
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvoAMtht from Methyloversatilis thermotolerans ovothiol biosynthesis is a bifunction enzyme: thiol oxygenase and sulfoxide synthase activities†
Ronghai
Cheng‡
a,
Andrew C.
Weitz‡
a,
Jared
Paris‡
b,
Yijie
Tang
b,
Jingyu
Zhang
c,
Heng
Song
a,
Nathchar
Naowarojna
a,
Kelin
Li
a,
Lu
Qiao
a,
Juan
Lopez
a,
Mark W.
Grinstaff
 a,
Lixin
Zhang
c,
Yisong
Guo
*b,
Sean
Elliott
*a and
Pinghua
Liu
*a
a,
Lixin
Zhang
c,
Yisong
Guo
*b,
Sean
Elliott
*a and
Pinghua
Liu
*a
aDepartment of Chemistry, Boston University, 590 Commonwealth Ave., Boston, MA 02215, USA. E-mail: pinghua@bu.edu; elliott@bu.edu
bDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 1521, USA. E-mail: ysguo@andrew.cmu.edu
cState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, 130 Meilong Rd, Shanghai, 200237, China
First published on 2nd March 2022
Abstract
Mononuclear non-heme iron enzymes are a large class of enzymes catalyzing a wide-range of reactions. In this work, we report that a non-heme iron enzyme in Methyloversatilis thermotolerans, OvoAMtht, has two different activities, as a thiol oxygenase and a sulfoxide synthase. When cysteine is presented as the only substrate, OvoAMtht is a thiol oxygenase. In the presence of both histidine and cysteine as substrates, OvoAMtht catalyzes the oxidative coupling between histidine and cysteine (a sulfoxide synthase). Additionally, we demonstrate that both substrates and the active site iron's secondary coordination shell residues exert exquisite control over the dual activities of OvoAMtht (sulfoxide synthase vs. thiol oxygenase activities). OvoAMtht is an excellent system for future detailed mechanistic investigation on how metal ligands and secondary coordination shell residues fine-tune the iron-center electronic properties to achieve different reactivities.
Introduction
Ergothioneine and ovothiol A (4, 8, Scheme 1) are naturally occurring thiol-histidine derivatives. Both of them have beneficial effects to human health. Recently, Ames proposed that ergothioneine is a longevity vitamin,1 and exhibits protective roles in many aging associated diseases, including dementia, depression, atherosclerosis, cardiovascular disorders, and nonalcoholic fatty liver disease.2,3 Ovothiol A inhibits cell proliferation with the concomitant activation of an autophagic process in human hepatocarcinoma cell lines, Hep-G2,4 suggesting its potential anti-cancer activities.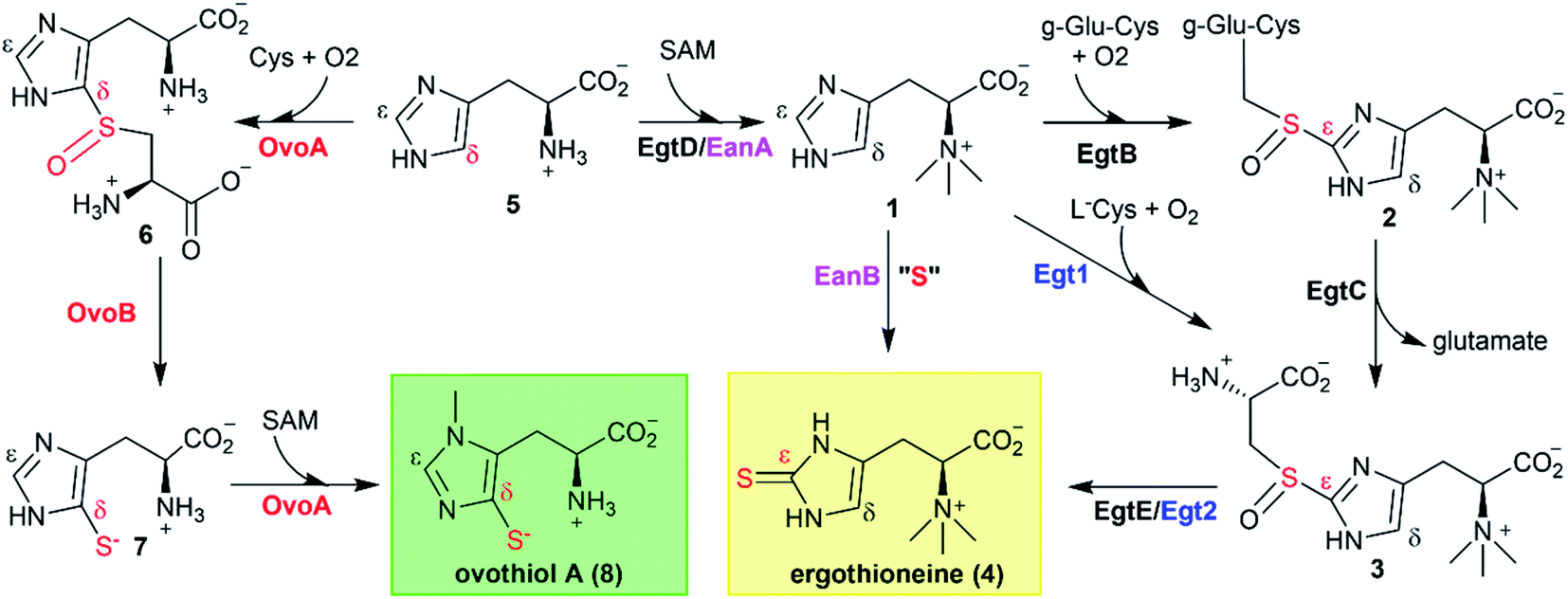 | ||
| Scheme 1 Ergothioneine and ovothiol biosynthetic pathways. Two aerobic ergothioneine biosynthetic pathways: the EgtB–EgtC–EgtE-catalysis in mycobacteria, and Egt1–Egt2-catalysis in fungi; An anaerobic ergothioneine biosynthetic pathway: EanB-catalysis in sulfur bacteria; The OvoA–OvoB catalysis in the aerobic ovothiol A biosynthetic pathway. | ||
Due to the growing interests in ergothioneine and ovothiol due to their biological activities,2,3,5 the discovery of the ergothioneine and ovothiol biosynthetic pathways (Scheme 1) provides a starting point for their production through a biosynthetic approach.6,7 Moreover, enzymes in these biosynthetic pathways are attractive from a mechanistic enzymology point of view because the C–S bond formation reactions in these two pathways are unprecedented transformations. Thus far, all attempts to trap reaction intermediates have failed because the reaction is fast and no intermediates could be observed by pre-steady state enzyme kinetics.
To address this issue, we decided to analyze and characterize enzymes from thermophilic and mesophilic organisms.8 When enzymes from these organisms are studied at ambient temperatures, they may have slower reaction rates, allowing us to trap reaction intermediates.
In this report, we characterized a mononuclear iron enzyme from Methyloversatilis thermotolerans (OvoAMtht), an organism with an optimum growth temperature of 30–37 °C. Besides having a significantly improved thermo-stability relative to the previously reported Erwinia tasmaniensis OvoAEta,9,10 OvoAMtht's biochemical properties are also distinct from the previously reported sulfoxide synthases.11–16 OvoAMtht is a bifunctional enzyme. When cysteine is provided as the only substrate, OvoAMtht catalyzes cysteine oxidation to cysteine sulfinic acid (the activity of cysteine dioxygenase, CDO). Further inclusion of histidine as the second substrate changes OvoAMtht from CDO-like activity to an oxidative coupling reaction between cysteine and histidine (a sulfoxide synthase). In addition, the variation of OvoAMtht activities between CDO-like and sulfoxide synthase could be modulated by either substrate/substrate analogs, or by a non-heme iron center secondary coordination shell tyrosine residue. Our subsequent biophysical characterizations using electron paramagnetic resonance (EPR) and Mössbauer spectroscopies indicate that the electronic properties of the active site iron are modulated by substrates, suggesting the change in reactivity is prompted by the substrates' close interaction with or binding to the iron center.
Results and discussion
Analysis of OvoA homologs from thermophilic/mesophilic organisms
In the last decade, one anaerobic and two aerobic12,14,15 ergothioneine biosynthetic pathways have been discovered (Scheme 1).5,17–19 One ovothiol biosynthetic pathway has also been biochemically characterized (Scheme 1).9,10 In these pathways, the key step is the activation of imidazole sp2 C–H bonds and replacing them with a C–S bond (catalyzed by EgtB,12 Egt1,14 EanB,17,18 and OvoA,9,20Scheme 1). In the aerobic ergothioneine biosynthetic pathways from Mycobacterium smegmatis and Neurospora crassa, a non-heme iron enzyme (EgtB12 or Egt1 (ref. 14)) catalyzes the oxidative coupling between hercynine 1 and cysteine or γ-glutamyl-cysteine to form a sulfoxide (2 or 3, Scheme 1). In the anaerobic ergothioneine biosynthetic pathway from green-sulfur bacterium Chlorobium limicola,17 a rhodanese catalyzes the key C–S bond formation step using polysulfide as the direct sulfur-source (EanB-catalysis, Scheme 1, pink).18,19 For ovothiol, thus far, only the Erwinia tasmaniensis ovothiol biosynthetic pathway has been biochemically characterized, with enzymes OvoA and OvoB (Scheme 1, red).9,10,20 A mononuclear non-heme iron enzyme (OvoAEta) catalyzes the oxidative coupling between histidine and cysteine to sulfoxide 6 (5 → 6, Scheme 1).9 Interestingly, in comparison to the ergothioneine biosynthetic enzymes (Egt1 & EgtB, Scheme 1), the OvoAEta enzyme is different in both substrate selectivity and product C–S bond regioselectivity.11,13 In addition, OvoAEta is a bi-functional enzyme, catalyzing the oxidative C–S bond formation reaction (5 → 6, Scheme 1) and the imidazole side-chain methylation reaction (7 → 8, Scheme 1).10 The second step in ovothiol biosynthesis (6 → 7, Scheme 1) is catalyzed by a PLP-dependent C–S lyase OvoB.Using Egt1 (Scheme 1) from N. crassa,14 OvoA (Scheme 1) from E. tasmaniensis,9 and EgtB (Scheme 1) from M. smegmatis12 as the query sequences, we searched for their homologs in thermophilic/mesophilic organisms. In total, we obtained 180 sequences. Using protein sequence similarity network analysis method,21 at an E-value cut-off of 10−60, these sequences segregate into two clusters (Fig. 1A). The larger cluster (light blue, Fig. 1A) has the biochemically characterized ergothioneine biosynthetic enzyme EgtB from M. thermoresistibile (EgtBMthr, dark blue).22 Interestingly, EgtBCth,15 an EgtB homolog from Candidatus Chloracidobacterium thermophilum (green, Fig. 1A), is located at a position bridging the major and the minor clusters, and EgtBCth has been reported to have both Egt1 and EgtB activity (Scheme 1).15 These results imply that the sequences in the small cluster (red, Fig. 1A) might be unique.
 | ||
| Fig. 1 Discovery of OvoA homologs through bioinformatic analysis. (A) At the E-value of 10−60, EgtB/OvoA/Egt1 homologs from thermophilic/mesophilic strains are separated into two clusters (blue and red clusters) by protein similarity network analysis. The dark-blue box represents EgtBMthr, and the green box represents EgtBCth, which has both Egt1 and EgtB activity; (B) phylogenetic analysis in MEGA7 for OvoAMtht, OvoAEta, and their homologs;27 (C) 1H-NMR assay of OvoAMtht when both cysteine and histidine are present, where OvoAMtht exhibits a sulfoxide synthase activity, producing sulfoxide 6 as the major product (>90%), while at the same time, producing a small amount (<10%) of sulfinic acid 9; the signals in 1H-NMR are from the hydrogen atoms at the corresponding positions in either histidine 5 or sulfoxide 6; (D) thermostability analysis of OvoAEta and OvoAMtht's sulfoxide synthase activity using histidine and cysteine as the substrates, the reaction was monitored by measuring the oxygen consumption rate. The enzyme was pre-incubated at different temperatures for 1 hour and then used for activity assays at room temperature. | ||
Domain structures of these five genes in the small cluster (red, Fig. 1A) were analyzed using the Pfam program (Fig. S1†).23 They all have the DinB_2 domain and FGE-sulfatase domain, and in recently reported X-ray crystal structures of two ergothioneine sulfoxide synthases, EgtBMthr and EgtBCth, the sulfoxide synthase active site is formed between these two domains.15,22 Among the five genes, we focused on Refseq ID: WP_018410809.1 from M. thermotolerans, named as OvoAMtht in this work.24 Similar to the bifunctional OvoAEta (Scheme 1), OvoAMtht also has a C-terminal methyl transferase domain (Pfam family: Methyltransf_31, Fig. S1†). Moreover, in OvoAMtht, residues important to its sulfoxide-synthase activity are conserved, including the mono-nuclear non-heme iron ligands (His68, His159, His163) and the catalytically-relevant tyrosine (Tyr405, Fig. S2†). Structural prediction using the Phyre2 program indicated that OvoAEta and OvoAMtht have similar protein folding and active site environments (Fig. S3†).25 These bioinformatic results imply that OvoAMtht might be an ovothiol biosynthetic enzyme.
To provide additional support for the above prediction, using the E. tasmaniensis C–S lyase, OvoBEta, as the query sequence, we searched for OvoB homologs in the M. thermotolerans genome. A gene (Refseq ID: WP_026224516.1, named as OvoBMtht in this work) with ∼55% similarity to OvoBEta was identified, and it is located adjacent to the OvoAMtht gene (Fig. S1B†). This bioinformatics analysis information highly suggests that these two genes in M. thermotolerans encode ovothiol biosynthetic enzymes.
After predicting OvoAMtht and OvoBMtht as the M. thermotolerans ovothiol biosynthetic genes, we conducted additional phylogenetic analysis of OvoA genes using the UPGMA method.26 Sequences were randomly picked from OvoA homologs among all taxa to create the phylogenetic tree shown in Fig. 1B. OvoAMtht and OvoAEta genes were manually added to the sequence pool to compare the evolutionary pathway between them. Results from this analysis imply that OvoAMtht is a comparatively more ancestral gene than OvoAEta (Fig. 1B).
Differences between OvoAMtht and the previously reported OvoAEta
The coding sequence of OvoAMtht, with codon-optimization for E. coli overexpression, was synthesized by Genscript and sub-cloned into pASK-IBA3+ vector. OvoAMtht was then overexpressed and purified using a protocol similar to what we have used in OvoAEta studies (Fig. S4A†).11,13 The purified OvoAMtht has 0.95 ± 0.05 equivalent of iron as determined by atomic emission spectroscopy (Fig. S4B†). After the pure OvoAMtht protein was obtained, we first evaluated its predicted ovothiol sulfoxide synthase activity. Because the ovothiol sulfoxide synthase uses cysteine and histidine as the two substrates, and O2 as the oxidant for the oxidative coupling process, OvoAMtht-catalysis was analyzed by three different assays: (a) 1H-NMR to monitor reactions on the histidine imidazole side-chain; (b) 13C-NMR assay for cysteine reactions using [β-13C]-cysteine as the substrate; (c) oxygen consumption rate analysis using NeoFoxy oxygen electrode.In an OvoAMtht reaction including L-cysteine and histidine as the substrates, the 1H-NMR-spectrum is indeed consistent with the predicted ovothiol sulfoxide synthase activity (Fig. 1C and S5†). In the 6–8 ppm region, the two signals are from the histidine imidazole hydrogens (∼7.6 ppm and ∼6.8 ppm). In the OvoAMtht reaction, a new signal appears at ∼7.8 ppm. Based on results reported previously on Egt1/EgtB/OvoAEta,11,14 the new signal at ∼7.8 ppm is from sulfoxide 6 imidazole ε-carbon C–H bond (Scheme 1), which provides the initial evidence indicating that OvoAMtht is indeed the ovothiol sulfoxide synthase.
OvoAMtht steady-state kinetic parameters were obtained by monitoring oxygen consumption rates using a NeoFoxy oxygen electrode. In order to rule out the oxygen consumptions caused by reductants auto-oxidation and uncoupled oxygen consumption, traces of reactions in absent of enzyme (Fig. S6A†) or cysteine substrate (Fig. S6B†) were recorded and were subtracted from the total oxygen consumption traces from enzymatic reactions. The kinetic parameters of OvoAMtht using histidine and cysteine as substrates at 20 °C are: kcat,O2 = 168.8 ± 4.5 min−1; KM, his = 630.1 ± 63.6 μM and KM, cys = 171.6 ± 18.6 μM (Fig. S6†). Under our assay condition, from 1H-NMR and 13C-NMR analysis (Fig. S5†), >90% of the activity is sulfoxide synthase activity. Therefore, these kinetic parameters obtained from oxygen consumption assay most likely represents the kinetic parameters for OvoAMtht's sulfoxide synthase activity. Because OvoAMtht is from a mesophilic organism, its thermostability was also examined. The thermostabilities of OvoAMtht and OvoAEta protein were analyzed by nanoDSF assay and the thermal unfolding curves of OvoAMtht and OvoAEta indicate that the Tm of OvoAMtht is ∼20 °C higher than that of OvoAEta (Fig. S4C†). Such a difference in thermostability between OvoAMtht and OvoAEta were also confirmed by the oxygen consumption assay after they were pre-incubated at a certain temperature for one hour. After incubation at 30 °C for one hour, OvoAEta lost most of its activity (Fig. 1D). When the temperature was further increased to 40 °C, after one hour, OvoAEta's activity was completely lost. However, for OvoAMtht, there is barely any loss of sulfoxide synthase activity between 20–40 °C after one hour pre-incubation (the red trace, Fig. 1D).
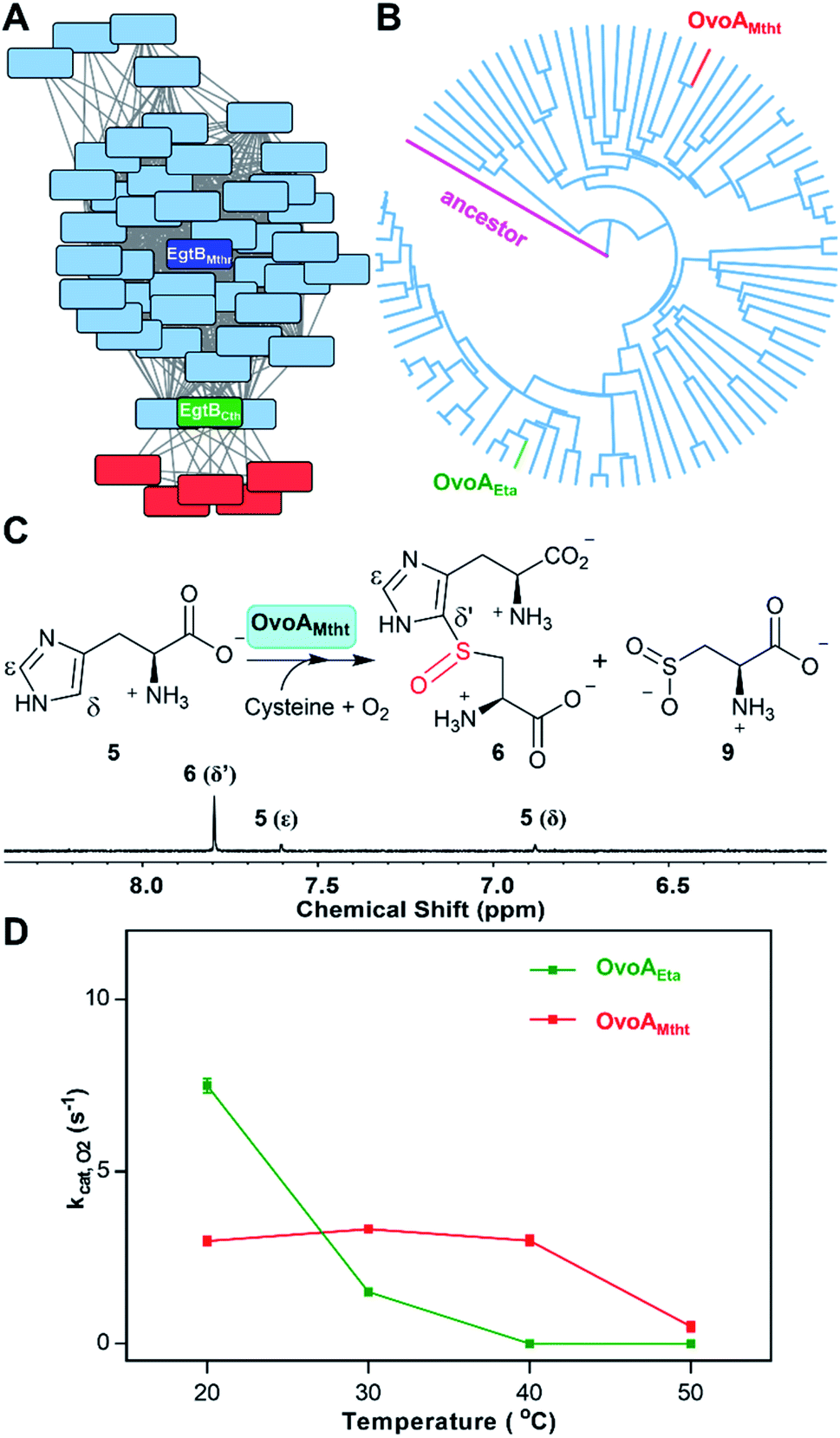
Besides an enhanced thermostability, biochemical analysis indicated that OvoAMtht is different from the previously reported OvoAEta in at least two aspects.17,25 First, when cysteine is the only substrate, OvoAEta and other sulfoxide synthases (Egt1/EgtB) have a very low O2 consumption activity (green trace, Fig. 2A),13,14 while OvoAMtht displays a very robust O2 consumption rate when cysteine is the substrate (red trace, Fig. 2A). Second, the cysteine oxidation product from OvoAMtht reaction is different from that in the OvoAEta reaction (Fig. 2Bvs.Fig. 2C). When cysteine is used as the only substrate, the OvoAEta reaction product is cystine, 10, as shown in 13C-NMR spectrum from the [β-13C]-cysteine reaction (OvoAEta reaction in Fig. 2B). In contrast, in the OvoAMtht reaction, cysteine sulfinic acid 9 is the product (Fig. 2C), which is the activity of cysteine dioxygenase enzymes (CDO). OvoAMtht's CDO-like activity was further confirmed under single-turnover conditions, where OvoAMtht and [β-13C]-cysteine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.9 in ratio) were mixed with an excess amount of O2. Under this condition, cysteine sulfinic acid 9 was detected as the only product (13C-NMR spectrum, Fig. S7†).
:0.9 in ratio) were mixed with an excess amount of O2. Under this condition, cysteine sulfinic acid 9 was detected as the only product (13C-NMR spectrum, Fig. S7†).
 | ||
| Fig. 2 Cysteine oxidation reactions by OvoAEta and OvoAMtht (A) oxygen consumption assay of OvoAEta and OvoAMtht using cysteine as the only substrate. (B) 13C-NMR analysis of OvoAEta reaction using [β-13C]-cysteine as the substrate, which shows cystine 10 as the major product. The 13C-NMR spectrum of [β-13C]-cysteine (top) is included as a control. (C) 13C-NMR analysis of OvoAMtht reaction using [β-13C]-cysteine as the substrate, which shows cysteine sulfinic acid 9 as the product. | ||
Detailed kinetic analysis of the OvoAMtht's CDO-like activity gives the kinetic parameters at 20 °C of: kcat,O2 = 16.2 ± 0.2 min−1 and KM, cys = 8.1 ± 0.6 mM (red trace in Fig. 2A and S8†). These kinetic parameters are at a level comparable to CDOs reported in literature.28 In the M. thermotolerans genome, our analysis indicated that besides OvoAMtht, it does not have another copy of the CDO gene, suggesting that OvoAMtht is a novel dual-function enzyme with both CDO-like and sulfoxide synthase activities.
OvoAMtht characterization by EPR and Mössbauer spectroscopies
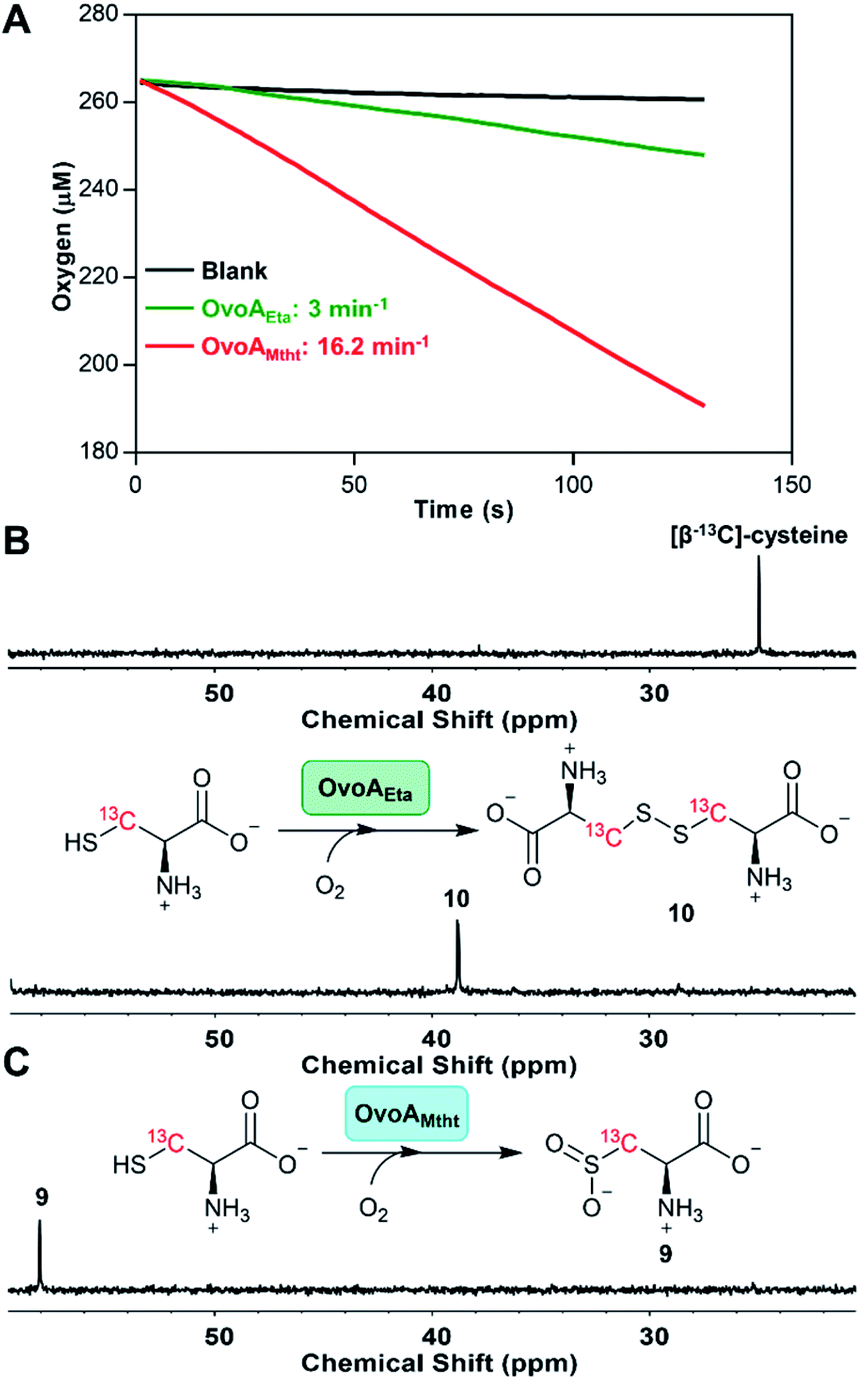
The presence of dual activities in OvoAMtht (CDO-like and sulfoxide synthase activities) immediately raises the next important question: how are these two activities in OvoAMtht controlled by the structural and electronic properties of the active site? As an initial step toward answering these questions, we characterized OvoAMtht using Mössbauer and EPR spectroscopies.For characterizations using Mössbauer spectroscopy, samples were prepared under anaerobic conditions by mixing 57Fe-loaded OvoAMtht with cysteine, histidine, or both for 5 minutes before the samples were frozen for analysis. The Mössbauer spectrum of the Fe(II)·OvoAMtht complex exhibits a quadrupole doublet with a broad linewidth, suggesting some level of structural inhomogeneity at the iron-center. This broad quadruple doublet can be simulated with two species having isomer shift values (δ) of 1.22 and 1.25 mm s−1 and quadrupole splitting values (ΔEQ) of 2.25 and 2.80 mm s−1 (species A & B, Fig. 3A and Table S1†), respectively. These Mössbauer parameters are indicative of a mononuclear high-spin (S = 2) ferrous iron. These parameters are also similar to those from the Fe(II)·CDO complex for cysteine dioxygenase, the Fe(II)·MDOAv complex for 3-mercaptopropionic acid dioxygenases (MDO), and Fe(II)·ADO reported in the literature.29,30 The Mössbauer spectrum of the Fe(II)·OvoAMtht·His complex also shows a broad quadruple doublet and could be simulated by including the quadruple doublet of the Fe(II)·OvoAMtht complex (∼70% of the total iron) and a new doublet having δ = 1.24 mm s−1 and ΔEQ = 3.13 mm s−1 (species C represented by grey trace, Fig. 3A and Table S1†). This new species is most likely due to the binding of histidine to the iron center. However, only a small fraction (∼20%) of iron is converted to this His-bound state, suggesting that histidine alone does not interact with the iron-center strongly. In contrast, the Fe(II)·OvoAMtht·Cys complex exhibited a much sharper quadrupolar doublet. The spectral simulation suggested that ∼25% of the total iron are from the Fe(II)·OvoAMtht complex, while the rest of the iron (∼75%) exhibited a different quadruple doublet with δ = 1.16 mm s−1 and ΔEQ = 3.36 mm s−1 (species D represented by the pink trace, Fig. 3A and Table S1†). The parameters of this additional doublet (species D, Fig. 3A) are in fact, similar to those of Cys-bound Fe(II) in CDO and in MDOAv, and thus could be similarly assigned as the Fe(II)·OvoAMtht·Cys complex.30,31 In the presence of both cysteine and histidine, the Mössbauer spectrum converted to a sharp quadrupolar doublet, which can be simulated with a single species having δ = 1.17 mm s−1 and ΔEQ = 3.12 mm s−1, suggesting a high structural homogeneity at the iron center in the Fe(II)·OvoAMtht·Cys·His complex (species E as represented by the orange trace, Fig. 3A and Table S1†).
 | ||
| Fig. 3 The effects of substrate binding on the OvoAMtht spectroscopic properties. (A) 4.2 K Mössbauer spectra of OvoA with various substrate combinations. The substrates used in these samples are labelled on the spectral traces. Samples were prepared by mixing 1 mM OvoA prepared anaerobically with 25 mM cysteine, histidine, or cysteine + histidine in 100 mM Tris buffer containing 500 mM NaCl 10% glycerol at pH 8.0. The resulting spectra have also been simulated with parameters listed in Table S1.† Black = experimental data, red: full simulation; green: species A in the Fe(II)·OvoAMtht complex; blue: species B in the Fe(II)·OvoAMtht complex, grey: species C for the putative Fe(II)·OvoAMtht·His complex; pink: species D for the putative Fe(II)·OvoAMtht·Cys complex; and orange: species E for the putative Fe(II)·OvoAMtht·Cys·His complex with Mössbauer parameters listed in Table S1.† (B) 15 K EPR spectra of NO-treated OvoAMtht. Trace I: enzyme only; trace II: enzyme with 0.2 mM cysteine; trace III: enzyme with 0.5 mM cysteine and 0.5 mM histidine. Black: experimental spectra; red: full simulations of experimental spectra; blue: component simulations of separate high spin (HS) and low spin (LS) iron nitrosyl species: (I/II/III-a) HS {FeNO}7 species. The minor g = 2 resonance of this species is omitted for clarity. See ESI† for full details. (I-b) Dinitrosyl iron complex impurity. (II/III-c) LS {FeNO}7 species associated with the presence of either Cys or Cys + His, respectively. (C) Schematic presentation of potential ligand environment changes under various conditions used in Mössbauer and EPR spectroscopic characterization. | ||
Results from these Mössbauer characterization suggest that Cys could bind to the iron center tightly, which is a prerequisite for the CDO-like reactivity. Histidine alone may not interact with the iron strongly, as shown by the presence of species C as only a minor species. However, Cys binding facilitates His binding, which may be the key for the change of OvoAMtht from CDO-like to sulfoxide synthase by promoting the C–S coupling reactivity (Fig. 2 and S5†).
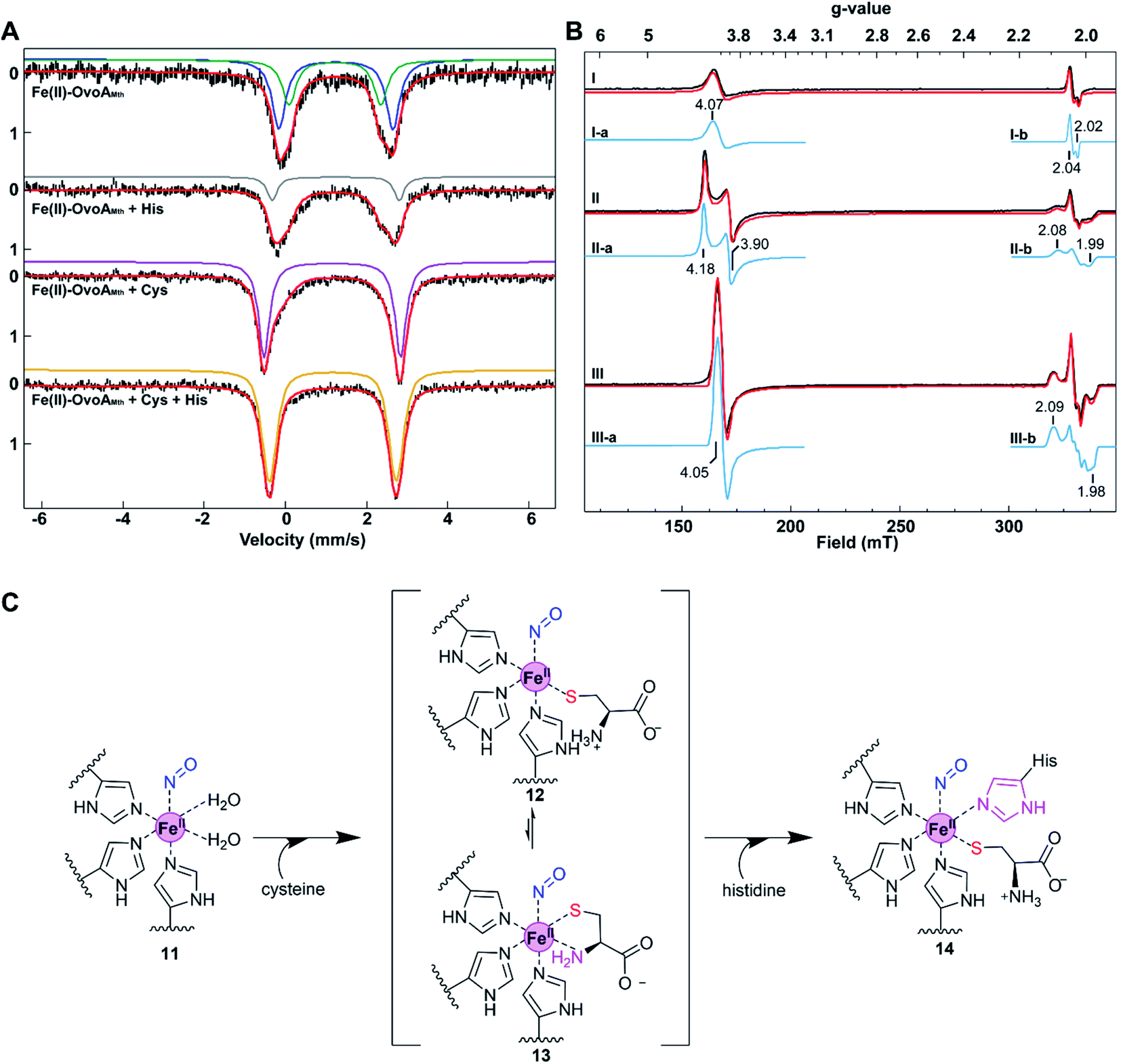
To provide further evidence on how the substrate modulates the electronic properties of the iron active site, we also characterized OvoAMtht using EPR spectroscopy. Because Fe2+ is EPR silent, nitric oxide (NO) is often used as a tool in EPR characterization of mononuclear non-heme iron enzymes, where the NO-bound complex becomes EPR-active, with the additional structural benefit of NO acting as an O2 mimic.32,33 The Fe(II)·OvoAMtht EPR spectra were collected after treating various enzyme–substrate combinations with NO from proliNONOate, including Fe(II)·OvoAMtht only, Fe(II)·OvoAMtht + Cys, or Fe(II)·OvoAMtht + Cys + His samples.
The Fe(II)·OvoAMtht + NO sample produced an EPR spectrum with two species (trace I, Fig. 3B). The broad axial signal at g = 4.07 (species I-a) is typical of a S = 3/2 {FeNO}7 species (with E/D = 0.008, σE/D = 0.005), where the electronic configuration of the iron nitrosyl species is described using the commonly-used Enemark/Feltham notation.34 The broad line shape can be attributed to the heterogeneity of active site, due to the lack of the bound substrate to better define the active site coordination.35 This heterogeneity is mirrored in the Mössbauer data of the Fe(II)·OvoAMtht complex in Fig. 3A. The low yield of this species (33 μM of 100 μM total enzyme) may be a function of poor NO binding in the absence of substrate, as observed in other oxygenases.32,36 The species near g = 2 (species I-b, 1.4 μM) is assigned as a non-enzymatic dinitrosyl iron species (DNIC).37,38 Its presence is common in NO adducts of Fe-containing enzymes and is a minor impurity in the OvoAMtht samples.39
Relative to the spectrum of the Fe(II)·OvoAMtht + NO sample, the resultant Fe(II)·OvoAMtht·Cys + NO sample's EPR spectrum (trace II) showed a clear change in the g = 4 region, as demonstrated by a more rhombic (E/D = 0.021, σE/D = 0.003) S = 3/2 species, having signals at g = 4.18, 3.90 (species II-a, Fig. 3B). This new EPR signal was generated in near full-yield (100 μM, Table S2†), which suggests that Cys binding facilitate O2/NO binding and this result is consistent with the presence of CDO-like activity in OvoAMtht. Additionally, a new low spin feature near g = 2 (species II-b) is present in the Fe(II)·OvoAMtht·Cys + NO sample as a minority species (7 μM, Table S2†). The g values of this feature (2.08, 2.03, 1.99) are reminiscent of those for the S = 1/2 {FeNO}7 species commonly associated CDO enzymes having bidentate-bound cysteine (Fig. S9A and Table S3†).30,32,40 There is a strong literature precedence that correlates low spin {FeNO}7 to bidentate (S/N) bound Cys (via thiolate S and amino N). Consequently, our EPR data suggest that the majority of Cys bound to OvoAMtht is most likely monodentate. Recently, with MDO, the yield of the low spin form was affected by the protonation state of tyrosine residues in the active site.30 Similar interactions with nearby amino acid residues may explain the presence of this species in OvoAMtht. For these reasons, when cysteine binds in OvoAMtht, we propose the Fe(II)·OvoAMtht·Cys adduct is a mixture of mono-dentate and bidendate complexes. In the reported crystal structures of ergothioneine sulfoxide synthases, the non-heme iron centers are coordinated by three protein histidine residues,15,22,28,32,41,42 which are also conserved in OvoAMtht. Based on this information, we propose the geometric model for the Fe(II)·OvoAMtht·Cys complex as shown in Fig. 3C, in which Cys binds to the iron-center as a mixture of mono-dendate and bi-dendate complexes.
When more than two equivalents of cysteine were added, the yield of the S = 3/2 species decreased while the yield of a S = 1/2 species near g = 2.04 increased (Fig. S10†). We attribute this additional species at g ∼2 to a DNIC species based on literature precedence,43–46 where the excess Cys may be binding adventitiously to the Fe, along with NO. To support this interpretation, we have independently formed this species by mixing free Fe, cysteine, and NO under anaerobic conditions (Fig. S11†). Therefore, the DNIC species with g = 2 region (Fig. S10†) is most likely irrelevant to OvoAMtht-catalysis.
Interestingly, upon introducing NO to the Fe(II)·OvoAMtht·Cys·His complex, both the g ∼4 and g ∼2 signals (trace III, Fig. 3B) change significantly from that observed in the Fe(II)·OvoAMtht·Cys + NO sample. The rhombic signal at g ∼4 in Fe(II)·OvoAMtht·Cys + NO sample (species II-a) changes to a sharper axial signal at g = 4.05 following the addition of histidine (species III-a). In addition, the Fe(II)·OvoAMtht·Cys·His + NO sample has a new S = 1/2 species (species III-b,19 μM, in Table S4†), having g-values 2.096, 2.029, 1.985 (Fig. 3B and S9B†). Samples of 0.05, 0.1, and 0.5 mM His in the presence of 0.5 mM Cys show that both species grow in a concentration-dependent manner (Fig. S12 and Table S4†).
Upon introduction of histidine into the Fe(II)·OvoAMtht·Cys complex, spectroscopic changes in both the high spin and low spin {FeNO}7 EPR signals are consistent with having both histidine and cysteine as iron-ligands as proposed in Fig. 3C. An OvoA crystal structure has yet to be reported, however crystal structures of two sulfoxide synthases in ergothioneine biosynthesis (EgtBMthr22 & EgtBCth15) are available. In the EgtB crystal structures, the mononuclear non-heme iron-center is coordinated by three protein histidine residues. Sequence alignments between EgtBMthr, EgtBcth and OvoAMtht indicated that the three iron histidine ligands are conserved in these three proteins (Fig. S2†). The EPR spectra of the Fe(II)·OvoAMtht·Cys + NO sample suggests the cysteine is predominantly bound in the HS/monodentate binding mode. Such a configuration would then allow for the additional histidine to bind at the active site as well. The remaining vacant site is presumably for O2 binding and activation (as evidenced by NO binding to the active site in the EPR samples). Together, the spectroscopic information obtained in our Mössbauer and EPR characterization of OvoAMtht support the second step of our OvoAMtht-schematic model in Fig. 3C, in which both cysteine and histidine bind to the iron-center as mono-dentate ligands.
Modulate OvoAMtht reaction by substrate analogs and Fe-secondary coordination shell residues
OvoAMtht is a bifunctional enzyme with both CDO-like and sulfoxide synthase activities (Fig. 1Cvs.Fig. 2C), which is very different from previously reported ergothioneine and ovothiol sulfoxide synthases (EgtB/Egt1/OvoAEta).47,48 Most of these sulfoxide synthases (EgtB/Egt1/OvoAEta) do not have CDO-like activity when cysteine is provided as the only substrate, while they do oxidize cysteine slowly to produce cystine as the oxidation product. In EgtBMthr, a very low level of sulfur oxidation activity (∼1% of OvoAMtht's CDO-like activity) was reported.22,49 In EgtB/Egt1/OvoAEta, upon mutating the active site tyrosine to a phenylalanine residue, the resulting mutants show ∼100% CDO-like activity.47,49 Therefore, in EgtB/Egt1/OvoAEta, the sulfoxide synthase and CDO-like activities are primarily modulated by an iron-center's secondary coordination shell residue, a tyrosine residue.In the absence of a OvoAMtht crystal structure, we created a homology model using the Phyre2 program (Fig. S3†).25 According to this model, Tyr405 in OvoAMtht is the corresponding active site tyrosine. The OvoAMtht,Y405F mutant was overexpressed and purified using a protocol similar to that used in wild type OvoAMtht. OvoAMtht activities were then examined under four different conditions (Fig. 4A):
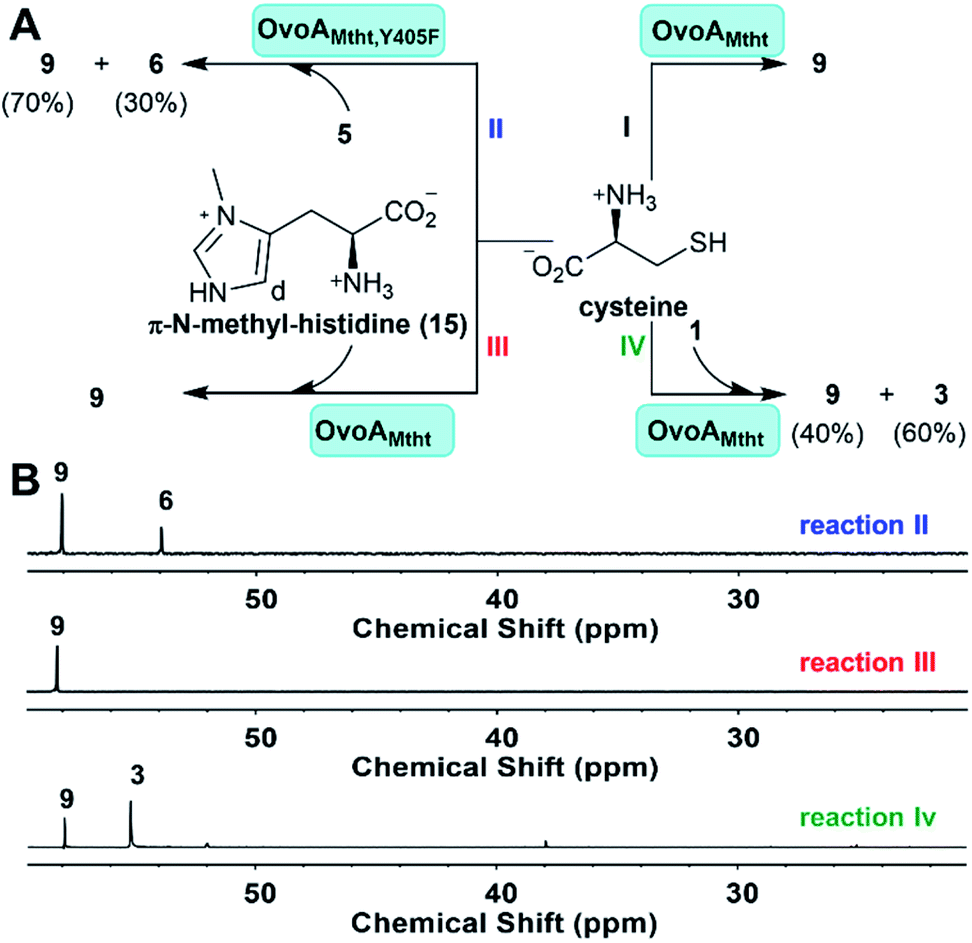 | ||
| Fig. 4 Examining factors controlling OvoAMtht's CDO-like and sulfoxide synthase activities. (A) Four reactions examined in this work; (B) 13C-NMR analysis of OvoAMtht reactions. Reaction II: OvoAMtht,Y405F mutant reaction using cysteine and histidine as substrates; reaction III has π-N-methyl-histidine and cysteine as the substrates; and reaction IV has hercynine 1 and cysteine as the substrates. | ||
(I) OvoAMtht using cysteine as the only substrate;
(II) OvoAMtht,Y405F mutant using cysteine and histidine as co-substrates;
(III) OvoAMtht using cysteine and π-N-methyl-histidine as co-substrates;
(IV) OvoAMtht using cysteine and hercynine as co-substrates.
For the OvoAMtht,Y405F mutant, when histidine and cysteine are co-substrates, the kinetic parameters from the O2 consumption assay are: kcat,O2 of 162.6 ± 2.1 min−1; KM, his = 152.4 ± 11.0 μM and KM, cys = 290.0 ± 16.1 μM. These OvoAMtht,Y405F mutant kinetic parameters (Fig. S13†) are similar to those of the wild type OvoAMtht (Fig. 1C). However, different from all previously reported EgtB/Egt1 or OvoA enzymes,13,47–49 whose active site tyrosine mutants show ∼100% CDO-like activity, the OvoAMtht,Y405F mutant exhibits sulfoxide synthase/CDO-like product formation in a ratio of 3:7 (reaction II, Fig. 4).
From the spectroscopic characterization (Fig. 3), it is clear that the iron-center's electronic properties are modulated by both substrates. To provide further evidence supporting the above conclusion, we also examined OvoAMtht activities using two histidine analogs, π-N-methyl-histidine 15, and trimethylhistidine (hercynine, 1). π-N-Methyl-histidine was synthesized according to the literature procedure (Fig. S14†).50 The reaction (reaction III, Fig. 4) was monitored by 1H-NMR, 13C-NMR, and the O2 consumption rate assay. The 1H-NMR spectrum indicates that when π-N-methyl-histidine and cysteine are co-substrates, OvoAMtht exhibits no sulfoxide synthase activity (Fig. S15†). However, the oxygen consumption rate of this reaction (reaction III, Fig. 4) is almost the same as the OvoAMtht native reaction (Fig. 1C), with kinetic parameters for reaction III in Fig. 4 of: kcat,O2 = 99.2 ± 2.7 min−1; KM, π-N-methyl-histidine = 112.2 ± 17.4 μM and KM, cys = 249.1 ± 28.3 μM (Fig. S16†). 13C-NMR indicated that the only product in this reaction is cysteine sulfinic acid 9. Therefore, in OvoAMtht, when π-N-methyl-histidine is used to replace histidine, OvoAMtht also changes from a sulfoxide synthase to a CDO. Intriguingly, π-N-methyl-histidine stimulates the OvoAMtht's CDO-like activity by nearly 200-fold (kcat,O2/KM for cysteine) relative to the cysteine-only reaction (reaction I, Fig. 4). As a matter of fact, under this condition, OvoAMtht's CDO-like activity is 1–2 order of magnitude greater than other CDOs reported in literature.28 The difference between these reactions (Fig. 1Cvs. reaction I & III in Fig. 4) indicate the histidine or its analogs modulate the OvoAMtht activities between CDO-like and sulfoxide synthase. Moreover, binding of histidine/histidine analogs also increases the O2 consumption rate by 1–2 orders of magnitude.
Different from π-N-methyl-histidine 15, whose methylation is on the side-chain, hercynine 1 is methylated at its α-amino group. Because the imidazole ring of histidine is likely an iron ligand, π-N-methyl-histidine 15 might modulate the electronic properties of the iron-center. Hercynine's methylation is on its α-amino group and any effects observed for hercynine reaction should be the secondary coordination shell effects (e.g., hydrogen bonding, hydrophobicity, or ionic interactions between the substrate α-amino group and its nearby residues). When hercynine and cysteine are used as the substrate, OvoAMtht exhibits a sulfoxide synthase/CDO-like ratio of 4:6 (reaction IV, Fig. 4). Interestingly, 1H-NMR analysis clearly indicated that OvoAMtht's sulfoxide synthase regioselectivity changes from the imidazole δ-position in the native reaction (Fig. 1C) to ε-position in Fig. 4 reaction IV (Fig. S17†). The kinetic parameters from the O2 consumption assay are: kcat,O2 = 47.8 ± 4.2 min−1; KM, hercynine = 185.9 ± 17.2 μM and KM, cys = 1060.2 ± 121.2 μM (Fig. S18†).
Conclusion
In this work, based on bioinformatics analysis of 180 potential sulfoxide synthases from thermophilic/mesophilic organisms,21 we selected OvoAMtht from M. thermotolerans for biochemical and spectroscopic characterization. OvoAMtht is biochemically distinct from the previously reported sulfoxide synthases (EgtB/Egt1/OvoAEta) in several aspects. First, for EgtB/Egt1/OvoAEta reported in previous studies, when cysteine is provided as the only substrate, the O2 consumption rate is very slow, and the oxidation product is cystine 10.13,47–49 However, when cysteine is the only substrate provided, the O2 consumption rate in the OvoAMtht-catalysis is at least 1–2 orders of magnitude faster than that of EgtB/Egt1/OvoAEta enzymes. Moreover, under this condition, cysteine sulfinic acid 9 instead of cystine 10 is the OvoAMtht reaction product. In the M. thermotolerans genome, our analysis does not identify an additional CDO gene, which suggests that OvoAMtht may satisfy the CDO requirements of M. thermotolerans. The kinetic parameters of OvoAMtht's CDO-like activity are comparable to those CDOs in the literature examples.28 The high cysteine KM for OvoAMtht is also consistent with the role of CDO as a detoxification enzyme to remove excess cysteine when its concentration reaches a high level.The second unique feature of OvoAMtht is that upon further introduction of histidine to the Fe(II)·OvoAMtht·Cys complex, OvoAMtht switches from a CDO-like to a sulfoxide synthase. As demonstrated by Mössbauer and EPR spectroscopies, the iron-center electronic properties are modulated by both substrates (Fig. 3). It has been reported that in EgtB/Egt1/OvoAEta, their CDO-like and sulfoxide synthase activities are controlled by a secondary coordination shell residue to the mononuclear non-heme iron center (Tyr377 in EgtBMthr and Tyr93 and Tyr94 in EgtBCth, Scheme 2).15,48,49 With the previously reported systems, mutation of the active site tyrosine residue(s) to phenylalanine, the sulfoxide synthase activity is abolished, and the mutant exhibits almost exclusively the CDO-like activity. In contrast, the OvoAMtht,Y405F still has sulfoxide synthase activity, displaying a sulfoxide synthase/CDO-like activity in a ratio of 3:7. Interestingly, upon replacing histidine by π-N-methyl-histidine to the Fe(II)·OvoAMtht·Cys complex, π-N-methyl-histidine changes OvoAMtht from sulfoxide synthase back to 100% CDO-like again. Moreover, π-N-methyl-histidine binding increases EgtBMthr's CDO-like reactivity by ∼200-fold than the case where cysteine is the only substrate (reaction III vs. reaction I, Fig. 4). Therefore, in OvoAMtht, the activities between sulfoxide synthase and CDO-like are modulated by iron-center ligands and secondary coordination shell tyrosine residue(s), while the iron-ligands seem to play a more dominant role in OvoAMtht in controlling the partition between sulfoxide synthase and CDO-like activities.
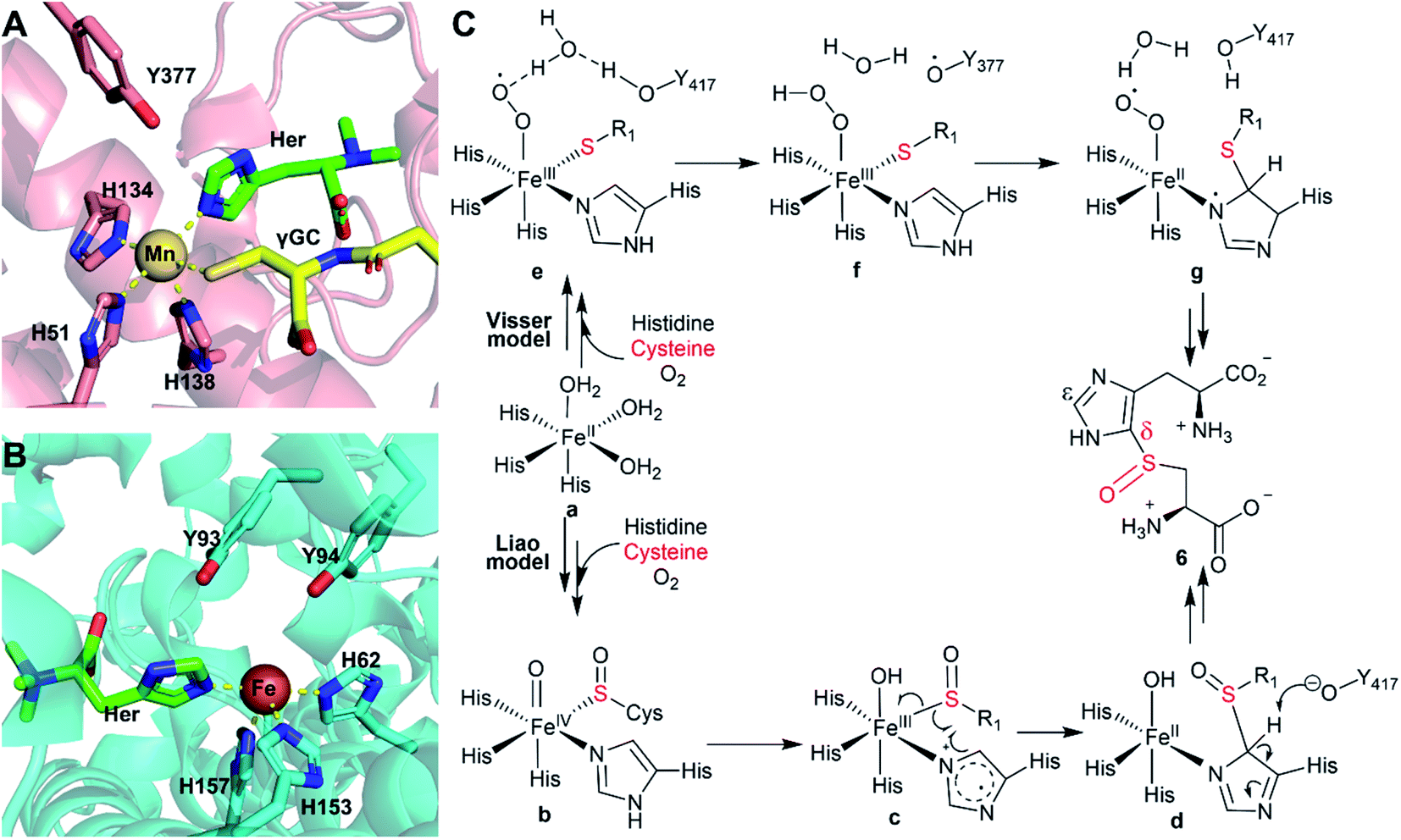 | ||
| Scheme 2 (A) Crystal structure of EgtBMthr with hercynine (Her) and gamma-gluatmyl-cysteine (γGC) binding to the active site (PDB ID: 4X8D).22 (B) Crystal structure of EgtBCth with hercynine binding to the active site (PDB ID: 6O6M).15 (C) Proposed mechanism for OvoAEta-catalysis based on information from structural information of EgtBMthr and EgtBCth and related computational studies. | ||
In recent years, a few mechanistic models have been proposed for ergothioneine and ovothiol sulfoxide synthases, with two representative models (Scheme 2) being suggested by Visser et al.51 and Liao et al.,52 respectively. The Visser model suggests that thioether formation is the first half of this reaction.51 In contrast, the Liao model52 proposes that sulfenic acid formation initiates the reaction. Both models involve the active site tyrosine for catalysis, while doing so with distinct functions. In the Visser model, the active site tyrosine plays a redox role and an inverse deuterium isotopic effect was predicted for 2H labelled histidine. The Liao model involves the active site tyrosine in acid/base catalysis and a primary 2H-labeled isotope effect as high as 5.7 was predicted for 2H-labeled histidine.52 With tyrosine analogs incorporated using the amber-suppressor method, we previously examined this relationship in OvoAEta-catalysis. Our observation of an inverse deuterium isotope effect is more consistent with the Visser model in OvoAEta-catalysis.48,51 However, in OvoMtht, we have found new experimental results where sulfoxide synthase activity is maintained in the Tyr → Phe mutant. As this active site Tyr is invoked in both mechanistic models, our results suggest there may be other interactions that guide product formation. In this paper, we have presented two new avenues to be explored further: (1) modulation of the Fe electronic properties (using π-N-methyl-histidine) and (2) noncovalent interactions between the substrate molecule and nearby amino acid residues (using hercynine).
Both Liao52 and Liu53 groups cautioned that the reaction pathway might be very sensitive to the detailed active site structure and the active site dynamics. Because OvoAMtht's sulfoxide synthase and CDO-like activities could be modulated by both iron-ligands and its secondary coordination shell residues, it offers an excellent system for future structure–function relationship studies to provide experimental evidence for refinement of these mechanistic models (Scheme 2).
Experimental
Detailed experimental procedures are included in the ESI.†Data availability
Data supporting this article have been uploaded as part of the ESI.†Author contributions
R. C., N. N., K. L., L. Q., and J. L., conducted biochemical studies. R. C., A. C. W., J. P., and Y. T. conducted spectroscopic studies. J. Z. and H. S. synthesized substrates and substrate analogues. R. C. A. C. W., M. W. G., Y. G., S. J. E., and P. L. wrote the manuscript with feedback from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by the National Science Foundation (CHE-2004109 to P. Liu, CHE-1654060 to Y. Guo) and National Institute of Health (R35-GM136294 to S. Elliott, and GM140040 to P. Liu).Notes and references
- B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10836–10844 CrossRef CAS PubMed.
- I. K. Cheah and B. Halliwell, Biochim. Biophys. Acta, 2012, 1822, 784–793 CrossRef CAS PubMed.
- I. K. Cheah and B. Halliwell, Antioxidants, 2020, 9(595) CAS.
- G. L. Russo, M. Russo, I. Castellano, A. Napolitano and A. Palumbo, Mar. Drugs, 2014, 12, 4069–4085 CrossRef CAS PubMed.
- N. Naowarojna, R. Cheng, L. Chen, M. Quill, M. Xu, C. Zhao and P. Liu, Biochemistry, 2018, 57, 3309–3325 CrossRef CAS PubMed.
- I. Castellano and F. P. Seebeck, Nat. Prod. Rep., 2018, 35, 1241–1250 RSC.
- N. Tanaka, Y. Kawano, Y. Satoh, T. Dairi and I. Ohtsu, Sci. Rep., 2019, 9, 1–10 Search PubMed.
- Y. Gumulya, J. M. Baek, S. J. Wun, R. E. S. Thomson, K. L. Harris, D. J. B. Hunter, J. B. Y. H. Behrendorff, J. Kulig, S. Zheng, X. M. Wu, B. Wu, J. E. Stok, J. J. De Voss, G. Schenk, U. Jurva, S. Andersson, E. M. Isin, M. Boden, L. Guddat and E. M. J. Gillam, Nat. Catal., 2018, 1, 878–888 CrossRef CAS.
- A. Braunshausen and F. P. Seebeck, J. Am. Chem. Soc., 2011, 133, 1757–1759 CrossRef CAS PubMed.
- N. Naowarojna, P. Huang, Y. Cai, H. Song, L. Wu, R. Cheng, Y. Li, S. Wang, H. Lyu, L. Zhang, J. Zhou and P. Liu, Org. Lett., 2018, 20, 5427–5430 CrossRef CAS PubMed.
- H. Song, M. Leninger, N. Lee and P. Liu, Org. Lett., 2013, 15, 4854–4857 CrossRef CAS PubMed.
- F. P. Seebeck, J. Am. Chem. Soc., 2010, 132, 6632–6633 CrossRef CAS PubMed.
- H. Song, A. S. Her, F. Raso, Z. Zhen, Y. Huo and P. Liu, Org. Lett., 2014, 16, 2122–2125 CrossRef CAS PubMed.
- W. Hu, H. Song, A. Sae Her, D. W. Bak, N. Naowarojna, S. J. Elliott, L. Qin, X. Chen and P. Liu, Org. Lett., 2014, 16, 5382–5385 CrossRef CAS PubMed.
- N. Naowarojna, S. Irani, W. Hu, R. Cheng, L. Zhang, X. Li, J. Chen, Y. J. Zhang and P. Liu, ACS Catal., 2019, 9, 6955–6961 CrossRef CAS PubMed.
- A. R. Stampfli, K. V. Goncharenko, M. Meury, B. N. Dubey, T. Schirmer and F. P. Seebeck, J. Am. Chem. Soc., 2019, 141, 5275–5285 CrossRef CAS PubMed.
- R. Burn, L. Misson, M. Meury and F. P. Seebeck, Angew. Chem., Int. Ed., 2017, 56, 12508–12511 CrossRef CAS PubMed.
- R. Cheng, L. Wu, R. Lai, C. Peng, N. Naowarojna, W. Hu, X. Li, S. A. Whelan, N. Lee, J. Lopez, C. Zhao, Y. Yong, J. Xue, X. Jiang, M. W. Grinstaff, Z. Deng, J. Chen, Q. Cui, J. Zhou and P. Liu, ACS Catal., 2020, 8981–8994, DOI:10.1021/acscatal.0c01809.
- R. H. Cheng, R. Lai, C. Peng, J. Lopez, Z. H. Li, N. Naowarojna, K. L. Li, C. Wong, N. Lee, S. A. Whelan, L. Qiao, M. W. Grinstaff, J. Y. Wang, Q. Cui and P. H. Liu, ACS Catal., 2021, 11, 3319–3334 CrossRef CAS PubMed.
- G. T. M. Mashabela and F. P. Seebeck, Chem. Commun., 2013, 49, 7714–7716 RSC.
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 CrossRef PubMed.
- K. V. Goncharenko, A. Vit, W. Blankenfeldt and F. P. Seebeck, Angew. Chem., Int. Ed., 2015, 54, 2821–2824 CrossRef CAS PubMed.
- J. Mistry, S. Chuguransky, L. Williams, M. Qureshi, G. A. Salazar, E. L. L. Sonnhammer, S. C. E. Tosatto, L. Paladin, S. Raj, L. J. Richardson, R. D. Finn and A. Bateman, Nucleic Acids Res., 2020, 49, D412–D419 CrossRef PubMed.
- N. V. Doronina, E. N. Kaparullina and Y. A. Trotsenko, Int. J. Syst. Evol. Microbiol., 2014, 64, 158–164 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- P. H. Sneath and R. R. Sokal, Numerical taxonomy. The principles and practice of numerical classification, 1973 Search PubMed.
- S. Kumar, G. Stecher and K. Tamura, Mol. Biol. Evol., 2016, 33, 1870–1874 CrossRef CAS PubMed.
- C. A. Joseph and M. J. Maroney, Chem. Commun., 2007, 3338–3349 RSC.
- Y. F. Wang, I. Davis, Y. Chan, S. G. Naik, W. P. Griffith and A. M. Liu, J. Biol. Chem., 2020, 295, 11789–11802 CrossRef CAS PubMed.
- S. Sardar, A. Weitz, M. P. Hendrich and B. S. Pierce, Biochemistry, 2019, 58, 5135–5150 CrossRef CAS PubMed.
- E. P. Tchesnokov, A. S. Faponle, C. G. Davies, M. G. Quesne, R. Turner, M. Fellner, R. J. Souness, S. M. Wilbanks, S. P. de Visser and G. N. L. Jameson, Chem. Commun., 2016, 52, 8814–8817 RSC.
- B. S. Pierce, J. D. Gardner, L. J. Bailey, T. C. Brunold and B. G. Fox, Biochemistry, 2007, 46, 8569–8578 CrossRef CAS PubMed.
- D. Arciero, A. Orville and J. Lipscomb, J. Biol. Chem., 1985, 260, 14035–14044 CrossRef CAS.
- J. Enemark and R. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- M. D. Krzyaniak, B. E. Eser, H. R. Ellis, P. F. Fitzpatrick and J. McCracken, Biochemistry, 2013, 52, 8430–8441 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- D. R. Truzzi, N. M. Medeiros, O. Augusto and P. C. Ford, Inorg. Chem., 2021, 60, 15835–15845 CrossRef CAS PubMed.
- C. E. Tinberg, Z. J. Tonzetich, H. Wang, L. H. Do, Y. Yoda, S. P. Cramer and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 18168–18176 CrossRef CAS PubMed.
- B. S. Pierce, B. P. Subedi, S. Sardar and J. K. Crowell, Biochemistry, 2015, 54, 7477–7490 CrossRef CAS PubMed.
- E. J. Blaesi, J. D. Gardner, B. G. Fox and T. C. Brunold, Biochemistry, 2013, 52, 6040–6051 CrossRef CAS PubMed.
- C. R. Simmons, Q. Liu, Q. Huang, Q. Hao, T. P. Begley, P. A. Karplus and M. H. Stipanuk, J. Biol. Chem., 2006, 281, 18723–18733 CrossRef CAS PubMed.
- S. Ye, X. Wu, L. Wei, D. M. Tang, P. Sun, M. Bartlam and Z. H. Rao, J. Biol. Chem., 2007, 282, 3391–3402 CrossRef CAS PubMed.
- T. R. Bryar and D. R. Eaton, Can. J. Chem., 1992, 70, 1917–1926 CrossRef CAS.
- A. R. Butler and I. L. Megson, Chem. Rev., 2002, 102, 1155–1166 CrossRef CAS PubMed.
- S. Lu, E. Libby, L. Saleh, G. Xing, J. M. Bollinger and P. Moënne-Loccoz, J. Biol. Inorg Chem., 2004, 9, 818–827 CrossRef CAS PubMed.
- N. Reginato, C. T. McCrory, D. Pervitsky and L. Li, J. Am. Chem. Soc., 1999, 121, 10217–10218 CrossRef CAS.
- L. Chen, N. Naowarojna, H. Song, S. Wang, J. Y. Wang, Z. X. Deng, C. M. Zhao and P. H. Liu, J. Am. Chem. Soc., 2018, 140, 4604–4612 CrossRef CAS PubMed.
- L. Chen, N. Naowarojna, B. Chen, M. Xu, M. Quill, J. Wang, Z. Deng, C. Zhao and P. Liu, ACS Catal., 2019, 9, 253–258 CrossRef CAS.
- K. V. Goncharenko and F. P. Seebeck, Chem. Commun., 2016, 52, 1945–1948 RSC.
- H. C. Beyerman, A. Vanzon and L. Maat, Recl. Trav. Chim. Pays-Bas, 1972, 91, 246–250 CrossRef CAS.
- A. S. Faponle, F. P. Seebeck and S. P. de Visser, J. Am. Chem. Soc., 2017, 139, 9259–9270 CrossRef CAS PubMed.
- W. J. Wei, P. E. M. Siegbahn and R. Z. Liao, Inorg. Chem., 2017, 56, 3589–3599 CrossRef CAS PubMed.
- G. Tian, H. Su and Y. Liu, ACS Catal., 2018, 8, 5875–5889 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05479a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |