode. The latter offered an order of magnitude of improvement in terms of limit of detection, at the cost of a slightly worse performance in terms of precision and linear range.36
Coming back to fundamental studies, in 2011 Laborda et al. devoted a work to the identification, characterization and determination of dissolved silver(I) and silver nanoparticles,37 exploiting and reinforcing all the concepts introduced in the works mentioned before, while Pace et al.38 published the most cited SP-ICP-MS article (425 citations so far, according to Scopus) to date, which was focused on methods to calculate the TE. These authors evaluated three different ways to calculate this parameter, which are represented in Fig. 3.
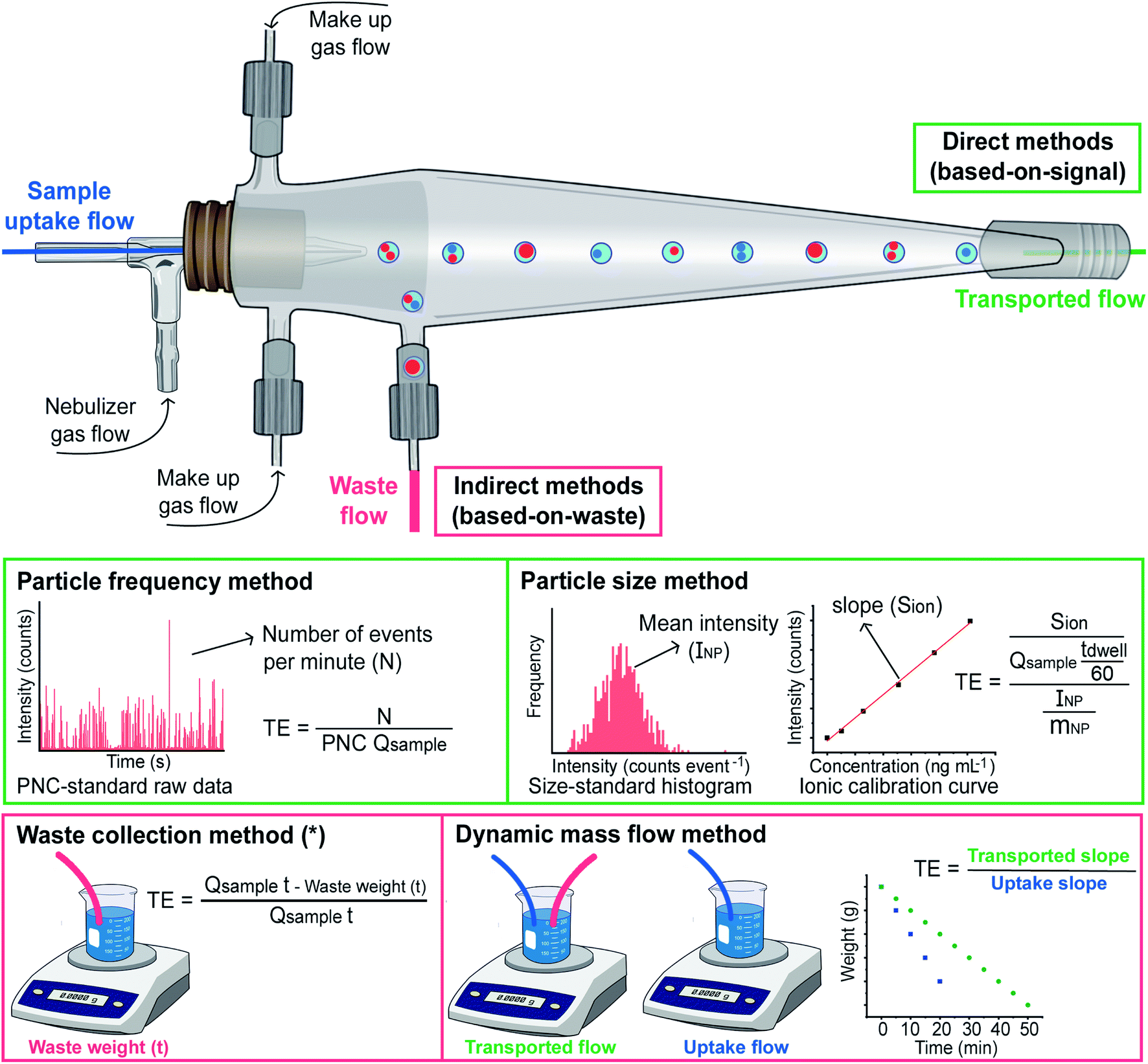 | ||
| Fig. 3 Scheme illustrating the four different approaches for calculating TE reported in the literature, where Qsample represents the sample flow uptake in mL min−1; mNP is the average particle mass in ng; and INP is expressed in counts. The dwell time (tdwell) is expressed in seconds, while the time indicated in the waste collection method (t) is expressed in minutes. *For the waste collection method, the weight of sample reaching the plasma can be calculated gravimetrically instead. | ||
The one labelled ‘waste collection’ is conceptually the simplest. It is based on an indirect approach, where both the volume of the waste exiting the spray chamber and the volume of the sample taken up are weighed after a sufficiently long measuring time. The weight or volume difference between the sample uptake and the waste stream is considered as the weight or volume of suspension reaching the plasma, and thus the TE can be calculated. However, this approach may not properly account for all the sources of potential losses (e.g., condensation) and, thus, is not widely used. Such approach was actually adapted from a previous work by Gustavsson.39 In practice, the sample uptake volume can also be calculated by multiplying the sample uptake flow by the measurement time, as shown in Fig. 3.
Pace et al. also proposed two novel approaches that are based on measuring what is actually entering the plasma. They rely on well-characterized nanoparticle reference samples and on the theory of SP-ICP-MS.38 One of these approaches is named ‘particle frequency’ determination, which requires a NP suspension with a known (preferably certified) PNC, such that by comparing the number of particles detected with the number present in the suspension consumed, the TE can be calculated when the sample flow rate is known. In principle, it needs to be noted that any type of NP can be used for this calibration (although this assumption needs to be further investigated), as the goal is not to establish the sensitivity (e.g., AgNPs can be used even if the goal is to characterize SiO2 NPs).
The other approach is called ‘particle size’. It is based on comparing the sensitivity for the target element using one (or more) monodisperse suspensions containing NPs of known size, shape, and chemical composition (so the mass can be calculated) containing the target analyte with that obtained for ionic standards of the same analyte. The difference should be related to the TE only. As discussed before, a NP will produce a signal or not, depending on whether it reaches the plasma or not, but for those NPs that do provide a signal, the TE can be considered as 100%.22 For the ionic flow, on the other hand, the TE will affect the mass flux into the plasma, and that is why, by comparing both sensitivities, this TE can be calculated.
The latter two are the most widely used approaches to this day. Which one should be preferred? This is not an easy question as, in fact, it is not so simple to differentiate between these two approaches in practice. The reason is the scarcity of reference materials with a certified PNC, among other reasons because their longer-term stability is limited. Thus, in many cases, authors apply the ‘particle frequency’ approach, but they determine the PNC based on the mass concentration and average particle size of the suspension. Therefore, this can hardly be considered as a true application of the ‘particle frequency’ method. For applying the ‘particle size’ approach, what is important is the availability of monodisperse suspensions of the target NPs of known size.
The personal experience of the authors is that the use of the ‘particle size’ approach can provide more accurate results in terms of sizing. The reason could be related to the fact that, in this approach, a calibration between ionic standards and NP suspensions is compared, and the difference is attributed to the TE, as discussed before. However, if other factors affect such difference (e.g., a difference in the ionization efficiency for ionic species and nanoparticles, which may occur depending on the measuring conditions, such as the sampling depth40), they will also be accounted for as a part of such TE factor, which will not occur when using the ‘particle frequency’ approach. Nevertheless, if that is the case, the TE obtained will not be a pure TE factor and, thus, determining the PNC via ‘particle size’ will render a biased result. From this point of view, using the ‘particle frequency’ approach for calculating the PNC seems recommended, as long as a suspension of NPs with a reliable PNC is available. Montaño et al. have previously discussed these aspects in detail in ref. 24.
Overall, the shortage of suitable certified reference NPs is a major issue affecting characterization via SP-ICP-MS. The uncertainties in the particle size and concentration of the standards affect the estimation of the TE and this is a critical value for single event-ICP-MS measurements, as it affects both the estimation of the PNC and of the mass, as can be seen in the equations displayed in Fig. 4. Table 1 displays the only NP reference materials available to date. It can be seen that, while a few of them provide reference size values, only one material reports a reference PNC (LGC5050). Thus, this can be seemed as one of the Achilles' heels of the technique, and the development of more CRMs with certified PNCs (and that offer sufficient stability) will help in reporting more accurate PNC values.
 | ||
| Fig. 4 Information provided by single event-ICP-MS after proper data processing and adequate calibrations. The symbols have the same meaning as used throughout the text (PNC, TE) or in Fig. 3 (N, tdwell, Qsample, Sion). INP and mNP have a slightly different meaning, as herein they refer to the intensity (in counts) and mass (in ng) of every particle, while in Fig. 3 (particle size method) they were used for the average of the distribution. Additional symbols are: MNP, representing the molar mass of the nanoparticle material; Mion, the molar mass of the analyte monitored; dP is the particle diameter, in nm; ρp is the particle density in g mL−1. | ||
| Manufacturer | Reference number | ENPs | Form | Main reference valuesa |
|---|---|---|---|---|
| a Only those related to size, PNC or chemical composition are indicated. b Reference materials 8012 (nominal size 30 nm) and 8013 (nominal size 60 nm) were formerly available, but they are currently out of stock. | ||||
| NIST (National Institute of Standards & Technology) | Standard Reference Material 1898 | Titanium dioxide nanomaterial | Dry agglomerated powder | Particle size (informational); elemental purity (information value); laser diffraction spectrometry particle size distribution for a water suspension |
| NIST | Reference Material 8011b | Gold nanoparticles (nominal 10 nm diameter) | Citrate-stabilized AuNPs nanoparticles in an aqueous suspension | Mean size (reference value); mass fraction (information values for Au, Cl and other species); particle size distribution values obtained with various techniques |
| NIST | Reference Material 8017 | Polyvinylpyrrolidone coated silver nanoparticles (nominal diameter 75 nm) | Lyophilized polyvinylpyrrolidone (PVP)-coated AgNP cake | Mean size (reference value); reference mass value for Ag; particle size distribution values for water suspensions obtained with various techniques, including SP-ICP-MS |
| LGC limited | Quality Control Material LGCQC5050 | Colloidal gold nanoparticles – nominal diameter 30 nm | Solution of colloidal spherical gold nanoparticles (citrate stabilised), suspended in water | Number particle concentration (assessed value); particle modal diameter and gold mass fraction (indicative values) |
| IRMM (Institute for Reference Materials and Measurements) | European Reference Material (ERM®) – FD100 | Colloidal silica water | Water suspension | Certified and indicative diameter values, obtained by various techniques |
| IRMM | ERM® – FD101b | Silica nanoparticles in aqueous solution | Water suspension | Certified and indicative diameter values, obtained by various techniques |
| IRMM | ERM® – FD102 | Mixture of silica nanoparticles in aqueous solution | Water suspension | Certified and indicative diameter values for two distributions, obtained with various techniques |
| IRMM | ERM® – FD304 | Colloidal silica in aqueous solution | Water suspension | Certified and indicative diameter values, obtained by various techniques |
It is fair to state that this lack of certified reference NP materials is not only a problem for SP-ICP-MS but for any technique deployed in this field. In fact, any comparison of the size distribution reported by every technique typically denotes some differences, as the results reported by each technique are based on some assumptions that are not always met. A conclusion of this is that attaining results by means of different techniques is always preferable, whenever possible. Moreover, for validating a new SP-ICP-MS method, comparing the results obtained with values reported via the same technique is very valuable for better understanding the effect that can be attributed to SP-ICP-MS assumptions only. This is again not easy, as such SP-ICP-MS values are reported for one reference material only (NIST 8017).
Finally, it needs to be mentioned that a novel alternative approach to determine the TE has been recently reported by the group of Goenaga-Infante.41 This method is termed dynamic mass flow approach and is based “on the direct continuous measurement of the sample mass flow reaching the plasma and the mass flow of the sample uptake by the ICP-MS nebulisation system”. Therefore, it is also a gravimetric method, as it requires weighing two vials sequentially for a period of time: one vial in which both the sample uptake and the waste tubings were simultaneously introduced, and another one in which only the sample uptake tubing was placed. Fig. 3 illustrates how through the measurement data obtained for both vials two linear regression plots can be fitted, and their slopes enable calculation of the TE. This approach is not very straightforward, but it does not require the availability of any reference NP, which makes it very relevant in a metrological context, as discussed in the previous paragraphs.
The papers by Laborda et al.37 and Pace et al.38 used and popularized the term “single particle”, which is the most widely used now when monitoring NPs or colloids. Later work focused on the proper separation between the signals from NPs and the BG signal, which is obviously of key importance. The need of using objective criteria (e.g., BG + 5σ) instead of just visual observation became apparent from the beginning.42 In another seminal article, Olesik and Gray discussed the effect of dwell time (time of acquisition of every individual signal intensity value) in detail, the probabilities of detecting one or more particles (double events, triple events, etc.) as a function of such dwell time, the optimal PNC, and the influence of the detector dynamic range,22 the latter being further assessed by Liu et al.43 A discussion on the importance and consequence of the dwell time selection will be presented in Section 3.1.
At this point, it could be considered that the main methodological aspects were already developed, opening possibilities for all the applications that came later on. Table 2 summarizes several selected examples, such that the reader can have a notion of the potential and application range of SP-ICP-MS. A recent review that focuses on applications of SP-ICP-MS can be consulted for more examples.44
| Year | Type of nanoparticle | Sample | Mass analyzer | Dwell time | LODsize or lowest size detected | Comments | Ref. |
|---|---|---|---|---|---|---|---|
| a Clay material is montmorillonite, where Al is 10.5% of the material. (K0.11Na0.07Ca0.49)1.16(Si7.82Al0.17)(Fe0.012+Mg0.85Fe0.333+Al3.5)O20(OH)4·n(H2O). | |||||||
| 2003 | Al2O3 | Alumina | q-ICP-MS | 10 ms | 30 nm | - First report of SP-ICP-MS | 28 |
| FEBEXa | Natural clay | 30 nm | |||||
| FeOOH | Goethite | 200 nm | |||||
| TiO2 | Rutile | 100 nm | |||||
| 2006 | Au | Monodisperse colloids of various sizes (80–250 nm) | q-ICP-MS | 10 ms | 25 nm | - Diluted colloid suspensions were introduced by a syringe driven by a linear motor | 31 |
| 2009 | Au | Serum | q-ICP-MS | 10 ms | 15 nm | - Au nanoparticles as tags to determine α-fetoprotein | 34 |
| 2011 | Ag | Aqueous suspensions of Ag NPs | q-ICP-MS | 5 ms | 18 nm | - Conventional pneumatic nebulization | 37 |
| - Study of the different behaviour of dissolved silver and silver NPs | |||||||
| 2012 | Ag | Aqueous suspensions of Ag NPs | q-ICP-MS | 10 ms | 20 nm | - Evaluation of different pneumatic and piezo-based sample introduction systems | 173 |
| - Simultaneous detection of silver NP and free silver ions | |||||||
| 2014 | Ag | Aqueous suspensions of Ag, Au and Au@Ag NPs; stream water | q-ICP-MS | Down to 0.1 ms | - The use of short dwell times enables detection of two elements from the same NP | 61 | |
| Au | |||||||
| Au@Ag | |||||||
| CeO2 | |||||||
| 2014 | Ag | River water, tap water and wastewater | q-ICP-MS | 10 ms | 21 ± 4 nm | - Estimation of LODsize using ionic standards for 40 elements and study of the influence of instrument sensitivity, nanoparticle density and BG noise on such value | 52 |
| CeO2 | 19 ± 8 nm | -Determination of the LODsize for 3 types of ENPs (Ag, CeO2 and TiO2) | |||||
| TiO2 | 130 ± 28 nm | ||||||
| Estimations: ≤10 nm (Ta, U, Ir, Rh, Th, Ce and Hf); 11–20 nm (Bi, W, In, Pb, Pt, Ag, Au, Tl, Pd, Y, Ru, Cd and Sb); 21–80 nm (Co, Sr, Sn, Zr, Ba, Te, Mo, Ni, V, Cu, Cr, Mg, Zn, Fe, Al, Li and Ti); >200 nm (Se, Ca and Si) | |||||||
| 2015 | Au | NIST Au NPs reference materials 8012 (30 nm) and 8013 (60 nm) | q-ICP-MS | 0.1–10 ms | - Post hoc inter laboratory comparison of SP ICP-MS for size measurements of Au NP reference materials | 54 | |
| 2015 | Ag | Aqueous suspensions of Ag and Au NPs | SF-ICP-MS | 10 ms | - The combination of MDG for signal calibration with a pneumatic nebulizer for NP measurements enables sizing NPs without the need to use matching reference materials | 89 | |
| Au | |||||||
| 2015 | Er2O3 | Suspensions of Er2O3 NPs in water | MC-ICP-MS | 200 ms | 130 nm | - Isotopic measurements in single particle mode for the isotope analysis of individual submicron-sized erbium oxide particles | 174 |
| 2016 | Ag | Plasma and blood of burn patients | q-ICP-MS | 5 ms | 16 nm | - Coupling of hydrodynamic chromatography to SP-ICP-MS plus use of home-made software for signal deconvolution to characterize dissolved Ag and Ag NPs from the same chromatogram | 175 |
| 2016 | Aqueous suspensions of the NPs investigated | q-ICP-MS | 6 ms | No gas/He | - Use of the collision cell filled with He for the characterization of NPs in cases of spectral interferences | 101 | |
| Ag | 30 nm/35 nm | ||||||
| Au | 18 nm/18 nm | ||||||
| Cr2O3 | N.A./39 nm | ||||||
| Fe2O3 | N.A./15 nm | ||||||
| ZnO | 18 nm/20 nm | ||||||
| 2016 | Ag | Aqueous suspensions of Ag NPs spiked with NaCl | q-ICP-MS | 10 ms | 30 nm | - Combination of isotopic dilution analysis with SP ICP-MS to overcome matrix effects | 85 |
| - 107Ag and 109Ag measured sequentially, not from the same NP | |||||||
| 2016 | Pt | Plant tissues | q-ICP-MS | 0.1 ms | - Characterization of the uptake and bioaccumulation of Pt NPs by plants | 176 | |
| - Enzymatic digestion to extract Pt NPs | |||||||
| 2016 | Ag | Surface and treated drinking water | q-ICP-MS | 0.1 ms | 21–23 nm | - Study of the fate of NPs during drinking water treatments | 177 |
| Au | 27–30 nm | ||||||
| TiO2 | 67–70 nm | ||||||
| 2016 | CeO2 | Surface and treated drinking water | q-ICP-MS | 0.1 ms | 18–20 nm | - Study of the fate of NPs during drinking water treatments | 178 |
| ZnO | 35–40 nm | ||||||
| 2017 | Cu | Copper foil | LA-SF-ICP-MS | 0.1 ms | 14 nm | - Characterization of the aerosol produced by femtosecond laser ablation using LA coupled to SP-ICP-MS and VBA data processing | 179 |
| 2017 | TiO2 (rutile) | River water samples | q-ICP-MS | 10 ms | 37 nm | - Study of the influence of dwell time for the sizing and quantification of NPs and dissolved Ti ions | 180 |
| TiO2 (anatase) | 37 nm | ||||||
| 2017 | Ag | Aqueous suspensions of the NPs investigated | q-ICP-MS | 0.05 ms | - Ag, Au and Au@Ag of same size elute together via FFF, but size and number concentration can be estimated via SP-ICP-MS, although the sizing accuracy is limited | 181 | |
| Au | |||||||
| Au@Ag | |||||||
| 2017 | La2O3 | Aqueous suspensions of La2O3 NPs, sometimes spiked with fulvic acid | q-ICP-MS | 0.5 ms | 17 nm | - Study of adsorptive losses | 182 |
| - The use of ion-exchange to remove dissolved La improved the accuracy for NP sizing | |||||||
| 2017 | CeO2 | Soil samples spiked with CeO2 ENPs | ICP-TOFMS | 0.3 ms | - Multi-element fingerprinting to discriminate ENPs from NNPs | 92 | |
| 2017 | Pt | Pt/SiO2 composite | q-ICP-MS | 10 ms | 17.2 nm | - Comparison of five different techniques for the determination of the content of Pt NPs in a Pt/SiO2 composite | 183 |
| 2017 | Au | Aqueous suspensions of Au nanorods | q-ICP-MS | 6 ms and 0.02 ms | - A new method is developed for detection and dimensional analysis of nanorod Au using only SP-ICP-MS with both ms-range and μs-range dwell times and evaluating the longest and shortest transit times | 51 | |
| 2017 | Au | Aqueous suspensions of Au NPs | q-ICP-MS | 2 ms | 10 nm | - Capillary electrophoresis coupled to SP-ICP-MS provides data with information on migration time, size, and number concentrations in a single run | 110 |
| 2017 | SiO2 | Aqueous suspensions of SiO2 NPs | ICP-MS/MS | 3 ms | 80 nm | - Use of ICP-MS/MS, in combination with deconvolution for 80–100 nm, enables characterization of SiO2 NPs ranging from 80–400 nm | 50 |
| 2018 | Pt | Road dust | ICP-MS/MS | 5 ms | 7.4 nm | - Use of ICP-MS/MS to overcome the potential spectral overlap from 155GdAr+ and 179HfO+ on 195Pt+ | 184 |
| 2018 | Fe (zero-valent) | Wastewater | ICP-MS/MS | 3 ms | 36 nm | - Interaction of Cd2+ and Fe NPs investigated by monitoring Cd in SP-ICP-MS | 185 |
| 2018 | Ag | Aqueous suspensions of the NPs investigated | q-ICP-MS | 6 ms and 0.02 ms | - Combination of normal (ms dwell time) and high resolution (μs dwell time) SP-ICP-MS for the characterization of bimetallic NPs | 64 | |
| Au | |||||||
| Au@Ag | |||||||
| 2018 | TiO2 | Candy products | ICP-MS/MS | 10 ms | 26 nm | - Use of SP-ICP-MS/MS to remove spectral interferences | 105 |
| 2018 | Ag | River and lake water before and after treatments | q-ICP-MS | 0.1 ms | - Study of the fate of NPs during water treatments | 186 | |
| Au | |||||||
| CeO2 | |||||||
| TiO2 | |||||||
| ZnO | |||||||
| 2018 | Se | Yeasts | q-ICP-MS | 0.1 ms | 18 nm | - Use of H2 in a collision/reaction cell to minimize spectral overlap | 102 |
| 2018 | Au@Ag (Bi0.5Na0.5)TiO3 | Suspensions of NPs and of nano steel composite (containing Cr, Fe, Mo, Ni) | q-ICP-MS | Various values | Values depend on analyte and type of ICP-MS | - Comparison of different types of ICP-MS devices for multi-element SP-ICP-MS | 106 |
| BiVO4 | ICP-TOFMS | ||||||
| CyTOF | |||||||
| 2019 | NbCN | Micro-alloyed steel | ICP-TOFMS | 1.8 ms | 26.5 nm | - Extraction of NPs from steel for analysis of their size and composition distributions via SP-ICP-MS and electron microscopy | 187 |
| TiNbCN | 46.6 nm | ||||||
| 2019 | ZnO | Pure water river water, rainwater | SF-ICP-MS | 0.05 ms | 8.2 nm | - Use of an ion exchange column for removal of the dissolved metal | 188 |
| 14.3 nm | |||||||
| 17.7 nm | |||||||
| 2019 | Ag | Sunscreen lotion, rainwater and swimming pool water | SF-ICP-MS and q-ICP-MS | 0.05 ms | 3.5 nm | - Use of dry aerosol introduction to improve the LODsize | 189 |
| TiO2 | 0.10 ms | 12.1 nm | |||||
| 2019 | TiO2 | Food samples containing E171 additive | q-ICP-MS | 0.1 ms | 28–36 nm (48Ti) | - Combination of 48Ti and 47Ti for the determination of TiO2 particle size distribution, expanding the working range | 104 |
| 67–85 nm (47Ti) | |||||||
| 2020 | Au | Aqueous suspensions of Ag@Au NPs, hollow Au NPs and porous silica particles | q-ICP-MS | 6 ms | 18.1 nm | - A new method for porosity determination for nano and sub-micron particles via SP-ICP-MS is proposed | 190 |
| Ag@Au | 6 ms | ||||||
| SiO2 | 3 ms | 292 nm | |||||
| 2020 | Ni | Aqueous suspensions of Ni NPs | q-ICP-MS | 0.5 ms | 16 nm (no PVP)/14 nm (with PVP) | - Comparison of TEM, SEM, XRD, SP-ICP-MS and flow injection coupled to SP-ICP-MS techniques | 191 |
| 2020 | Fe3O4 | Aqueous suspensions of Fe3O4 NPs | q-ICP-MS | 0.05 ms | 28 nm | - Different ICP-MS devices and approaches evaluated to cope with spectral overlap, including SF-ICP-MS operating on pseudo medium resolution | 95 |
| ICP-MS/MS | 0.1 ms | 19 nm | |||||
| SF-ICP-MS | 0.05 ms | 19 nm | |||||
| 2020 | AuNPs (as proxy to determine thrombin) | Serum | q-ICP-MS | 5 ms | - AuNPs modified with thrombin aptamers were adsorbed onto the surface of graphene oxide | 192 | |
| - In the presence of thrombin, the AuNPs desorb. The desorbed AuNPs, which are proportional to the concentration of thrombin, are quantified by SP-ICP-MS | |||||||
| 2021 | Pt | Human urine and blood serum | q-ICP-MS | 5 ms | 21.6 nm | - 1% TMAH for extracting the NPs | 193 |
| 2021 | Ag | Roots of sunflower | q-ICP-MS | 0.1 ms | - LA-SP-ICP-MS used for imaging, providing sizing and counting information from every map pixel | 194 | |
| -AgNPs can be ablated selectively using low laser fluence | |||||||
But what type of information can SP-ICP-MS provide? As displayed in Fig. 4, the technique can deliver the average (or median or mode value) NP size, but also the size distribution and the PNC, and all of this after only a few minutes of measurement and, often, after minimal sample pretreatment (e.g., a simple dilution in appropriate media), providing the sample is in liquid form. Otherwise, for solids, proper extraction, or digestion (hopefully, not affecting the analyte's chemical form or the integrity of the particles) will be needed. Finally, if the goal is to also determine the analyte present in ionic form, it can be simultaneously quantified because, as discussed earlier, the temporal behavior of the corresponding signal (quasi stable) will differ from that of the NPs (highly transient).
The benefits of this approach are therefore clear, as probably no other technique can provide all this information in such a simple way. However, there are several drawbacks that affect the performance of SP-ICP-MS. The scarcity of suitable certified reference NPs has already been mentioned and the need to carry out rather complex calculations will be discussed into detail in Section 3.2. Another one is related to the analytes that can be detected. ICP-MS can, in principle, monitor most of the elements of the periodic table, but some show poorer sensitivity because of inefficient (or even inexistent) ionization in the Ar-based plasma. F, for instance, cannot be monitored due to its high ionization potential. Non-metals, in general, are more challenging as, for several reasons,45 the sensitivity is lower for such elements. In SP-ICP-MS, this translates into more analyte mass needed to detect the NPs, and thus, in a higher limit of detection in terms of NP size (LODsize).
The same applies to elements suffering from strong spectral interference, which can sometimes be resolved only at the cost of sensitivity.46,47 In short, elements with a m/z ≤ 80 (which corresponds to the signal of 40Ar2+) often suffer from this problem, as well as from a lower sensitivity mainly due to space-charge effects in the interface between the plasma and the MS discriminating against the lighter ions.48
The isotopic composition of the analyte also plays a role. If an analyte shows several stable isotopes, the total signal intensity is “distributed” among them. Unless a simultaneous mass analyzer is deployed, in which it becomes possible to sum the signal intensities for these isotopes49 (beware that the signal-to-noise ratio for the less abundant nuclides can be rather poor), only one nuclide is monitored in SP-ICP-MS during every run, thus losing the signals corresponding to the other isotopes. Again, this results in higher LODsize values. Nevertheless, there are also some positive aspects deriving from the existence of various stable isotopes, such as the higher possibility of finding at least one of them free from spectral overlap, the potential to carry out isotope dilution for calibration and the extra confidence that can be achieved when validating a result using different isotopes from the same element.
The composition of the NP needs also to be considered and needs to be known in advance for proper calculations. Moreover, the more elements composing the NP, the lower the signal intensity will be for a particular constituting element (or, more accurately, the analyte nuclide monitored), leading to increasing LODsize values. On the positive side, however, this brings about the opportunity for cross-validation by monitoring different elements. Also, ENPs are less complex than NNPs (naturally occurring NPs) in composition, so the monitoring of the multiple elements present can help to distinguish between both NP types.
Another aspect affecting SP-ICP-MS monitoring is the occurrence of ionic species of the target element in the matrix. As discussed before, it is in principle possible to differentiate the signal originating from a NP and from an ion, but only to some extent. If the concentration of ionic species becomes very high, it will then become challenging to appreciate small NPs. Two solutions, however, can be deployed for this problem: use of deconvolution (mathematical) approaches, or just increasing the dilution factor, as the latter will only affect the sensitivity of the ionic species. The latter solution, however, will not be possible if such ionic species are present in the solvent, as it may occur for ubiquitous elements.50 Finally, it is necessary to indicate that some of these ionic species may come from the dissolution of the own target NPs, thus resulting in a bias in the final values. It is therefore of the utmost importance to use the appropriate treatment and the right media for dilution to guarantee the stability of the NPs subjected to analysis, following the recommendations of the manufacturers, if available.
Finally, there is the issue of the shape. It needs to be stressed that most publications to date studied spherical NPs. These probably are the most important type of ENPs, but there are also NPs of other shapes. The issue here is that SP-ICP-MS actually provides one single output: analyte mass. Thus, based on the chemical composition of the NP and its density, and assuming sphericity, the corresponding diameter can be derived (Fig. 4d). If the NP shows a different (known) shape and the volume also depends on one geometrical parameter (e.g., for a cube), then the corresponding representative value (the edge, in such case) can also be derived. If, however, the volume depends on several geometrical parameters (e.g., both width and length), they can, in principle, not be calculated by this technique. An exception to this rule will be discussed below, referring to the work of Kálomista et al.51 for Au nanorods, which opens new ways for other shapes as well.
In short, monometallic, spherical NPs, composed of one element of medium or high m/z that show only one or few stable isotopes are the best targets for this technique, particularly in terms of LODsize values. It is thus no coincidence that Ag and Au are the most targeted analytes, further strengthened by their obvious relevance in terms of applications. For these elements, state-of-the-art ICP-MS devices can characterize NPs of sizes down to 10 nm. For other elements/compositions, this limit will typically be higher (worse). Lee et al. published a work reporting on LODsize values for 40 elements,52 which represents the most comprehensive comparison of this aspect thus far, although it needs to be mentioned that these values were estimated using ionic standards (not NPs). It should also be noted that lower sizes can currently be determined, as this paper dates from 2014 and the ICP-MS instrumentation has improved significantly in various aspects (e.g., data acquisition speed, use of high-performance ICP-MS instrumentation to overcome spectral overlap) positively affecting such values.
At this point, it is necessary to highlight the publication of a number of interlaboratory comparisons that have established how well the method performs in real life, targeting Ag, Au and TiO2 NPs, in comparison with alternative techniques.53–58 In short, SP-ICP-MS tends to perform reasonably well for size characterization, and its potential for sample screening, checking for the presence or absence of NPs to properly comply with regulations, is clear. Calculating accurate PNC values seems more challenging, but this is mostly attributed to problems associated with sample preparation, stabilization and ageing, which are not solely affecting SP-ICP-MS. It is also interesting to notice that the dispersity of the size distribution populations obtained via SP-ICP-MS has been deemed to be higher than for other techniques in the case of AuNPs,54,56 which is a topic that requires further investigation as the technique becomes more mature.
Besides applications, other fundamental aspects of SP-ICP-MS have been investigated since the earlier works discussed above, and the results thereof will be covered in the following subsections.
3.1. Improvements in the data acquisition speed
The first articles were carried out with instrumentation that could not go below the millisecond level concerning the data acquisition time (dwell time). The strategy at that time was to use a dwell time high enough to ensure that every single event could be completely monitored in a time window (see Fig. 5a). Since a typical NP pulse lasts for 300–500 μs (see Fig. 1b), using 3–5 ms was the preferred choice at the time. Fig. 6 represents different situations that can occur during single event-ICP-MS measurements. Please note that selecting a shorter dwell time (e.g., 1 ms) would be risky, as it may result in the recording of only a portion of the whole event, leading to biased results (see Fig. 5a). In this case, a single particle event recorded in two consecutive dwell times would result in an overestimation of the PNC and an underestimation of the particle size.22 The risk of monitoring several events together can be minimized by sufficient dilution. To some extent, it could also later be corrected for mathematically when evaluating the distribution of results: for monodisperse NP suspensions, those distributions found at intensities that are double, triple, etc. as high as that corresponding to one nanoparticle could be identified and deconvoluted.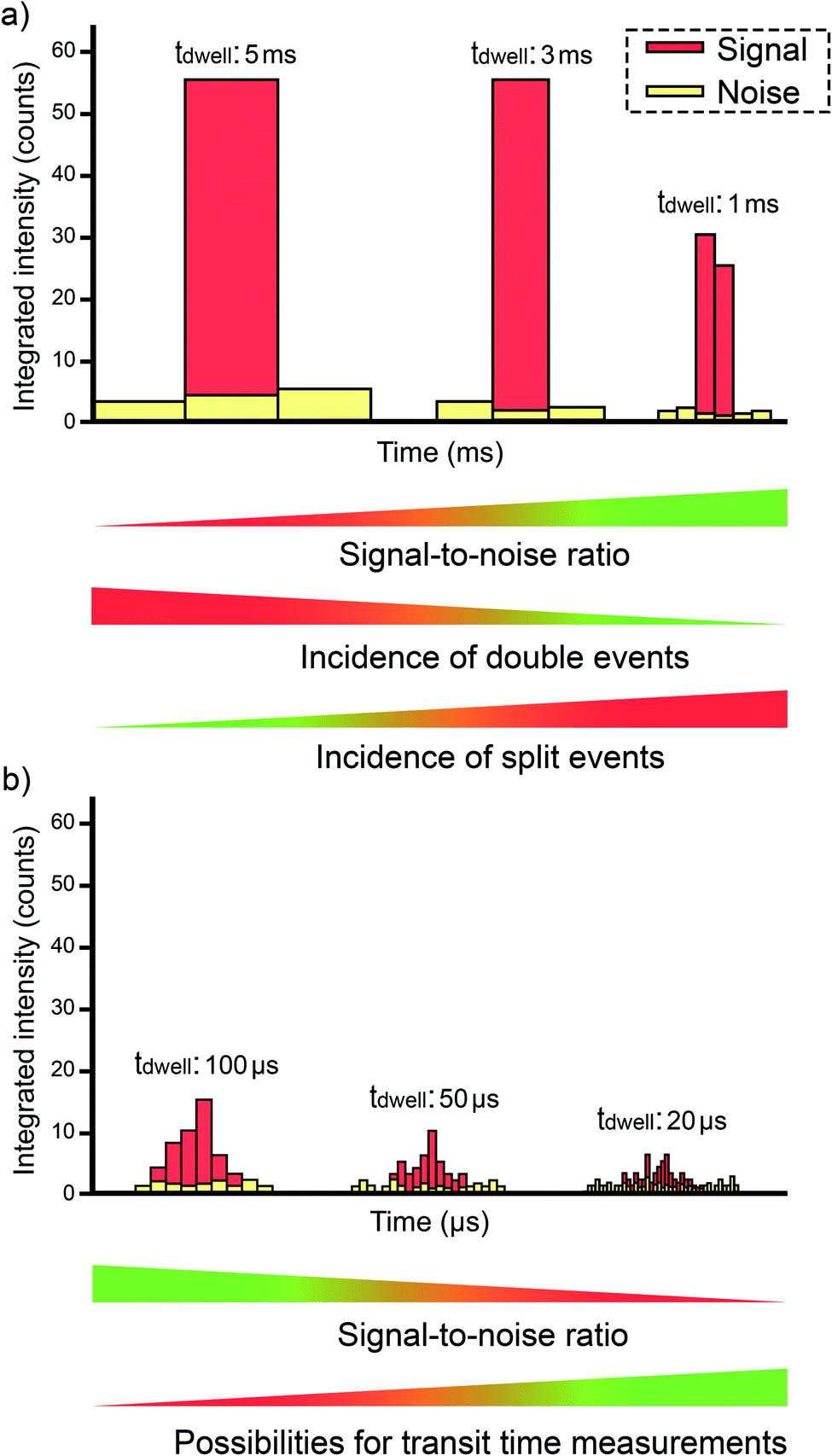 | ||
| Fig. 5 Examples of data (counts) acquired from an individual entity as a function of the dwell time used: (a) using millisecond dwell time; (b) using microsecond dwell times. | ||
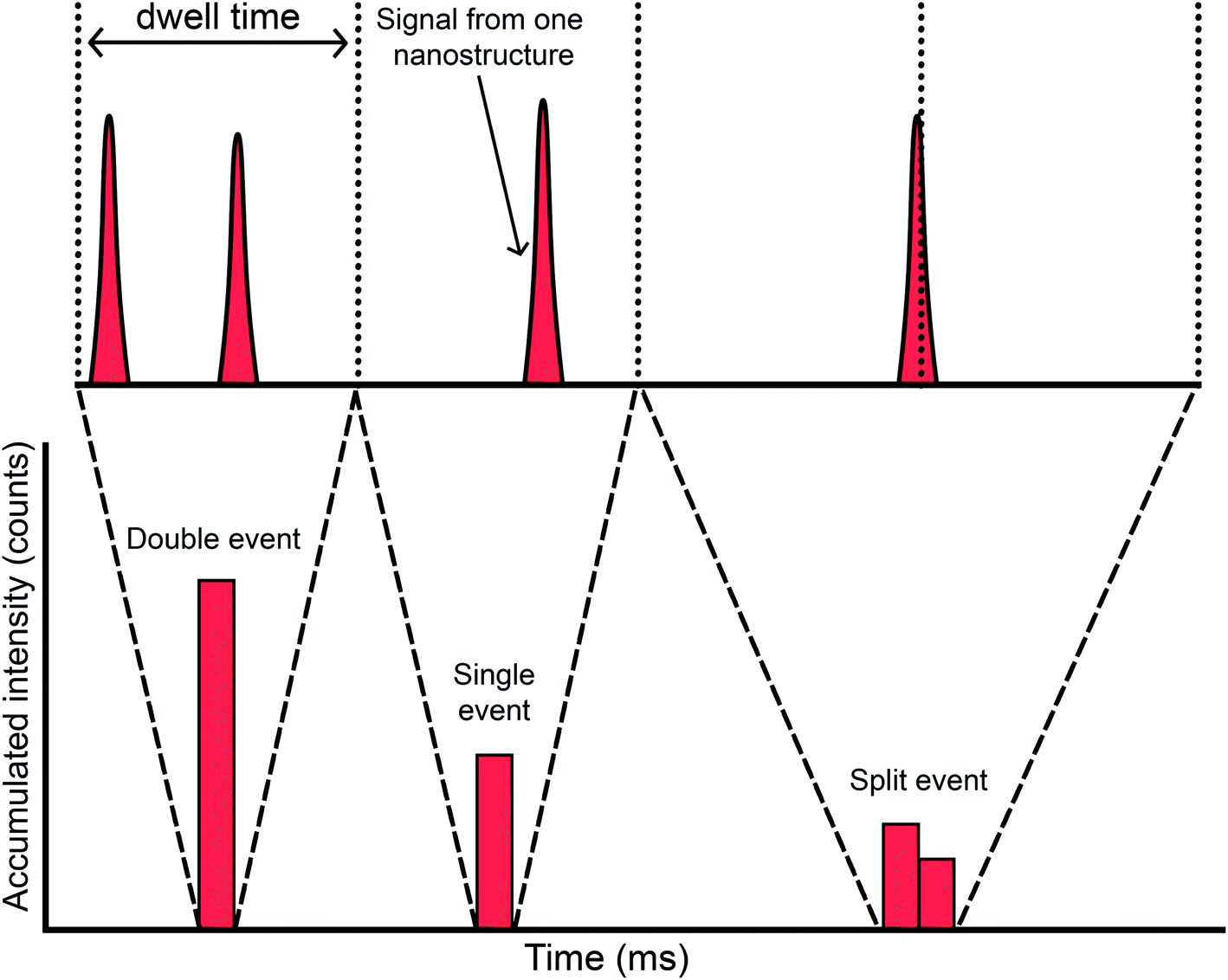 | ||
| Fig. 6 Potential situations encountered when performing ICP-MS measurements in single event mode for analysis of discrete entities. | ||
Some authors evaluated the use of external data acquisition units for improving this aspect,59 but a next generation of ICP-MS instrumentation with a detector set-up allowing a dwell time as low as 10 μs became commercially available in the mid 2010s.60–62 This represents an obvious advantage, because the signal intensity for the BG (practically constant with time) will decrease proportionally as the acquisition time decreases, but that of the NP pulse will not (see Fig. 5b). Thus, an improvement in the signal-to-BG ratio is achieved, paving the way to the detection of smaller NPs and/or to minimization of the effects of noise, spectral interference (unless such interference originates from elements found in the own NP), and dissolved analyte. Moreover, the possibility of monitoring two events together also decreases. However, if the dwell time is much lower than the duration of the single pulse, then the signal recorded for such pulse will be distributed over consecutive acquisition intervals and thus, the signal intensity over one dwell time will decrease significantly, further complicating the detection of NP events.63 As a compromise, a value of 50–100 μs is often preferred. The difference is that, under such conditions, every individual signal is now described by a set of data points instead of only one. This means that data processing is more elaborate, as every signal profile corresponding to every single event needs to be properly identified, such that its overall signal can be appropriately estimated (by summing all the data points that correspond to such single event). For a more detailed discussion on data processing, we refer to the next section.
Recent works from the group of Galbács have explored the possibility to obtain further information via microsecond temporal resolution (dwell time of 20 μs only), demonstrating the linear relation between transit time and particle size.64 It was also demonstrated that, by observing average signal profiles, it is possible to differentiate Ag–Au bimetallic NPs from core@shell Ag@Au NPs, with the possibility of figuring out which of the two element comprises the core. The molar ratio of both components can be determined using “conventional” (meaning not ultra-fast) SP-ICP-MS and, eventually, the core diameter and shell thickness can be calculated. This study was entirely executed using a q-ICP-MS unit, thus requiring separate measurements for Au and Ag. Furthermore, this group also tackled an even more compelling challenge in a prior work,51 making use of the same temporal resolution: the characterization of nanorods, instead of just spherical or cubical NPs. For such Au nanorods, these authors demonstrated that their high temporal resolution provided signal profiles that varied between two extreme values, one related to the nanorods being introduced lengthwise into the ICP and the other related to the nanorods being introduced in a perpendicular fashion. If the longitudinal axis of the nanorod is perpendicular to the direction of propagation, the pulse will be short and will provide information on the width, and when such axis is horizontally aligned, the pulse will show the longest duration and will provide information on the length.51 The aspect ratio of the nanorods can thus be calculated, which represents the second output needed, besides the total mass, to fully characterize nanorods using SP-ICP-MS only, opening possibilities for other shapes as well.
Finally, a recent work by Duffin et al.65 has shown the potential benefits in acquiring data even faster. The authors modify a MC-ICP-MS with a time-to-digital converter capable of generating timestamps to 0.5 ns of accuracy, and then used no predetermined integration windows. They developed an approach to identify each NP by the timing between successive ion arrivals at the detector, which are much shorter than when ionic species are sampled, so the overall signal corresponding to each NP can be subsequently calculated. The authors report on the benefits of this approach for small NPs (AuNPs of 5 to 20 nm), as for larger ones the linear response was lost.
It is likely that this trend towards exploring the potential of faster data acquisition continues, particularly as the sensitivity of the instruments increases, but for the moment a dwell time value of 50–100 μs can be considered as standard for most applications.
3.2. Improvements in data processing approaches
Another aspect that has received considerable attention is data processing, as was needed for a wider use of the methodology. Carrying out all the calculations required for processing the large data sets originating from SP-ICP-MS measurements was originally not straightforward. The goal of this review is not to provide an in-depth discussion of the data treatment protocol (see the paper by Peters et al.66 instead and/or the ISO protocol ISO/TS 19590:2017(E),67 which describes in detail the methodology for calculating size, size distribution, PNC, the ionic concentration, as well as the limit of detection both in terms of size and of particle concentration via SP-ICP-MS), but to provide some insight into the most important considerations. In short, it is necessary to properly detect every individual pulse and calculate its integrated intensity (Fig. 4a), establish the BG (for later subtraction), calculate the sample uptake flow rate, calibrate the instrument sensitivity (typically, with an ionic solution of the same analyte) and the TE (as discussed before). A frequency vs. integrated intensity distribution is then constructed for the nanoparticles (and the BG in case the determination of the ionic content is also intended), as shown in Fig. 4b. This graphical representation can then be converted into a frequency vs. analyte mass distribution after adequate calibration (Fig. 4c). In the case of NPs, the density, the analyte fraction (which depends on the stoichiometry of the NP), and the volume equation (normally, a spherical shape is assumed), are used to obtain the frequency vs. size distribution (Fig. 4d). Overall, size (average, mode or whatever parameter is needed), size distribution and concentration (in mass per L but also in particles per L for mass and particle number concentrations, respectively) are provided via SP-ICP-MS.Peters et al.66 published a key paper describing all these calculations into detail, and made two spreadsheets (one for calibration, one for the samples) designed to perform all the requested computations freely available. It was a very timely release. With time, however, some constraints appeared for the use of such spreadsheets. These were mostly associated with the software used (MS Excel), because there is a limit in the number of rows (1048576 rows) and such limit began to be exceeded as new instruments with dwell times below 1 ms (see discussion in Section 3.1) were deployed. Moreover, the spreadsheet was programmed for situations where each NP correspond to a single data point, which again is not the case when using dwell times of tens of μs. When working with these very short dwell times, a suitable approach to reconstruct the signal for every individual NP is required. Anyway, many authors used these spreadsheets as a basis and modified and/or complemented them at need.
At the same time, different manufacturers of ICP-MS instrumentation began to release specific software packages for SP-ICP-MS, also helping in rendering the methodology more user-friendly and more widely available. A detailed explanation of how a certain software package works was published by Newman et al.68 However, particularly in the context of research, it is important to know exactly how such calculations are performed. For instance, the criteria used to identify the signals originating from every NP42 first, and later on, those used to differentiate the NP distribution from that of the BG69 need to be well understood. Thus, some authors rely on their own scripts, usually using alternative software more powerful than spreadsheets (e.g., Python-based42,70). Fig. 7 represents an example of an in-house software that was originally developed for post-processing of mass spectrometry data in the context of elemental mapping (imaging) using LA-ICP-MS, but that was further also tuned for NP characterization by (1) the identification of the transient signal peaks randomly originating from the introduction of NPs, (2) the calculation of their integrated signal intensity and (3) the documentation of the NP event duration.71
 | ||
| Fig. 7 SP-ICP-MS data treatment using the in-house (UGent) developed Hyper Dimensional Image Processing (HDIP) software. (A and B) Show the raw data (intensity vs. time) from one measurement replicate of a Au NP standard. (C) Illustrates the identification of the signal spikes corresponding to single NP events. (D) Shows the average peak profile and the results obtained for the complete data set (peak profile, signal duration, number of peaks found, area average, etc.). The red triangles highlight the peak maxima, while the integration intervals are visualized by green triangles. Reproduced with permission from Elsevier (DOI: 10.1016/j.aca.2019.05.077).71 | ||
Finally, it is worth noticing that, in case of overlap between the distribution of NPs and the BG, the use of deconvolution approaches can help in resolving challenging situations.50,72 Of course, this further complicates the data treatment, as the signal distributions need to be fitted to a suitable model, such as normal (Gaussian) or Poisson distributions.73
3.3. Improvements in TE
One of the ever-pending aspects of ICP-MS is the poor sample introduction efficiency when using “traditional” pneumatic nebulization. Thus, a plethora of alternative approaches have been investigated since the early days of the technique. The case of SP-ICP-MS is unique in the sense that a better TE will not increase the sensitivity, as discussed in Section 2. A NP will be transported to the plasma or not, but those that do will show the same signal regardless of the TE. In other words, a higher TE will not have a significant impact on the LODsize.As indicated before, one of the key advantages of single event-ICP-MS, in comparison with other techniques, is the possibility to obtain the PNC simply by counting the events detected in a period of time, knowing the sample flow and the TE. The use of sampling approaches that reach 100% efficiency (or something acceptably close) will thus eliminate the requirement to carry out additional calibration measurements to determine the TE. Here it needs to be pointed out that such value cannot assumed to be constant for pneumatic nebulization, as it will most likely be affected by the matrix. Thus, metrologically, it would be advantageous to reach quantitative sample introduction into the plasma.
Beside this aspect, a higher transport efficiency also means less time needed to record a sufficiently high number of events for reliable characterization of NPs. Thus, not the sensitivity, but sample throughput is the parameter that could be improved in this way. However, that is not really the case in practice, and this is because the main way to improve the TE is to work at lower sample flows. When pneumatic nebulizers are operated at sample uptake rates around 10 μL min−1, instead of the traditional 200–1000 μL min−1, most of the water evaporates from the droplets before these enter the ICP, significantly improving the TE to values that can reach 60 to 80%.22 Again, the use of a low flow is not detrimental for the sensitivity in single event-ICP-MS, so the use of total consumption or near-to-total consumption introduction devices based on micro-nebulizers and low-volume spray chambers have been proposed in this context.74 These spray chambers often also deploy a sheath gas to focus the particles on-axis and thus, to prevent particles from colliding with the walls of the spray chamber.22Fig. 1b shows one of these commercially available devices, different from the more conventional cyclonic spray chamber shown in Fig. 1a. Heating the spray chamber is another way to improve the TE, as again, this favors water evaporation from the droplets before their introduction into the ICP.75
Using one of these total-consumption (no drain) sample introduction systems (a home-designed system that was termed as modified HECIS, standing for high-efficiency cell introduction system), Miyashita et al.76 reported TE values as high as 93.3 ± 0.9% for an 8.6 μL min−1 sample uptake rate, which compares with a value of 22.6 ± 0.4% for a conventional system at an uptake of 107 μL min−1 value (which is already a low flow, and thus already provides a high TE) when targeting 70 nm Pt NPs. However, lower values of 82.9 ± 1.3% and 87.2 ± 1.8% were obtained for 60 and 100 nm Ag NPs, respectively.
There are other relatively similar devices that have been reported on in literature for other ICP-MS applications and could be deployed for SP-ICP-MS.77 However, it is not clear that a sufficiently high TE can be achieved consistently, such that quantitative sample uptake can be assumed. These devices, in any case, are much more important when targeting single cell analysis, as will be discussed in the next section, so their further exploration can be anticipated.
In addition to a high TE, as close to 100% as possible, achieving a homogenous droplet size is also important in terms of precision. Continuing earlier research on microdroplet introduction in ICP,78,79 the group of Günther proposed the use of a microdroplet generator (μDG) based on a “commercial dispenser head consisting of a piezoelectrically actuated quartz capillary with a specified inner nozzle diameter of 30 μm and an internal, annular carrier gas supply”.80 This paper was published in 2011 and at that time, an oscilloscope had to be used for fast signal acquisition, as commercially available instruments were not capable of that (see Section 3.1). However, the use of this device is not only relevant to avoid the calculation of TE, but also shows other important benefits in terms of calibration and correction for matrix effects, as will be discussed in the next section.
3.4. Minimizing matrix effects
The occurrence of matrix effects is a general problem inherent to ICP-MS, thus not restricted to single event-ICP-MS.81 However, its effect in the latter case may be stronger. On the one hand, the matrix may affect the TE, thus leading to a biased PNC and size distribution (see equations in Fig. 4). On the other hand, the matrix may also affect the sensitivity, thus further affecting the size distribution result.Matrix effects can be often mitigated because of the high dilution factor typically used for SP-ICP-MS. However, some matrix is typically always present due to the addition of citrate or similar compounds for securing NP stability. The presence of organics is well known to both affect the TE (often increasing it due to the lower surface tension of the solutions, thus leading to smaller droplets) and the sensitivity for some elements.82 Besides, also introduction of the NP itself can, depending on its size and nature, affect the plasma conditions, thus leading to unavoidable matrix effects.
Therefore, accurate characterization of real samples should always consider this aspect and make use of suitable strategies to deal with it. That means that, unless some separation is performed, matrix-matching is recommendable,83 whenever possible. Alternative approaches to counteract the influence of matrix effects on the analytical results, such as calibration via isotope dilution, have also been reported in the literature, but seldom.84,85 One limitation to such approach (besides the fact that some relevant elements, such as Au, are mono-isotopic) is that, when a non-simultaneous ICP-MS is deployed, the analyte isotopes are not really measured from the same NPs. Very recently, the use of an ICP-TOF-MS device for this purpose has been reported, discussing also the modest precision that characterizes this approach when targeting NPs (owing to low count statistics, as also reported for multicollector ICP-MS instrumentation)86 and the effect of the concentration of the spike on the results.87
The use of other strategies, such as internal standardization, are also affected by the sequential character of the monitoring. In this regard, the group of Günther extensively tested a prototype ICP-TOF-MS instrument88 in the context of NP characterization, very often in combination with the μDG described in the previous section. Instead of just using the μDG for sample introduction, it can be connected via a “T piece” to the exit of the spray chamber.89 This configuration opened different options for introducing the sample and calibrants, as reported by Rosenkranz et al.90 One key aspect is that the mass flow of the droplets introduced via the μDG can be known, as they show a relatively constant size, are quantitatively introduced into the plasma, and can be prepared with known concentrations of the analyte. Thus, these droplets can be used as NP proxies73 for calibration instead of relying on NPs of known size in suspensions with known PNC. This is important because there is a clear shortage of well-characterized NPs in the market for such purpose, as discussed before.
The use of ICP-TOF-MS instrumentation in this context enables various approaches, such as doping the droplets (with an internal standard or an enriched stable isotopic spike) so their signals can be differentiated from those originating from the same analyte but found inside the NPs. Notice that, in this case, both signals are highly transient and such differentiation would otherwise be difficult. The benefits of this approach to deal with matrix effects are discussed in detail by Hendriks et al.49 and by Gundlach-Graham and Mehrabi,91 in a recent review. Other benefits of using ICP-TOF-MS for the simultaneous acquisition of (almost) the whole m/z elemental spectrum include the possibility to differentiate between ENPs and NNPs via multi-element fingerprinting.92 The classification of NPs can be further enhanced and automated by suitable data processing strategies,93 including the use of machine learning.94
It is obvious that single event-ICP-MS is one of the application fields in which the use of ICP-TOF-MS makes more sense. However, the somewhat limited sensitivity of such type of instrumentation (especially in the low mass range) affects the LODsize attainable.
3.5. Minimizing the effect of spectral overlap
The occurrence of spectral interferences is another well-known problem for ICP-MS. Besides some eminent isobaric overlaps, this mainly occurs as a result of the formation of polyatomic ions from species ubiquitously present at high levels in the ICP (e.g., Ar, C, N and/or O) or in the matrix (e.g., Cl or S). As discussed above, spectral interferences strongly affect those elements with an m/z ≤ 80.There are some widely used ENPs, the characterization of which using single event-ICP-MS suffers from spectral interference, such as Fe2O3, SiO2 or TiO2. To overcome this problem, different approaches can be used. Besides isolation of the NPs from matrix elements (an approach that is only useful in the case of matrix-related interferences), the increase in either physical or chemical resolution can be seen as the most effective strategy.95
The use of ICP-MS instrumentation that can be operated at a higher mass resolution is well-established,96 and its advantages and disadvantages are well-known within the field. For such approach, an ICP-MS instrument equipped with a double-focusing sector field mass spectrometer is needed. Unfortunately, operating this device at a higher mass resolution involves a reduction in the ion transmission efficiency, and thus in sensitivity. It is noteworthy that such type of instrument, when operated at lower resolution, can possibly provide the highest detection efficiency of all the types of ICP-MS, when the so-called Jet interface and μDG are deployed, as discussed recently by Kocic et al.,97 who reported LODsizes below 10 nm for several NP types. However, that is only feasible for elements not affected by spectral overlap. In addition, former models of SF-ICP-MS instrumentation did not offer ultra-fast data acquisition speeds, although this problem has already been solved for state-of-the-art instruments.95,98
The other approach is based on selective ion–molecule reactions in a pressurized multipole cell that is located before the quadrupole mass filter. Nowadays, most q-ICP-MS devices are equipped with such collision/reaction cell (CRC) system. Also, the combination of pressurizing the cell with an inert gas and kinetic energy discrimination can sometimes be used. The idea behind the latter approach is that, when the cell is pressurized with such an inert gas (e.g., He), the polyatomic ions giving rise to the interference will collide more than the analyte atomic ions due to their larger collisional cross-section, thus losing more kinetic energy in their journey through the cell than the analyte atomic ions.99 At the end of such cell, a voltage barrier is established to prevent these lower-energy polyatomic ions from entering the quadrupole mass filter. This can be seen as an effective way to filter out this type of species, although it always comes at the cost of losing some sensitivity as well, as the transport efficiency of the analyte atomic ions is ultimately also negatively affected, at least to some extent.
A more powerful approach is based on chemical reactions. In this case, the cell is filled with a reaction gas (e.g., NH3 or O2) instead of with an inert gas. Such a reactive gas can either interact with the interfering species, leaving the analyte free from the overlap at its original m/z (measurement on-mass), or else, react with the analyte atomic ions giving rise to a new species (reaction product ion) that can be measured free from spectral interference at a different m/z (mass-shift). The full power of this approach is accomplished when two quadrupoles (one before and one after the reaction cell) are deployed, as is the case in tandem ICP-MS instrumentation (ICP-MS/MS). This relatively novel technique was commercially introduced by Agilent Technologies in 2012 and was evaluated for the first time in the same year by Diaz Fernández et al.100 In principle, no loss of sensitivity should accompany this approach, although that ultimately depends on the efficiency of the reactions taking place in the CRC. In any case, this setup combines the robustness of q-ICP-MS instrumentation with a much higher resolution. It needs to be noted that the first of the two quadrupole mass analyzers in ICP-MS/MS filters out all concomitant matrix ions with different m/z ratio than that of the target analyte ion, thus resulting in a better control over the in-cell chemistry and an improvement of the ion–molecule processes occurring within the CRC system.
It is beyond the scope of the current work to discuss such approach in detail, and thus, the reader is referred to dedicated tutorial reviews.46,47 Instead, the particularities of these approaches in the context of SP-ICP-MS(/MS) will be discussed. Currently, not a lot of information on this topic is available already. There are probably two reasons for this. One is that the effect of spectral interference is expected to be less severe when aiming at single event-ICP-MS in comparison with bulk analysis, as discussed at the beginning of this section. This is simply because the analyte is delivered in short but very intense bursts, and the interfering species (unless it is related to some elements found in the targeted particles themselves) will provide a constant signal, much like the ionic species of the analyte present in the sample suspension. Thus, there is a temporal resolution, permitting the signals corresponding to the discrete entities and those of the interferences to be resolved, at least to some extent. The other reason is that the occurrence of gas reactions in the CRC may significantly increase the duration of the signals originating from every NP, with consequences that will be discussed in more detail below.71
Kálomista et al.101 evaluated the use of He in a collision cell to characterize suspensions of Ag, Au, Cr2O3, Fe2O3, and ZnO NPs. LODsize values for Cr2O3 (39 nm) and Fe2O3 (15 nm) were reported with a He-pressurized cell, whereas without this collision gas analysis was not possible owing to strong spectral overlap. For the other elements, the use of CRC technology only showed a moderate decrease in size detection limits: Ag (30 nm in no gas mode, 35 nm in He mode), Au (18 nm in no gas mode, 18 nm in He mode) and ZnO (18 nm in no gas mode, 20 nm in He mode). This is of additional interest if attempting to measure several NPs simultaneously (e.g., with an ICP-TOF-MS unit equipped with a CRC system).
Jiménez-Lamana et al. pressurized a CRC with H2 to monitor Se NPs.102 Rush et al. evaluated the benefits of pressurizing the cell in detail, not with the intention to deal with spectral interference, but to extend the dynamic range,103 an approach that was reported previously by Liu et al.43 This indicates another problem of SP-ICP-MS. Since the number of counts is usually low, one may forget that the count rate (counts per s) can be very high. This may trigger a change in the detector (electron multiplier) from pulse counting to analog mode and, unless these modes are perfectly cross-calibrated, monitoring part of the signal corresponding to every event in one mode and the remainder in the other may result in inaccurate values. Pressurizing the cell to decrease the sensitivity may help in such situation to properly characterize larger NPs, although it is more adequate to name them colloids in this case, as this term is typically used for particles that are larger than 100 nm. Another approach to solve such problem consists in monitoring various nuclides of different isotopic abundance, if available, as shown by Bucher and Auger for TiO2 NPs.104 It must be stressed that, besides this issue, the characterization of large particles may still be affected by other problems, such as incomplete digestion in the plasma.
Continuing with the use of a pressurized CRC, Bolea-Fernández et al. used chemical resolution in ICP-MS/MS for the characterization of SiO2 NPs.50 In this case, the challenge for ICP-MS was not only the occurrence of strong spectral interference, but also the ubiquitous presence of Si species, which were to be found in dissolved form, thus increasing the BG signal. The authors compared various CRC gases and different approaches and concluded that the use of H2 (monitoring on-mass) and of CH3F (mass-shift) provided the best results in term of LODsize values. In particular, the use of H2 in combination with a deconvolution approach allowed for the characterization of 80 nm SiO2 NPs. It needs to be noted that only millisecond detection was available for the ICP-MS/MS instrumentation used at that time, but nowadays dwell times in the range of 100 μs and even lower are feasible, so that this method can be currently further improved for the characterization of smaller SiO2 NPs.
Candás-Zapico et al.105 evaluated ICP-MS/MS for characterizing TiO2 NPs in candy products. They also demonstrated the utility of this technique, compared various reaction gases, and attained the best results with O2 and NH3. The use of O2 provided the best LODsize values (26 nm), but NH3 was preferred for real samples containing Ca, to avoid overlap with the signals from Ca-based polyatomic species (e.g., 48Ca16O+ and 48Ti16O+). Again, the instrument used allowed a dwell time in the ms range only, so even better figures of merit may be possible nowadays. Rua-Ibarz et al.95 compared the performance of state-of-the-art SF-ICP-MS and ICP-MS/MS for the characterization of Fe3O4 NPs of approx. 50 nm size, achieving satisfactory results in both cases. This work demonstrated for the first time the potential of a new generation of SF-ICP-MS instruments capable of operating at higher mass resolution (pseudo medium resolution) in the context of single event analysis (dwell times in the microsecond range), providing a suitable alternative to q-ICP-MS instruments and chemical resolution for NPs affected by the occurrence of spectral interference.
However, also one negative effect accompanying the use of CRC technology in SP-ICP-MS analysis was revealed by Bolea-Fernandez et al.71 In this work, it was demonstrated that the interaction between the package of ions generated from a single NP and the gases in the CRC may increase the duration of the NPs signals, as mentioned before. This effect is more prominent for heavier gases (e.g., NH3) than for lighter ones (such as H2 or He) and it was also found to be more pronounced for larger particles. As a result of this effect, LODsize values deteriorate due to poorer signal-to-background ratios, while the potential for double event monitoring escalates as well. A positive aspect, however, can also be derived from this issue, as a longer signal duration may allow for the monitoring of more than one nuclide/isotope from every event when deploying a sequential ICP-MS instrument. In fact, a few reports on monitoring of two isotopes from the same nanoparticle event with q-ICP-MS have been published already,61,106 and the two most recent ones have made use of gas addition in the cell for stretching the signal corresponding to each NP.107,108 In any case, it is important to keep this aspect in mind when optimizing the method to ensure conditions in which the integrity of the signal corresponding to every individual event can be preserved and recorded.
Other examples of coping with spectral interferences are listed in Table 2. To conclude this section, it can be said that SP-ICP-MS remains an area of high research activity, with the number of applications developed supporting the relevance of this technique in various fields (e.g., environmental or biomedical17). There is, however, still a need to further enhance the detection power, thus improving the LODsize, to be able to characterize nanoclusters, and that calls for further increase of the instrumental sensitivity. In this regard, the proposal of a new torch design (conical) can be mentioned. It has not been proved for ICP-MS so far but only for optical emission, but the authors state that such conical torch is able to complete the vaporization, atomization, and ionization of particles more rapidly, therefore reducing ion diffusion inside the plasma and leading to concentrated ion clouds, thus potentially leading to higher peak intensities for single particle monitoring (3.5–8 improvement for peak intensity and 2–4 in peak area for optical emission).109 In addition, further improvements in terms of simultaneous multi-element monitoring, and effective strategies to cope with spectral and non-spectral interferences are also still very welcome. Finally, the coupling to other techniques can provide additional information, as shown by Franze et al.110 for capillary electrophoresis SP-ICP-MS.
4. Interrogating single cells. The development of mass cytometry
As already discussed in Section 2, the concept behind single event-ICP-MS can be applied to different types of entities, not only to NPs/colloids. And there is no structure more important for life than the cell. Since inter-cell variations can be significant, the interest of achieving information from every individual cell is rather obvious, and single cell analysis has become the goal of “omics” during the last decade.111 Naturally, this need goes beyond elemental analysis, but it must be kept in mind that, actually, SC-ICP-MS can provide information on different cellular compounds via proper tagging, as will be discussed below, while many enzymes have a metal as co-factor.A great amount of the concepts and the procedures discussed for SP-ICP-MS can be transferred directly to SC-ICP-MS. In fact, the latter could be perceived as an evolution of the former. There is some truth to that in the sense that some of the research groups that mastered SP-ICP-MS have extended their expertise to SC-ICP-MS, but, in reality, the development of SC-ICP-MS is mostly parallel to that of SP-ICP-MS, as can be appreciated from Fig. 2, although much more moderate in terms of number of publications so far, until the last couple of years in which these numbers started to rise more steeply. This upwards trend can be expected to continue.
Moreover, it must be stressed that these values are not really accurate because the vast majority of articles using mass cytometry (which is a single cell-ICP-MS technique based on a dedicated ICP-TOF-MS device) do not use the SC-ICP-MS terminology. Mass cytometry works have been reported to surpass the number of 1000.14 These publications are updated elsewhere,112 and Fig. 2b shows that this has become a separate, very important biomedical field. For reviews on mass cytometry the reader is referred elsewhere.113,114
Due to the obvious similarities between SP-ICP-MS and SC-ICP-MS, this section attempts to stress the main differences and the most important challenges associated with the latter and will refer to more detailed reviews where appropriate.
4.1. To tag, or not to tag. The origin of SC-ICP-MS and the rise of mass cytometry
The first manuscript proposing the use of what now is termed as SC-ICP-MS was published by Li et al. in 2005.115 The authors investigated bacteria suspensions (Bacillus subtilis) in TRA mode (4 ms dwell time) and demonstrated that the short U spike signals originated from U compounds incorporated into individual cells, which behave like large particles in the ICP-MS. This is the same concept already explained in detail in Section 2.On the other hand, a group of researchers led by Tanner were working on the use of elemental tags for the labeling of antibodies and proposing this strategy to perform multiplexed bio-assays.116,117 The authors, in cooperation with other research groups, realized the importance of high-throughput single cell assay and noticed that the limitations of flow cytometry (which is based on tags producing fluorescence – fluorophores) could be overcome via mass spectrometry.118 While the possibilities for multiplexing in fluorescence spectrometry were rather limited due to the occurrence of spectral overlap, they realized that mass spectrometry offered a much higher degree of multiplexing with ≥100 m/z ratios available. To realize this, two developments were required: (i) “purpose-specific element (or isotope) tag reagents”, making antibodies visible for elemental mass spectrometry owing to the presence of a metal nuclide, and (ii) “a high-sensitivity, rapid (microsecond) simultaneous detection-and-reporting multielement detector having a sample introduction system that provides for reliable introduction of single cells or particles in a manner consistent with efficient ionization in the ICP”.119 This research group managed to develop both aspects, as proved by the success of the approach and the company erected (see Fig. 2b), coining the term mass cytometry. The progress of this group was reported in a series of papers120,121 and the characteristics of the ICP-TOF-MS device developed for this particular purpose were described in ref. 122. The basic concept is still preserved in later versions of the instrumentation. A scheme illustrating this approach is shown in Fig. 8. The technique consists of an ICP-TOF-MS device that only monitors the mid-to high-mass area of the spectrum (above m/z = 75), which is not a problem as the tags are normally rare earth nuclides. It also needs to be noted that elements with low mass would not be appropriate tags for mass cytometry, as those are essential mineral elements that can already be present in the target cells (see Section 4.3 for additional discussion on the quantification of elements inherently present in cells). The sample introduction system consists of a heated spray chamber, not so different from those described in Section 3.3. The topic of sampling introduction in single cell-ICP-MS will be addressed in more detail below. This instrument operates at a data acquisition frequency of 76.8 kHz. That means that the entire (75–209 amu) spectrum is acquired every 13 μs.123 This instrument is referred to as CyTOF, and the last model has been labelled as XT™.
 | ||
| Fig. 8 Scheme highlighting the main steps of analysis via mass cytometry. | ||
Again, more details of this approach, which is extremely powerful as it offers the current potential to quantify nearly 50 different markers with single-cell resolution and at high sample throughput, are provided elsewhere,113,114 while Table 3 lists some selected applications. Please note that it is also possible to couple LA to this type of instrumentation to carry out direct high-resolution elemental mapping measurements on tissue samples using metal-tagged antibodies (the commercial name of this technology is the Hyperion Imaging System). This approach allows to distinguish cell structural features in tissue with high spatial resolution and documents the quantitative distribution of biomolecules across such tissue. For additional details, the reader is referred to specific literature about this technology.20,124
| Year | Type of cell | Analyte(s) | Mass analyzer | Dwell time/sampling rate | Comments | Ref. |
|---|---|---|---|---|---|---|
| 2005 | Bacillus subtilis (bacteria) | U | SF-ICP-MS | 4 ms | - Microconcentric nebulizer | 115 |
| - First report on SC-ICP-MS | ||||||
| 2007 | Myeloblast cells | Au, Rh (used as labels) | q-ICP-MS | 10 μs (with an oscilloscope) | - Study for establishing mass cytometry possibilities | 119 |
| 2008 | Myeloid leukemia cells (KG1a) | Eu, La, Nd, Tb, Tm, Yb (used as labels) | CyTOF prototype | 55 kHz TOF sampling rate | - First studies on cell tagging protocols with metal-containing reagents for multiparametric mass cytometry | 120 |
| - Concentric nebulizer plus low volume spray chamber results in at least 50% cell disruption | ||||||
| 2009 | Leukemia cells | Diverse lanthanide isotopes as labels (Dy, Er, Eu, Gd, Ho, La, Nd, Pr, Sm, Tb, Tm, Yb) | CyTOF prototype | 76.8 kHz TOF sampling rate (1 value every 13 μs) | - Simultaneous detection of 20 surface antigens tagged with different lanthanide isotopes | 122 |
| I, Ir | - Concentric nebulizer plus a custom-made heated spray chamber to avoid cell disruption | |||||
| 2010 | Chlorella vulgaris (alga) | Cu (detected) | q-ICP-MS | 10 ms | - Quantitative calibration with metal oxide particles. Semiquantitative calibration with aqueous standards | 135 |
| Cr (sorption study) | - V-groove nebulizer plus Scott-type double-pass spray chamber (2°C) | |||||
| Mg (determined) | ||||||
| Mn (detected) | ||||||
| 2011 | Helicobacter pylori | Bi (for tracking antiulcer drug uptake) | q-ICP-MS | 10 ms | - Study for tracking Bi antiulcer drug uptake in H. pylori | 195 |
| Mg (for cell counting) | - Microconcentric nebulizer plus single pass spray chamber | |||||
| 2011 | Human bone marrow | Diverse stable transition element isotopes as tags for antibodies | CyTOF | - Comparison of mass cytometry and traditional fluorescence cytometry | 118 | |
| - 34 parameters measured simultaneously via SC-ICP-MS | ||||||
| 2013 | Saccharomyces cerevisiae (yeast) | Ca, Cu, Fe, Mg, Mn, P, Zn | q-ICP-MS | 10 ms | - High-efficiency cell introduction system (HECIS): high-performance concentric nebulizer and a low volume (15 mL) on axis spray chamber using a sheath gas flow. TE of 75.0 ± 4.7% | 134 |
| ICP-TOFMS | 34 μs | |||||
| 2013 | Single selenized yeast (Saccharomyces cerevisiae) | Cu, Se, Zn | SF-ICP-MS | 100 μs | - Micro droplet generator producing single droplets of 23 μm diameter at a fixed rate of 50 Hz to embed the cells | 141 |
| - TE of 100% | ||||||
| 2013 | RAW 264.7 cell line (mouse leukemic monocyte macrophage) | Cd as a proxy for carboxyl CdSeS quantum dots | q-ICP-MS | 5 ms | - Micro-concentric nebulizer with a quartz impact bead spray chamber. TE of 2.9% | 156 |
| 2014 | Saccharomyces cerevisiae (yeast) | Al, C, Ca, Cr, Fe, K, Mg, Mn, P, S, Zn (not all analytes were detected in all cells) | q-ICP-MS | 50–100 μs, via external ion pulse counting unit | - Modified HECIS system134 with a large-bore central capillary, combined with a narrower injector for improved TE (from 86 to ca. 100%, depending on the cell size) | 128 |
| Synechocystis sp. PCC 6803 (bacteria) | ||||||
| Cyanidioschyzon merolae 10 D (red alga) | ||||||
| Galdieria sulphuraria (red alga) | ||||||
| Chlamydomonas reinhardtii CC-125 (green alga) | ||||||
| 2014 | Bovine red blood cells | Fe | q-ICP-MS | 10 ms (plus 3 ms settling time) | - Microfluidic system generating droplets of 50 μm to embed the cells | 145 |
| 2015 | Human pancreatic cancer cell line BxPC-3 | I, Pt | CyTOF | - Study on the uptake, intratumoral distribution and cell cycle effects of cisplatin | 196 | |
| Cervical cancer cell line ME-180 | ||||||
| 2015 | Cervical cancer cells (HeLa) | Cu, Fe, Mn, P, S, Zn | q-ICP-MS | 5 ms | - Concentric PFA-ST nebulizer and a baffled quartz cyclonic spray chamber | 197 |
| Lung carcinoma cell (A549) | -Reaction cell used to minimize spectral overlaps | |||||
| Normal human bronchial epithelial cell (16HBE) | ||||||
| 2015 | Cervical cancer cells (HeLa) | Gd, Pt | q-ICP-MS | 5 ms | - Study of cisplatin and Gd@C82(OH)22 treatment | 198 |
| Normal human bronchial epithelial cells (16HBE) | - Micro-concentric nebulizer with a quartz impact bead spray chamber | |||||
| 2016 | Cervical cancer cells (HeLa) | Cr | q-ICP-MS | 10 ms | - Study on Cr(III) and Cr(VI) uptake by cancer cells | 199 |
| Breast cancer cells (MCF-7) | - V-groove nebulizer and water-cooled Scott-type double-pass spray chamber | |||||
| 2017 | Human ovarian carcinoma. Cisplatin resistant (A2780cis) and sensitive (A2780) cell lines | Fe (for counting cells) | q-ICP-MS | 10 ms | - EnyaMist nebulizer plus a total consumption spray chamber. TE of 25% | 200 |
| Pt (for tracking cisplatin uptake) | ||||||
| Tb (for tracking Tb-DTPA uptake) | ||||||
| 2017 | Pseudomonas putida KT2440 (bacteria) | Ag (to study Ag NP/ionic Ag toxicity) | CyTOF | - Study of a mass cytometry method for counting bacteria and differentiating live from dead bacterial cells | 201 | |
| Pt (for tracking cisplatin uptake) | - Study on Ag NP and ionic Ag uptake in single cells and their toxicity | |||||
| Ru (used as label for tracking all cells) | ||||||
| Ag to study Ag NP/ionic Ag toxicity | ||||||
| 2017 | Human T-lymphocyte cell line Jurkat (ATCC TIB-152) | Ag | CyTOF | - Study on cellular binding and uptake of Ag in NP-exposed cells | 202 | |
| 2017 | Chlorella vulgaris (green alga) | Cr (to track Cr uptake) | q-ICP-MS | 10 ms | - Study of heterogeneous cellular responses to environmental stresses | 203 |
| Mg (as biomarker of growth and physiological state of the cells) | - V-groove nebulizer plus Scott-type double-pass spray chamber (2°C) | |||||
| 2017 | Cervical cancer cells (HeLa) | Co, Cu, Fe, Mn, P, S, Zn | q-ICP-MS | 4 ms | - HECIS introduction system.128 Microconcentric nebulizer plus single-pass heated spray chamber | 136 |
| Human lung carcinoma cells (A549) | ||||||
| Normal human bronchial epithelial cell line (16HBE) | ||||||
| 2017 | Human hepatocyte carcinoma cell line (HepG2) | Zn (for tracking ZnO NPs uptake) | q-ICP-MS | 5 ms | - Microfluidic device droplet generator for cell encapsulation in hexanol, coupled to a PFA nebulizer (20–35 μL min−1). TE of 2.96% | 146 |
| 2017 | Melanoma mouse model (B16-OVA) | Au (for quantitation of Au NP biodistribution) | CyTOF | - Label-free method to track inorganic nanoparticles in tandem with highly multivariate cellular phenotyping, thus enabling quantitative analysis of the in vivo fate of inorganic nanomedicines. Au NPs selected as example | 204 | |
| Diverse metals (as tags for antibodies allowing cellular phenotyping) | ||||||
| 2017 | Leukemia cell lines (HL60 and NB4) | As, Dy, Nd, Pt | q-ICP-MS | 5 ms | - Examination of As2O3 cytotoxicity in leukemia cells, using cisplatin as viability dye and lanthanide tags | 205 |
| - Microconcentric nebulizer plus single-pass spray chamber | ||||||
| 2018 | Cryptomonas ovata, Chroomonas sp, Gonyostomum semen (algae) | Au | q-ICP-MS | 50 μs | - Quantification of Au NPs uptake and distribution in freshwater algae | 129 |
| - Asperon introduction system: high-efficiency glass concentric nebulizer plus liner pass spray chamber. TE of 30% | ||||||
| 2018 | Human epithelial lung adenocarcinoma cells (A549) | As (for tracking As uptake after incubation with arsenite) | ICP-MS/MS | 3 ms | - Micromist 400 μL nebulizer plus Scott-type spray chamber. TE of 0.5% | 206 |
| S and P (for cell counting) | ||||||
| 2018 | Human THP-1 monocytes | Ag | SF-ICP-MS | 100 μs | - Study of Ag NPs uptake by THP-1 monocytes and their partially differentiated macrophages | 157 |
| - Micromist concentric nebulizer plus conical quartz spray chamber with an impact bead. TE of 2–3% | ||||||
| 2018 | Human breast carcinoma (MCF-7) cells | Au, Y | q-ICP-MS | 10 ms | - Quantification of Au NPs and NaYF4:Yb,Er in single cells | 148 |
| - Microdroplet generator (MicroCross) with hexanol and dimethyl carbonate as organic phase for encapsulating single cells | ||||||
| 2019 | Cyclotella meneghiniana (marine diatom alga) | Mg | q-ICP-MS | 5 ms | - HPLC system for online removal of ionic Mg (inline filter) coupled to an ICP-MS in TRA mode for SC determination | 151 |
| - Cylindrical single-pass spray chamber (volume 8 mL) with a sheath gas port mounted in an aluminum block placed in a customized heater. TE of 1% | ||||||
| 2019 | Human breast cancer cells of different malignancy (MCF7 and MDA-MB 231) | Nd, P | ICP-MS/MS | 5 ms | - Nd as tag to quantify transferrin receptor 1 on the surface of cell cultures of breast cancer | 137 |
| - High-efficiency nebulizer and spray chamber.128 TE of 50–55% | ||||||
| 2019 | Escherichia coli, Staphylococcus aureus, Listeria monocytogenes, Shigella dysenteriae, Vibrio parahemolyticus | Eu (as tag for counting bacteria) | q-ICP-MS | 50–100 μs | - SC-ICP-MS for counting and recognizing single bacterial cells | 207 |
| Gd, La, Nd, Pr, Sm (as tags for specific bacterial identification) | - Asperon introduction system. TE of 15–36% | |||||
| - Study on cell-to-cell variability in the response of Staphylococcus aureus to vancomycin or Ag NPs exposure | ||||||
| 2019 | Chlorella vulgaris (alga) | Ag (for tracking Ag NP exposure) | q-ICP-MS | 10 ms | - Dual mass detection mode (two isotopes detected sequentially in the same run) | 208 |
| Mn | - Micromist nebulizer plus Scott-type spray chamber (2°C) | |||||
| Mg (for counting cells) | ||||||
| 2019 | Chlamydomonas reinhardtii CC-1690 | As | q-ICP-MS | 50 μs | - Complementary use of SC-ICP-MS and ambient molecular MS allowed for the study of arsenate uptake by cells | 209 |
| - Asperon introduction system. TE of 9.9 ± 0.9% | ||||||
| 2019 | Microcystis aeruginosa (toxic alga) | Cu (for tracking uptake of copper-based algaecides) | q-ICP-MS | 100 μs | - Meinhard nebulizer plus quartz single cell spray chamber heated at 29–32 °C. TE of 46–64% | 210 |
| Mg (for counting cells) | ||||||
| 2019 | Human breast cancer cells (MCF7) | 59Co for tracking cellular uptake and excretion of Co-based drugs | q-ICP-MS | 10 ms | - Standard concentric nebulizer and spray chamber. TE of 1% | 211 |
| Human hepatocyte carcinoma cells (HepG2) | ||||||
| 2019 | HeLa cells | Au (for tracking Au NP uptake) | q-ICP-MS | 5 ms | - Microfluidic device droplet generator described in ref. 146 | 147 |
| Zn (for counting cells) | ||||||
| 2019 | Human leukemia cells (K562) | Au | q-ICP-MS | 1 ms | - Spiral-Helix (3D) tubing array for single cell focusing/aligning coupled to a laboratory-made direct injection nebulization device. TE of 42.1 ± 7.2% | 139 |
| - Quantification of Au NP uptake | ||||||
| 2019 | Human hepatocyte carcinoma cells (HepG2) | Ag (for tracking Ag+ and Ag NP uptake) | q-ICP-MS | 5 ms | - 3D droplet based microfluidic device for encapsulating the cells in hexanol single droplets coupled to a Burgener nebulizer. Detection efficiency of 24% | 149 |
| Zn (for counting cells) | ||||||
| 2020 | Human red blood cells | Cu | q-ICP-MS | 100 μs | - High-efficiency introduction system for SC composed of a flow cell, a visual contrast calibration device, a customized nebulizer and a fabricated spray chamber. TE of 100% | 140 |
| 2020 | Saccharomyces cerevisiae (yeast) | P, Se | ICP-MS/MS | 5 ms | - SC-ICP-MS/MS performed first to detect intracellular Se. After mechanic lysis of the cells, SP-ICP-MS performed for proving the presence of SeNPs | 138 |
| - HECIS introduction system134 with a large-bore high-performance concentric nebulizer. TE of 69% | ||||||
| 2020 | Staphylococcus aureus and Escherichia coli (bacteria) | P, Te | ICP-MS/MS | 5 ms | - SC-ICP-MS/MS performed first to detect TeNP uptake. After mechanic lysis of the cells, SP-ICP-MS performed for characterization of the TeNPs changes after interaction with the cells | 130 |
| - High-performance concentric nebulizer combined with a total consumption spray chamber. TE (74 ± 6)% and (85 ± 7)% for E. coli and S. aureus, respectively | ||||||
| 2020 | Tetrahymena (protozoa) | Hg | q-ICP-MS | 3 ms | - Home-made capillary online introduction system directly coupled to the ICP-MS nebulizer. TE of 10.2% | 212 |
| 2020 | Single yeast | Fe, Mg, P, S, Zn | ICP-MS/MS | 100 μs | - Conventional concentric glass nebulizer and total consumption spray chamber (SC-SIS). TE ca. 10% | 131 |
| Green alga | ||||||
| Red blood cells | ||||||
| 2020 | Five diatom species: Cyclotella meneghiniana, Phaeodactylum tricornutum, Stephanodiscus minutulus, Thalassiosira weissflogii, Thalassiosira pseudonana | Fe, Mg, P, Si, Zn | ICP-TOF-MS | 1 full spectrum every 2.82 ms | - HPLC system for online algal cleanout coupled to an ICP-MS in TRA mode for SC determination | 213 |
| 2021 | Saccharomyces cerevisiae (yeast) | Cu, K, Mg, Mn, P, Zn | q-ICP-MS | 3 ms | - SC-ICP-MS to study changes in the elemental composition of yeast cells during growth | 214 |
| - Conventional sample introduction system with cyclonic spray chamber. TE of 0.3% | ||||||
| 2021 | Ovarian carcinoma cells sensitive and resistant to cisplatin (A2780 and A2780cis, respectively) | Fe, P, Pt | ICP-MS/MS | 5 ms | - Cellular uptake of Fe2O3 NPS transporting a cisplatin prodrug | 215 |
| - High-efficiency nebulizer and spray chamber137 | ||||||
| 2021 | Human hepatocyte carcinoma cells (HepG2) | Ag | q-ICP-MS | 100 μs | - Dean flow-assisted vortex capillary cell sampling unit to focus and align the flow of cells directly coupled to the nebulizer. TE ca. 30% | 216 |
| - Double coupling to a laser-induced fluorescence detector |
Tagging target compounds is obviously not limited to the use of such instrument. It was already mentioned in Section 3 that, in one of the earliest SP-ICP-MS articles, Hu et al. reported the use of Au NPs as tags to determine α-fetoprotein in serum.34 It is not infrequent to use NPs as tags, as NPs offer a high amplification power owing to the large number of atoms they are composed of,35,125 as will be further discussed in Section 4.3. Thus, SP-ICP-MS and SC-ICP-MS are clearly intertwined.
4.2. Sampling and sample introduction
This is an aspect where differences between SP-ICP-MS and SC-ICP-MS become more obvious. Self-evidently, there are some similarities between these two approaches, as discrete entities need to be introduced one-by-one into the ICP. To achieve this type of introduction, the samples need to be sufficiently diluted to avoid two cells monitored simultaneously, as in the case of NPs. However, the stability of NPs and cells is not the same. NPs are not very stable per se, so addition of a stabilizing agent to the suspensions (e.g., citrate) is usually recommended. But with cells, the situation is far more complex, and it also strongly depends on the type of cell. Some cells, such as bacteria, are quite robust, and it is possible to work with them in simple aqueous solutions. Other cells, however, are not, and the use of proper media is needed to prevent their disruption. A high osmotic pressure gradient between the cell and the solvent could end up in their lysis, with the subsequent release of the analytes into the suspension, thus leading to a constant signal (i.e., the analyte is present in dissolved form) instead of to a temporally resolved one. Please consider that the media used (e.g., a buffer, ethanol, etc.) may induce matrix effects that need to be effectively addressed when attempting quantification. Moreover, some cells tend to heavily adhere to surfaces, resulting in poor transport efficiencies and memory effects.126 This issue can be minimized by a procedure named trypsinization,14 adding an enzyme named trypsin that digests the proteins responsible for the adherence of the cells to the surfaces. To improve the robustness of some cells and to improve sample storage, cell fixation approaches using different types of fixatives (e.g., paraformaldehyde) are common practice for single cell analysis. Cell sedimentation is also to be prevented, which can be avoided via agitation. Some dedicated autosamplers are already equipped for the latter, and may also control the temperature, all of which is of high relevance when analyzing fresh cells. Finally, washing the cells to remove superficial contamination may also be required, as otherwise not only the elements present within the cell will be determined. For details on sample preparation prior to SC-ICP-MS, the recent reviews by Theiner et al.14 and Corte-Rodríguez et al.126 are recommended.The other important problem related to SC-ICP-MS concerns sample introduction into the ICP. The conventional spray chambers are designed to filter out the larger particles, thus preventing them from entering the plasma. Some cells are larger than the droplet sizes allowed by such spray chambers. For instance, mammalian cells typically show diameters between 8 and 30 μm.14 It is already well-established that their TE will be very poor when using conventional sample introduction systems. In fact, Nomizu et al. already reported analysis of cell suspensions via ICP-OES in 1994,127 and the TE for mouse and human cell lines was found to be of less than 0.1% only.
Therefore, what in SP-ICP-MS can be considered as optional, (1) use of an alternative type of spray chamber or (2) direct injection via microdroplets, is often compulsory in SC-ICP-MS, and several examples of both approaches can be found in the literature.
Concerning the first approach, it can be mentioned that the spray chamber displayed in Fig. 1b and 3 was actually designed for single cell analysis. It is a commercially available (under the name Asperon™) high-efficiency sample introduction setup comprising a low-pressure nebulizer (to ensure that cells survive the nebulization process intact) and a low volume spray chamber. As discussed in Section 3.3, these spray chambers are often equipped with a sheath gas to shape the nebulized aerosol plume and reduce cell deposition on the spray chamber walls.128 With such device, for example, a TE of approx. 30% for Au NPs and 2.5 μm lanthanide-doped polystyrene beads (used as cell proxies) has been reported.129 A similar device has been produced by Glass Expansion under the name Single-Cell Sample Introduction System (SC-SIS). With such device, Gomez-Gomez et al. reported TE values of 74 ± 6% and 85 ± 7% for E. coli and S. aureus, respectively, attributing this discrepancy to the difference in size of the target samples: 2.14 ± 0.52 μm for E. coli versus 0.74 ± 0.12 μm for S. aureus.130 Tanaka et al. used the same device but reported much lower TE values between 8 and 13% for cells (single yeast, green alga, and red blood cells), while for silica NPs (200 nm) it was 55%.131 They attributed this difference to the larger size of the cells investigated (approx. 5 μm). These TEs reported for cells using the SC-SIS seem to be in good agreement with those reported recently by Liu et al. (approx. 15%) for three different types of human cell lines.132 Finally, as mentioned earlier, CyTOF devices also make use of a similar total consumption spray chamber with an Ar make up gas, but in this case the spray chamber is also heated to stimulate solvent evaporation. TE values in the 20–30% range have been published for such approach when using a 60 μL min−1 sample introduction flow rate.133
Besides these now commercially available systems, home-made alternatives have been explored for years. One of these has already been mentioned in Section 3.3, when applied to NPs, but it was in fact designed for cell analysis. It is known as the HECIS and was proposed first by Groombridge et al. in 2013,134 followed the year after by a modified version128 to further improve the TE. The latter device (shown in Fig. 9a) consists of a large-bore central capillary and a small-volume (15 cm3) on-axis spray chamber utilizing a sheath gas flow. It is a total consumption spray chamber. The authors reported TE values ranging from 86% for C. reinhardtii CC-125 (mean cell diameter of 6.4 μm) to ca. 100% for other microbes with mean cell diameters of 2.0–3.0 μm,128 while for the original version the TE was 75 ± 4.7% (for S. cerevisiae). The authors also emphasize the benefit of using a torch injector with smaller diameter (1.5 mm instead of 2.5 mm) to better focus the ion plume and enable a more reproducible plasma sampling by the cone, which was already recommended by Ho and Chan,135 in one of the first articles devoted to SC-ICP-MS. This strategy based on the use of a smaller diameter injector is now common in all single event-ICP-MS applications.
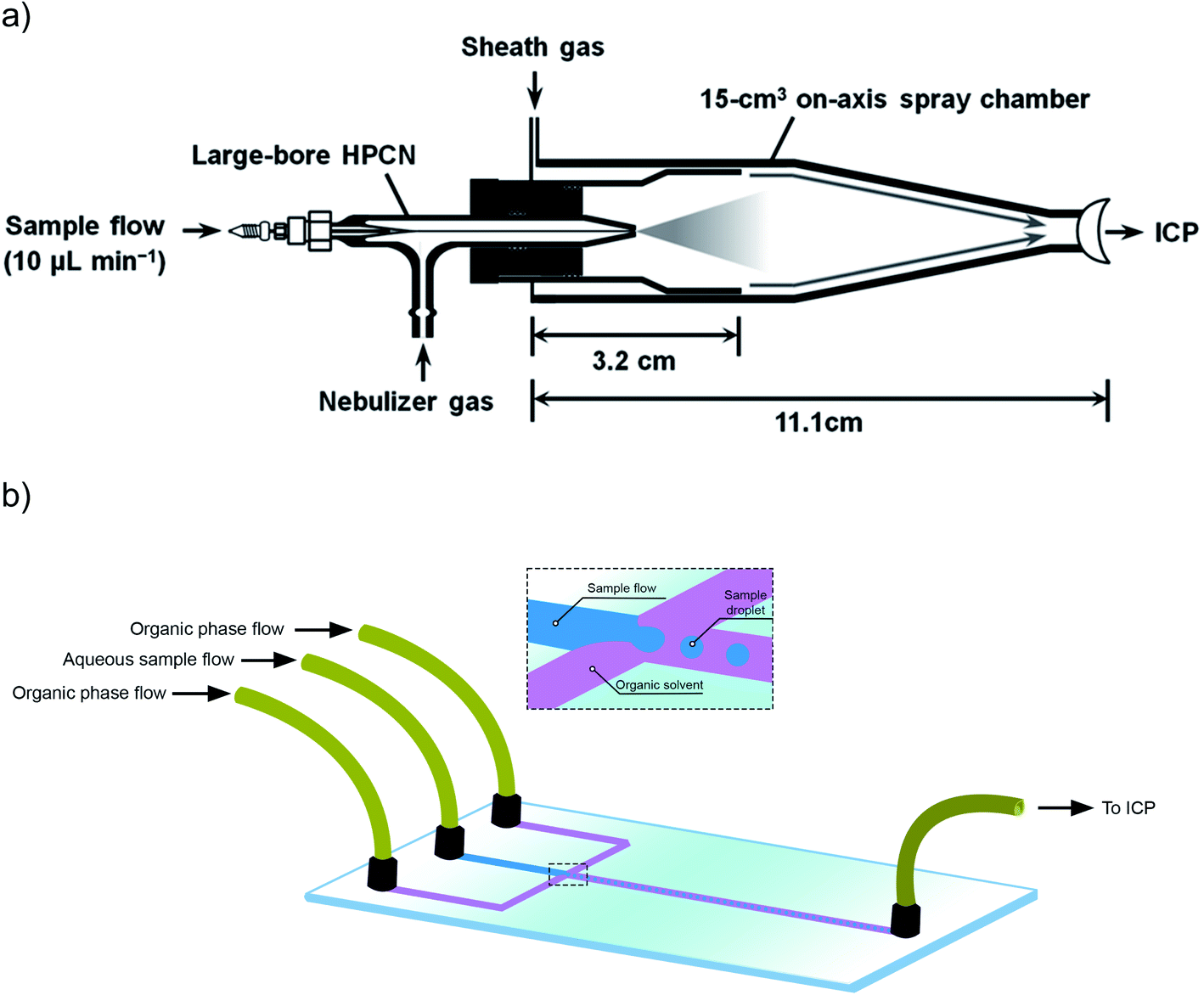 | ||
| Fig. 9 (a) Schematic diagram of a modified High-Efficiency Cell Introduction System (HECIS). Reproduced with permission from RSC (DOI: 10.1039/C4JA00040D);128(b) schematic diagram illustrating the operation of a microfluidic chip for sample introduction into an ICP. | ||
Other authors have also made use of this HECIS device. For instance, Wang et al. deployed a HECIS with a heating cover.136 The authors indicate that, for In-containing solutions, analyte introduction was up to 10-fold more efficient in comparison to the set-up with a PFA cyclonic spray chamber (take also into account that for such set-up the sample uptake rate was approximately 300 μL min−1, in comparison with the 10 μL min−1 of the HECIS). Corte-Rodríguez et al. reported the use of a system similar to the modified HECIS with TEs up to 55% for their method of determination of transferrin receptor 1 in breast cancer cells,137 while for 30 nm Au NPs they reported a TE of 70%. In another work, Álvarez-Fernández García et al. reported a value of 69 ± 3% for yeast cells using a similar device.138
Another direct injection device with a custom-made concentric nebulizer and an almost total consumption spray chamber was proposed by Wei et al.,139 providing a TE value of 42.1 ± 7.2%, as evaluated with 30 nm Au NPs. Furthermore, this work couples the system to a spiral tubing array (named Helix) to facilitate single cell focusing and proper cell spacing to measure them at time intervals of 0.97 ± 0.41 ms, reporting a sample throughput of 40000 cells per minute. Finally, Cao et al. introduced another similar total consumption spray chamber device and reported 100% TE for red blood cells, but at a flow of 0.8 μL min−1 for the cell suspension, leading to the monitoring of 120 cells per minute “only”.140
Overall, there is a large variation in the TE values that are being reported in the literature. For larger cells, it is clearly more challenging to achieve values close to 100%, which is in fact not so surprising. Moreover, increasing the sample throughput while keeping a high transport efficiency is a key issue that will probably remain a topic of future research.
Less popular to date, but an interesting alternative to pneumatic nebulization, is the encapsulation of cells into larger droplets. Two different types of systems have been deployed for such purpose: a piezoelectrical μDG (already described in Section 3.3 for SP-ICP-MS) and microfluidic-based dispensers. The first paper reporting on the use of the piezoelectrical μDG encapsulating cells (single selenized yeast cells) was published by Shigeta et al. in 2013.141 The authors used a dwell time of 100 μs and reported a duration of the events in the range of 400 to 500 μs, all very similar to SP-ICP-MS conditions. As an advantage, the authors state that “the fixed droplet generation rate of 50 Hz produced equidistant signals in time of each droplet event and this was advantageous to separate contributions from noise, background and blank from the analytical signal.” In addition, TE values of 100% were reported and absolute amounts of femtogram to tens of femtogram were determined for Cu, Fe, Mg, Na, Se and Zn. Further improvements in terms of signal acquisition and data processing for that system were discussed in ref. 142.
Finally, also using μDG, a recent work of Vonderach et al.143 proposed a novel orientation for the ICP, i.e., downwards instead of horizontal, as a gravity-assisted sampling approach to improve the TE of larger cells and droplets, which are obviously more challenging to transport in conventional, horizontally oriented ICP-MS devices. The authors report linearity when increasing the frequency up to 1000 Hz for droplets up to sizes of approx. 70 μm. This work was followed by another one in which the quadrupole mass filter used in the ICP instrument was replaced by a TOF-MS detector, enabling the multi-element monitoring of individual droplets containing single cells.144
An alternative approach for cell encapsulation is based on the design of microfluidic chips. These normally consists of two micro channels, that carry an organic compound, which intersect with the sample stream (aqueous phase) micro channel, as shown in Fig. 9b, resulting in the production of droplets which can transport a single cell. Verboket et al.,145 were the first to use microfluidics in the context of SC-ICP-MS, achieving more than 50% TE. Several similar devices have been proposed, and one key difference is related with the organic compound used for the encapsulation, as it is well-known that the ICP is not impervious to the introduction of such compounds. Verboket et al. proposed perfluorohexane, while the group of Hu, which has been particularly active in this field, proposed hexyl alcohol (due to the lower C content than other compounds146,147), and Wei et al.148 used hexanol in combination with dimethyl carbonate, the latter being added to facilitate the oxidation of the alcohol, thus minimizing carbon deposition on the cones.
Moreover, these are all 2-D microfluidic chips, while Yu et al., proposed the use of a novel 3-D chip that can be assembled from commercially available modules,149 thus being very simple to manufacture. The authors also used hexyl alcohol, as in their previous works, and hydroxypropyl methyl cellulose as suspending agent, to keep the cells evenly dispersed. TE values of approx. 25% were reported. Yu et al. provided a detailed review on sample introduction strategies for SC-ICP-MS.150
A final aspect related with sample introduction that can be mentioned is the possibility to couple SC-ICP-MS with other techniques. Von der Au et al. proposed the coupling of HPLC to ICP-MS to perform on-line purification of the sample (thus, reducing the ionic BG) followed by SC-ICP-MS, in an automated way for the determination of Mg in C. meneghiniana.151
4.3. Calculations
The calculations for SC-ICP-MS are similar to those discussed before for SP-ICP-MS. In fact, some aspects can even be seen as easier. For example, there is no need to convert the mass distributions into size distributions (spherical equivalent diameters) as is typically done for SP-ICP-MS. In the case of SC-ICP-MS, the target information is the mass distribution (Fig. 4c) of a cell population. The number of cells per unit volume or cell concentration (also sometimes referred to as cell density) can also be calculated, but this information is less useful in this context, because there are other techniques routinely deployed in cell studies that can provide such data.14 Very often, this cell concentration is known in advance and calculating the cell concentration via SC-ICP-MS is useful for quality control purposes only.It also needs to be mentioned that in case of multiplexed bioassay (mass cytometry), the calculations are much more complex, as multiparametric values are obtained, and the reader is thus referred to Kimball et al.152 for more information on this topic.
The calculations for preparing a diluted suspension prior to SC-ICP-MS are similar to those discussed for SP-ICP-MS in Section 3. The recommended number of events is similar, because the duration of a single event generated by a cell is about the same as that generated by an ENP. In this sense, Liu et al. also evaluated the effect of CRC technology on the signal duration of single cell events and results similar to those obtained for SP-ICP-MS were reported in the case of pressurization of the CRC with gases like O2 and NH3.132 This means that also in the case of SC-ICP-MS, special attention needs to be paid to the dilution factors applied when SC-ICP-MS is deployed for analytes affected by interferences and chemical resolution is used to overcome spectral overlap.
As for the sensitivity, cells are very often much larger entities, but, unlike the case for NPs, the target element will not be a major component of the structure. The number of atoms of such analyte present in every cell (or in the tag(s)) is the key parameter that determines if a signal will be detected or not. This also depends on the analyte of interest (mass number, isotopic abundance, ionization energy, occurrence of spectral interference) and on the type of ICP-MS unit deployed, but again it should be kept in mind that at least 10000–1000000 ions are needed to produce a signal. This determines the single cell limits of detection, which are in the fg per cell level for several target analytes. Nowadays, different labelling strategies can be used to amplify the analytical signal, thus improving the limits of detection even further. Such signal amplification is achieved by increasing the number of ICP-MS detectable atoms attached to the target species. For this purpose, different types of exogenous metal tags, such as chelates, metal-containing compounds, inorganic nanoparticles, and polymer-based elemental tags, have been used for the quantification of biological markers. However, the tagging of antibodies with larger entities, such as various polymer-based elemental tags, each containing a number of atoms, could affect the behavior of the antibodies, so that tagging with a single metal atom might be preferred, although at the cost of much more limited signal amplification. Furthermore, special attention should be paid to the homogeneity of the labelling probes, as appropriate quantification depends on the reproducibility of the ICP-MS signals. For example, in the case of using NPs, low polydispersity is required to ensure a homogeneous population, but these conditions cannot always be fulfilled. For more details about amplification strategies, the reader is referred elsewhere.153
SC-ICP-MS shows a wide range of different applications. It allows for the quantification of the absolute amounts (masses) of endogenous and exogenous metal(loid)s, as well as of some important non-metals (e.g., P, S). In the first case, SC-ICP-MS focuses on analytes that are found at relatively high levels in the cells owing to their cellular role (e.g., Cu, Fe, K, Na, or Zn), as many proteins and metabolites need the aid of metals to exert their biological functions. Secondly, cells can be exposed to metals in the context of different applications, such as metallodrug studies (e.g., cis-Pt as a chemotherapeutic drug), exposure to ENPs (that are often used as carriers for drug delivery) or use of metal tags (mass cytometry). However, the determination of elements found at trace levels in cells remains challenging. To have a better idea of the applications reported, the reader is referred to Table 3.
As indicated above, calibration is needed in a similar way as for SP-ICP-MS. If the goal is to determine both the cell density and the mass per cell distribution, then a “double” calibration is needed, one to establish the TE of cells, and the other to determine the sensitivity. For the first one, reference materials are not available. Polystyrene microspheres spiked with known contents of four elements (Ce, Eu, Ho and Lu) can be used as a cell proxy, as mentioned before.129 Such a standard was originally developed for mass cytometry calculations154 and is commercially available.155 The manufacturers provide a reference value for the tags (Ce, Eu, Ho and Lu atoms) per bead, with ±15% as uncertainty.
However, the TE determined for highly stable, monodisperse, and relatively hard polymeric particulate material cannot always be compared to that obtained for relatively fragile cell entities, and thus, the TE obtained for polymer beads tends to be higher than that for cells, thus leading to an underestimation of the cell density. Again, it could be that the cell density is already known by means of other techniques (e.g., using a hemocytometer), so the TE can be directly estimated by counting the number of events detected and comparing this value with the number of cells injected during a period of time. Please note that “bulk” ICP-MS analysis after acid digestion of a well-known number of cells is traditionally used in SC-ICP-MS to validate the results, but this requires previous knowledge of the cell density. This is an important difference compared to SP-ICP-MS, where other independent techniques (e.g., microscopic techniques), rather than ICP-MS, can be used to validate the size distribution results.
For assessing the sensitivity, ionic solutions are often used, assuming the same response from ions and intracellular species.156 Matrix effects could, however, occur, as discussed in Section 3.4, and in this case, they may be even more significant than for SP-ICP-MS. In fact, if it is large enough, the cell itself can affect the plasma,141 and the conditions used need to ensure that the cell is completely “digested”, as otherwise biased results will be obtained. An appropriate calibration always requires the use of a correction factor for the TE of ionic standard solutions, as can be seen in eqn (2) accompanying Fig. 4. However, there is a difference here in comparison with SP-ICP-MS. In the latter case, the TE of NPs is assumed to be the same as that of the ionic standard solutions typically used for calibration purposes, so only one TE value needs to be calculated (both TE values appearing in eqn (1) and (2) of Fig. 4 are equal). However, the TE of cells is different (much lower) than that of the corresponding solutions used for calibration, as discussed before. Thus, most authors make use of well-established NP reference standards (e.g., NIST Au NPs) to calculate a TE that is later applied to calculate the absolute amounts (masses) of a given element per cell.
In other words, if both absolute mass per cell distribution and cell concentration need to be determined by SC-ICP-MS, it is better to establish two different TE values for the calculations shown in eqn (1) and (2). This could be seen as one of the most important differences between SP and SC. But, in practice, it is often unnecessary to calculate the cell concentration via single event-ICP-MS as discussed before, and the actual TE for cells is only estimated by researchers to assess the efficiency of their sampling device, and not for further calculations.
As an alternative to the use of ionic standard solutions for calibration, particles (e.g., MgO instead of Mg ions) have been proposed as their signals were observed to be more similar to those obtained from the cells.135 Calibration with NP suspensions when targeting the quantification of the cellular uptake of Ag NPs157 has also been suggested.
It could be finally mentioned that a recent paper by Degueldre has proposed a method for the counting and identification of single viruses, based on the measuring of C, N, P and S and the calculation of N/C, P/C and S/C ratios. This is, however, so far a theoretical proposal only, the realization of which would require instrumentation with high-mass resolution and simultaneous multi-element detection, besides other modifications to reduce the BG signal of the most abundant elements (e.g., placing the torch in an Ar atmosphere, instead of operating under atmospheric conditions).158
A timely and alternative approach for the simultaneous detection of both SARS-CoV-2 and influenza A, has been reported by Xu et al.159 The method is based on the addition of AuNPs and AgNPs, which, in the presence of SARS-CoV-2 (in the case of Au) or influenza A (in the case of Ag), form larger aggregates that can be monitored using SP-ICP-MS. There are no mutual interferences, so both viruses can be detected simultaneously. The authors stress that this type of approach can, in principle, be adapted for the detection of multiple nucleic acids, proteins, cells and other biological molecules by changing different modification sequences and using different NP probes.
Overall, this a vibrant field as illustrated by the upwards trend in publications observed during the last couple of years, and many of the developments made could be beneficial for all single event-ICP-MS types of application.
5. Quantification of micro/nanoplastics. A new hope
This application represents another interesting turn of events. It was a priori considered that single event-ICP-MS was not appropriate for monitoring carbon-based materials. In fact, in the first review on this approach, Laborda et al. wrote that “Single particle ICPMS is able to provide information about the elemental chemical composition of noncarbon nanomaterials (carbon-based nanomaterials are excluded due to the intrinsic low sensitivity of this element in ICPMS)…”.160There are certainly reasons for such statement because carbon is characterized by a high ionization potential (and it is thus poorly ionized in an ICP) and, moreover, a typical ICP does not operate in a closed environment, such that contamination from ubiquitous C-sources (e.g., CO2 in the Ar plasma gas and the atmosphere) will increase the BG. Still, results have proved that there is enough potential to measure carbon-based nanomaterials or, at least, micromaterials with ICP-MS.
Some early attempts for carbon nanotubes (CNTs) were reported,161 although metals were used as proxy to detect the CNTs. This is of course not ideal, as it requires an a priori knowledge on the target CNTs (known content of some metallic impurities), besides a homogeneous distribution of the target metal.
But, more recently, Bolea-Fernandez et al. published the first article on the detection of microplastics based on C-monitoring using ICP-MS operated in single-event mode.162 The work is based on the same principles as described before in Fig. 1b. Carbon (as major constituent of polymeric particles) was monitored as 13C+ (relative abundance = 1.1%), due to the high BG intensity observed for the most abundant C isotope, (12C, relative abundance = 98.9%). This indicates that the key towards monitoring of even smaller structures is to reduce such BG (e.g., removing potential carbon sources).
By using a sample introduction device similar to that shown in Fig. 1b and described in Section 4.2 and with a dwell time of 100 μs, the signals shown in Fig. 10 were obtained. This figure shows the signals resulting from the monitoring of either 13C+ or 165Ho+ for the 2.5 μm lanthanide-doped polystyrene beads mentioned in Section 4.2. The histograms clearly show that a much more favorable signal-to-background ratio is obtained when measuring the lanthanide, but detecting C is still possible. The comparison between the number of signals detected for C and Ho using different dilution factors demonstrated accurate PNC results. Furthermore, the authors also measured 1 μm polystyrene microspheres and showed that the signal intensities from both polymeric particles are proportional, as expected if the particles are fully “digested” by the plasma, thus demonstrating the potential of this approach for the characterization of microplastics.
 | ||
| Fig. 10 Transient signals (a and c) and integrated intensity distributions (b and d) obtained for 2.5 μm lanthanide-doped polystyrene microspheres measured using ICP-MS operated in single-event mode via the monitoring of 13C+ or 165Ho+. Reproduced with permission from the Royal Society of Chemistry (DOI: 10.1039/C9JA00379G).162 | ||
It is important to state that the signal obtained in this measurement mode will be proportional to the amount of C present in each plastic. That means that, for accurate quantification, the exact composition of every target plastic needs to be known in advance, like in the case of ENPs that are composed of more than one element. The shape does not need to be known if the goal is just to provide the mass of each plastic particle, but it is also possible to convert that value into the diameter if microspheres are measured, in the same way as discussed for NPs.
This work was followed up by an article by Laborda et al.,163 confirming the potential for studying the particle size of polymer microspheres. Quantification using aqueous dissolved carbon standards was demonstrated. The authors defined their working range between 1 μm (due to the limitations for C monitoring discussed above) to 5–6 μm. It was also reported that larger particles showed lower TE, which is not surprising as the same trend was already discussed in the previous section for cells of different sizes.
Another work by Gonzalez de Vega et al.164 has explored the possibilities to use ICP-MS/MS in this context (see Section 3.5 for a discussion on this approach46,47). In this work, the authors recommend the monitoring of 12C+ instead of 13C+, leading to size detection limits of 0.62 μm (measured on-mass, with H2 in the cell) and 0.96 μm (measured on-mass, without any gas in the cell), in pure water and in seawater, respectively, for polystyrene-based microspheres. The authors discussed matrix effects and the need to correct for them via matrix-matching in the case of seawater. Moreover, they have also demonstrated the utility of this approach (measuring C in single-event mode) to monitor single cells (5 different algae species). In this case, to minimize spectral overlap, O2 was used as reaction gas, thus monitoring the reaction product ion 12C16O+via ICP-MS/MS at m/z 28. Knowledge about the C mass fraction per cell allowed the calculation of cell sizes.
Very recently, Liu et al.165 relied on the 13C+ monitoring to quantitatively analyze the microplastics ageing process (UV-light accelerated ageing dynamics) with a wide particle size range (800 nm to 5 μm) and a PNC at an environmentally relevant value (down to 7.1 × 106 particles per L). The ecotoxicological risk of microplastics during ageing was evaluated by exposing Daphnia magna to pristine and aged microplastics.
While the first paper on the use of single-event ICP-MS for microplastics characterization based on C-monitoring dates from 2020 only, this is an emerging field for which a fast development can already be anticipated. To some extent, this can be attributed to the lack of suitable analytical techniques for the characterization of low μm-range microplastics (1–10 μm) and nanoplastics (1 nm to 1 μm).19 To characterize microplastics down to 10 μm size, other techniques, such as Raman microscopy (μRaman), micro-Fourier Transform Infrared Spectroscopy (μFTIR) and Laser Direct Infrared Spectroscopy (LDIR), are being extensively used. However, the smaller plastic particles might be more harmful to human health and other life forms, and despite this, they have been consistently neglected in quantification studies. Since it is expected that, in many real-life samples, the number of particles increases steeply for smaller particle sizes, it is in this size range that this novel approach can find its application niche.
It needs to be mentioned as well that, besides direct monitoring of C and C-species, tagging is always an option to amplify the signal,166 thus enabling the characterization of even smaller plastic particles (nanoplastics), as demonstrated by Jiménez-Lamana et al.167 for polystyrene particles conjugated with functionalized Au NPs, for which a size detection limit of 135 nm was reported. Lai et al. have also described a new approach based on the separation of the nanoplastics found in environmental waters by cloud-point extraction, followed by in situ growth of Au NPs onto their surface, thus enabling the use of SP-ICP-MS for their detection. This way, different types of nanoplastics with sizes down to 50 nm can be counted.168 Furthermore, the development of new metal-doped (e.g., Ir and Pd) micro- and nano-plastics169,170 and of nanoparticle–nanopolymer composites171,172 further contribute to the extension of the application range of this methodology, as it can play an important role in exposure and risk assessment studies.
6. Conclusions and outlook
Single event-ICP-MS is an example of how, by pushing forward the speed of data acquisition, a mature technique like ICP-MS can still produce new types of information, thus fulfilling scientific and societal demands.It can be anticipated that single event-ICP-MS is here to stay, and new applications beyond nanoparticles and cells will be explored in the coming years (e.g., micro/nanoplastics). There is still a need to improve the detection power to be able to detect events created by a smaller number of ions. In fact, an ideal ICP-MS device should be able to carry out simultaneous multi-element monitoring, without sacrificing sensitivity and with high (physical or chemical) resolution. At this point, there is not an ideal type of ICP-MS that meets all these requirements. However, considering the advances observed over the last decade in the field of ICP-MS (e.g., speed of data acquisition, introduction of ICP-MS/MS, improvements in ICP-TOF-MS instrumentation) and the rapid evolution of single event-ICP-MS, the old adagio comes to mind: when there is a need, there is a way.
List of abbreviations
| BG | Background |
| CRC | Collision/reaction cell |
| ENP | Engineered nanoparticle |
| FFF | Field flow fractionation |
| ICP-MS/MS | Inductively coupled plasma-mass spectrometer configured with a collision reaction cell located in-between two quadrupole-based mass analyzers. Also often referred to as triple quadrupole-ICP-MS |
| ICP-TOF-MS | Time-of-flight inductively coupled plasma-mass spectrometer |
| LA | Laser ablation |
| LODsize | Size detection limit |
| NNP | Naturally occurring nanoparticle |
| NP | Nanoparticle |
| PNC | Particle number concentration |
| q-ICP-MS | Quadrupole-based inductively coupled plasma-mass spectrometer |
| SC-ICP-MS | Single cell-inductively coupled plasma-mass spectrometry |
| SF-ICP-MS | High-resolution sector field inductively coupled plasma-mass spectrometer |
| SP-ICP-MS | Single particle-inductively coupled plasma-mass spectrometry |
| TE | Transport efficiency |
| TRA | Time-resolved analysis |
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Author contributions
M. Resano: conceptualization; funding acquisition; project administration; resources; visualization; writing – original draft; writing – review & editing. M. Aramendía: visualization; writing – review & editing. E. García-Ruiz: visualization; writing – review & editing. A. Bazo: visualization; writing – review & editing. E. Bolea-Fernández: writing – review & editing. F. Vanhaecke: funding acquisition; project administration; resources; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the European Regional Development Fund for financial support through the Interreg POCTEFA EFA 176/16/DBS, as well as to project PGC2018-093753-B-I00 (MCIU/AEI//FEDER, UE) and the Aragon Government (Construyendo Europa desde Aragón, Grupo E43_20R). E. Bolea-Fernández thanks FWO-Vlaanderen for his postdoctoral grant (12ZA320N). A. Bazo thanks the Aragon Government for his doctoral grant.References
- M. Resano, M. A. Belarra, E. García-Ruiz, M. Aramendía and L. Rello, TrAC, Trends Anal. Chem., 2018, 99, 75–87 CrossRef CAS.
- L. Delahaye, H. Veenhof, B. C. P. Koch, J.-W. C. Alffenaar, R. Linden and C. Stove, Ther. Drug Monit., 2021, 43, 310–321 CrossRef PubMed.
- G. Nys, M. G. M. Kok, A.-C. Servais and M. Fillet, TrAC, Trends Anal. Chem., 2017, 97, 326–332 CrossRef CAS.
- K. Kanaki and S. A. Pergantis, J. Anal. At. Spectrom., 2016, 31, 1041–1046 RSC.
- M. Trojanowicz and K. Kołacińska, Analyst, 2016, 141, 2085–2139 RSC.
- P. J. Sylvester and S. E. Jackson, Elements, 2016, 12, 307–310 CrossRef CAS.
- M. Aramendía, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2009, 648, 23–44 CrossRef PubMed.
- E. Mavrakis and S. A. Pergantis, Anal. Chim. Acta, 2021, 1179, 338830 CrossRef CAS PubMed.
- E. Bolea-Fernández, K. Phan, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2016, 941, 1–9 CrossRef PubMed.
- S. Capiau, E. Bolea-Fernandez, L. Balcaen, C. Van Der Straeten, A. G. Verstraete, F. Vanhaecke and C. P. Stove, Talanta, 2020, 208, 120055 CrossRef CAS PubMed.
- C. T. Driscoll, R. P. Mason, H. M. Chan, D. J. Jacob and N. Pirrone, Environ. Sci. Technol., 2013, 47, 4967–4983 CrossRef CAS PubMed.
- N. B. Coffman and J. J. Fernandes, J. Am. Osteopath. Assoc., 1992, 92, 907–909 CAS.
- J. Picot, C. L. Guerin, C. Le Van Kim and C. M. Boulanger, Cytotechnology, 2012, 64, 109–130 CrossRef PubMed.
- S. Theiner, K. Loehr, G. Koellensperger, L. Mueller and N. Jakubowski, J. Anal. At. Spectrom., 2020, 35, 1784–1813 RSC.
- H. M. Davey and D. B. Kell, Microbiol. Rev., 1996, 60, 641–696 CrossRef CAS PubMed.
- D. D. Carlo and L. P. Lee, Anal. Chem., 2006, 78, 7918–7925 CrossRef PubMed.
- X. Huang, H. Liu, D. Lu, Y. Lin, J. Liu, Q. Liu, Z. Nie and G. Jiang, Chem. Soc. Rev., 2021, 50, 5243–5280 RSC.
- A. B. Silva, A. S. Bastos, C. I. L. Justino, J. P. da Costa, A. C. Duarte and T. A. P. Rocha-Santos, Anal. Chim. Acta, 2018, 1017, 1–19 CrossRef CAS PubMed.
- M. Velimirovic, K. Tirez, S. Verstraelen, E. Frijns, S. Remy, G. Koppen, A. Rotander, E. Bolea-Fernandez and F. Vanhaecke, J. Anal. At. Spectrom., 2021, 36, 695–705 RSC.
- P. A. Doble, R. Gonzalez de Vega, D. P. Bishop, D. J. Hare and D. Clases, Chem. Rev., 2021, 121, 11769–11822 CrossRef CAS PubMed.
- R. Thomas, Spectroscopy, 2001, 16, 38–42 Search PubMed.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155 RSC.
- M. Tanner and D. Günther, Anal. Chim. Acta, 2009, 633, 19–28 CrossRef CAS PubMed.
- M. D. Montaño, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Anal. Bioanal. Chem., 2016, 408, 5053–5074 CrossRef PubMed.
- T. Nomizu, S. Kaneco, T. Tanaka, T. Yamamoto and H. Kawaguchi, Anal. Sci., 1993, 9, 843–846 CrossRef CAS.
- S. Kaneco, T. Nomizu, T. Tanaka, N. Mizutani and H. Kawaguchi, Anal. Sci., 1995, 11, 835–840 CrossRef CAS.
- T. Nomizu, H. Hayashi, N. Hoshino, T. Tanaka, H. Kawaguchi, K. Kitagawa and S. Kaneco, J. Anal. At. Spectrom., 2002, 17, 592–595 RSC.
- C. Degueldre and P. Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- C. Degueldre, Talanta, 2004, 62, 1051–1054 CrossRef CAS PubMed.
- C. Degueldre, P.-Y. Favarger and C. Bitea, Anal. Chim. Acta, 2004, 518, 137–142 CrossRef CAS.
- C. Degueldre, P.-Y. Favarger and S. Wold, Anal. Chim. Acta, 2006, 555, 263–268 CrossRef CAS.
- C. Degueldre, P. Y. Favarger, R. Rossé and S. Wold, Talanta, 2006, 68, 623–628 CrossRef CAS PubMed.
- M. H. P. Yau and W.-T. Chan, J. Anal. At. Spectrom., 2005, 20, 1197 RSC.
- S. Hu, R. Liu, S. Zhang, Z. Huang, Z. Xing and X. Zhang, J. Am. Soc. Mass Spectrom., 2009, 20, 1096–1103 CrossRef CAS PubMed.
- A. Lores-Padín, P. Menero-Valdés, B. Fernández and R. Pereiro, Anal. Chim. Acta, 2020, 1128, 251–268 CrossRef PubMed.
- R. Liu, Z. Xing, Y. Lv, S. Zhang and X. Zhang, Talanta, 2010, 83, 48–54 CrossRef CAS PubMed.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- A. Gustavsson, Spectrochim. Acta, Part B, 1984, 39, 743–746 CrossRef.
- K.-S. Ho, W.-W. Lee and W.-T. Chan, J. Anal. At. Spectrom., 2015, 30, 2066–2073 RSC.
- S. Cuello-Nuñez, I. Abad-Álvaro, D. Bartczak, M. E. del Castillo Busto, D. A. Ramsay, F. Pellegrino and H. Goenaga-Infante, J. Anal. At. Spectrom., 2020, 35, 1832–1839 RSC.
- J. Tuoriniemi, G. Cornelis and M. Hassellöv, Anal. Chem., 2012, 84, 3965–3972 CrossRef CAS PubMed.
- J. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Anal. Chem., 2014, 86, 3405–3414 CrossRef CAS PubMed.
- E. Bolea, M. S. Jimenez, J. Perez-Arantegui, J. C. Vidal, M. Bakir, K. Ben-Jeddou, A. C. Gimenez-Ingalaturre, D. Ojeda, C. Trujillo and F. Laborda, Anal. Methods, 2021, 13, 2742–2795 RSC.
- M. Resano, M. Aramendía, F. V. Nakadi, E. García-Ruiz, C. Alvarez-Llamas, N. Bordel, J. Pisonero, E. Bolea-Fernández, T. Liu and F. Vanhaecke, TrAC, Trends Anal. Chem., 2020, 129, 115955 CrossRef CAS.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2015, 894, 7–19 CrossRef CAS PubMed.
- E. Bolea-Fernández, L. Balcaen, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 1660–1679 RSC.
- S. D. Tanner, Spectrochim. Acta, Part B, 1992, 47, 809–823 CrossRef.
- L. Hendriks, B. Ramkorun-Schmidt, A. Gundlach-Graham, J. Koch, R. N. Grass, N. Jakubowski and D. Günther, J. Anal. At. Spectrom., 2019, 34, 716–728 RSC.
- E. Bolea-Fernández, D. Leite, A. Rua-Ibarz, L. Balcaen, M. Aramendia, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 2140–2152 RSC.
- I. Kálomista, A. Kéri, D. Ungor, E. Csapó, I. Dekany, T. Prohaska and G. Galbács, J. Anal. At. Spectrom., 2017, 32, 2455–2462 RSC.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS PubMed.
- T. P. J. Linsinger, R. Peters and S. Weigel, Anal. Bioanal. Chem., 2014, 406, 3835–3843 CrossRef CAS PubMed.
- A. R. M. Bustos, E. J. Petersen, A. Possolo and M. R. Winchester, Anal. Chem., 2015, 87, 8809–8817 CrossRef PubMed.
- S. Weigel, R. Peters, K. Loeschner, R. Grombe and T. P. J. Linsinger, Anal. Bioanal. Chem., 2017, 409, 4839–4848 CrossRef CAS PubMed.
- A. R. Montoro Bustos, K. P. Purushotham, A. Possolo, N. Farkas, A. E. Vladár, K. E. Murphy and M. R. Winchester, Anal. Chem., 2018, 90, 14376–14386 CrossRef CAS PubMed.
- E. J. Petersen, A. R. Montoro Bustos, B. Toman, M. E. Johnson, M. Ellefson, G. C. Caceres, A. L. Neuer, Q. Chan, J. W. Kemling, B. Mader, K. Murphy and M. Roesslein, Environ. Sci.: Nano, 2019, 6, 2876–2896 RSC.
- O. Geiss, I. Bianchi, C. Senaldi, G. Bucher, E. Verleysen, N. Waegeneers, F. Brassinne, J. Mast, K. Loeschner, J. Vidmar, F. Aureli, F. Cubadda, A. Raggi, F. Iacoponi, R. Peters, A. Undas, A. Müller, A. K. Meinhardt, E. Walz, V. Gräf and J. Barrero-Moreno, Food Control, 2021, 120, 107550 CrossRef CAS PubMed.
- I. Strenge and C. Engelhard, J. Anal. At. Spectrom., 2016, 31, 135–144 RSC.
- A. Hineman and C. Stephan, J. Anal. At. Spectrom., 2014, 29, 1252–1257 RSC.
- M. D. Montaño, H. R. Badiei, S. Bazargan and J. F. Ranville, Environ. Sci.: Nano, 2014, 1, 338–346 RSC.
- I. Abad-Alvaro, E. Pena-Vazquez, E. Bolea, P. Bermejo-Barrera, J. R. Castillo and F. Laborda, Anal. Bioanal. Chem., 2016, 408, 5089–5097 CrossRef CAS PubMed.
- A. Kaňa, M. Loula, R. Koplík, M. Vosmanská and O. Mestek, Talanta, 2019, 197, 189–198 CrossRef PubMed.
- A. Kéri, I. Kálomista, D. Ungor, Á. Bélteki, E. Csapó, I. Dékány, T. Prohaska and G. Galbács, Talanta, 2018, 179, 193–199 CrossRef PubMed.
- A. M. Duffin, E. D. Hoegg, R. I. Sumner, T. Cell, G. C. Eiden and L. S. Wood, J. Anal. At. Spectrom., 2021, 36, 133–141 RSC.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, J. Anal. At. Spectrom., 2015, 30, 1274–1285 RSC.
- International Organization for Standardization-ISO, ISO/TS 19590:2017(E). Nanotechnologies—Size distribution and concentration of inorganic nanoparticles in aqueous media via single particle inductively coupled plasma mass spectrometry, 2017 Search PubMed.
- K. Newman, C. Metcalfe, J. Martin, H. Hintelmann, P. Shaw and A. Donard, J. Anal. At. Spectrom., 2016, 31, 2069–2077 RSC.
- J. Tuoriniemi, G. Cornelis and M. Hassellöv, J. Anal. At. Spectrom., 2015, 30, 1723–1729 RSC.
- T. E. Lockwood, R. Gonzalez de Vega and D. Clases, J. Anal. At. Spectrom., 2021, 36, 2536–2544 RSC.
- E. Bolea-Fernandez, D. Leite, A. Rua-Ibarz, T. Liu, G. Woods, M. Aramendia, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2019, 1077, 95–106 CrossRef CAS PubMed.
- G. Cornelis and M. Hassellov, J. Anal. At. Spectrom., 2014, 29, 134–144 RSC.
- A. Gundlach-Graham, L. Hendriks, K. Mehrabi and D. Günther, Anal. Chem., 2018, 90, 11847–11855 CrossRef CAS PubMed.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2020, 35, 1740–1783 RSC.
- A. Williams, A. Al Hejami and D. Beauchemin, J. Anal. At. Spectrom., 2020, 35, 2165–2170 RSC.
- S. Miyashita, H. Mitsuhashi, S. Fujii, A. Takatsu, K. Inagaki and T. Fujimoto, Anal. Bioanal. Chem., 2017, 409, 1–15 CrossRef PubMed.
- A. Cañabate, E. García-Ruiz, M. Resano and J. L. Todolì, J. Anal. At. Spectrom., 2017, 32, 78–87 RSC.
- C. C. Garcia, A. Murtazin, S. Groh, V. Horvatic and K. Niemax, J. Anal. At. Spectrom., 2010, 25, 645–653 RSC.
- J. W. Olesik, Appl. Spectrosc., 1997, 51, 158A–175A CrossRef CAS.
- S. Gschwind, L. Flamigni, J. Koch, O. Borovinskaya, S. Groh, K. Niemax and D. Günther, J. Anal. At. Spectrom., 2011, 26, 1166–1174 RSC.
- C. Agatemor and D. Beauchemin, Anal. Chim. Acta, 2011, 706, 66–83 CrossRef CAS PubMed.
- M. Loula, A. Kaňa and O. Mestek, Talanta, 2019, 202, 565–571 CrossRef CAS PubMed.
- I. Abad-Alvaro, D. Leite, D. Bartczak, S. Cuello-Nunez, B. Gomez-Gomez, Y. Madrid, M. Aramendia, M. Resano and H. Goenaga-Infante, J. Anal. At. Spectrom., 2021, 36, 1180–1192 RSC.
- L. Telgmann, C. D. Metcalfe and H. Hintelmann, J. Anal. At. Spectrom., 2014, 29, 1265–1272 RSC.
- C. A. Sötebier, D. J. Kutscher, L. Rottmann, N. Jakubowski, U. Panne and J. Bettmer, J. Anal. At. Spectrom., 2016, 31, 2045–2052 RSC.
- S. Yamashita, M. Ishida, T. Suzuki, M. Nakazato and T. Hirata, Spectrochim. Acta, Part B, 2020, 169, 105881 CrossRef CAS.
- D. P. Leite, New methods for nanoparticle characterization by means of single particle inductively coupled plasma mass spectrometry, Universidad de Zaragoza, 2021 Search PubMed.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- B. Ramkorun-Schmidt, S. A. Pergantis, D. Esteban-Fernández, N. Jakubowski and D. Günther, Anal. Chem., 2015, 87, 8687–8694 CrossRef CAS PubMed.
- D. Rosenkranz, F. L. Kriegel, E. Mavrakis, S. A. Pergantis, P. Reichardt, J. Tentschert, N. Jakubowski, P. Laux, U. Panne and A. Luch, Anal. Chim. Acta, 2020, 1099, 16–25 CrossRef CAS PubMed.
- A. Gundlach-Graham and K. Mehrabi, J. Anal. At. Spectrom., 2020, 35, 1727–1739 RSC.
- A. Praetorius, A. Gundlach-Graham, E. Goldberg, W. Fabienke, J. Navratilova, A. Gondikas, R. Kaegi, D. Günther, T. Hofmann and F. von der Kammer, Environ. Sci.: Nano, 2017, 4, 307–314 RSC.
- K. Mehrabi, R. Kaegi, D. Günther and A. Gundlach-Graham, Environ. Sci.: Nano, 2021, 8, 1211–1225 RSC.
- T. R. Holbrook, D. Gallot-Duval, T. Reemtsma and S. Wagner, J. Anal. At. Spectrom., 2021, 36, 2684–2694 RSC.
- A. Rua-Ibarz, E. Bolea-Fernandez, G. Pozo, X. Dominguez-Benetton, F. Vanhaecke and K. Tirez, J. Anal. At. Spectrom., 2020, 35, 2023–2032 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739–1763 CrossRef.
- J. Kocic, D. Günther and B. Hattendorf, J. Anal. At. Spectrom., 2021, 36, 233–242 RSC.
- P. Shaw and A. Donard, J. Anal. At. Spectrom., 2016, 31, 1234–1242 RSC.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, Part B, 2002, 57, 1361–1452 CrossRef.
- S. Diez Fernández, N. Sugishama, J. Ruiz Encinar and A. Sanz-Medel, Anal. Chem., 2012, 84, 5851–5857 CrossRef PubMed.
- I. Kálomista, A. Kéri and G. Galbács, J. Anal. At. Spectrom., 2016, 31, 1112–1122 RSC.
- J. Jiménez-Lamana, I. Abad-Álvaro, K. Bierla, F. Laborda, J. Szpunar and R. Lobinski, J. Anal. At. Spectrom., 2018, 33, 452–460 RSC.
- L. A. Rush, M. C. Endres, M. Liezers, J. D. Ward, G. C. Eiden and A. M. Duffin, Talanta, 2018, 189, 268–273 CrossRef CAS PubMed.
- G. Bucher and F. Auger, J. Anal. At. Spectrom., 2019, 34, 1380–1386 RSC.
- S. Candás-Zapico, D. J. Kutscher, M. Montes-Bayón and J. Bettmer, Talanta, 2018, 180, 309–315 CrossRef PubMed.
- S. Naasz, S. Weigel, O. Borovinskaya, A. Serva, C. Cascio, A. K. Undas, F. C. Simeone, H. J. P. Marvin and R. J. B. Peters, J. Anal. At. Spectrom., 2018, 33, 835–845 RSC.
- N. D. Donahue, S. Kanapilly, C. Stephan, M. C. Marlin, E. R. Francek, M. Haddad, J. Guthridge and S. Wilhelm, Nano Lett., 2022, 22, 294–301 CrossRef CAS PubMed.
- K.-H. Chun, J. T.-S. Lum and K. S.-Y. Leung, Anal. Chim. Acta, 2022, 1192, 339389 CrossRef CAS PubMed.
- S. Alavi, X. Guo, S. M. Javid, A. Ebrahimi and J. Mostaghimi, Anal. Chem., 2020, 92, 11786–11794 CrossRef CAS PubMed.
- B. Franze, I. Strenge and C. Engelhard, J. Anal. At. Spectrom., 2017, 32, 1481–1489 RSC.
- D. Wang and S. Bodovitz, Trends Biotechnol., 2010, 28, 281–290 CrossRef CAS PubMed.
- Fluidigm Canada Inc, Fluidigm CyTOF Publications, https://www.fluidigm.com/publications/cytof--helios, accessed September 11, 2021.
- S. C. Bendall, G. P. Nolan, M. Roederer and P. K. Chattopadhyay, Trends Immunol., 2012, 33, 323–332 CrossRef CAS PubMed.
- A. Delgado-Gonzalez and R. M. Sanchez-Martin, Anal. Chem., 2021, 93, 657–664 CrossRef CAS PubMed.
- F. Li, D. W. Armstrong and R. S. Houk, Anal. Chem., 2005, 77, 1407–1413 CrossRef CAS PubMed.
- V. I. Baranov, Z. Quinn, D. R. Bandura and S. D. Tanner, Anal. Chem., 2002, 74, 1629–1636 CrossRef CAS PubMed.
- V. I. Baranov, Z. A. Quinn, D. R. Bandura and S. D. Tanner, J. Anal. At. Spectrom., 2002, 17, 1148–1152 RSC.
- S. C. Bendall, E. F. Simonds, P. Qiu, E. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696 CrossRef CAS PubMed.
- S. D. Tanner, O. Ornatsky, D. R. Bandura and V. I. Baranov, Spectrochim. Acta, Part B, 2007, 62, 188–195 CrossRef.
- O. I. Ornatsky, X. Lou, M. Nitz, S. Schäffer, W. S. Sheldrick, V. I. Baranov, D. R. Bandura and S. D. Tanner, Anal. Chem., 2008, 80, 2539–2547 CrossRef CAS PubMed.
- S. D. Tanner, D. R. Bandura, O. Ornatsky, V. I. Baranov, M. Nitz and M. A. Winnik, Pure Appl. Chem., 2008, 80, 2627–2641 CrossRef CAS.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed.
- Fluidigm Canada Inc, Helios, a CyTOF System. User Guide, https://www.fluidigm.com/binaries/content/documents/fluidigm/consumables/pages/mass-cytometry/mass-cytometry/fluidigm%3Aresources%5B2%5D/helios-user-guide0400250/fluidigm%3Afile, accessed September 15, 2021 Search PubMed.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS PubMed.
- J. Hu, D. Deng, R. Liu and Y. Lv, J. Anal. At. Spectrom., 2018, 33, 57–67 RSC.
- M. Corte-Rodríguez, R. Álvarez-Fernández, P. García-Cancela, M. Montes-Bayón and J. Bettmer, TrAC, Trends Anal. Chem., 2020, 132, 116042 CrossRef.
- T. Nomizu, S. Kaneco, T. Tanaka, D. Ito, H. Kawaguchi and B. T. Vallee, Anal. Chem., 1994, 66, 3000–3004 CrossRef CAS.
- S. Miyashita, A. S. Groombridge, S. Fujii, A. Minoda, A. Takatsu, A. Hioki, K. Chiba and K. Inagaki, J. Anal. At. Spectrom., 2014, 29, 1598–1606 RSC.
- R. C. Merrifield, C. Stephan and J. R. Lead, Environ. Sci. Technol., 2018, 52, 2271–2277 CrossRef CAS PubMed.
- B. Gomez-Gomez, M. Corte-Rodríguez, M. T. Perez-Corona, J. Bettmer, M. Montes-Bayón and Y. Madrid, Anal. Chim. Acta, 2020, 1128, 116–128 CrossRef CAS PubMed.
- Y. Tanaka, R. Iida, S. Takada, T. Kubota, M. Yamanaka, N. Sugiyama, Y. Abdelnour and Y. Ogra, ChemBioChem, 2020, 21, 3266–3272 CrossRef CAS PubMed.
- T. Liu, E. Bolea-Fernandez, C. Mangodt, O. De Wever and F. Vanhaecke, Anal. Chim. Acta, 2021, 1177, 338797 CrossRef CAS PubMed.
- O. Ornatsky, D. Bandura, V. Baranov, M. Nitz, M. A. Winnik and S. Tanner, J. Immunol. Methods, 2010, 361, 1–20 CrossRef CAS PubMed.
- A. S. Groombridge, S. Miyashita, S. Fujii, K. Nagasawa, T. Okahashi, M. Ohata, T. Umemura, A. Takatsu, K. Inagaki and K. Chiba, Anal. Sci., 2013, 29, 597–603 CrossRef CAS PubMed.
- K.-S. Ho and W.-T. Chan, J. Anal. At. Spectrom., 2010, 25, 1114–1122 RSC.
- H. Wang, M. Wang, B. Wang, L. Zheng, H. Chen, Z. Chai and W. Feng, Anal. Bioanal. Chem., 2017, 409, 1415–1423 CrossRef CAS PubMed.
- M. Corte-Rodríguez, E. Blanco-González, J. Bettmer and M. Montes-Bayón, Anal. Chem., 2019, 91, 15532–15538 CrossRef PubMed.
- R. Álvarez-Fernández García, M. Corte-Rodríguez, M. Macke, K. L. LeBlanc, Z. Mester, M. Montes-Bayón and J. Bettmer, Analyst, 2020, 145, 1457–1465 RSC.
- X. Wei, X. Zhang, R. Guo, M.-L. Chen, T. Yang, Z.-R. Xu and J.-H. Wang, Anal. Chem., 2019, 91, 15826–15832 CrossRef CAS PubMed.
- Y. Cao, J. Feng, L. Tang, C. Yu, G. Mo and B. Deng, Talanta, 2020, 206, 120174 CrossRef CAS PubMed.
- K. Shigeta, G. Koellensperger, E. Rampler, H. Traub, L. Rottmann, U. Panne, A. Okino and N. Jakubowski, J. Anal. At. Spectrom., 2013, 28, 637–645 RSC.
- T. Iwai, K. Shigeta, M. Aida, Y. Ishihara, H. Miyahara and A. Okino, J. Anal. At. Spectrom., 2015, 30, 1617–1622 RSC.
- T. Vonderach, B. Hattendorf and D. Günther, Anal. Chem., 2021, 93, 1001–1008 CrossRef CAS PubMed.
- T. Vonderach and D. Günther, J. Anal. At. Spectrom., 2021, 36, 2617–2630 RSC.
- P. E. Verboket, O. Borovinskaya, N. Meyer, D. Guenther and P. S. Dittrich, Anal. Chem., 2014, 86, 6012–6018 CrossRef CAS PubMed.
- H. Wang, B. Chen, M. He and B. Hu, Anal. Chem., 2017, 89, 4931–4938 CrossRef CAS PubMed.
- H. Wang, B. Chen, M. He, X. Li, P. Chen and B. Hu, Talanta, 2019, 200, 398–407 CrossRef CAS PubMed.
- X. Wei, D.-H. Zheng, Y. Cai, R. Jiang, M.-L. Chen, T. Yang, Z.-R. Xu, Y.-L. Yu and J.-H. Wang, Anal. Chem., 2018, 90, 14543–14550 CrossRef CAS PubMed.
- X. Yu, B. Chen, M. He, H. Wang and B. Hu, Anal. Chem., 2019, 91, 2869–2875 CrossRef CAS PubMed.
- X. Yu, M. He, B. Chen and B. Hu, Anal. Chim. Acta, 2020, 1137, 191–207 CrossRef CAS PubMed.
- M. von der Au, M. Schwinn, K. Kuhlmeier, C. Büchel and B. Meermann, Anal. Chim. Acta, 2019, 1077, 87–94 CrossRef PubMed.
- A. K. Kimball, L. M. Oko, B. L. Bullock, R. A. Nemenoff, L. F. van Dyk and E. T. Clambey, J. Immunol., 2018, 200, 3–22 CrossRef CAS PubMed.
- A. L. Larraga-Urdaz, M. L. F. Sanchez, J. R. Encinar and J. M. Costa-Fernandez, Anal. Bioanal. Chem., 2022, 414, 53–62 CrossRef CAS PubMed.
- A. I. Abdelrahman, O. Ornatsky, D. Bandura, V. Baranov, R. Kinach, S. Dai, S. C. Thickett, S. Tanner and M. A. Winnik, J. Anal. At. Spectrom., 2010, 25, 260–268 RSC.
- Fluidigm Canada Inc, EQ Four Element Calibration Beads—100 mL, https://store.fluidigm.com/Cytometry/ConsumablesandReagentsCytometry/MaxparBuffersAndSolutions/EQ%20Four%20Element%20Calibration%20Beads%E2%80%94100%20mL, accessed September 15, 2021 Search PubMed.
- L.-N. Zheng, M. Wang, B. Wang, H.-Q. Chen, H. Ouyang, Y.-L. Zhao, Z.-F. Chai and W.-Y. Feng, Talanta, 2013, 116, 782–787 CrossRef CAS PubMed.
- A. L.-S. Oliver, S. Baumgart, W. Bremser, S. Flemig, D. Wittke, A. Grützkau, A. Luch, A. Haase and N. Jakubowski, J. Anal. At. Spectrom., 2018, 33, 1256–1263 RSC.
- C. Degueldre, Talanta, 2021, 228, 122211 CrossRef CAS PubMed.
- Y. Xu, B. Chen, M. He and B. Hu, Anal. Chim. Acta, 2021, 1186, 339134 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- R. B. Reed, D. G. Goodwin, K. L. Marsh, S. S. Capracotta, C. P. Higgins, D. H. Fairbrother and J. F. Ranville, Environ. Sci.: Processes Impacts, 2013, 15, 204–213 RSC.
- E. Bolea-Fernandez, A. Rua-Ibarz, M. Velimirovic, K. Tirez and F. Vanhaecke, J. Anal. At. Spectrom., 2020, 35, 455–460 RSC.
- F. Laborda, C. Trujillo and R. Lobinski, Talanta, 2021, 221, 121486 CrossRef CAS PubMed.
- R. Gonzalez de Vega, S. Goyen, T. E. Lockwood, P. A. Doble, E. F. Camp and D. Clases, Anal. Chim. Acta, 2021, 1174, 338737 CrossRef CAS PubMed.
- Z. Liu, Y. Zhu, S. Lv, Y. Shi, S. Dong, D. Yan, X. Zhu, R. Peng, A. A. Keller and Y. Huang, Environ. Sci. Technol. Lett., 2022, 9, 50–56 CrossRef CAS.
- L. Marigliano, B. Grassl, J. Szpunar, S. Reynaud and J. Jiménez-Lamana, Molecules, 2021, 26, 7093 CrossRef CAS PubMed.
- J. Jiménez-Lamana, L. Marigliano, J. Allouche, B. Grassl, J. Szpunar and S. Reynaud, Anal. Chem., 2020, 92, 11664–11672 CrossRef PubMed.
- Y. Lai, L. Dong, Q. Li, P. Li, Z. Hao, S. Yu and J. Liu, Environ. Sci. Technol., 2021, 55, 4783–4791 CrossRef CAS PubMed.
- Q. Gao, Y. Wang, Y. Ji, X. Zhao, P. Zhang and L. Chen, J. Hazard. Mater., 2022, 424, 127628 CrossRef CAS PubMed.
- D. M. Mitrano, A. Beltzung, S. Frehland, M. Schmiedgruber, A. Cingolani and F. Schmidt, Nat. Nanotechnol., 2019, 14, 362–368 CrossRef CAS PubMed.
- A. Barber, S. Kly, M. G. Moffitt, L. Rand and J. F. Ranville, Environ. Sci.: Nano, 2020, 7, 514–524 RSC.
- R. J. Rauschendorfer, K. M. Whitham, S. Summer, S. A. Patrick, A. E. Pierce, H. Sefi-Cyr, S. Tadjiki, M. D. Kraft, S. R. Emory, D. A. Rider and M. D. Montaño, Front. Toxicol., 2021, 3, 752296 CrossRef PubMed.
- B. Franze, I. Strenge and C. Engelhard, J. Anal. At. Spectrom., 2012, 27, 1074–1083 RSC.
- S. Yongyang, W. Wei, L. Zhiming, D. Hu, Z. Guoqing, X. Jiang and R. Xiangjun, J. Anal. At. Spectrom., 2015, 30, 1184–1190 RSC.
- M. Roman, C. Rigo, H. Castillo-Michel, I. Munivrana, V. Vindigni, I. Mičetić, F. Benetti, L. Manodori and W. R. L. Cairns, Anal. Bioanal. Chem., 2016, 408, 5109–5124 CrossRef CAS PubMed.
- J. Jiménez-Lamana, J. Wojcieszek, M. Jakubiak, M. Asztemborska and J. Szpunar, J. Anal. At. Spectrom., 2016, 31, 2321–2329 RSC.
- A. R. Donovan, C. D. Adams, Y. Ma, C. Stephan, T. Eichholz and H. Shi, Chemosphere, 2016, 144, 148–153 CrossRef CAS PubMed.
- A. R. Donovan, C. D. Adams, Y. Ma, C. Stephan, T. Eichholz and H. Shi, Anal. Bioanal. Chem., 2016, 408, 5137–5145 CrossRef CAS PubMed.
- A. Donard, F. Claverie, F. Pointurier, C. B. Frayret, B. Svatosova and C. Pécheyran, Anal. Chem., 2017, 89, 8791–8799 CrossRef CAS PubMed.
- J. Vidmar, R. Milačič and J. Ščančar, Microchem. J., 2017, 132, 391–400 CrossRef CAS.
- R. C. Merrifield, C. Stephan and J. R. Lead, Talanta, 2017, 162, 130–134 CrossRef CAS PubMed.
- L. Fréchette-Viens, M. Hadioui and K. J. Wilkinson, Talanta, 2017, 163, 121–126 CrossRef PubMed.
- A. Sápi, A. Kéri, I. Kálomista, D. G. Dobó, Á. Szamosvölgyi, K. L. Juhász, Á. Kukovecz, Z. Kónya and G. Galbács, J. Anal. At. Spectrom., 2017, 32, 996–1003 RSC.
- K. Folens, T. Van Acker, E. Bolea-Fernández, G. Cornelis, F. Vanhaecke, G. Du Laing and S. Rauch, Sci. Total Environ., 2018, 615, 849–856 CrossRef CAS PubMed.
- J. Vidmar, P. Oprčkal, R. Milačič, A. Mladenovič and J. Ščančar, Sci. Total Environ., 2018, 634, 1259–1268 CrossRef CAS PubMed.
- A. R. Donovan, C. D. Adams, Y. Ma, C. Stephan, T. Eichholz and H. Shi, Chemosphere, 2018, 195, 531–541 CrossRef CAS PubMed.
- A. Hegetschweiler, O. Borovinskaya, T. Staudt and T. Kraus, Anal. Chem., 2019, 91, 943–950 CrossRef CAS PubMed.
- L. Fréchette-Viens, M. Hadioui and K. J. Wilkinson, Talanta, 2019, 200, 156–162 CrossRef PubMed.
- M. Hadioui, G. Knapp, A. Azimzada, I. Jreije, L. Frechette-Viens and K. J. Wilkinson, Anal. Chem., 2019, 91, 13275–13284 CrossRef CAS PubMed.
- A. Kéri, A. Sápi, D. Ungor, D. Sebők, E. Csapó, Z. Kónya and G. Galbács, J. Anal. At. Spectrom., 2020, 35, 1139–1147 RSC.
- R. P. Lamsal, M. S. E. Houache, A. Williams, E. Baranova, G. Jerkiewicz and D. Beauchemin, Anal. Chim. Acta, 2020, 1120, 67–74 CrossRef CAS PubMed.
- Y. Xing, J. Han, X. Wu, D. T. Pierce and J. X. Zhao, Analyst, 2021, 145, 7932–7940 RSC.
- S. Fernández-Trujillo, M. Jiménez-Moreno, Á. Ríos and R. del C. R. Martín-Doimeadios, Talanta, 2021, 231, 122370 CrossRef PubMed.
- D. Metarapi, J. T. van Elteren, M. Šala, K. Vogel-Mikuš, I. Arčon, V. S. Šelih, M. Kolar and S. B. Hočevar, Environ. Sci.: Nano, 2021, 8, 647–656 RSC.
- C.-N. Tsang, K.-S. Ho, H. Sun and W.-T. Chan, J. Am. Chem. Soc., 2011, 133, 7355–7357 CrossRef CAS PubMed.
- Q. Chang, O. I. Ornatsky, C. J. Koch, N. Chaudary, D. T. Marie-Egyptienne, R. P. Hill, S. D. Tanner and D. W. Hedley, Int. J. Cancer, 2015, 136, 1202–1209 CrossRef CAS PubMed.
- H. Wang, B. Wang, M. Wang, L. Zheng, H. Chen, Z. Chai, Y. Zhao and W. Feng, Analyst, 2015, 140, 523–531 RSC.
- L.-N. Zheng, M. Wang, L.-C. Zhao, B.-Y. Sun, B. Wang, H.-Q. Chen, Y.-L. Zhao, Z.-F. Chai and W.-Y. Feng, Anal. Bioanal. Chem., 2015, 407, 2383–2391 CrossRef CAS PubMed.
- X. Wei, L.-L. Hu, M.-L. Chen, T. Yang and J.-H. Wang, Anal. Chem., 2016, 88, 12437–12444 CrossRef CAS PubMed.
- M. Corte Rodríguez, R. Álvarez-Fernández García, E. Blanco, J. Bettmer and M. Montes-Bayón, Anal. Chem., 2017, 89, 11491–11497 CrossRef PubMed.
- Y. Guo, S. Baumgart, H.-J. Stärk, H. Harms and S. Müller, Front. Microbiol., 2017, 8, 1–9 CAS.
- A. Ivask, A. J. Mitchell, C. M. Hope, S. C. Barry, E. Lombi and N. H. Voelcker, Anal. Chem., 2017, 89, 8228–8232 CrossRef CAS PubMed.
- W.-Y. Lau, K.-H. Chun and W.-T. Chan, J. Anal. At. Spectrom., 2017, 32, 807–815 RSC.
- Y.-S. S. Yang, P. U. Atukorale, K. D. Moynihan, A. Bekdemir, K. Rakhra, L. Tang, F. Stellacci and D. J. Irvine, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- Y. Zhou, H. Li and H. Sun, Chem. Commun., 2017, 53, 2970–2973 RSC.
- S. Meyer, A. López-Serrano, H. Mitze, N. Jakubowski and T. Schwerdtle, Metallomics, 2018, 10, 73–76 CrossRef CAS PubMed.
- Y. Liang, Q. Liu, Y. Zhou, S. Chen, L. Yang, M. Zhu and Q. Wang, Anal. Chem., 2019, 91, 8341–8349 CrossRef CAS PubMed.
- J. T.-S. Lum and K. S.-Y. Leung, Anal. Chim. Acta, 2019, 1061, 50–59 CrossRef CAS PubMed.
- E. Mavrakis, L. Mavroudakis, N. Lydakis-Simantiris and S. A. Pergantis, Anal. Chem., 2019, 91, 9590–9598 CrossRef CAS PubMed.
- X. Shen, H. Zhang, X. He, H. Shi, C. Stephan, H. Jiang, C. Wan and T. Eichholz, Anal. Bioanal. Chem., 2019, 411, 5531–5543 CrossRef CAS PubMed.
- Q.-X. Sun, X. Wei, S.-Q. Zhang, M.-L. Chen, T. Yang and J.-H. Wang, Anal. Chim. Acta, 2019, 1066, 13–20 CrossRef CAS PubMed.
- J. Shi, X. Ji, Q. Wu, H. Liu, G. Qu, Y. Yin, L. Hu and G. Jiang, Anal. Chem., 2020, 92, 622–627 CrossRef CAS PubMed.
- M. von der Au, O. Borovinskaya, L. Flamigni, K. Kuhlmeier, C. Büchel and B. Meermann, Algal Res., 2020, 49, 101964 CrossRef.
- W. Qin, H.-J. Stärk, S. Müller, T. Reemtsma and S. Wagner, Metallomics, 2021, mfab032 CrossRef PubMed.
- D. Turiel-Fernández, L. Gutiérrez-Romero, M. Corte-Rodriguez, J. Bettmer and M. Montes-Bayón, Anal. Chim. Acta, 2021, 1159, 338356 CrossRef PubMed.
- C. Wu, X. Wei, X. Men, X. Zhang, Y.-L. Yu, Z.-R. Xu, M.-L. Chen and J.-H. Wang, Anal. Chem., 2021, 93, 8203–8209 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |