
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalised nanopores: chemical and biological modifications
Dominic F.
Cairns-Gibson
and
Scott L.
Cockroft
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: scott.cockroft@ed.ac.uk
First published on 22nd December 2021
Abstract
Nanopore technology has established itself as a powerful tool for single-molecule studies. By analysing changes in the ion current flowing through a single transmembrane channel, a wealth of molecular information can be elucidated. Early studies utilised nanopore technology for sensing applications, and subsequent developments have diversified its remit. Nanopores can be synthetic, solid-state, or biological in origin, but recent work has seen these boundaries blurred as hybrid functionalised pores emerge. The modification of existing pores and the construction of novel synthetic pores has been an enticing goal for creating systems with tailored properties and functionality. Here, we explore chemically functionalised biological pores and the bio-inspired functionalisation of solid-state pores, highlighting how the convergence of these domains provides enhanced functionality.
 Dominic F. Cairns-Gibson | Dominic Cairns-Gibson obtained an MChem in Medicinal and Biological Chemistry from the University of Edinburgh in 2018. As part of his studies, he undertook an international exchange to the University of Connecticut, working with Dr Jessica Rouge to study DNA nanoparticles. On returning to Edinburgh, he completed his final-year research project on photoswitchable transmembrane channels under the supervision of Prof. Scott Cockroft. He continued in the Cockroft group as a PhD student, researching transmembrane protein nanopores. |
 Scott L. Cockroft | Scott Cockroft is professor of supramolecular chemistry at the University of Edinburgh. Scott conducted his PhD and postdoctoral work under the supervision of Profs. Christopher A. Hunter FRS (University of Sheffield, now University of Cambridge, UK) and M. Reza Ghadiri (The Scripps Research Institute, California), respectively. The Cockroft group investigate the origins of molecular recognition using synthetic model systems, while seeking to harness these principles in the construction of molecular devices assembled from synthetic and biological molecules. |
Introduction
The origins of nanopore technology can be traced back to the development of patch-clamp electrophysiology in the 1970s.1 During patch-clamp electrophysiology, a micropipette containing a counter electrode and electrolyte solution is sealed against a small section of the cell membrane.2 If this ‘patch’ of membrane contains a single ion channel, then the application of a voltage across the membrane results in a measurable flow of ions through the channel (positive ions towards the cathode and negative ions towards the anode, Fig. 1A). Since sub-picoamp changes to the flow of ions can be detected, single-channel electrophysiology enables the opening and closing of single ion channels to be monitored in real time. This breakthrough, arguably the first single-molecule biophysics technique, led to Neher and Sakmann being awarded the 1991 Nobel Prize in Physiology or Medicine for their contributions to the understanding of ion channel function.1 | ||
| Fig. 1 The principle of nanopore sensing, illustrated with the example of an α-hemolysin protein nanopore spanning a lipid bilayer. (A) A consistent free-pore current (Io) is observed when a voltage is applied across the membrane-spanning nanopore separating two electrolyte-containing wells. (B) When an analyte molecule (purple) enters the pore, an ion current blockage (Ib) is observed, which is related to the size and charge of the analyte. The resulting ion current trace can reveal the kinetics and mechanisms of chemical reactions and molecular recognition events occurring at the single-molecule level. | ||
Contemporary nanopore technology extends beyond examining biological ion channel function. In contrast with the original intention of single-channel electrophysiology to monitor the dynamics of channel opening and closing, nanopore sensing instead exploits solid-state or biological nanopores with stable open channels. The most widely employed biological nanopores are toxin proteins secreted by pathogenic bacteria, whose stable open channels often provide low-noise ion currents (Io in Fig. 1A) when an applied potential difference (voltage) is applied across the membrane.3 Such proteins have been widely repurposed for sensing applications; most notably for single-molecule DNA sequencing.4 The archetypical repurposed biological pore is α-hemolysin (α-HL), which remains a workhorse for nanopore investigations (Fig. 1).5 The central principle of nanopore sensing is that the entry of an analyte molecule into a nanopore reduces the ability of ions to flow through the channel, giving rise to characteristic blockages in the ion current (Ib in Fig. 1B), which depend on the size and charge of the species causing the blockage. As patch-clamp amplifiers can record ion currents with high sensitivity (<0.1 pA) on the >100 kHz timescale, nanopore sensing can reveal information about the dynamics of molecular interactions and reactions occurring within the pore. By virtue of being a single-molecule technique, the ordering of events, short-lived intermediates, and the temporal distribution of events can be resolved that would otherwise be obscured in bulk ensemble measurements.
The turn of the 21st century saw patch-clamp amplifiers being used in combination with nanopores constructed from inorganic materials and nanotubes.6,7 Solid-state nanopores are often fabricated with diameters in the 1–20 nm range in materials such as silicon oxide or silicon nitride.8 Since effective single-molecule sensing requires the diameter of the nanopore to be slightly larger than the analyte molecule, diameters on the lower end of this scale are often desired.9 Solid-state nanopores tend to be much more thermally and chemically stable than their biological counterparts. However, the top-down approaches used in their formation, such as laser- or electron-beam etching, limit the control over the structure at the atomic level. This limited precision, combined with the relatively inert substrates, often means that solid-state nanopores lack handles for synthetic modification. In contrast, biological nanopores tend to be less robust, but have atomically precise dimensions encoded by the protein sequences and present numerous handles for chemical and genetic modification.
Research is now turning to the amalgamation of biological and synthetic channels to achieve the ‘best of both worlds’ and circumvent these issues of tunability and stability. This perspective explores the coalescence of the synthetic and biological components in the development of nanopore-based technologies (Fig. 2). The chemical and genetic modification of biological nanopores and the introduction of biomolecules to enhance the functionality of solid-state pores will be discussed. Future prospects of fully synthetic channels are only briefly covered here and readers are instead directed to excellent reviews on this topic.10,11
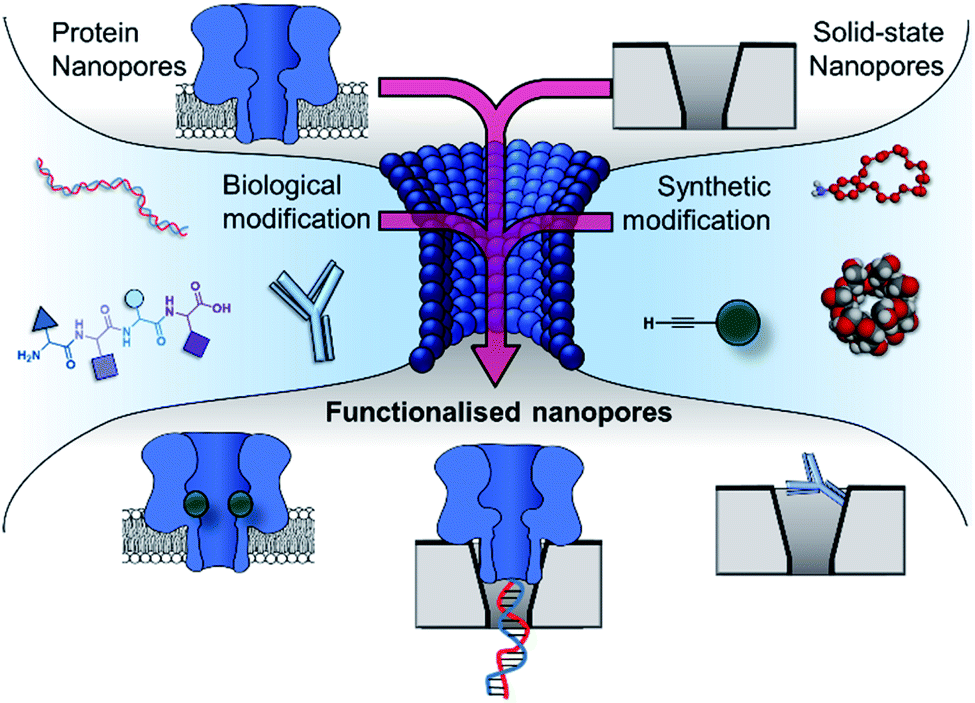 | ||
| Fig. 2 The incorporation of biological and synthetic modifications into protein and solid-state nanopores creates functionalised nanopores with enhanced functionality. Modification with biomolecules such as DNA, peptides and antibodies can endow otherwise inert solid-state nanopores with the ability to engage in specific (bio)molecular interactions. Meanwhile, biological pores can be modified with synthetic components such as crown ethers, cyclodextrins and reactive small molecules, enabling new modes of detection and the real-time observation of molecular interactions and chemical reactions at the single-molecule level. Similarly, biological and solid-state nanopores can themselves be combined to yield hybrid nanopores with increased robustness while retaining molecular recognition properties arising from the atomically precise three-dimensional structure of the biomolecular component. | ||
Solid-state nanopores functionalised with biomolecules
Solid-state nanopores are typically constructed from robust but relatively inert matrices derived from silicon or other inorganic materials.8 Unlike their biological counterparts, the absence of atomically precise dimensions and positioned functional groups render most solid-state nanopores incapable of engaging in specific molecular recognition processes. However, one of the major challenges in nanopore studies using biological pores is the inherent lack of stability – often arising from the membrane the pore is anchored within and not the protein itself. By utilising the reliable framework of a solid-state pore challenges to stability can be overcome. Hence, the incorporation of biomolecules with solid-state pores provides a means of overcoming the intrinsic limitations and challenges of both biological and solid-state nanopores.One approach to introducing reactive handles into solid-state pores is to functionalise the surface with a more reactive material. For example, the Rant group showed that solid-state silicon nitride nanopores could be coated with a thin gold film to enable functionalisation with a self-assembled thiol monolayer (blue in Fig. 3).12 The combination of the gold treatment and monolayer assembly reduced the diameter of the nanopore channel to 12–39 nm, thereby making the nanopore a suitable size for the detection of individual protein molecules. Alkane-thiols modified with nitrilotriacetic acid ligands (purple in Fig. 3) were included during monolayer formation. In the presence of Ni2+, the nitrilotriacetic groups could bind His6-tagged proteins, which allowed stochastic sensing of protein association/dissociation by monitoring ion currents flowing through the nanopore. By increasing the number of nitrilotriacetic acid head groups in the synthetic linker to three, the authors found that they were able to bind His6-tagged proteins for hour-long durations. This robust anchoring further enabled the detection of protein–protein interactions between the anchored protein (red in Fig. 3) and immunoglobulin G antibodies (orange in Fig. 3). Hence, the authors demonstrated the construction of a selective single-molecule biosensor via the biochemical modification of a previously inert solid-state nanopore. Reliable and rapid biosensing systems such as this are invaluable tools, particularly in research and clinical settings. Future developments may see a wider range of proteins being detected, potentially for early detection of disease or high sensitivity analyte detection in the field.
 | ||
| Fig. 3 Cross section of a silicon nitride solid-state nanopore coated with a thin gold film, with enlargement shown below. Synthetic nitrilotriacetic acid receptors (purple) were anchored to the gold surface within a self-assembled monolayer (blue). These receptors were then able to bind a His6-tagged protein molecule (red) via Ni2+ chelation. By increasing the number of nitrilotriacetic acid groups in the receptor, protein molecules could be bound for hour-long durations enabling the further detection of protein–protein interactions, such as recognition of the red protein by immunoglobulins (orange).12 | ||
Non-covalent modification can be employed to instil new functionality into solid-state nanopores.13 A demonstration of this approach was the addition of a lipid bilayer to a solid-state pore to enhance protein-sensing capabilities. The Mayer group demonstrated that lipid-coated solid-state nanopores could be used to fingerprint the approximate shape and charge of individual protein molecules.14 As a single protein molecule translocates the pore it will rotate and perturb the ion flow, so proteins with different sizes and shapes give rise to characteristic ion current ‘fingerprints’ (Fig. 4A). The fluid lipid bilayer prevents non-specific adhesion of the protein. To achieve sufficient time resolution of the translocation events, a range of protein tethers were embedded in the lipid bilayer (Fig. 4B). The flexibility and length of the tether permitted free rotation of the protein in the pore, such that all orientations could be accessed. An elegant analysis of the magnitude and distribution of the ion current fingerprints enabled the shape and volume of several different proteins to be elucidated. Meanwhile, the net charge of the protein could be determined from the dwell times of the translocation events. In subsequent work, the Mayer group demonstrated a tether-free method for the analysis of proteins translocating lipid-coated solid-state pores.15 A more sophisticated experimental set-up brought about a 40% reduction in the current noise, which enhanced the signal-to-noise ratio and alleviated the need to further slow the translocation of the protein with a tether. Lipid-functionalised solid-state nanopores have the potential for a wide scope of applications beyond protein fingerprinting. Many conventional biophysical methods require labelling to study proteins and few techniques allow the study of native wild-type proteins. Since labelling can influence structure and activity, caution is required when interpreting results obtained using labelled proteins. Such ambiguity can therefore be avoided by using lipid-functionalised solid-state nanopores to study native proteins in biologically relevant buffers.
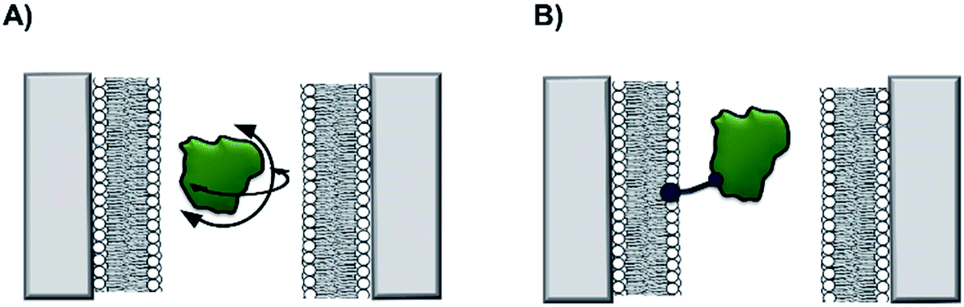 | ||
| Fig. 4 (A) A silicon nitride nanopore coated with synthetic lipids enabled the shape and size of different proteins to be determined from ion current fluctuations as they tumbled through the nanopore. (B) Tethers in the lipid bilayer binding the target protein further slowed rotational dynamics to reveal the characteristics of the translocating protein in even greater detail.14,15 | ||
As solid-state nanopore technology has developed, the potential of biomimetic pores has been realised. The Siwy group fabricated a potassium ion selective solid-state nanopore using surface functionalisation.16 The authors modified a silicon nitride-coated pore with triethoxysilylpropylmaleamic acid (TESPMA) as a reactive handle for further functionalisation (Fig. 5A). The terminal carboxylic acid of TESPMA was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), allowing functionalisation of the pore with a crown ether via an amide linkage (Fig. 5A). Functionalisation of the pore with only crown ether (Fig. 5B) gave rise to notable potassium ion selectivity. Potassium ion selectivity decreased exponentially with an increase in pore diameter and was non-existent for pores with diameters > 3 nm. When a pore with an appropriate diameter was used, transit of potassium ions was facilitated due to binding/unbinding of the crown-ether modification. Drawing inspiration from biological voltage-gated ion channels, the authors theorised that appending single-stranded DNA to the membrane surface might provide a means of further modulating ion selectivity (Fig. 5C). They hypothesised that the high negative charge density on one side of the pore would increase the local concentration of positively charged ions, and thereby promote the passage of cations without affecting ion selectivity. This was indeed shown to be the case, and significantly higher potassium ion transport was observed when the silicon wafer was additionally modified with DNA.
 | ||
| Fig. 5 (A) A solid-state silicon pore coated with silicon nitride (blue) and further functionalised with TESPMA (triethoxysilylpropylmaleamic acid). The TESPMA acts as a reactive handle for controlled synthetic modification. (B) 18-Crown-6 (red) coupled to the pore via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling. (C) Additional single-stranded DNA (ssDNA, purple) modification on one side of a silicon nitride membrane further modulated the ion selectivity of the nanopore.16 | ||
Solid-state nanopores are an attractive platform for sensing and single-molecule studies due to their chemical robustness and stability. The addition of a reactive coating or lipid membrane to the surface allows new functionality or reactive handles to be introduced. By taking inspiration from, and co-opting biology, sophisticated channels with novel functionality can be created. Clearly, we have only sampled some of the possibilities enabled by modification of solid-state pores with biologically derived molecules and synthetic chemical modification further broadens the horizons.13,17
Engineered protein nanopores and site-specific modification
Side-chain modification of membrane proteins has long been used to gain insight into the structure and activity of native transmembrane proteins.18–21 However, the modification of protein nanopores to add new functionality is a more contemporary pursuit. A vast range of modifications involving mutagenesis and chemical modification have been demonstrated,22 and only select examples are presented in this perspective.Mutagenesis is a widely applied method for modifying proteins due to the high degree of control and selectivity that can be achieved. An excellent example of nanopore engineering comes from the Maglia group, who used cytolysin A (ClyA) channels to study the post-translational ubiquitination of a protein (Fig. 6).23 The authors selected the E2 Ubc4 enzyme from Saccharomyces cerevisiae for their investigation due to its ability to self-ubiquitinate. Moreover, single E2 enzyme molecules can be trapped within the lumen of ClyA under an applied transmembrane potential (Fig. 6). The authors used a genetically engineered ClyA pore in which a ring of glutamine residues was mutated to tryptophan on the inner surface of the channel (yellow in Fig. 6). This mutation increased the dwell time of captured E2 protein molecules by increasing the hydrophobicity of the pore interior. By using these pores, different isomeric E2-ubiquitin conjugates could be resolved and the ubiquitination of E2 enzymes could be monitored in real time. Real-time measurements, particularly those that can be conducted in biologically relevant conditions, are invaluable for the study of fundamental protein interactions. In a further work, the Maglia group have also introduced charged residues into the beta barrel of an α-HL channel to slow the translocation of single-stranded DNA (Fig. 7).24 A range of homo- and hetero-heptameric α-HL mutants with varying numbers of positive charges within the channel were produced and found to influence the kinetics of DNA translocation (Fig. 7B). However, these charged mutations also greatly reduced the flow of ions through the channel, resulting in near-complete blockage of the ion current during DNA translocation. Such poor signal-to-noise ratios therefore rendered the pores unsuitable for DNA sequencing applications that rely on an ion-current readout. Nonetheless, an effective strategy for slowing the translocation of DNA was demonstrated, and the authors proposed that similar channels might be exploited in combination with other means of nucleotide readout where slower movement of DNA is required.
 | ||
| Fig. 6 Cytolysin A (ClyA) engineered with a ring of hydrophobic tryptophan residues in place of glutamine residues within the lumen of the pore (yellow). This modification increases the hydrophobicity of the pore and increases the dwell time of individual E2 enzyme molecules (orange). Captured E2 enzymes that were post-translationally self-ubiquitinated (orange + pink) gave distinct ion current traces compared to the unmodified E2 enzyme.23 | ||
 | ||
| Fig. 7 (A) Unconstrained translocation of single-stranded DNA (ssDNA, blue) through a wild-type α-hemolysin pore is too rapid for much sequence information to be elucidated from ion currents. (B) Translocation of ssDNA through a protein channel can be slowed by engineering a ring of positively charged lysine resides into the nanopore.24 | ||
The aforementioned example highlights the limitations of modifying residues within a protein channel, since varying the charges within a nanopore can affect ion currents and the associated electro-osmotic flow.25,26 Nonetheless, such changes can be exploited to attain beneficial characteristics. For example, the Luchian group demonstrated that pH-tuning enabled balancing of the electro-osmotic force against the usually dominant electrophoretic force.27 The fine balancing of these forces then enabled a single charged peptide to be captured within the pore. Similarly, varying the pH and the charged residues within fragaceatoxin C (FraC) nanopore has enabled the detection of differently charged peptides that would otherwise not enter the pore.26
The chemical tailoring of protein channels is not limited to mutagenesis in isolation, since some amino acids can be further modified using bioconjugation chemistry.28 Early examples include the modification of α-HL cysteine mutants with single stranded DNA (see also DNA-modified nanopore section below),29,30 and a photocleavable group that blocked pore assembly until it was removed by UV light.31 Indeed, genetic engineering provides a powerful method for incorporating handles for subsequent functionalisation in desired positions.22,28 As there are no naturally occurring cysteine residues with the α-HL protein, introducing cysteine residues affords absolute control of the positioning of the reactive site.32 The Bayley group have made extensive use of the powerful pairing of cysteine mutagenesis and the subsequent chemical modification of α-HL nanopores. In 2001, they examined the viability of using chemical modification reactions to probe the internal three-dimensional structure of a transmembrane channel.33 α-HL was used as a model protein as the crystal structure of the assembled protein is already known.32 Seven homoheptameric mutants were produced that contained rings of cysteine residues positioned in different locations within the nanopore channel. The approximate location of the constriction site at the mid-point of the nanopore was determined by analysing the relative rate of disulfide exchange when each of the mutants nanopores reacted with monomethoxypoly(ethylene glycol)-o-pyridyl disulfide polymers of differing lengths. In addition, larger blockages in the ion current were observed when reactions occurred at the narrowest points in the channel, providing further insight into the internal dimensions of the transmembrane ion channel.
The ability to assemble heteroheptameric α-HL mutants provides additional control over chemical modification. Indeed, α-HL nanopores assembled with a single cysteine in just one of the seven constituent proteins have been particularly fruitful (Fig. 8). Since the assembly of single-mutant pores requires mixing the mutant and wild-type proteins, a means of separating the desired single mutant from those assembled with zero or more than one mutant protein is required (Fig. 8B and C). Following assembly of a mixture of heptameric pores in rabbit red blood cell membranes, separation via gel electrophoresis is enabled by the additional presence of a genetically encoded 8-residue aspartic acid tail on the C-terminus of the mutant monomer protein (D8 in Fig. 8B).35 The isolated mutant protein can then be reconstituted in a lipid bilayer for nanopore experiments. For example, a single cysteine handle within such a mutant protein has been used to monitor the stepwise growth of a polymer chain (Fig. 8C and D).34 When 5,5′-dithiobis-(2-nitrobenzoic acid) and (mercaptoethyl)-ether were placed on opposite sides of a suspended lipid bilayer containing a single cysteine mutant pore, sequential disulfide exchange reactions occurring within the channel to form a polymer chain could be monitored by the progressive blockage of the ion channel current.34 The same mutant nanopore has also been used to probe the formation of the thioarsenic bonds at the single-molecule level (Fig. 8E).35 By measuring the durations of the events and the concentration dependency, the kinetics of bond formation and disassociation were determined. Later work used a similar approach to probe pyramidal inversion of thioarsenic chiral centres36 and multi-component reaction networks.37
 | ||
| Fig. 8 Production of α-hemolysin homoheteroheptameric nanopores modified at a single cysteine residue before (bottom panel) or after nanopore assembly (top panel). (A) α-Hemolysin monomers synthesised using in vitro transcription/translation from a plasmid encoding the mutant protein. (B) The mutant protein monomer contains an octa-aspartic acid tail (D8) at the c-terminus to enable separation by gel electrophoresis. (C) A mutant pore containing a single cysteine residue modified after reconstitution in a lipid bilayer to monitor (D) stepwise polymer growth arising from disulfide exchange reactions,34 and (E) reversible thioarsenic chemistry.35–38 (F) to (G) Cysteine mutant monomers have been functionalised with a wide range of modifications prior to assembly into heptameric nanopores, purification, and reconstitution in a lipid bilayer. (H) Modification with β-cyclodextrin enabled the discrimination of individual nucleoside bases.39,40 (I) Attachment of phenanthroline enabled the detection of chelating groups (pink) such as neurotransmitters41 and amino acid enantiomers42 in the presence of Cu(II). (J) Photoinitiated degradation of a 3,4-dimethoxy-6-nitrobenzylcarbamate group bond to the pore.43 (K) A maleimide-based linker used to attach functional groups enabled attachment and monitoring of subsequent cycloaddition addition reactions involving tetrazine, cyclooctene, and cyclooctyne functional groups.44 | ||
Taking the approach a step further, the Bayley group have constructed α-hemolysin nanopores containing one mutant protein that contains a track of up to six cysteine footholds along the membrane-spanning β-barrel (Fig. 9A).38,45 The first such study exploited the reversible thioarsenic chemistry introduced above to create a molecule capable of walking along the cysteine track. The walking occurred via alternating intra- and inter-molecular As–S bond-making/breaking reactions via a hand-over-hand mechanism, reminiscent of the way in which dynein and kinesin motor proteins walk along microtubules. The thioarsenic walker was found to stochastically walk towards a thermodynamic sink position, but more recent work based on disulfide exchange has developed a molecular hopper in which the direction of movement along the cysteine track could be controlled (Fig. 9B and C).45 A trapavidin-biotin complex served as a bulky stopper enable capture of the loading molecule within the nanopore, while the length of the carrier linker was tuned to target loading (via disulfide exchange) onto the desired cysteine residue within the pore (Fig. 9C). The externally applied transmembrane voltage could then be used to control the direction of hopping since the electric field exerts a force on the negatively charged single-stranded DNA (ssDNA) cargo. In a more recent application of this work, the Bayley group have demonstrated that single nucleobase variations in a DNA sequence can be detected using the chemical stepping of the molecular hopper.46
 | ||
| Fig. 9 (A) α-Hemolysin nanopore constructed from six wild-type proteins and one mutant protein containing a track of cysteine residues. Such proteins have been used as tracks for arsenic-based38 and (B) disulfide-based molecular walkers.45 (C) Molecule used to load the disulfide walker onto the cysteine track. Due to the conformational restrictions of the β-barrel, disulfide bonds cannot form between the cysteine resides of the track. Therefore, it is highly improbable that the hopper will dissociate due to the formation of a new protein–protein disulfide bond. Instead, the hopper will move from reside to residue and carry its payload with it until it reaches the end of the track. The voltage can then be inverted to send the hopper in the opposite direction. | ||
Contrasting with the approach of modifying cysteine residues after assembly into heptameric nanopores (i.e.Fig. 8A–E and 9), singly modified α-HL nanopores may also be generated by functionalising cysteine mutant monomers prior to assembly and purification (Fig. 8B and F–K). The Bayley group have modified cysteine-mutant α-HL monomers with a diverse selection of synthetic groups and found that this does not prevent assembly into the desired singly functionalised heteroheptameric nanopores (Fig. 8H–K). For example, β-cyclodextrin has been covalently attached to α-HL monomers prior to assembly to generate a nanopore capable of continuous read-out of molecular recognition events occurring within the β-cyclodextrin cavity, and even found capable of discriminating the bases of individual nucleosides (Fig. 8H).39,40 Phenanthroline-modified nanopores have also been generated using the pre-assembly modification approach (Fig. 8I). The presence of copper(II) as a bridging chelating agent enabled the binding and resolution of different neurotransmitters41 and amino acid enantiomers based on changes in the ion current.42 The same pre-assembly modification strategy has also been used to assemble α-HL nanopores modified with a single azobenzene unit to observe light-induced configurational switching between E and Z isomers.47
The ability to readily modify α-HL has facilitated its use as a nanoreactor to observe chemical reactions at the single-molecule level. Monitoring ion currents allows reactions to be monitored down to the high microsecond timescale, thereby enabling the observation of transient intermediates. For example, a nanopore pre-modified with 3,4-dimethoxy-6-nitrobenzylcarbamate was observed to undergo photoinitiated degradation via a multi-step process in which the kinetics of individual steps could be elucidated (Fig. 8J).43 Maleimide has also served as a useful reagent for the modification of cysteine-modified α-HL monomers (Fig. 8K).44 α-HL nanopores were pre-modified with three different maleimide linkers containing additional terminal sites for further bioconjugation; a tetrazine, a cyclo-octene, and a cyclo-octyne. When these modified pores were reconstituted in a lipid bilayer, the ion current facilitated the observation of cycloaddition reactions with reagents added to the solution on one side of the membrane. Five different cycloaddition reaction combinations were examined, but no intermediates were observed on the timescale of the ion current recordings, meaning that the intermediates these reactions must have lifetimes of <80 μs. The Bayley group has also employed cysteine-maleimide bioconjugation to modify pre-assembled α-HL pores (i.e.Fig. 8, top), rather than prior to assembly as in the preceding work.48 This in situ modification approach avoided the hydrolysis of the reactive nitrile group in nanopores modified with 2-cyanobenzothiazole that would otherwise occur during gel purification. Contrasting with the bioconjugation studies noted above, several intermediates were observed along the course of bioorthogonal condensation reactions occurring between the 2-cyanobenzothiazole functionality and different aminothiols.
As is evident, extensive work has been carried out on the functionalisation of α-HL via mutagenesis. This can most likely be attributed to the relatively low cost and high availability of the protein, along with its comprehensive characterisation. However, a growing number of alternative pores have emerged in previous years. Aerolysin, for example, has a similar heptameric, β-barrel structure to α-hemolysin, albeit with a narrower diameter of 13.7 Å.49 This makes aerolysin an attractive alternative to α-HL as enhanced signal-to-noise ratios can be achieved. Nonetheless, similar problems with rapid translocation of analytes may persist. A rational design approach has been used to produce a series of aerolysin single mutants, in which residues were substituted with alanine or tryptophan (to vary the size of the channel), or lysine, glutamine or glutamate (to vary the charge).50 Experimental and simulation data showed that the translocation and capture of DNA and protein could be modulated by varying residues at the constriction points of the pore. Similarly, the Maglia group has shown that the introduction of charged residues has no effect on peptide retention in a fragaceatoxin (FraC) nanopore, whereas the incorporation of an aromatic residue increased the peptide capture capabilities of the pore.51
Chemical functionalisation of protein nanopores is not limited to the modification of genetically engineered variants. Recent work has seen the in situ modification of wild-type α-HL at both lysine and methionine residues.52,53 α-HL presents several solvent-exposed lysine residues that could serve as potential sites for bioconjugation (Fig. 10A). Initially, Borsley and Cockroft used reversible iminoboronate chemistry to map the reactivity of these residues (Fig. 10B).52 Ion current recordings revealed a surprisingly simple reactivity pattern that could be attributed to iminoboronate formation at one of the three rings of lysine residues lining the interior of the channel (Fig. 10A), indicating that reactions occurring on any other external lysine residues did not influence the ion current. Moreover, the central-most ring of lysine residues was found to be substantially more reactive than the other lysine sites. Irreversible modification by amide bond formation was the demonstrated using 3-sulfo-N-succinimidyl benzoate sodium salt (Fig. 10C).
 | ||
| Fig. 10 (A) Locations of lysine residues in the α-hemolysin nanopore (blue). (B) Reversible modification of lysine with a boronic acid was used to probe the reactivity of the solvent-exposed lysine residues. Only reactions occurring on the three rings of lysine residues on the interior of the channel were found to modulate the transmembrane ion current. The central ring of lysine residues were found to be the most reactive. (C) Irreversible amide modification of lysine residues within the channel interior was achieved using 3-sulfo-N-succinimidyl benzoate sodium salt.52 (D) α-Hemolysin contains one solvent-exposed ring of seven methionine residues within the lumen of the nanopore. Methionine (red) irreversibly reacts with oxaziridines to yield nanopores functionalised with (E) alkynes or (F) azides, which can be further functionalised using click chemistry. Ion current changes also allowed short-lived reactive intermediates to be resolved and assigned.53 | ||
The same group went on to selectively modify methionine residues in wild-type α-HL using a similar in situ approach (Fig. 10D–F).53 Methionine is a highly desirable target for protein modification as it is one of the least common canonical amino acids and is rarely solvent exposed due to its high hydrophobicity, meaning that a higher degree of selectivity can be achieved. Indeed, α-HL presents only one solvent-exposed methionine residue in each of its subunits (Fig. 10D). Bioorthogonal azide and alkyne groups were introduced into α-HL using oxaziridine reagents originally developed by Toste (Fig. 10E and F).54 Subsequent click reactions enabled the decoration of the alkyne- and azide-modified pores with a range of substrates, including an acid, a base, a nucleotide and even a single-stranded DNA oligonucleotide (Fig. 10E).53 Notably, characteristic ion current traces were observed during the Cu(I)-catalysed azide–alkyne [3 + 2] cycloaddition (CuAAC) modifications of the alkyne-modified nanopore. A series of experiments in which the Cu(I) concentration and applied potential was varied enabled the complete assignment of all the observed ion current levels including short-lived copper-bound reaction intermediates. The Bayley group similarly investigated CuAAC reactions in α-HL channels in earlier work, but using the more challenging strategy of incorporating an unnatural alkyne amino acid.55
DNA-modified nanopores
The pursuit of single-molecule DNA sequencing has been a major driver in the development of nanopore technologies. Detection of specific oligonucleotides has a range of applications, particularly for the detection of disease and gene abnormalities involving single-nucleotide polymorphism. Both solid-state48,56 and biological nanopores29,30 have been modified with DNA for the detection of complementary counter strands. Even single-nucleotide mismatches within an otherwise fully complementary stand could be detected using ion currents.29,30,57 Bashir56 and Valbusa57 used 1,4-phenylene diisothiocyanate to cross-link amino-terminated DNA to solid-state nanopores coated with (3-aminopropyl)trimethoxysilane (Fig. 11).56 Meanwhile, Howorka and Bayley used pre-assembly cysteine modification (Fig. 8G) to attach single-stranded DNA to α-HL protein nanopores (Fig. 12A).29,30 Bayley and Dekker subsequently used DNA-modified α-HL to enable electrophoretic capture in a solid-state nanopore.58 Post-assembly modification was employed (Fig. 8C) to attach a thiol-terminated 12-mer DNA sequence to a cysteine residue within an extended loop at the β-barrel opening. The 12-mer oligonucleotide acted as a handle for hybridisation to a 2 kb double-stranded plasmid DNA with a complementary sticky end (blue/green in Fig. 12). Under an applied potential, the negatively charged DNA tail guided the α-HL into a solid-state pore that was large enough to accommodate the narrow β-barrel of α-HL, but too small for the cap domain to pass though, thereby trapping the protein (Fig. 12B). Ion-current traces of the nanopore within a nanopore showed that free single-stranded DNA molecules were still able to transit through the α-HL channel. The insertion of the biological nanopore into a solid-state one draws benefits from both approaches. The solid-state components provide enhanced stability, with the hybrid pores lasting for several days, which is generally unobtainable using lipid-membrane systems. Meanwhile, the biological component provides atomically precise dimensions and the potential for further functionalisation that would otherwise be difficult for a typical solid-state pore. Such developments herald the wider application of solid-state/biological hybrid nanopores. | ||
| Fig. 11 Solid-state nanopores modified using 1,4-phenylene diisothiocyanate as a cross-linking agent to attach amino-terminated (A) DNA hairpins56 and (B) single-stranded DNA.57 The passage of complementary strands through such pores was slowed, resulting in prolonged ion current drops. | ||
 | ||
| Fig. 12 (A) Membrane-spanning α-hemolysin nanopore modified with an 8-mer single-stranded DNA via a disulfide linkage.29,30 (B) α-Hemolysin modified with a 2kb DNA tether enabled threading and capture of the protein nanopore within a solid-state nanopore.58 | ||
In addition to decorating existing nanopores with simple oligonucleotides, larger DNA origami constructs can also be used to tune the properties of solid-state pores. The pairing of DNA origami and nanopore technology has been explored in numerous contexts.59 DNA origami can be fabricated into precise and intricate structures through the utilisation of molecular self-assembly.60 DNA nanoplates are an example of one of the many architectures possible. Dietz has shown that nanoplates can be captured at the entrance of solid-state nanopores to enhance their function and give greater control over the dimensions of the pore (Fig. 13A/B).61 Like the preceding example in presented in Fig. 12B, the dangling DNA in Fig. 13A enables the DNA origami nanoplates to be captured and held in place by the applied voltage. The nanoplate approach constitutes an interesting platform for developing biological-synthetic hybrid nanopores with enhanced sensing capabilities. In the absence of the nanoplate, the addition of the small protein, streptavidin caused fleeting transient blockades in the ion current as it entered the SiN nanopore. However, once the DNA nanoplate was in place, prolonged translocation events were detected. The addition of the much larger immunoglobulin G gave no translocation events when the nanoplate was in place, demonstrating size-selective capabilities of the portal within the DNA nanoplate. In addition to having tuneable size selectivity, DNA nanoplates were constructed that position a DNA “bait” sequence at the opening of the pore (Fig. 13A). When the target “prey” sequence hybridises with the bait sequence this prevents or slows the passage of the analyte of interest for detection purposes.
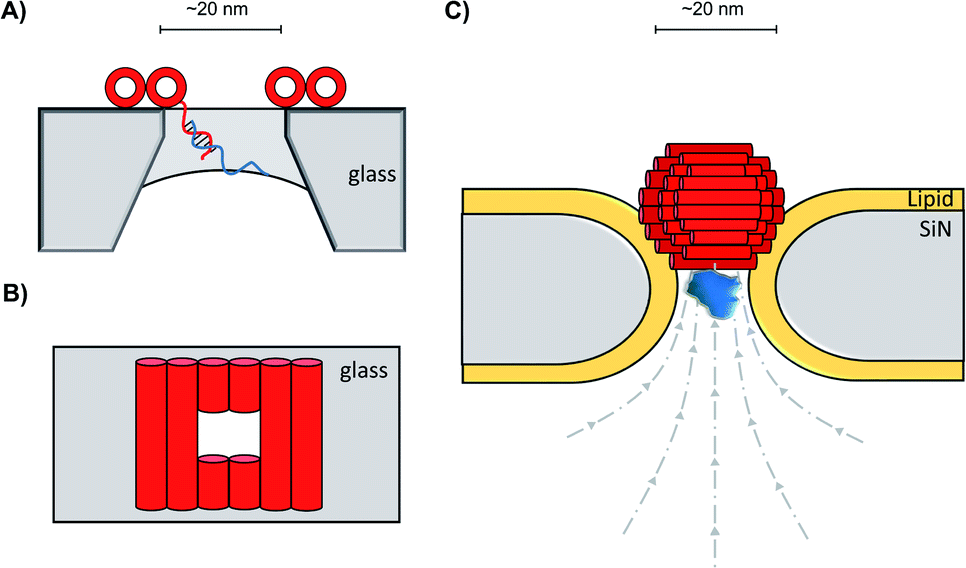 | ||
| Fig. 13 (A) Single-stranded DNA was used to guide the voltage-driven capture of a DNA-origami nanoplate (red cylinders) at the opening of a SiN nanopore. Nanoplates containing a terminal single-stranded DNA strand could be used to detect the binding of complementary sequences.61 (B) Top-down view of the nanoplate. The size of the opening can be controlled by the design of the sequences used to assemble the nanoplate.61 (C) Voltage-driven capture of a DNA nanosphere (red) in a lipid-coated SiN nanopore enabled the electro-osmotic trapping (dashed lines) and detection of several different proteins (blue).62 | ||
A major advantage of DNA origami is the ability to fabricate complex 3D architectures. Alongside the DNA nanoplate, Dietz and Dekker have demonstrated that DNA nanospheres can be captured within a lipid-coated solid-state nanopore under an applied potential (Fig. 13C).62 The DNA sphere was permeable to ions, so a total blockage of ions was not observed. Indeed, an electro-osmotic hydrodynamic flow was facilitated that enabled long-duration trapping of protein molecules. Proteins varying in size and mass were trapped. Not only did each protein give a distinct signal, but differences in the orientation and conformations of the individual proteins could be resolved. This presents a novel approach where a lipid-functionalised nanopore further functionalised with DNA origami exhibits enhanced protein trapping and identification properties. Future development of this work may lead to significant advances in label-free protein analysis. The combination of DNA nanotechnology and solid-state nanopores provides platforms with wide-reaching biosensing applications, as a near limitless range of modifications are accessible through DNA assembly and existing synthetic DNA modifications.
Synthetic protein nanopores
The functionalisation of existing biological and solid-state nanopores has drastically reshaped the remit and application of nanopore approaches. Nanopore electrophysiology has evolved from a technique used to study pores themselves, to a method that is sensitive enough to resolve single amino acid and nucleotide variations.46,63 However, the modification of existing pores is not without its challenges and limitations. An emerging area that holds great promise is the development of de novo synthetic protein nanopores.Taking a step in this direction, Woolfson and Bayley designed and constructed a semi-synthetic self-assembled α-helical transmembrane protein based on the structure of Wza, an Escherichia coli polysaccharide transporter (Fig. 14A).64 An initial BLAST search (http://blast.ncbi.nlm.nih.gov/) was performed using the membrane-spanning residues of the Wza sequence as the input query to reveal an evolutionarily well-conserved consensus sequence. This 35-residue sequence (cWza) was synthesised using solid-state peptide synthesis. Ion current recordings indicated that the peptide formed discrete transmembrane channels, which (based on crystallographic evidence) were expected to assemble as an octamer in the lipid bilayer (Fig. 14B). The channels were also seen to undergo reversible conformational changes between a low-conductance state and a high-conductance state (Fig. 14B to C). This channel, therefore, does not afford the same structural rigidity and reproducibility as a biological pore such as α-HL. However, the semi-synthetic cWza pore was remarkably more stable than the original Wza query sequence, which only formed noisy and unstable pores. The orientation of the peptides within the membrane and the octameric nature of the pore was confirmed using several clever experiments. Firstly, ion currents showed that cysteine-mutant channels reacted with polyethyleneglycol (PEG) pyridyl-disulfide derivatives at different rates depending on which side of the bilayer the reagent was added. Secondly, the addition of dithiothreitol (DTT) reopened the pore in a stepwise manner by cleaving the PEG-disulfide bonds, but this always occurred with eight or less steps (Fig. 14C and D). A pyridyl disulfide-derivatised octasaccharide, γ-cyclodextrin could also be used to template the formation of cysteine-mutant pores, with the addition of DTT resulting in the release of the cyclodextrin (Fig. 14E to C). In addition, the binding of amine-derivatised hexa-, hepta- and octa-saccharides (α, β and γ-cyclodextrin) was compared, with γ-cyclodextrin being found to bind most favourably (Fig. 14E). The work presents an exciting premise for the development of custom-designed protein channels.
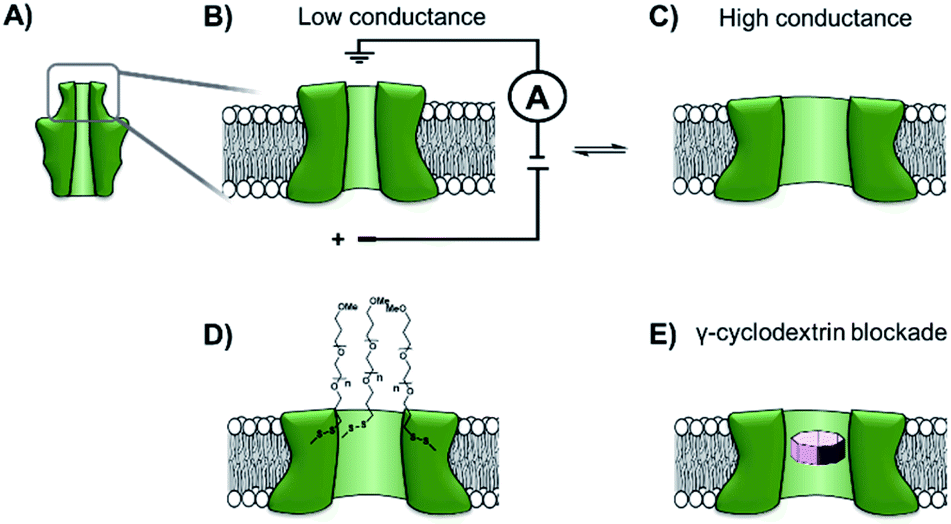 | ||
| Fig. 14 (A) The D4 domain of E. coli Wza protein. (B) The evolutionarily conserved, 35-amino acid consensus sequence (cWza) of this membrane-spanning domain was determined via a BLAST sequence search. The sequence was synthesised using solid-state peptide synthesis and found to form pores that were much more stable than those assembled using the original Wza sequence. (C) Ion currents showed that the cWza pores were in conformational exchange between low and high conductance states. (D) Nanopores constructed using a cysteine-containing peptide were modified with polyethylene glycol groups (PEG) via disulfide bond exchange. The disulfide bonds were subsequently cleaved using dithiothreitol (DTT) revealing ≤ eight steps in the ion current to return to the unmodified form, thereby confirming the octameric nature of the pore. (E) Octasaccharide γ-cyclodextrin derivatives were found both to bind to and template the formation of the octameric semi-synthetic cWza pores.64 | ||
While pore-forming sections of larger proteins have been exploited as monodisperse channels for nanopore sensing, multiple proteins can also be combined in a modular fashion to attain functional nanpores. The Maglia group genetically modified α-HL with loop sequences from the GroES protein that bind the GroEL chaperonin protein (Fig. 15A).65 The modified pore was found to retain pore-forming ability while also being able to bind a single-ring version of the GroEL chaperonin protein. Remarkably, GroEL-assisted protein folding by the GroES nanopore was found to be on par with the native GroES-GroEL co-chaperonin complex. This presents a novel concept where protein pores with a specific function can be fabricated via a bottom-up approach from pre-existing components. In this case, the use of α-HL as a scaffold to construct multi-protein assemblies provides a promising approach for the assembly of novel functionalised pores.
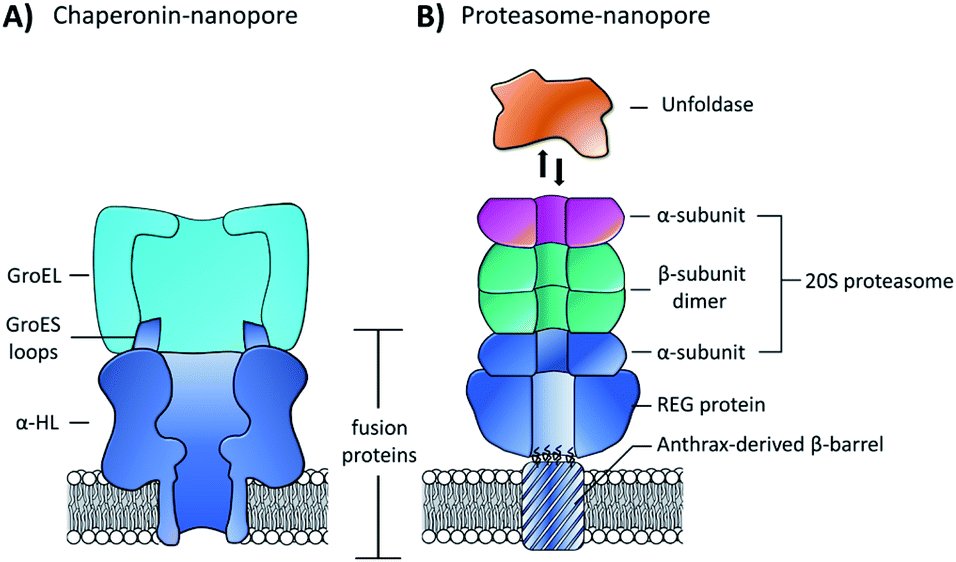 | ||
| Fig. 15 (A) Chimeric protein nanopore formed by the genetic fusion of α-HL and loops from the GroES co-chaperonin protein (dark blue). The fused GroES loops endows the fusion protein with the ability to bind a single-ring variant of the GroEL protein-folding chaperonin protein (turquoise). The chimeric protein formed stable channels in membranes, while also catalysing protein refolding in the presence of ATP and GroEL.65 (B) Representation of a proteasome nanopore formed by the fusion of three proteins. The anthrax-derived β-barrel was genetically fused to both a REG protein and an α-subunit of a 20S proteasome (dark blue). The rest of the 20S proteasome was assembled non-covalently by the back-to-back stacking of two β-subunits (teal) and an additional α-subunit (purple). The whole assembly was furthermore capable of binding an unfoldase (orange), which could catalyses the unfolding and translocation of proteins into the proteosome nanopore. The whole assembly was shown to be capable of coupling protein translocation and degradation when assembled using a proteolytically active proteasome.66 | ||
Building on this precedent, the Maglia group went on to demonstrate the assembly of another modular nanopore constructed from several different proteins (Fig. 15B).66 The channel was constructed from the genetically encoded fusion of three proteins; a membrane spanning β-barrel derived from Bacillus anthracis, part of the proteasome activator 28α (REG), and the α-subunit of the 20S proteasome from T. acidophilum. By replacing the disordered region of REG with the transmembrane β-barrel a synthetic multi-component nanopore was constructed. The multi-protein assembly retained the ability to insert into a lipid bilayer and form a stable channel. However, at voltages beyond +20 mV the channel tended to gate. Interestingly, the isolated β-barrel domain would not insert into the membrane by itself, meaning that the larger protein framework was required to maintain integrity. The outermost α-subunit of the assembly retained its ability to reversibly bind an unfoldase enzyme from T. acidophilum (orange, Fig. 15B). Remarkably, the complete assembly enabled ATP-driven unfolding and proteolytic digestion of green fluorescent protein as it was threaded through the nanopore (depending upon whether a proteolytically active or inactive variant of the proteasome–nanopore was used). While single amino acid detection based on the resulting ion currents was not demonstrated, this may become a possibility in future work. Indeed, the authors propose that single amino acid resolution might be achieved by engineering a similar system with a smaller pore diameter. While further work is required for single-molecule fingerprinting, this modular approach for incorporating enzymatic nanomachinery from biology into transmembrane nanopores presents an exciting new direction.
Advances in the computational design of protein architectures,67 mean that we are entering an era where it becomes possible to design entirely synthetic protein pores with a specific function in mind. For example, building on established understanding of protein structure and facilitated by molecular dynamics (MD) simulations, the Kawano group have developed a β-hairpin peptide capable of forming a monodisperse transmembrane channel.68 An initial sequence design contained an amphiphilic β-sheet backbone, stabilised by strategically placed tyrosine residues. A negatively charged β-turn sequence and positively charged terminal region afforded control over the orientation of assembly within the bilayer. While this initial sequence was successful in forming stable channels, the sizes ranged from 1.7–6.3 nm. The subsequent incorporation of a glycine-kink in the transmembrane region of the peptide gave a monodisperse channel with a diameter of 1.7 nm. This channel could then be applied for the detection of a poly-L-lysine. Indeed, the capability of the synthetic channel to detect the poly-L-lysine outstripped that of α-HL. While some refinement is required, this presents an exciting future avenue for the future of nanopore technology where bespoke pores can be designed for specific processes.
Conclusions and outlook
Early ventures in nanopore research saw the adoption of naturally occurring transmembrane protein channels as nanoscale sensors. In the intervening years, the incorporation of non-native functionalities, reactive handles, and molecular adaptors has helped to realise this potential. The scope of these modifications, the technological requirements that they have fulfilled, and their accompanying challenges and future potential are summarised in Table 1.| Pore type | Modification | Technological requirements fulfilled | Challenges and limitations | Future perspective | Ref. |
|---|---|---|---|---|---|
| Biological | -Introduction of charged or hydrophobic residues via mutagenesis | -Dwell time control | -Changes to pore stability and electro-osmotic flow | -Label-free, real-time detection of proteins and charged molecules -Fingerprinting and detection for proteins | 23, 24, 26, 50 and 51 |
| -Signal differentiation | -Limited by viable mutations | ||||
| -Limited by membrane stability | |||||
| -Introduction of reactive handle via mutagenesis | -Dwell time control | -Limited by viable mutations and inherent pore instability | -Design of molecular machines, observations of reaction mechanisms, enhanced substrate detection | 34, 35, 38–46 and 48 | |
| -Signal differentiation | |||||
| -Multiple reads | |||||
| -Capture of molecules | |||||
| -Observation of reactive intermediates | -The use of unnatural amino acids is underexplored | ||||
| -Introduction of reactive handles via chemical modification | -Signal differentiation | -Limited to available solvent exposed reactive handles -Potential lack of selectivity | -Real-time observations of reaction mechanics at the single-molecule level | 52 and 53 | |
| -Observation of reactive intermediates | |||||
| Solid state | -Surface functionalisation with reactive inorganic coating | -Signal differentiation | -Challenging to create a monodisperse layer and achieve consistent pore geometry | -High stability biomimetic channels -Applications for on-site, high-sensitivity measurements | 12 and 16 |
| -Dwell time control | |||||
| -Channel selectivity | |||||
| -Surface functionalisation lipid bilayer | -Capture of molecules | -Relies on non-specific interactions between lipid and analyte | -Label-free, and real-time detection of native proteins | 13 and 62 | |
| -Signal differentiation | |||||
| -Surface functionalisation with DNA | -Capture of molecules | -Design of DNA sequences. -Attachment to solid matrixes | -High sensitivity detection of target DNA and RNA sequences for medical applications | 56, 57, 61 and 62 | |
| -Channel selectively | |||||
| -Dwell time control | |||||
| Hybrid/semi-synthetic | -Combination of biological and solid-state pores | -Enhanced stability | -Challenging to fabricate | -Extremely high-stability pores with atomically precise geometries | 4 and 58 |
| -Signal differentiation | |||||
| -Sequestering of membrane active sections of known proteins | -Capture of molecules | -Less-reliable stability compared to native pores -Difficult to predict structure | -Custom pores with tailored size and functionality | 64 | |
| -Combination of existing proteins into a bespoke channel | -Signal differentiation | -Similar issues to native pores with rapid translocation beyond the limit of resolution | -Utilisation of properties from multiple proteins for new sensing and detection applications | 65 and 66 | |
| -Capture of molecules | |||||
| de novo | -Synthesis of synthetic transmembrane nanopore | -Signal differentiation | -Challenges in the design of viable channels -Difficult to predict structure | -Custom-designed bespoke channels | 68 |
| -Capture of molecules |
The modification of protein nanopores via site-directed mutagenesis has focussed on the incorporation of the 20 canonical amino acids. Indeed, cysteine modification of nanopores has proven to be particularly powerful when paired with bioconjugation reactions.22 In contrast, unnatural amino acids have rarely been used to modify protein nanopores.55,69,70 This may be due to the increased technical challenge and difficulty in predicting protein structure that comes with the use of unnatural amino acids.71,72 It may also be that the ability to directly modify,52,53 or to pair conventional mutagenesis with post-translational modification28 renders the more challenging use of unnatural amino acids unnecessary. Nonetheless, future advances in synthetic molecular biology may lead to greater exploitation of non-natural amino acids to access to their diverse chemical functionalities and properties. More generally, the modification of biological pores, whether through mutagenesis or chemical methods is not without caveats. Changes to the structure of the protein may affect the ability of a protein to fold and insert into a membrane. Even if a modified pore is successfully formed, functionality can be difficult to predict, and unwanted behaviours are entirely possible. For example, the introduction of positive charges to a protein pore may increase the likelihood of trapping DNA but may also alter the ion flow through the channel and decrease the signal-to-noise ratio.
The development of solid-state nanopore sensors followed shortly after the initial exploitation of biological pore proteins. Solid-state nanopores present more robust, but relatively inert, frameworks for nanopore technology. Mirroring the strategies of modifying biological nanopores, solid-state pores have been functionalised with biological molecules both to control the aperture size and to enhance molecular recognition capabilities. For example, functionalised solid-state channels have been applied as biomimetic ion selective channels and scaffolds for high throughput oligonucleotide sensors.16,56 Amalgamating chemical functionalisation with biological and solid-state components maximises the potential of nanopore-based approaches. Indeed, Oxford Nanopore's DNA sequencing devices represent a pinnacle example of the convergence of biology and man-made nanopore technology, and feature several hundred mutant protein nanopores within stabilised membranes arrayed in a solid-state framework.4 Indeed, the greatest achievement of nanopore technology to date has undoubtedly been the attainment of fast and affordable single-molecule DNA sequencing. Hybrid nanopore systems in handheld devices can also provide rapid and sensitive diagnostic information and could prove vital in future healthcare applications. Indeed, Oxford Nanopore's MinIon was deployed during the SARS-CoV-2 pandemic for genome analysis.73
While the functionalisation and amalgamation of biological and solid-state pores has proved fruitful, the design of de novo synthetic channels provides the potential for gaining unrivalled control over channel properties.10,11,74 The stability and channel-forming abilities of synthetic nanopores are rapidly improving by exploiting, adapting, or taking inspiration from biomolecules. The design of stable entirely synthetic channels remains challenging, meaning that the modification or re-engineering of existing proteins presents a more accessible alternative. A particularly exciting avenue is presented by the ability to combine multiple biological proteins in a modular fashion to integrate biological nanomachines into novel functional nanopores.65,66
Nanopore technology is a rapidly growing field. From its inception, it has proved a valuable tool for sensing applications. This perspective demonstrates how the coalescence of the synthetic and the biological has enabled the development of systems with bespoke functionalities and robustness suited to a particular application; solid-state nanopores have been functionalised with biological elements, while biological pores have been subjected to synthetic modification. Such modifications have progressed nanopore technology from being a tool for sensing, to a platform for the study of biophysics, reaction mechanisms and beyond.44,53,55,75–77 Whether we see a growing number of chemically modified biological pores, biologically modified solid-state pores, or wholly synthetic biological pores in the future, the full potential of the technology is only just beginning to be explored.
Author contributions
Both authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC, the ERC (Grant No. 336935), and The Leverhulme Trust (Philip Leverhulme Prize to SLC) for funding. We acknowledge Dr Rebecca Burns and Daniel Edwards for their assistance in the preparation of this manuscript.References
- E. Neher and B. Sakmann, Nature, 1976, 260, 799–802 CrossRef CAS PubMed.
- B. Sakmann and E. Neher, Ann. Rev. Physiol., 1984, 46, 455–472 CrossRef CAS.
- Y.-L. Ying, C. Cao and Y.-T. Long, Analyst, 2014, 139, 3826–3835 RSC.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS PubMed.
- J. Li, D. Stein, C. McMullan, D. Branton, M. J. Aziz and J. A. Golovchenko, Nature, 2001, 412, 166–169 CrossRef CAS PubMed.
- L. Sun and R. M. Crooks, J. Am. Chem. Soc., 2000, 122, 12340–12345 CrossRef CAS.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mat., 2020, 5, 931–951 CrossRef CAS.
- A. J. Storm, J. H. Chen, X. S. Ling, H. W. Zandbergen and C. Dekker, Nat. Mat., 2003, 2, 537–540 CrossRef CAS PubMed.
- N. Sakai and S. Matile, Langmuir, 2013, 29, 9031–9040 CrossRef CAS PubMed.
- G. W. Gokel and I. A. Carasel, Chem. Soc. Rev., 2007, 36, 378–389 RSC.
- R. Wei, V. Gatterdam, R. Wieneke, R. Tampé and U. Rant, Nat. Nanotechnol., 2012, 7, 257–263 CrossRef CAS PubMed.
- G. Pérez-Mitta, L. Burr, J. S. Tuninetti, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, Nanoscale, 2016, 8, 1470–1478 RSC.
- E. C. Yusko, B. R. Bruhn, O. M. Eggenberger, J. Houghtaling, R. C. Rollings, N. C. Walsh, S. Nandivada, M. Pindrus, A. R. Hall, D. Sept, J. Li, D. S. Kalonia and M. Mayer, Nat. Nanotechnol., 2017, 12, 360–367 CrossRef CAS PubMed.
- J. Houghtaling, C. Ying, O. M. Eggenberger, A. Fennouri, S. Nandivada, M. Acharjee, J. Li, A. R. Hall and M. Mayer, ACS Nano, 2019, 13, 5231–5242 CrossRef CAS PubMed.
- E. T. Acar, S. F. Buchsbaum, C. Combs, F. Fornasiero and Z. S. Siwy, Sci. Adv., 2019, 5, eaav2568 CrossRef CAS PubMed.
- M. Wanunu and A. Meller, Nano Lett., 2007, 7, 1580–1585 CrossRef CAS PubMed.
- H. Tokunaga, M. Tokunaga and T. Nakae, J. Biol. Chem., 1981, 256, 8024–8029 CrossRef CAS.
- R. P. Darveau, R. E. W. Hancock and R. Benz, Biochim. Biophys. Acta, Biomembr., 1984, 774, 67–74 CrossRef CAS.
- J. A. Mindell, H. Zhan, P. D. Huynh, R. J. Collier and A. Finkelstein, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5272–5776 CrossRef CAS PubMed.
- M. Krishnasastry, B. Walker, O. Braha and H. Bayley, FEBS Lett., 1994, 356, 66–71 CrossRef CAS.
- M. Ayub and H. Bayley, Curr. Opin. Chem. Biol., 2016, 34, 117–126 CrossRef CAS PubMed.
- C. Wloka, V. van Meervelt, D. van Gelder, N. Danda, N. Jager, C. P. Williams and G. Maglia, ACS Nano, 2017, 11, 4387–4394 CrossRef CAS PubMed.
- M. Rincon-Restrepo, E. Mikhailova, H. Bayley and G. Maglia, Nano Lett., 2011, 11, 746–750 CrossRef CAS PubMed.
- E. C. Yusko, R. An and M. Mayer, ACS Nano, 2010, 4, 477–487 CrossRef CAS PubMed.
- G. Huang, K. Willems, M. Soskine, C. Wloka and G. Maglia, Nat. Commun., 2017, 8, 935 CrossRef PubMed.
- A. Asandei, I. Schiopu, M. Chinappi, C. H. Seo, Y. Park and T. Luchian, ACS Appl. Mater. Interfaces, 2016, 8, 13166–13179 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- S. Howorka, L. Movileanu, O. Braha and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12996–13001 CrossRef CAS PubMed.
- S. Howorka, S. Cheley and H. Bayley, Nat. Biotechnol., 2001, 19, 636–639 CrossRef CAS PubMed.
- C.-Y. Chang, B. Niblack, B. Walker and H. Bayley, Chem. Biol., 1995, 2, 391–400 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1865 CrossRef CAS PubMed.
- L. Movileanu, S. Cheley, S. Howorka, O. Braha and H. Bayley, J. Gen. Physiol., 2001, 117, 239–252 CrossRef CAS PubMed.
- S.-H. Shin and H. Bayley, J. Am. Chem. Soc., 2005, 127, 10462–10463 CrossRef CAS PubMed.
- S.-H. Shin, T. Luchian, S. Cheley, O. Braha and H. Bayley, Angew. Chem., Int. Ed., 2002, 41, 3707–3709 CrossRef CAS PubMed.
- S.-H. Shin, M. B. Steffensen, T. D. W. Claridge and H. Bayley, Angew. Chem., Int. Ed., 2007, 46, 7412–7416 CrossRef CAS PubMed.
- M. B. Steffensen, D. Rotem and H. Bayley, Nat. Chem., 2014, 6, 603–607 CrossRef CAS PubMed.
- G. S. Pulcu, E. Mikhailova, L.-S. Choi and H. Bayley, Nat. Nanotechnol., 2015, 10, 76–83 CrossRef CAS PubMed.
- H.-C. Wu, Y. Astier, G. Maglia, E. Mikhailova and H. Bayley, J. Am. Chem. Soc., 2007, 129, 16142–16148 CrossRef CAS PubMed.
- J. Clarke, H.-C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS PubMed.
- A. J. Boersma, K. L. Brain and H. Bayley, ACS Nano, 2012, 6, 5304–5308 CrossRef CAS PubMed.
- A. J. Boersma and H. Bayley, Angew. Chem., Int. Ed., 2012, 51, 9606–9609 CrossRef CAS PubMed.
- T. Luchian, S.-H. Shin and H. Bayley, Angew. Chem., Int. Ed., 2003, 42, 1926–1929 CrossRef CAS PubMed.
- Y. Qing, G. S. Pulcu, N. A. W. Bell and H. Bayley, Angew. Chem., Int. Ed., 2018, 57, 1218–1221 CrossRef CAS PubMed.
- Y. Qing, S. A. Ionescu, G. S. Pulcu and H. Bayley, Science, 2018, 361, 908–912 CrossRef CAS PubMed.
- Y. Qing and H. Bayley, J. Am. Chem. Soc., 2021, 143, 18181–18187 CrossRef CAS PubMed.
- S. Loudwig and H. Bayley, J. Am. Chem. Soc., 2006, 128, 12404–12405 CrossRef CAS PubMed.
- Y. Qing, M. D. Liu, D. Hartmann, L. Zhou, W. J. Ramsay and H. Bayley, Angew. Chem., Int. Ed., 2020, 59, 15711–15716 CrossRef CAS PubMed.
- I. Iacovache, S. De Carlo, N. Cirauqui, M. Dal Peraro, F. G. van Der Goot and B. Zuber, Nat. Commun., 2016, 7, 12062 CrossRef CAS PubMed.
- C. Cao, N. Cirauqui, M. J. Marcaida, E. Buglakova, A. Duperrex, A. Radenovic and M. Dal Peraro, Nat. Commun., 2019, 10, 4918 CrossRef PubMed.
- F. L. R. Lucas, K. Sarthak, E. M. Lenting, D. Coltan, N. J. van Der Heide, R. C. A. Versloot, A. Aksimentiev and G. Maglia, ACS Nano, 2021, 15, 9600–9613 CrossRef PubMed.
- S. Borsley and S. L. Cockroft, ACS Nano, 2018, 12, 786–794 CrossRef CAS PubMed.
- M. M. Haugland, S. Borsley, D. F. Cairns-Gibson, A. Elmi and S. L. Cockroft, ACS Nano, 2019, 13, 4101–4110 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- J. Lee and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13768–13773 CrossRef CAS PubMed.
- S. M. Iqbal, D. Akin and R. Bashir, Nat. Nanotechnol., 2007, 2, 243–248 CrossRef CAS PubMed.
- V. Mussi, P. Fanzio, L. Repetto, G. Firpo, S. Stigliani, G. P. Tonini and U. Valbusa, Biosens. Bioelectron., 2011, 29, 125–131 CrossRef CAS PubMed.
- A. R. Hall, A. Scott, D. Rotem, K. K. Mehta, H. Bayley and C. Dekker, Nat. Nanotechnol., 2010, 5, 874–877 CrossRef CAS.
- S. Hernández-Ainsa and U. F. Keyser, Nanoscale, 2014, 6, 14121–14132 RSC.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Met., 2011, 8, 221–229 CrossRef CAS PubMed.
- R. Wei, T. G. Martin, U. Rant and H. Dietz, Angew. Chem., Int. Ed., 2012, 51, 4864–4867 CrossRef CAS PubMed.
- S. Schmid, P. Stömmer, H. Dietz and C. Dekker, Nat. Nanotechnol., 2021, 16, 1244–1250 CrossRef CAS PubMed.
- H. Brinkerhoff, S. W. Kang Albert, J. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef PubMed.
- K. R. Mahendran, A. Niitsu, L. Kong, A. R. Thomson, R. B. Sessions, D. N. Woolfson and H. Bayley, Nat. Chem., 2017, 9, 411–419 CrossRef CAS PubMed.
- C.-W. Ho, V. van Meervelt, K.-C. Tsai, P.-J. de Temmerman, J. Mast and G. Maglia, Sci. Adv., 2015, 1, e1500905 CrossRef PubMed.
- S. Zhang, G. Huang, R. C. A. Versloot, B. M. H. Bruininks, P. C. T. de Souza, S.-J. Marrink and G. Maglia, Nat. Chem., 2021, 13, 1192–1199 CrossRef CAS PubMed.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- K. Shimizu, B. Mijiddorj, M. Usami, I. Mizoguchi, S. Yoshida, S. Akayama, Y. Hamada, A. Ohyama, K. Usui, I. Kawamura and R. Kawano, Nat. Nanotechnol., 2021 DOI:10.1038/s41565-021-01008-w.
- A. Banerjee, E. Mikhailova, S. Cheley, L.-Q. Gu, M. Montoya, Y. Nagaoka, E. Gouaux and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8165–8170 CrossRef CAS PubMed.
- D. L. Beene, D. A. Dougherty and H. A. Lester, Curr. Opin. Neurobiol., 2003, 13, 264–270 CrossRef CAS PubMed.
- S. Howorka, Nat. Nanotechnol., 2017, 12, 619–630 CrossRef CAS PubMed.
- J. Zhang, J. Cao, W. Jia, S. Zhang, S. Yan, Y. Wang, P. Zhang, H.-Y. Chen, W. Li and S. Huang, ACS Sensors, 2021, 6, 2449–2456 CrossRef CAS PubMed.
- R. A. Bull, T. N. Adikari, J. M. Ferguson, J. M. Hammond, I. Stevanovski, A. G. Beukers, Z. Naing, M. Yeang, A. Verich, H. Gamaarachchi, K. W. Kim, F. Luciani, S. Stelzer-Braid, J.-S. Eden, W. D. Rawlinson, S. J. van Hal and I. W. Deveson, Nat. Commun., 2020, 11, 6272 CrossRef CAS PubMed.
- L. E. Bickerton, T. G. Johnson, A. Kerckhoffs and M. J. Langton, Chem. Sci., 2021, 12, 11252–11274 RSC.
- M. Li, A. Rauf, Y. Guo and X. Kang, ACS Sensors, 2019, 4, 2854–2857 CrossRef CAS PubMed.
- K. Willems, V. van Meervelt, C. Wloka and G. Maglia, Phil. Trans. Roy. Soc. B, 2017, 372, 20160230 CrossRef PubMed.
- L. Ma and S. L. Cockroft, ChemBioChem, 2009, 11, 25–34 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |