
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEDOT-based conjugated polymers accessed via C–H direct arylation for efficient photocatalytic hydrogen production†
Zhi-Rong
Tan
a,
Yu-Qin
Xing
a,
Jing-Zhao
Cheng
a,
Guang
Zhang
b,
Zhao-Qi
Shen
a,
Yu-Jie
Zhang
a,
Guangfu
Liao
 c,
Long
Chen
b and
Shi-Yong
Liu
*a
c,
Long
Chen
b and
Shi-Yong
Liu
*a
aJiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry, Department of Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, China. E-mail: chelsy@zju.edu.cn; chelsy@jxust.edu.cn
bDepartment of Chemistry, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University, Tianjin 300072, China
cEngineering Research Center of Nano-Geomaterials of Ministry of Education, China University of Geosciences, Wuhan 430074, China
First published on 14th January 2022
Abstract
3,4-Ethylene dioxythiophene (EDOT), as a monomer of commercial conductive poly(3,4-ethylene dioxythiophene) (PEDOT), has been facilely incorporated into a series of new π-conjugated polymer-based photocatalysts, i.e., BSO2–EDOT, DBT–EDOT, Py–EDOT and DFB–EDOT, through atom-economic C–H direct arylation polymerization (DArP). The photocatalytic hydrogen production (PHP) test shows that donor–acceptor (D–A)-type BSO2–EDOT renders the highest hydrogen evolution rate (HER) among the linear conjugated polymers (CPs) ever reported. A HER up to 0.95 mmol h−1/6 mg under visible light irradiation and an unprecedented apparent quantum yield of 13.6% at 550 nm are successfully achieved. Note that the photocatalytic activities of the C–H/C–Br coupling-derived EDOT-based CPs are superior to those of their counterparts derived from the classical C–Sn/C–Br Stille coupling, demonstrating that EDOT is a promising electron-rich building block which can be facilely integrated into CP-based photocatalysts. Systematic studies reveal that the enhanced water wettability by the integration of polar BSO2 with hydrophilic EDOT, the increased electron-donating ability by O–C p–π conjugation, the improved electron transfer by D–A architecture, broad light harvesting, and the nano-sized colloidal character in a H2O/NMP mixed solvent rendered BSO2–EDOT as one of the best CP photocatalysts toward PHP.
Introduction
Photocatalytic water splitting is considered as one of the most promising technologies for converting solar energy into clean hydrogen fuels.1 Since Fujishima and Honda reported the photocatalytic hydrogen production (PHP) from TiO2-based photoelectrodes,2 many inorganic semiconducting materials have been explored for photocatalysis.3 However, some inherent drawbacks of inorganic photocatalysts still remain, which mainly involve poor response toward visible light and limited tuneability of energy gaps owing to the rigid structures. In recent years, π-conjugated polymers (CPs), as a kind of “soft” photocatalyst that contains only lightweight elements,4 have attracted extensive attention for photocatalysis due to their broad flexibility in molecular structures, synthetic methods, light absorption, and energy levels.5–13 Exploiting high-performance CP-based photocatalysts which can be facilely constructed from commonly used building blocks is still a key task waiting to be tackled.Thiophene and its derivatives are among the most important building blocks in organic electronics, e.g., organic photovoltaics (OPVs) and organic field effect transistors (OFETs),14,15 owing to their appropriate aromatic stabilization energy (ASE) and electron-donating ability. Among them, 3,4-ethylene dioxythiophene (EDOT) is the monomer of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS), one of the most widely utilized conductive polymers16,17 in organic electronics.18–22 As a typical electron-donating unit, EDOT has not yet been used in CP-based photocatalysts. On the other hand, CPs are usually constructed via classical Suzuki or Stille C–M/C–Br couplings (M = B or Sn) which require additional preparation of C–M precursors. In recent years, tin- & boron-free C–H direct arylation (i.e., C–H/C–Br coupling) has emerged as an atom-economic protocol for synthesizing π-conjugated materials.23–27 Our group has synthesized new CPs by C–H direct arylation polymerization (DArP) for PHP applications,28 which, however, still suffer from lower photocatalytic activities than their counterparts derived from the classical Stille coupling owing to the relatively lower polymerization efficiency of DArP.29
In this work, a facile DArP synthetic strategy was employed to synthesize a series of EDOT-based CPs, i.e., BSO2–EDOT, DBT–EDOT, Py–EDOT and DFB–EDOT. PHP performances of these CPs were systematically investigated. Among them, donor–acceptor (D–A)-type BSO2–EDOT achieved a remarkable hydrogen evolution rate (HER) up to 0.95 mmol h−1/6 mg under visible light irradiation. Both the balloon-inflating and water-displacing experiments visually demonstrated that 3 mg of ETSO2–EDOT can produce 100 ml of H2 gas in 3 h. Encouragingly, the PHP activities of CPs from DArP were superior to the analogues synthesized via Stille coupling, demonstrating that DArP is more advantageous to prepare EDOT-based CPs than the classical C–M/C–Br couping. Detailed analysis of the chemical and electronic properties reveals that EDOT has several beneficial effects on CPs: the regioselective α-C–H arylation, enhanced hydrophilicity of the CPs and increased electron-donating ability by C–O polar bonds and p–π conjugation. In contrast to previously exploited polycyclic pyrene7,29 and electron-accepting sulfones,30–34 the electron-donating moieties, e.g., thiophene derivatives, have often been overlooked. In this context, we believe that EDOT here reported will become an attractive and universal electron-donating π unit which can be facilely integrated into CPs via the atom-economic C–H/C–Br coupling, and paves the way for the green synthesis of high-performance polymeric photocatalysts.
Results and discussion
Synthesis, structural characterization, and morphologies
Four new CPs (Scheme 1), i.e. BSO2–EDOT, DBT–EDOT, Py–EDOT and DFB–EDOT, were smoothly synthesized via Pd-catalyzed DArP28,35 from EDOT and different aryl bromides, including 3,7-dibromodibenzothiophene-5,5-dioxide (BTSO2), 3,7-dibromodibenzo[b,d]thiophene (BTS), 1,3,6,8-tetrabromo-pyrene (TBPy) and 1,4-dibromo-2,5-difluorobenzene (DBFB). Note that both the C–H bonds on EDOT can be effectively arylated with various aryl bromides. The ethylene dioxy group at the 3,4-positions of EDOT allows α-C–H bonds to be the exclusive sites toward arylation, which affords regioregular CPs in excellent yields (see the Experimental section). The as-prepared CPs are insoluble in common organic solvents such as MeOH, toluene, dichloromethane and chloroform (Fig. S1†) owing to the lack of alkyl side chains. But impressively, they all can be well dispersed in N-methyl pyrrolidone (NMP) to form stable colloidal solutions (Fig. S2†), which is in line with our previous report on NMP-dispersed colloidal CPs.36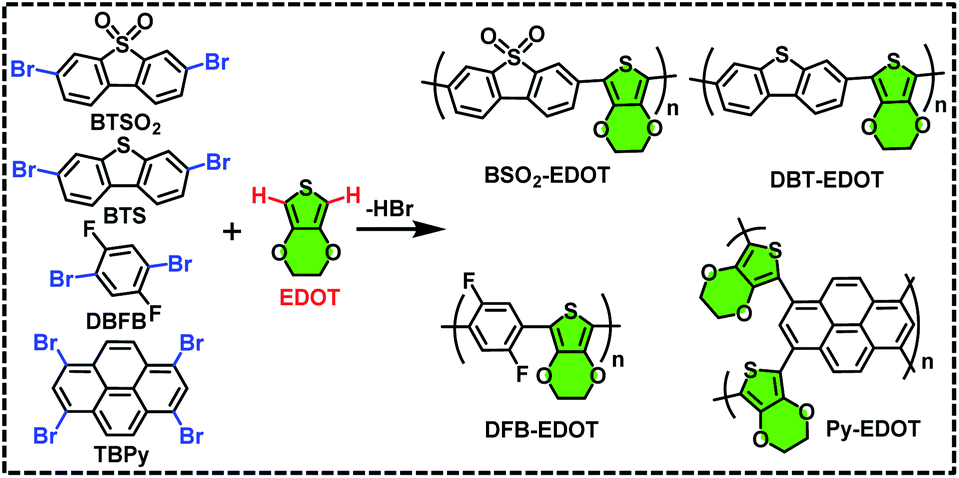 | ||
| Scheme 1 Synthetic routes for BSO2–EDOT, DBT–EDOT, Py–EDOT, and DFB–EDOT via C–H direct arylation polymerization. | ||
The chemical structures of these CPs were confirmed by Fourier transformed infrared (FT-IR) and solid-state 13C NMR spectra (Fig. 1). For FT-IR spectra, all CPs display similar peaks at ∼1091 and ∼1632 cm−l, which are assigned, respectively, to the skeletal vibrations of aromatic rings and the C–O–C stretching vibrations of EDOT units. BSO2–EDOT showed a typical peak at 1154 cm−1, corresponding to the stretching mode of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO groups. DFB–EDOT has a characteristic peak at 1280 cm−1 which corresponds to the vibration of the C–F bonds. All CPs evidently retained the characteristic peak of EDOT at ∼1632 cm−1 (Fig. 1a), suggesting the successful incorporation of EDOT units into CPs. Meanwhile, the peak corresponding to the C–Br characteristic stretching mode of aryl bromides at ∼610 cm−1 disappeared in all CPs (Fig. S3†), implying that the C–Br bonds have been completely transformed into the C–C bonds by reacting with EDOT.
SO groups. DFB–EDOT has a characteristic peak at 1280 cm−1 which corresponds to the vibration of the C–F bonds. All CPs evidently retained the characteristic peak of EDOT at ∼1632 cm−1 (Fig. 1a), suggesting the successful incorporation of EDOT units into CPs. Meanwhile, the peak corresponding to the C–Br characteristic stretching mode of aryl bromides at ∼610 cm−1 disappeared in all CPs (Fig. S3†), implying that the C–Br bonds have been completely transformed into the C–C bonds by reacting with EDOT.
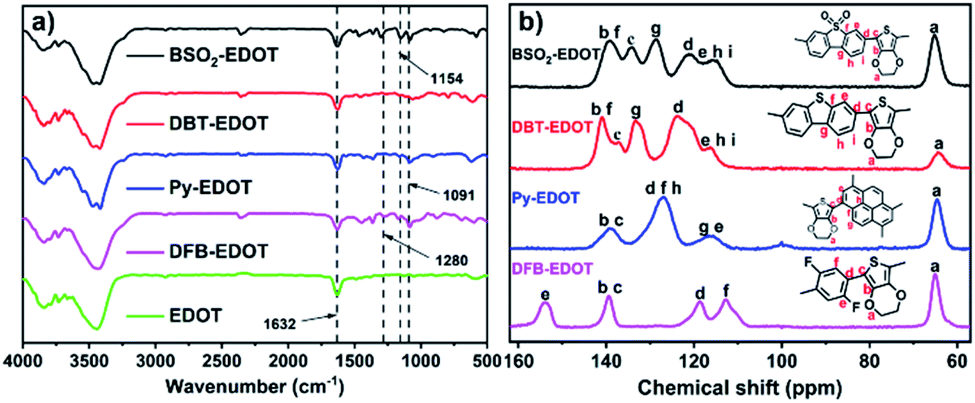 | ||
| Fig. 1 (a) FT-IR spectra and (b) solid-state 13C NMR spectra of the as-prepared CPs. | ||
Fig. 1b shows the solid-state 13C NMR spectra of the CPs. Typically, the signals at 106–118 and 118–154 ppm are attributed to the aromatic 3° and 4° carbons, respectively. DBT–EDOT and BSO2–EDOT have the same number of carbons and very similar chemical shifts, except that BSO2–EDOT exhibited a slight shift to high fields caused by the uneven distribution of electrons arising from the electron-withdrawing sulfone group. For DFB–EDOT, the signal at 154 ppm is attributed to the carbons adjacent to F atoms. Note that all as-prepared CPs exhibit an evident peak at ∼65 ppm corresponding to the methylene carbon signals of EDOT, which further proves that EDOT has been successfully integrated into all CPs via DArP.
The morphology and microstructure of the as-prepared CPs were probed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images (Fig. 2) showed that BSO2–EDOT, DBT–EDOT, and DFB–EDOT exhibited similar layer-stacked morphologies with smooth surfaces, while Py–EDOT exhibited an interlaced rod-like structure.
 | ||
| Fig. 2 SEM images of (a) BSO2–EDOT, (b) DBT–EDOT, (c) Py–EDOT, and (d) DFB–EDOT. | ||
For comparison, two types of samples, i.e., bulk dispersions in MeOH (Fig. S1†) and colloidal dispersions in NMP (Fig. S2†), were subjected to TEM measurements, respectively (Fig. S4†). In contrast to the bulk particles, the NMP-dispersed colloidal CPs exhibit morphologies of semi-transparent ultra-thin sheets with a size up to hundreds of nanometers. Atomic force microscopy (AFM) further verified the sheet-like morphologies of NMP-dispersed colloidal CPs with a thickness of ∼10 nm and width >100 nm (Fig. S5†), implying that NMP exerts an exfoliation effect on CPs.36 The NMP-dispersed colloidal CPs checked by the dynamic light scattering (DLS) method revealed size distributions of hundreds to thousand nanometers (Fig. S6†), which are in the same order of magnitude with the diameters of nanosheets observed by TEM and AFM. Typically, compared with bulk particles, the colloidal nanoparticles have better dispersibility which will facilitate light harvesting and exciton dissociation on the photocatalyst surface.37–41 The NMP-exfoliated nanosheets with enlarged specific surface areas and shortened exciton diffusion pathways should be beneficial to expose surface-active sites and improve charge dissociation, thereby enhancing the photocatalytic activity.
Optical and electrochemical properties
The optical and electrochemical properties of the EDOT-based CPs (Fig. 3) were investigated by UV-vis diffuse reflectance spectroscopy (DRS), cyclic voltammetry (CV), photoluminescence (PL), transient photocurrent response (TPR), and calculations based on density functional theory (DFT). All CPs exhibited a broad light response between 300 and 600 nm (Fig. 3a), which is beneficial to visible light harvesting and charge transfer between frontier molecular orbitals (FMOs), and thus promote photocatalysis. Among the CPs, Py–EDOT has the widest light absorption, followed by DFB–EDOT, BSO2–EDOT, and DBT–EDOT. This absorption trend is directly reflected by the colors of CPs: deeper colors corresponding to better absorbance toward longer wavelengths (insets in Fig. 3a). Correspondingly, the optical bandgaps (Eg) of BSO2–EDOT, DBT–EDOT, Py–EDOT, and DFB–EDOT derived from the Tauc plots (Fig. S7†) are 2.13, 2.29, 2.03, and 2.07 eV, respectively. Note that the π skeletons of DBT–EDOT and BSO2–EDOT are much alike except that BSO2–EDOT has an electron-withdrawing OSO group instead of the S atom. This minor modification on DBT–EDOT, however, endows BSO2–EDOT with a red-shift and broader light absorption (Fig. 3a) owing to the improved D–A (EDOT → BSO2) intramolecular charge transfer (ICT).
 | ||
| Fig. 3 (a) UV-vis DRS (inset: photographs of CPs). (b) Energy band diagrams. (c) Steady state PL spectra. (d) Transient photocurrents of CPs under visible light irradiation. | ||
The FMOs of CPs were estimated by CV measurements (Fig. S8†). The highest occupied molecular orbital (HOMO) energy levels were obtained from the oxidation potential in CV curves, and the lowest unoccupied molecular orbital (LUMO) energy levels were calculated based on Eg and HOMOs: ELUMO = EHOMO + Eg. Fig. 3b shows the corresponding energy band diagrams of the CPs. The LUMO levels of all CPs intrinsically have sufficient driving forces for proton reduction.
To investigate the photogenerated charge recombination, the steady-state PL spectra of CPs were checked (Fig. 3c). Among the CPs, BSO2–EDOT has the lowest PL intensity, followed by Py–EDOT, DBT–EDOT and DFB–EDOT, implying that BSO2–EDOT with a D–A architecture can inhibit radiative recombination more effectively. Furthermore, the photoelectric responses of CPs were evaluated by transient photocurrent response (TPR). The photocurrent–time (I–t) curves under visible light irradiation (Fig. 3d) show that BSO2–EDOT exhibits the highest photocurrent response: ∼1.1 μA cm−2 with several on–off cycles, suggesting that the photogenerated charge carriers can be efficiently transported.
DFT calculations were performed to reveal the distributions of FMOs (Table S1†). The HOMOs of all CPs are mainly distributed on the EDOT unit owing to its intrinsic electron-donating character. Among them, BSO2–EDOT has a more prominent HOMO–LUMO separation compared to other CPs. The LUMO of BSO2–EDOT is mainly localized on the electron-accepting BSO2 unit, which leads to less overlap between the HOMO and LUMO, and thus reduces the probability of electron–hole recombination.
Photocatalytic H2 production and mechanism analysis
The outstanding light absorption, appropriate FMO levels and colloidal character of CPs in NMP encouraged us to investigate their PHP performance, which was carried out under visible light irradiation (λ > 420 nm) in the absence of the Pt co-catalyst, wherein ascorbic acid (AA) was employed as a sacrificial electron donor (SED) and water-soluble NMP worked as a co-solvent to improve the dispersibility of CPs.29,36Fig. 4 shows the PHP performances of CPs and corresponding apparent quantum yield (AQY) values. As shown in Fig. 4a and Table S2,† all CPs (6 mg) dispersed in H2O/NMP/AA mixed solution exhibited promising performance, among which, BSO2–EDOT exhibited the highest HER up to 0.95 mmol h−1, followed by DBT–EDOT (0.39 mmol h−1), DFB–EDOT (0.13 mmol h−1) and Py–EDOT (0.07 mmol h−1), corresponding to the normalized values of 158.4, 65.7, 22.4, and 11.0 mmol h−1 g−1, respectively. Fig. 4b shows that the CPs dispersed in AA/H2O/NMP exhibit 1.5–44 times higher HER than the dispersions in AA/H2O/MeOH, which are mainly ascribed to the exfoliation effect in NMP (Fig. S2, S4 and S5†),37 yielding colloidal CPs that possess more exposed active sites and a shorter migration distance of charge carriers compared to their bulk counterparts in MeOH. Meanwhile, NMP can also serve as a medium to create a favorable environment for the PHP reaction. As an aprotic bipolar solvent having a hydrogen bond-accepting group of carbonyls (CO), NMP exerts a hydrogen bond non-covalent interaction with protons (Fig. S9†), which facilitates the dissociation of the O–H bond, thus decreasing the activation energy of the PHP process.28,29,42
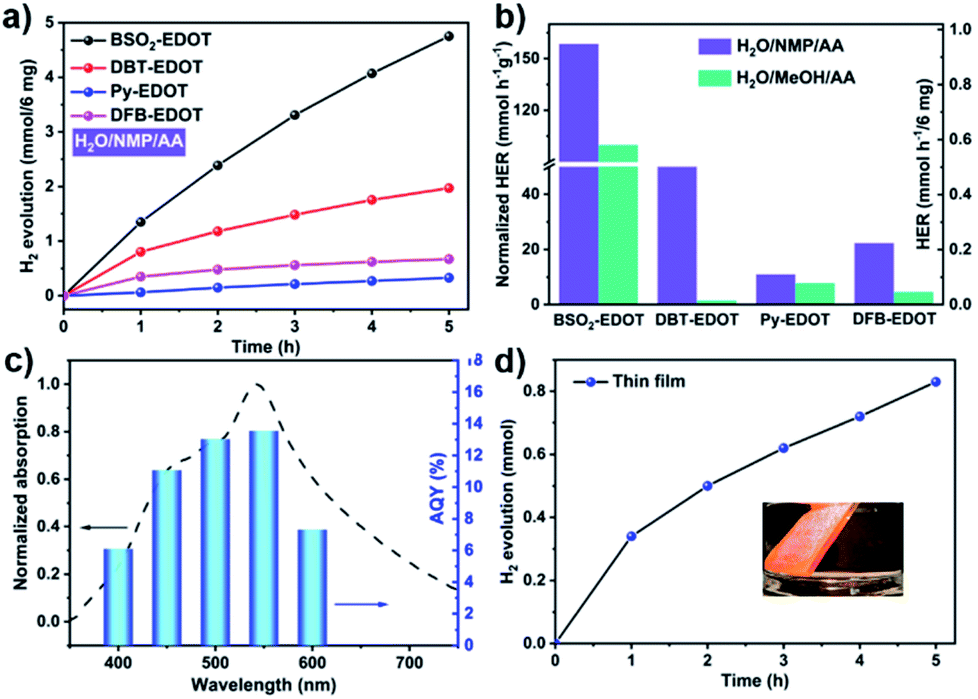 | ||
| Fig. 4 (a) PHP as a function of time for 6 mg CPs under visible light irradiation. (b) Normalized HERs of CPs dispersed in AA/H2O/NMP and AA/H2O/MeOH. (c) AQYs of PHP for BSO2–EDOT at five different incident light wavelengths. (d) PHP from H2O for the BSO2–EDOT film (inset: film photograph) [typical conditions for PHP: 6 mg CPs dispersed in a mixed solution containing 30 ml H2O, 5 g SED, and 3 ml NMP under visible light irradiation at 25 °C]. | ||
Note that BSO2–EDOT achieves the highest HER among the linear CP photocatalysts, and outperforms most of the reported organic semiconductor photocatalysts (Table S3, ESI†). To unravel the reason behind this, first, a comparison study was made between our D–A copolymer BSO2–EDOT and the homopolymers, i.e., PEDOT and PBSO2 (with exclusive donor unit EDOT and acceptor unit BSO2, respectively). BSO2–EDOT showcased a much higher HER than the reported PBSO2,43–45 while PEDOT under the same experimental conditions had no PHP activity (Table S4†). The effectiveness of this D–A structure is also reflected by the comparison between BSO2–EDOT and DBT–EDOT, both of which have closely similar π skeletons (Scheme 1), but BSO2–EDOT exhibited a HER 2.5 times higher than that of D1–D2 type DBT–EDOT (Fig. 4a). These comparisons evidently demonstrate that D–A type BSO2–EDOT with EDOT as the donor and BSO2 as the acceptor is highly favorable for PHP, owing to the presence of push–pull D–A interaction,46–50 improved exciton diffusion, and enhanced electron transfer for proton reduction. Besides, BSO2–EDOT with the D–A ICT effect led to a much broader absorption toward visible light compared to either D1–D2 type DBT–EDOT (Fig. 3a) or A-type PBSO2,43–45 which also accounts for its excellent HER.
Secondly, considering the long-term stability of the NMP-based colloidal solutions of CPs (Fig. S2†), we also investigated their dispersibility in H2O/NMP (5/1, v/v) mixed solvents (Fig. S10†) which were the same as those used for PHP tests. All the as-prepared CPs can be well dispersed in H2O/NMP upon ultrasonication. After three days of standing, BSO2–EDOT can still retain the colloidal state (Fig. S10a†), which may be due to the polar sulfone groups in BSO2–EDOT. We proposed that sulfone (OSO), as a hydrogen bond-acceptor group, can form hydrogen bonds with H2O, and thus have a solvation stabilization effect on colloidal BSO2–EDOT (Fig. S10b†), which will promote the PHP process between the catalyst surface and H2O. Meanwhile, DLS measurement revealed that BSO2–EDOT has the smallest diameter (Fig. S6†), which may increase its total available surface area for proton reduction. To sum it up, the excellent light harvesting, D–A architecture, and the nano-sized colloidal character in H2O/NMP mixed solvent rendered BSO2–EDOT as one of the best photocatalysts toward PHP.
To understand the correlation between PHP and light absorbance, the apparent quantum yields (AQYs) were measured for the BSO2–EDOT/H2O/NMP/AA mixture under various mono-chromatic light irradiations (Fig. 4c). The AQYs of BSO2–EDOT (6 mg) were 6.1%, 11.0%, 13.0%, 13.6%, and 7.3% at 400, 450, 500, 550 and 600 nm, respectively, which matched well with the UV-vis spectrum of the colloidal solution, indicating that the reduction of proton is driven by light irradiation. And a record AQY value of 13.6% at 550 nm was achieved for BSO2–EDOT. Most of the previously reported highest AQYs were below 500 nm wavelengths (Table S3†). However, the light wavelength window between 500–600 nm constitutes the largest fraction of energy in the solar spectrum. BSO2–EDOT exhibits the highest AQY in this region and thus yields excellent HERs.37 Correspondingly, BSO2–EDOT under visible light irradiation had a comparable HER with the one under full-arc irradiation (0.95 vs. 1.06 mmol h−1, Fig. S11†), because the absorbance of BSO2–EDOT is mainly located in the visible light region (Fig. 3a), and thus UV fraction, i.e. λ < 420 nm, has a limited contribution to its light harvesting ability (Fig. 4c).
The stability of BSO2–EDOT for PHP was assessed by cycling experiments lasting for 20 h with each cycle of 5 h (Fig. S12†). More impressively, the PHP activity of BSO2–EDOT after each cycle can be facilely recovered by simple sonication of the colloidal dispersion and replenishment of AA. After the fourth run over a period of 20 h, a HER of 0.66 mmol h−1 can be obtained, which was still higher than that of most of the organic photocatalysts (Table S3†). The control experiments on BSO2–EDOT/NMP/AA in the absence of H2O exhibited no hydrogen evolution, implying that the H2 produced in the above PHP tests did come from H2O, instead of AA or NMP.
The PHP process of BSO2–EDOT was visually demonstrated by both balloon-inflating and water-displacing experiments. In the balloon-inflating experiment, 3 mg colloid-like BSO2–EDOT can even generate 6 × 5 × 5 cm3 hydrogen gas in 5 h from H2O under visible light irradiation (ESI Video 1†), and the actual volume of H2 may be larger than this value because of the tension of the balloon. Concurrent with the balloon-inflating experiment, the water-displacing experiment revealed that 100 ml H2 can be amazingly produced by 3 mg BSO2–EDOT within 3 h, and such a surprisingly high HER can be maintained for at least 3 cycles for 9 h (ESI Video 2†). All the above experiments unambiguously demonstrate that BSO2–EDOT does feature excellent PHP activity. Inspired by the widely used PEDOT-based conductive films,16–22 here, BSO2–EDOT was processed into a film by drop casting the NMP-based colloidal dispersion onto a glass substrate, which still retained the intrinsic activity of the photocatalyst (Fig. 4d). Under visible light irradiation in the presence of H2O and AA, the BSO2–EDOT-based film is initially covered with small bubbles, which then stably overflow from the surface (ESI Video 3†). A respectable HER of 132.2 mmol h−1 m−2 was successfully achieved. Considering that the oxygen generation half-reaction is a four-electron pathway that is relatively difficult to achieve by a single semiconductor, BSO2–EDOT here developed may be used to construct heterojunctions with a second semiconductor51,52 that have a deep-lying valence band, which will provide opportunity for efficient overall water splitting.
C–H direct arylation vs. classical Stille coupling
In parallel with DArP, tin-functionalized EDOT, i.e. 2,5-bis(trimethylstannyl)-3,4-ethylenedioxythiophene (bi-Sn-EDOT), together with BTSO2 or DBT was subjected to C–Sn/C–Br Stille coupling polymerization reactions, yielding St–BSO2–EDOT and St–DBT–EDOT, respectively (Scheme S1† and Fig. 5) for the comparison study. The reaction rates for C–H/C–Br coupling of EDOT were surprisingly fast with obvious colour changes even in ten minutes (Fig. 5a and c) and were comparable to those of the classical Stille couplings (Fig. 5b and d). | ||
| Fig. 5 Color changes of reaction mixtures with time: BSO2–EDOT synthesized via (a) DArP and (b) Stille coupling and DBT–EDOT synthesized via (c) DArP and (d) Stille coupling. | ||
More impressively, the PHP activities of both DArP-derived BSO2–EDOT and DBT–EDOT are superior to those of their Stille-derived counterparts, i.e., 0.95 vs. 0.91 mmol h−1 for BSO2–EDOT and St–BSO2–EDOT, and 0.39 vs. 0.26 mmol h−1 for DBT–EDOT and St–DBT–EDOT (Fig. S13†), which were contrary to our previous results.30 These findings demonstrate that EDOT has high reactivity toward DArP, yielding high-performance CPs with sufficient polymerization degrees. Moreover, DArP without the inclusion of Sn precursors also contributes to the superior PHP compared with the Stille-derived counterparts, because tin residues in photocatalysts might have a negative effect on PHP.53
EDOT vs. thiophene
To gain deeper insights into the effectiveness of EDOT for DArP, BTSO2 with thiophene (Th) or 2,5-bis(trimethylstannyl) thiophene (bi-Sn-Th) was subjected to DArP and Stille polymerization, yielding BSO2–Th and St–BSO2–Th, respectively (Scheme S2†). PHP tests showed that DArP-derived BSO2–Th had a much lower HER than that of the Stille-derived St–BSO2–Th (0.08 vs. 0.38 mmol h−1, Fig. S14†), which was contrary to the above EDOT-based CPs, owing to the relatively low reactivity of thiophene toward DArP, and the undesired reaction on β-C–H bonds of thiophene to form branched side products.Although the chemical structures of thiophene-based BSO2–Th and EDOT-based BSO2–EDOT are closely similar, BSO2–Th exhibited much lower HERs than that of BSO2–EDOT (0.39 vs. 0.95 mmol h−1, Fig. S14†). UV-vis DRS showed that BSO2–EDOT has an obvious red-shift and broader visible light absorption compared with BSO2–Th (Fig. S15†), which means that BSO2–EDOT can capture more photons in the visible region. The red shift in UV-vis DRS of BSO2–EDOT is mainly ascribed to its stronger ICT arising from the stronger electron-donating ability of EDOT. The p–π conjugation between the O–C bonds at the 3,4-positions endows EDOT with a 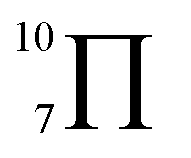 electron-rich system (i.e., 7 atoms share 10 electrons), and thus a stronger electron-donating ability than the thiophene unit (with a
electron-rich system (i.e., 7 atoms share 10 electrons), and thus a stronger electron-donating ability than the thiophene unit (with a 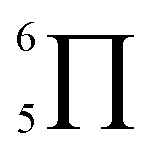 conjugation system).
conjugation system).
Moreover, considering that the surface wettability is essential to aqueous photocatalysis, especially for organic photocatalysts,30,31,41,54–56 here, the water wettability of BSO2–Th and BSO2–EDOT was investigated. As shown in Fig. 6, the water contact angle of BSO2–EDOT (42.3°) was much smaller than that of BSO2–Th (96.7°) owing to the presence of the polar C–O bond41,54,55 from the ethylene-dioxy substituent, which stressed the pivotal role of EDOT to enhance the hydrophilicity of CPs. As a result, a better interfacial water wettability is achieved for EDOT-based CPs (Fig. 6a), facilitating the accessibility of active sites toward proton reduction.
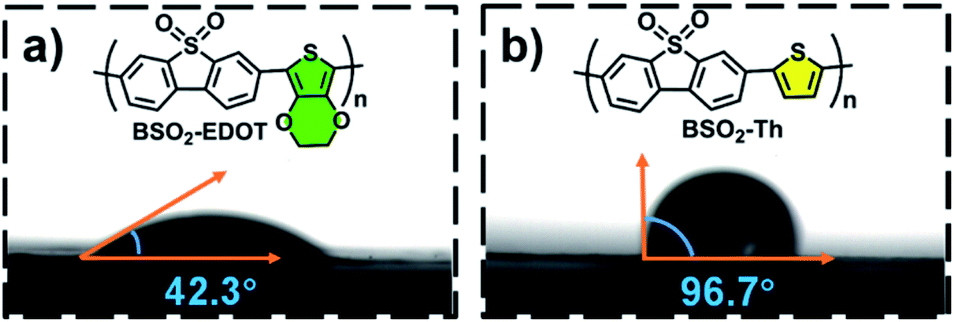 | ||
| Fig. 6 Water contact angles of (a) BSO2–EDOT and (b) BSO2–Th. | ||
Effect of residual Pd on PHP performance
Owing to the insolubility of BSO2–EDOT, the removal of Pd via post-treatments by gel permeation chromatography57,58 or a chelating agent59 was not possible. To study the effect of residual Pd, five batches of BSO2–EDOT were synthesized, respectively, by using 0.5, 1.5, 2.5, 3.5 and 5 mol% Pd2(dba)3 catalyst for DArP. The residual Pd determined by ICP-OES was in the range of 0.16 to 1.24 wt%. The corresponding HERs of these five BSO2–EDOTs showed no linear relationship with the residual Pd contents (Fig. S16†). The lowest HER of BSO2–EDOT via 0.5 mol% Pd2(dba)3 was mainly ascribed to the insufficient degree of DArP by too low amount of Pd-catalysts. With the increase of the Pd2(dba)3 amount, the degree of DArP became normal, while the HERs fluctuated in the range of 0.76 to 1.08 mmol h−1 (Fig. S16†) instead of linear enhancement, indicating that there might be a threshold for the content of Pd.57,58,60 Considering the facts that EDOT-based CPs dispersed in NMP/H2O have a much higher HER than the ones dispersed in MeOH/H2O (Fig. 4b), and BSO2–EDOT outperforms other EDOT-counterparts (Fig. 4a) and BSO2–Th, we proposed that the following three factors, i.e., the combination of polar sulfone groups61 with hydrophilic EDOT (Fig. 6), the presence of NMP–H2O hydrogen bond non-covalent interaction (Fig. S9†),29,42 and the colloidal character of BSO2–EDOT in NMP/H2O,36 should have more positive effects on the best-performing BSO2–EDOT than residual Pd.Conclusions
Four EDOT-based new CPs have been facilely prepared via an atom-economic DArP strategy. PHP tests showed that all these EDOT-based CPs mediate considerable HERs under visible light irradiation without additional Pt co-catalysts, among which BSO2–EDOT with a D–A architecture exhibited the highest HER up to 0.95 mmol h−1, and unprecedented AQY values of 11.1–13.6% within 450–550 nm. The remarkable HERs were both demonstrated instrumentally using standard devices and visually using home-made setups, which showed that 3 mg of colloidal BSO2–EDOT even produced 100 ml of H2 (ESI Videos†). Comprehensive comparative studies62 demonstrated that simple EDOT features many outstanding merits (Fig. S17†), including high stability, excellent reactivity toward DArP with high regioselectivity, water wettability, and enhanced electron-donating ability by O–C p–π conjugation, which make it an attractive electron-donating unit for CP photocatalysts. We envision that more EDOT-based CPs will be synthesized and applicable to all kinds of photocatalytic reactions beyond PHP, such as CO2 reduction, N2 fixation, and organic transformations. Meanwhile, the EDOT-based CPs might be combined with other semiconductors to construct heterojunctions for overall water splitting.Experimental
Materials
TBPy (98%), DBFB (98%), EDOT (98%), thiophene (98%), Cs2CO3 (99.9%), P(o-MeOPh)3 (98%), Pd2(dba)3 (98%), PivOH (99%), AA (98%), sodium ascorbate (SA) (98%), DMF (AR), and NMP (AR) were purchased from Energy Chemical Ltd (Shanghai, China). BTSO2 (98%) was purchased from TCI. BTS (98%) was purchased from Alfa Aesar. bi-Sn-EDOT (98%) and bi-Sn-Th (98%) were purchased from SunaTech Inc. (Suzhou, China) and Derthon Co., Ltd (Shenzhen, China), respectively. MeOH (AR) and toluene (AR) were purchased from XiLong Scientific (Shanghai, China).Typical procedure for the synthesis of CPs via DArP28,35
A Schlenk tube was charged with the monomers, P(o-MeOPh)3, Cs2CO3, pivalic acid, and Pd2(dba)3. The mixed solid in the tube was purged by repetitions of vacuum and argon filling (×3). Then anhydrous toluene was added into the tube via a syringe. The reaction mixture was put through freeze–vacuum–thaw cycles three times to remove dissolved gases, and then vigorously stirred at 100 °C for 48 h under an argon atmosphere. After cooling to room temperature, the mixture was poured into CH2Cl2. The precipitate was collected by filtration and washed with CH2Cl2, MeOH, and distilled water. Further purification of the polymers was carried out by soaking with MeOH and CH2Cl2, respectively, and the final powder products were obtained by drying in a vacuum oven at 70 °C for 12 h.Typical procedure for the synthesis of CPs via Stille coupling
A Schlenk tube was charged with the monomers, Pd2(dba)3 and P(o-MeOPh)3. The mixed solid in the tube was purged by repetitions of vacuum and argon filling (×3). Then anhydrous toluene together with a small amount of anhydrous DMF were added into the tube via a syringe, respectively. The reaction mixture was put through freeze–vacuum–thaw cycles three times to remove dissolved gases, and then vigorously stirred at 120 °C for 48 h under an argon atmosphere. After cooling to room temperature, the mixture was poured into CH2Cl2. The precipitate was collected by filtration and washed with CH2Cl2, MeOH, saturated NaF solution, distilled water, and MeOH. Further purification of the polymers was carried out by soaking with CH2Cl2, MeOH, saturated NaF solution, distilled water, and MeOH, respectively, and the final powder products were obtained by drying in a vacuum oven at 70 °C for 12 h.Synthesis of BSO2–EDOT via DArP
BTSO2 (0.535 mmol, 200.0 mg), EDOT (1 equiv., 0.535 mmol, 76.0 mg), Cs2CO3 (2 equiv., 1.07 mmol, 348.6 mg), PivOH (30 mol%, 16.4 mg), Pd2(dba)3 (1.5 mol%, 7.3 mg), P(o-MeOPh)3 (3 mol%, 5.7 mg), and 7 ml anhydrous toluene were used to synthesize BSO2–EDOT via DArP. The product was obtained as an orange-red powder (176.5 mg, 93.2%). Elem. Anal for (C18H10O4S2)n (%): C 61.00; H 2.84; S 18.09. Found: C 58.35; H 3.22; S 19.28.Synthesis of DBT–EDOT via DArP
BTS (0.584 mmol, 200.0 mg), EDOT (1 equiv., 0.584 mmol, 83.2 mg), Cs2CO3 (2 equiv., 1.168 mmol, 381.0 mg), PivOH (30 mol%, 17.9 mg), Pd2(dba)3 (1.5 mol%, 10.7 mg), P(o-MeOPh)3 (3 mol%, 8.2 mg), and 7 ml anhydrous toluene were used to synthesize DBT–EDOT via DArP. The product was obtained as a yellow powder (172.5 mg, 91.4%). Elem. Anal for (C18H10O2S2)n (%): C 67.06; H 3.13; S 19.89. Found: C 63.16; H 3.07; S 20.05.Synthesis of Py–EDOT via DArP
TBPy (0.386 mmol, 200.0 mg), EDOT (2 equiv., 0.772 mmol, 109.8 mg), Cs2CO3 (2 equiv., 1.544 mmol, 503.1 mg), PivOH (30 mol%, 23.6 mg), Pd2(dba)3 (1.5 mol%, 14.1 mg), P(o-MeOPh)3 (3 mol%, 10.9 mg), and 7 ml anhydrous toluene were used to synthesize Py–EDOT via DArP. The product was obtained as a dark-red powder (174.3 mg, 94.3%). Elem. Anal for (C28H16O4S2)n (%): C 69.98; H 3.36; S 13.34. Found: C 68.43; H 3.06; S 12.56.Synthesis of DFB–EDOT via DArP
DBFB (0.736 mmol, 200.0 mg), EDOT (1 equiv., 0.736 mmol, 104.6 mg), Cs2CO3 (2 equiv., 1.472 mmol, 479.4 mg), PivOH (30 mol%, 22.5 mg), Pd2(dba)3 (1.5 mol%, 13.5 mg), P(o-MeOPh)3 (3 mol%, 10.4 mg), and 7 ml anhydrous toluene were used to synthesize DFB–EDOT via DArP. The product was obtained as a red powder (168.1 mg, 90.6%). Elem. Anal for (C12H6F2O2S)n (%): C 57.14; H 2.40; S 12.71. Found: C 54.08; H 2.29; S 12.06.Synthesis of St–BSO2–EDOT via Stille coupling
BTSO2 (0.535 mmol, 200.0 mg), bi-Sn-EDOT (1 equiv., 0.535 mmol, 250.3 mg), Pd2(dba)3 (2 mol%, 9.8 mg), P(o-MeOPh)3 (4 mol%, 7.5 mg), 7 ml anhydrous toluene, and 0.1 ml anhydrous DMF were used to synthesize St–BSO2–EDOT via Stille coupling. The product was obtained as an orange-red powder (148.9 mg, 78.5%). Elem. Anal for (C18H10O4S2)n (%): C 61.00; H 2.84; S 18.09. Found: C 58.26; H 2.64; S 17.52.Synthesis of St–DBT–EDOT via Stille coupling
BTS (0.585 mmol, 200.0 mg), bi-Sn-EDOT (1 equiv., 0.585 mmol, 273.7 mg), Pd2(dba)3 (2 mol%, 10.7 mg), P(o-MeOPh)3 (4 mol%, 8.2 mg), 7 ml anhydrous toluene, and 0.1 ml anhydrous DMF were used to synthesize St–DBT–EDOT via Stille coupling. The product was obtained as a yellow powder (144.0 mg, 76.3%). Elem. Anal for (C18H10O2S2)n (%): C 67.06; H 3.13; S 19.89. Found: C 64.63; H 3.01; S 18.45.Synthesis of BSO2–Th via DArP
BTSO2 (0.535 mmol, 200.0 mg), thiophene (1 equiv., 0.535 mmol, 45.0 mg), Pd2(dba)3 (2 mol%, 9.8 mg), P(o-MeOPh)3 (4 mol%, 7.5 mg), 7 ml anhydrous toluene, and anhydrous 0.1 ml DMF were used to synthesize BSO2–Th via DArP. The product was obtained as a yellow green powder (90.6 mg, 57.2%). Elem. Anal for (C16H8O2S2)n (%): C 64.85; H 2.72; S 21.64. Found: C 60.82; H 2.34; S 19.76.Synthesis of St–BSO2–Th via Stille coupling
BTSO2 (0.535 mmol, 200.0 mg), bi-Sn-Th (1 equiv., 0.535 mmol, 219.2 mg), Pd2(dba)3 (2 mol%, 9.8 mg), P(o-MeOPh)3 (4 mol%, 7.5 mg), 7 ml anhydrous toluene, and 0.1 ml anhydrous DMF were used to synthesize St–BSO2–EDOT via Stille coupling. The product was obtained as an orange-red powder (144.6 mg, 91.2%). Elem. Anal for (C16H8O2S2)n (%): C 64.85; H 2.72; S 21.64. Found: C 63.01; H 2.58; S 20.52.Hydrogen evolution tests
The typical PHP test was performed in a photocatalytic online analysis system (LabSolar-III AG, Beijing Perfect Light) linked with a gas chromatograph (GC9790, FuLi). First, the as-prepared photocatalysts (6 mg) were ultrasonically dispersed in 6 ml NMP for 30 min, and then the suspension was transferred into a mixed aqueous solution containing 30 ml H2O, 2.5 g AA, and 2.5 g SA. SA was used to adjust the pH to 4.0, a similar value to the pKa of AA.30 The mixture was degassed to remove the dissolved air before irradiation using an oil pump. Then, the reactor was irradiated by using a 300 W Xe lamp (Beijing Perfect Light, PLS-SXE300) equipped with a 420 nm cut-off filter; the reaction temperature was fixed at 25 °C using a flow of cooling water. The amount of hydrogen produced was analyzed by using a gas chromatograph equipped with a thermal conductive detector (TCD) using argon as the carrier gas.Balloon-inflating and water-displacing experiments were carried out with a 25 ml Schlenk tube. The Schlenk tube contained 3 mg photocatalysts and 3 ml NMP, which were ultrasonically dispersed for 30 min, and then the mixed aqueous solution containing 15 ml H2O, 1.3 g AA and 1.3 g SA was added. The Schlenk tube was put through freeze–vacuum–thaw cycles three times to remove dissolved gases and lastly filled with 1 atm pressure of argon, and then stirred for the PHP test under visible light irradiation. The detailed set-up can be found in ESI Videos 1 and 2.†
Hydrogen evolution test of the BSO2–EDOT film
BSO2–EDOT (3 mg) powder was added into NMP (1 ml) and ultrasonically dispersed for 30 min. The obtained dispersion was drop cast onto glass and dried at 75 °C for 3 h. Then, the yielded BSO2–EDOT-based film was immersed in a mixed aqueous solution containing 30 ml H2O, 2.5 g AA, and 2.5 g SA for the PHP test under visible light irradiation (ESI Video 3†). After that, the film can be dried and recycled.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or ESI.†Author contributions
Z.-R. T. performed and analyzed the experiments and contributed to the writing of the original draft. Y.-Q. X. J.-Z. C., Z.-Q. S. and Y. J. Z. performed part of the experiments. Z.-R. T., G. Z., G. L., L. C. and S.-Y. L. analyzed the data and prepared the manuscript. S.-Y. L. supervised and conceptualized the project, funding acquisition, provided guidance during all stages, and contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (Nos 21374075 and 22169009), Jiangxi Provincial Natural Science Foundation (No. 20212ACB204007) and the Jiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry (20212BCD42018) are acknowledged for financial support. The authors thank Prof. Ai-Guo Zhong for DFT calculations.Notes and references
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photon., 2012, 6, 511–518 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2020, 6, 168–190 CrossRef.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- Y. O. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. W. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS.
- Y. Li, Y. Wang, C. L. Dong, Y. C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huang, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633–3643 RSC.
- X. Li, P. M. Maffettone, Y. Che, T. Liu, L. Chen and A. I. Cooper, Chem. Sci., 2021, 12, 10742–10754 RSC.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2020, 35, 115–130 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Muller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- H. Shi, C. C. Liu, Q. L. Jiang and J. K. Xu, Adv. Electron. Mater., 2015, 1, 1500017 CrossRef.
- L. V. Kayser and D. J. Lipomi, Adv. Mater., 2019, 31, 1806133 CrossRef PubMed.
- Y. Y. Jiang, T. F. Liu and Y. H. Zhou, Adv. Funct. Mater., 2020, 30, 2006213 CrossRef CAS.
- Y. Liang, A. Offenhausser, S. Ingebrandt and D. Mayer, Adv. Healthcare Mater., 2021, 10, 2100061 CrossRef CAS PubMed.
- Y.-Q. Zheng, Y. Liu, D. Zhong, S. Nikzad, S. Liu, Z. Yu, D. Liu, H.-C. Wu, C. Zhu, J. Li, H. Tran, J. B.-H. Tok and Z. Bao, Science, 2021, 373, 88–94 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- S.-Y. Liu, M.-M. Shi, J.-C. Huang, Z.-N. Jin, X.-L. Hu, J.-Y. Pan, H.-Y. Li, A. K. Y. Jen and H.-Z. Chen, J. Mater. Chem. A, 2013, 1, 2795–2805 RSC.
- J. R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupre and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- N. S. Gobalasingham and B. C. Thompson, Prog. Polym. Sci., 2018, 83, 135–201 CrossRef CAS.
- T. Bura, S. Beaupre, M. A. Legare, J. Quinn, E. Rochette, J. T. Blaskovits, F. G. Fontaine, A. Pron, Y. Li and M. Leclerc, Chem. Sci., 2017, 8, 3913–3925 RSC.
- W.-Y. Huang, Z.-Q. Shen, J.-Z. Cheng, L.-L. Liu, K. Yang, X.-R. Chen, H.-R. Wen and S.-Y. Liu, J. Mater. Chem. A, 2019, 7, 24222–24230 RSC.
- J.-Z. Cheng, L.-L. Liu, G. Liao, Z.-Q. Shen, Z.-R. Tan, Y.-Q. Xing, X.-X. Li, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J. X. Jiang, Adv. Mater., 2021, 33, 2008498 CrossRef CAS PubMed.
- C. M. Aitchison, M. Sachs, M. A. Little, L. Wilbraham, N. J. Brownbill, C. M. Kane, F. Blanc, M. A. Zwijnenburg, J. R. Durrant, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 8744–8756 RSC.
- C. Han, P. Dong, H. Tang, P. Zheng, C. Zhang, F. Wang, F. Huang and J. X. Jiang, Chem. Sci., 2021, 12, 1796–1802 RSC.
- S.-Y. Liu, D.-G. Wang, A.-G. Zhong and H.-R. Wen, Org. Chem. Front., 2018, 5, 653–661 RSC.
- J.-Z. Cheng, Z.-R. Tan, Y.-Q. Xing, Z.-Q. Shen, Y.-J. Zhang, L.-L. Liu, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2021, 9, 5787–5795 RSC.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- C. H. Dai, Y. T. Pan and B. Liu, Adv. Energy Mater., 2020, 10, 2002474 CrossRef CAS.
- C. M. Aitchison and R. S. Sprick, Nanoscale, 2021, 13, 634–646 RSC.
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Chem. Sci., 2019, 10, 3796–3801 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2021, 2105007 CrossRef PubMed.
- W.-C. Lin, J. Jayakumar, C.-L. Chang, L.-Y. Ting, M. H. Elsayed, M. Abdellah, K. Zheng, A. M. Elewa, Y.-T. Lin, J.-J. Liu, W.-S. Wang, C.-Y. Lu and H.-H. Chou, Appl. Catal., B, 2021, 298, 120577 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- G. Shu, Y. Li, Z. Wang, J.-X. Jiang and F. Wang, Appl. Catal., B, 2020, 261, 118230 CrossRef CAS.
- Z. A. Lan, W. Ren, X. Chen, Y. F. Zhang and X. C. Wang, Appl. Catal., B, 2019, 245, 596–603 CrossRef CAS.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- Z. Q. Sheng, Y. Q. Xing, Y. Chen, G. Zhang, S. Y. Liu and L. Chen, Beilstein J. Nanotechnol., 2021, 12, 607–623 CrossRef CAS PubMed.
- J. Zhao, J. Ren, G. Zhang, Z. Zhao, S. Liu, W. Zhang and L. Chen, Chem.–Eur. J., 2021, 27, 10781–10797 CrossRef CAS PubMed.
- Q. Xie, Y. Yang, W. Zhang, Z. Gao, X. Li, J. Tang, C. Pan and G. Yu, Chem. Sci., 2021, 12, 5631–5637 RSC.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef CAS PubMed.
- G. Liao, C. Li, S.-Y. Liu, B. Fang and H. Yang, Trends Chem., 2022, 4(2), 111–127 CrossRef.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- Y. Bai, Z. Hu, J. X. Jiang and F. Huang, Chem.–Asian J., 2020, 15, 1780–1790 CrossRef CAS PubMed.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Angew. Chem., Int. Ed., 2020, 59, 6007–6014 CrossRef CAS PubMed.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- J. Kosco and I. McCulloch, ACS Energy Lett., 2018, 3, 2846–2850 CrossRef CAS.
- K. T. Nielsen, K. Bechgaard and F. C. Krebs, Macromolecules, 2005, 38, 658–659 CrossRef CAS.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francas, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- Comparisons including BSO2–EDOT vs. PEDOT & PBSO2, BSO2–EDOT vs. DBT–EDOT, DArP vs. Stille coupling, and BSO2–EDOT vs. BSO2–Th, see Fig. 5 and 6, Tables S3 and S4 and Fig. S13 and S15.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05784g |
| This journal is © The Royal Society of Chemistry 2022 |