
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From a nanoparticular solid-state material to molecular organo-f-element-polyarsenides†‡
Niklas
Reinfandt§
,
Adrian
Hauser§
 ,
Luca
Münzfeld
and
Peter W.
Roesky
*
,
Luca
Münzfeld
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, D-76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 4th February 2022
Abstract
A convenient pathway to new molecular organo-lanthanide-polyarsenides in general and to a f-element complex with the largest polyarsenide ligand in detail is reported. For this purpose, the activation of the solid state material As0nano (nanoscale gray arsenic) by the multi electron reducing agents [K(18-crown-6)][(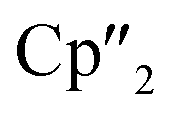 Ln+II)2(μ-η6:η6-C6H6)] (Ln = La, Ce, Cp′′ = 1,3-bis(trimethylsilyl)cyclopentadienyl anion) and [K(18-crown-6)]2[(
Ln+II)2(μ-η6:η6-C6H6)] (Ln = La, Ce, Cp′′ = 1,3-bis(trimethylsilyl)cyclopentadienyl anion) and [K(18-crown-6)]2[( Ln+II)2(μ-η6:η6-C6H6)] (Ln = Ce, Nd) is shown. These non-classical divalent lanthanide compounds were used as three and four electron reducing agents where the product formation can be directed by variation of the applied reactant. The obtained Zintl anions As33−, As73−, and As144− were previously not accessible in molecular 4f-element chemistry. Additionally, the corresponding compounds with As144−-moieties represent the largest organo-lanthanide-polyarsenides known to date.
Ln+II)2(μ-η6:η6-C6H6)] (Ln = Ce, Nd) is shown. These non-classical divalent lanthanide compounds were used as three and four electron reducing agents where the product formation can be directed by variation of the applied reactant. The obtained Zintl anions As33−, As73−, and As144− were previously not accessible in molecular 4f-element chemistry. Additionally, the corresponding compounds with As144−-moieties represent the largest organo-lanthanide-polyarsenides known to date.
Introduction
Zintl anion and Zintl cluster chemistry is a traditional topic in inorganic chemistry.1 However, recent spectacular new findings did not only result in new structural motifs but also shed some light into the formation of nanomaterials.2–4 For group 15 Zintl ions, the research on molecular coordination compounds is heavily focused on phosphorus and transition metals or main group elements.5–8 This can be ascribed to the availability of the molecular and soluble phosphorus allotrope P4 (white phosphorus), which allows the synthesis of often unpredictable and diverse phosphorus-containing compounds.9 Examples for the activation of P4 are the formation of P42− and other rings such as P5− and P6.5–7,10 Also the opening of one edge of the P4 tetrahedron to form butterfly-type structures is known.11–15 While most of these reactions were reported with main group and transition metals, f-elements were less investigated and focused more on the lighter homologue nitrogen.16–19 In contrast, the heavier molecular congener As4 is inconvenient to synthesise, highly prone to decompose into its thermodynamically stable modification (gray arsenic) and extremely photosensitive.20 Therefore, only freshly prepared As4 solutions can be used for synthetic purposes.21 Unfortunately, even the latter suffer from rapid decomposition to gray arsenic and – as a consequence – a non-quantifiable As4 concentration. This is a major drawback for the synthesis and reproduction of unexpected polyarsenides. To circumvent these drawbacks, materials that can capture, stabilize, and release intact As4 tetrahedra were extensively investigated and designed.22–24 Scheer and co-workers developed a storage system for white phosphorus and yellow arsenic. This system uses porous activated carbon, in which intact As4 molecules were reversibly captured and released for the synthesis of transition metal complexes.25 With the synthesis of arsenic nanoparticles by reduction of AsI3 with lithium naphthalenide, we presented a different approach for this problem only recently, which allows the stoichiometric usage of an elemental As source.26 Starting from the nanoscale solid-state material (As0nano, d = 7.2 ± 1.8 nm) a variety of new f-element arsenic compounds as well as aluminium arsenic clusters have been successfully synthesised so far.26–28In general, molecular arsenic Zintl ions of the lanthanides were only reported for Sm. In all these reactions the SET pathway from Sm(II) to Sm(III) was applied for the synthesis.29 Considering potential applications (lanthanide pnictide compounds have been discussed as potential thermoelectrical devices, transparent electrical contacts or solar cells)30–32 and in terms of possible optical and magnetic properties, this restriction to samarium compounds within 4f-element chemistry is a strong limitation.
Herein, we report a new pathway towards organo-f-element arsenic Zintl ions beyond Sm by combining the solid-state material As0nano with the high redox potentials of various molecular non-classical divalent lanthanide compounds. For this purpose, the non-classical divalent lanthanide three electron reducing agents ([K(18-crown-6)][(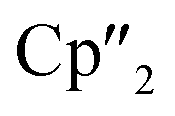 Ln)2(μ-η6:η6-C6H6)], Ln = La, Ce) (A) and four electron reducing agents ([K(18-crown-6)]2[(
Ln)2(μ-η6:η6-C6H6)], Ln = La, Ce) (A) and four electron reducing agents ([K(18-crown-6)]2[(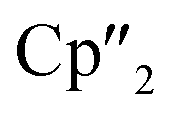 Ln)2(μ-η6:η6-C6H6)], Ln = Ce, Nd) (B) were employed (Fig. 1).33–35
Ln)2(μ-η6:η6-C6H6)], Ln = Ce, Nd) (B) were employed (Fig. 1).33–35
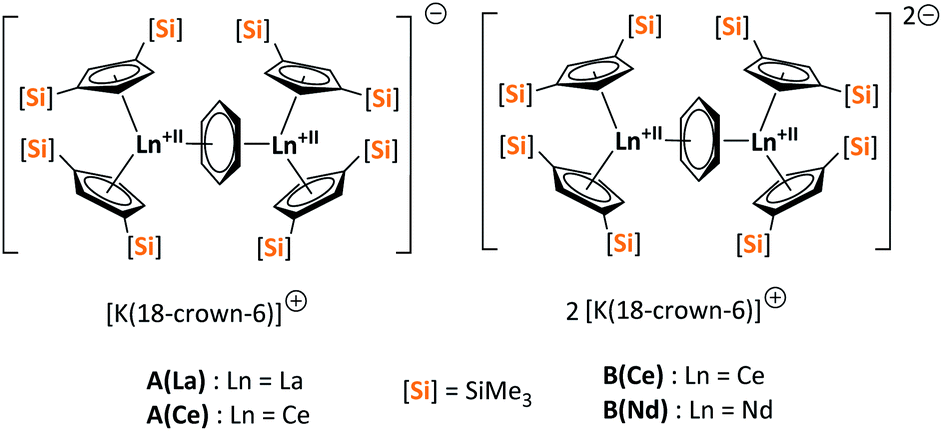 | ||
| Fig. 1 Three- (A(La), A(Ce)) and four- (B(Ce), B(Nd)) electron reducing agents, featuring non-classical divalent lanthanides ([Si] = SiMe3).33,34 | ||
Results and discussion
Reduction of As0nano with the three electron reducing agents A(La) and A(Ce) resulted in the organo-lanthanide-polyarsenides [{K(18-crown-6)}(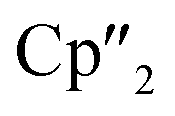 Ln)2(μ3-η2:η2:η2-As7)] (Scheme 1, Ln = La (1), Ce (2)) in yields of 33% for 1 and 28% for 2. Within these compounds an As73− Zintl anion with a nortricyclene structure forms the core of compounds 1 (Fig. S6‡) and 2 (Fig. 2). For charge balance, [K(18-crown-6)]+ and two
Ln)2(μ3-η2:η2:η2-As7)] (Scheme 1, Ln = La (1), Ce (2)) in yields of 33% for 1 and 28% for 2. Within these compounds an As73− Zintl anion with a nortricyclene structure forms the core of compounds 1 (Fig. S6‡) and 2 (Fig. 2). For charge balance, [K(18-crown-6)]+ and two 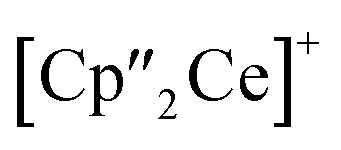 fragments are coordinated by the equatorial arsenic atoms As2, As3 and As4 of the polyanion, each of which is bonded to two neighboring As atoms and thus negatively charged according to the 8-N rule. While a norbornadiene like As73− structure is known for Sm compounds, 1 and 2 featuring a nortricyclic structural motif in molecular f-element chemistry for the first time.36 A comparison of the 3-electron reducing agents A(Ce) and A(La) with the 1-electron reducing agent
fragments are coordinated by the equatorial arsenic atoms As2, As3 and As4 of the polyanion, each of which is bonded to two neighboring As atoms and thus negatively charged according to the 8-N rule. While a norbornadiene like As73− structure is known for Sm compounds, 1 and 2 featuring a nortricyclic structural motif in molecular f-element chemistry for the first time.36 A comparison of the 3-electron reducing agents A(Ce) and A(La) with the 1-electron reducing agent 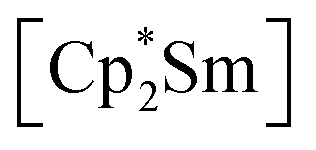 at room temperature shows fundamental differences. While treatment of As0nano with
at room temperature shows fundamental differences. While treatment of As0nano with 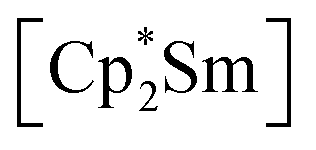 leads to an As22− moiety compounds 1 and 2 feature the As73− scaffold.26 Furthermore, while the product formation in the presence of
leads to an As22− moiety compounds 1 and 2 feature the As73− scaffold.26 Furthermore, while the product formation in the presence of 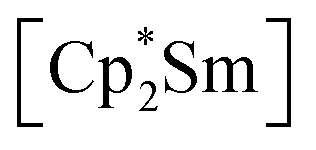 is temperature dependent,26 the formation of 1 and 2 occurs also at elevated temperature.
is temperature dependent,26 the formation of 1 and 2 occurs also at elevated temperature.
 | ||
| Scheme 1 Synthesis of compounds 1 and 2. | ||
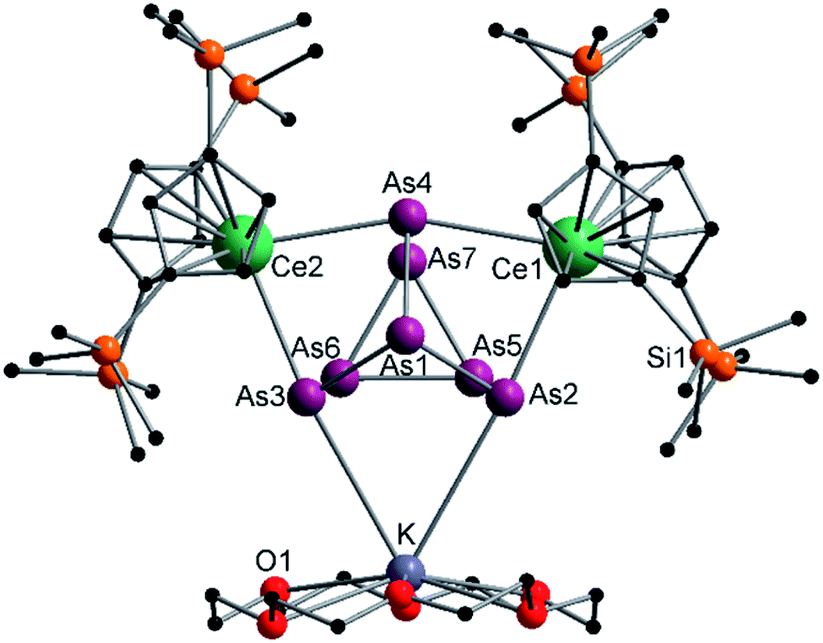 | ||
| Fig. 2 Molecular structure of 2 in the solid state. Solvent molecules, and hydrogen atoms are omitted for clarity. For bond lengths and angles see ESI (Fig. S9‡). | ||
It should be kept in mind that the solid-state material As0nano is gray arsenic, which features a polymeric structure. Thus, the selective formation of the As73− cages is a complicated process, which requires several steps of bond breaking and bond formation.
Since compound 1 and 2 exhibit the same structural motif, only the molecular solid-state structure of compound 2 is discussed in detail here. The As–As bond lengths within the As73− unit are longest on average for the uncharged basal arsenic atoms As5, As6 and As7 (As5–As6 2.4654(5), As5–As7 2.4717(5), As6–As7 2.4805(5) Å). Compared to these, the bonds towards the charged arsenic atoms tend to be shortened, with those towards the apical and uncharged As1 being in average slightly longer than those towards the As5–As6–As7 plane (As1–As2 2.4101(5), As1–As3 2.4129(5), As1–As4 2.4777(5) Å vs. As2–As5 2.3877(5), As3–As6 2.3845(5), As4–As7 2.4010(5) Å). In contrast to symmetrically coordinated As73− units, e.g., [(Li{dme})3As7] or K3As7,37,38 there are slight deviations in the bonding parameters from a symmetrical setup in the As73− core of compounds 1 and 2 due to the asymmetrical coordination with the various cationic fragments. The observed Ce–As bond lengths (Ce1–As2 3.0460(4), Ce1–As4 3.1053(4), Ce2–As3 3.0412(4), Ce2–As4 3.0897(4) Å) match with previous observations.33 The K–As distances are relatively long at ∼3.76 Å, which suggests weak coordination by the [K(18-crown-6)]+ fragment. Nevertheless, they are in a range observed for other potassium polyarsenides (e.g. 3.19–3.84 Å in [K(2.2.2-cryptand)]2(KAs7)).39,40
After the successful application of the 3-electron reducing agents A(La) and A(Ce) in the activation of nanoscale arsenic, we felt challenged to treat As0nano with 4-electron reducing agents in the reduction process. For comparison, we reacted the closely related compounds B(Ce) and B(Nd) with As0nano. This allows to maintain the same steric influence of the reducing agents to have an unobstructed view of the influence of the different reduction processes (three vs. four electrons). The reaction of both with As0nano at room temperature resulted in a mixture of different products in the subsequent crystallization. However, by changing the reaction conditions upon usage of B(Nd) (excess of As0nano and prolonged heating) it was possible to isolate [{K(18-crown-6)}2( Nd)2(μ4-η2:η2:η2:η2-As14)] (3) exclusively but in low yields of 7% (Scheme 2).
Nd)2(μ4-η2:η2:η2:η2-As14)] (3) exclusively but in low yields of 7% (Scheme 2).
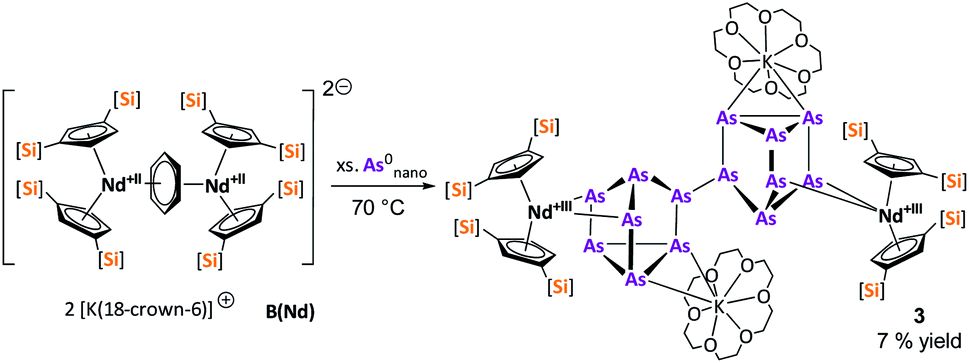 | ||
| Scheme 2 Clean isolation of compound 3. | ||
Compound 3 represents the largest organo-lanthanide-polyarsenide known to date. The central As144− unit formally consists of two covalently linked As72− units (Fig. 3).
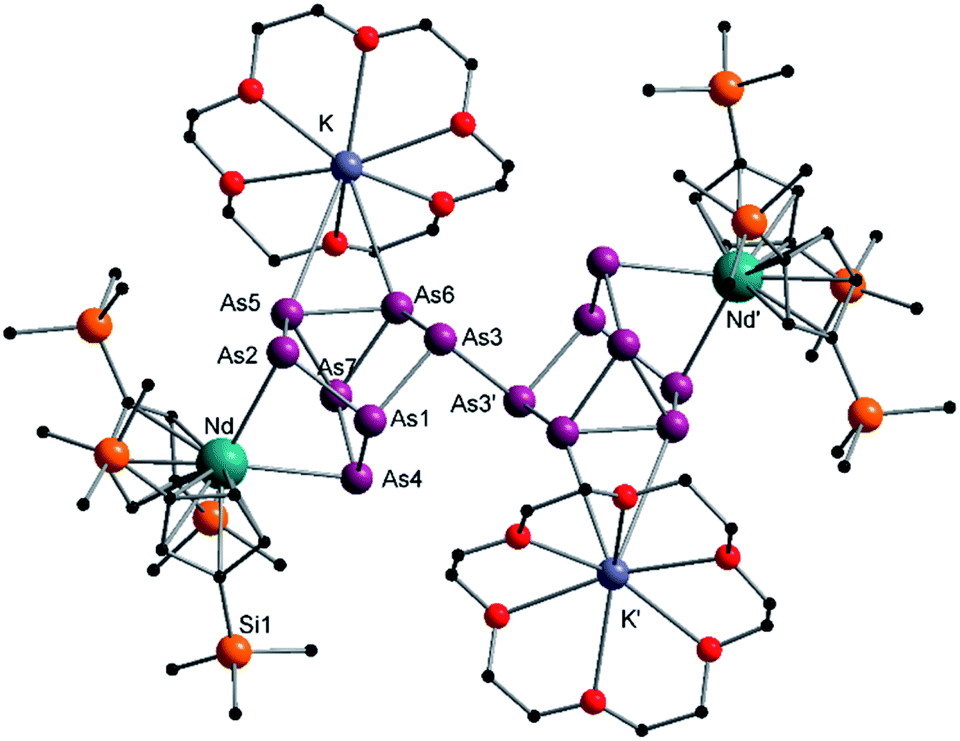 | ||
| Fig. 3 Molecular structure of 3 in the solid state. Solvent molecules and hydrogen atoms are omitted for clarity. Only one part of the positional disordered [K(18-crown-6)]+ is depicted. For both parts, bond lengths and angles see ESI (Fig. S10‡). | ||
Considering that As72− radicals are a known species,41 the formation of 3 can be explained via the radical recombination of two As72− units. The newly formed As–As bond, linking the two As72− units, of 2.4522(14) Å is in the range of a single bond (e.g. ca. 2.44 Å in carbene-stabilized diarene)42 and comparable to the bonds between the basal arsenic atoms As5, As6, and As7 (2.4516(10)–2.4580(11) Å). Analogous to the previously obtained As73− units, the As–As bonds involving the charged equatorial arsenic atoms As2 and As4 are the shortest. However, in contrast to the nortricyclene structures of 1 and 2, the [K(18-crown-6)]+ fragments are not coordinated by the equatorial and charged As atoms but by the basal ones, presumably for steric reasons. Additionally, they are disordered over two positions each (see Fig. S8‡). The  cations are coordinated by the two respective charged equatorial arsenic atoms (Nd–As2 3.0506(7) Å, Nd–As4 3.0673(7) Å), with bond lengths comparable to the literature.33 In general, As144− Zintl anions are uncommon. There is one iron species, [K(dme)2]2[(Cp*Fe)2(μ,η2:2:2:2-As14)], which formally consists of two As7-norbonadiene motifs connected by means of an additional As–As bond.43 This is in contrast to 3, where two covalently linked As7-nortricyclene units form the As144− scaffold. The central motif of 3 is just reported from the solid state compound [Rb(18-crown-6)]4As14·6 NH3 and not known in molecular f-element chemistry.44
cations are coordinated by the two respective charged equatorial arsenic atoms (Nd–As2 3.0506(7) Å, Nd–As4 3.0673(7) Å), with bond lengths comparable to the literature.33 In general, As144− Zintl anions are uncommon. There is one iron species, [K(dme)2]2[(Cp*Fe)2(μ,η2:2:2:2-As14)], which formally consists of two As7-norbonadiene motifs connected by means of an additional As–As bond.43 This is in contrast to 3, where two covalently linked As7-nortricyclene units form the As144− scaffold. The central motif of 3 is just reported from the solid state compound [Rb(18-crown-6)]4As14·6 NH3 and not known in molecular f-element chemistry.44
As an interim conclusion, we note that the formation and isolation of the As73− and As144− species by activation of nanoscale gray arsenic depends on the reducing agent used (3 vs. 4 electron reducing agents) and the reaction conditions. Moreover, the formation of the As73− and As144− species requires several steps of bond breaking and bond formation of gray arsenic. Thus, the formation of the well-characterized species 1–3 in one step seems to be unlikely. This can be seen by either using B(Ce) as a reducing reagent or by carrying out the reaction with B(Nd) at room temperature (note: 3 was formed at elevated temperature). In both cases mixtures of products were obtained. These could not be fully characterized as their separation failed due to similar solubility and identical appearance. However, their formation allows further insights into the complicated reduction process of As0nano as a solid-state material. Therefore, the hereby obtained compounds should be briefly discussed in the following section.
The activation of As0nano with B(Ce) at room temperature resulted in two different products (Scheme 3). In addition to crystals of compound 2, which was already obtained when A(Ce) was used, crystals of the As144− species [{K(18-crown-6)}2( Ce)2(μ4-η2:η2:η2-As14)] (4) (Fig. 4) were isolated, representing the Ce analogue of 3. As observed for 3, the newly formed As–As bond of 2.4468(13) Å is in the range of the bonds of the also uncharged basal arsenic atoms As5, As6, and As7 (2.4490(10)–2.4661(9) Å).
Ce)2(μ4-η2:η2:η2-As14)] (4) (Fig. 4) were isolated, representing the Ce analogue of 3. As observed for 3, the newly formed As–As bond of 2.4468(13) Å is in the range of the bonds of the also uncharged basal arsenic atoms As5, As6, and As7 (2.4490(10)–2.4661(9) Å).
 | ||
| Scheme 3 Synthesis of compounds 2 and 4. | ||
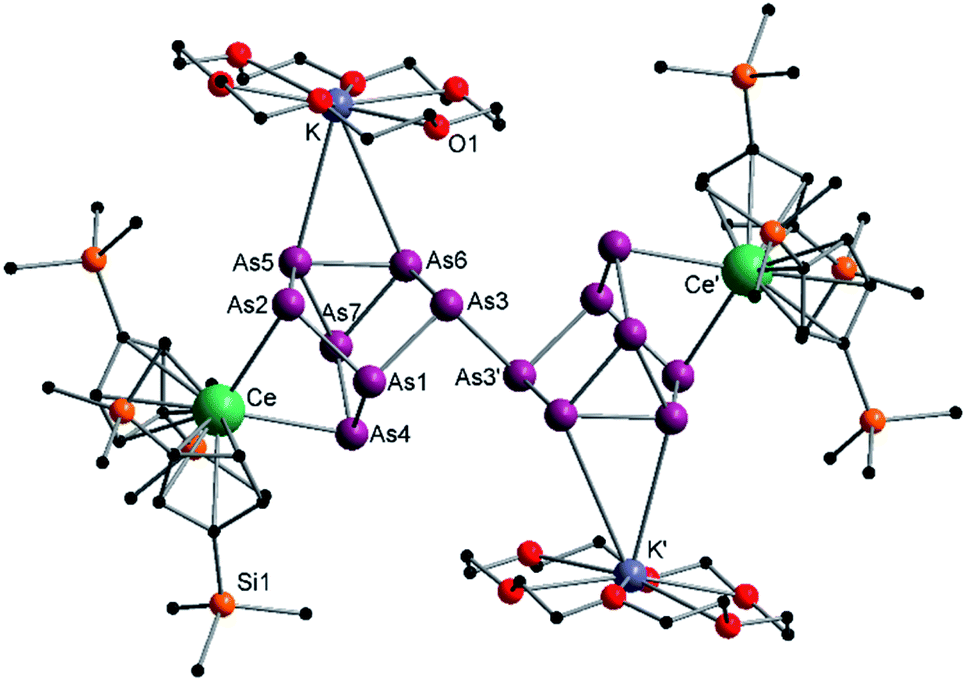 | ||
| Fig. 4 Molecular structure of 4 in the solid state. Solvent molecules and hydrogen atoms are omitted for clarity. Only one part of the positional disordered [K(18-crown-6)]+ is depicted. For both parts, bond lengths and angles see ESI (Fig. S11‡). | ||
All other bond lengths are in accordance with 3 as well. The 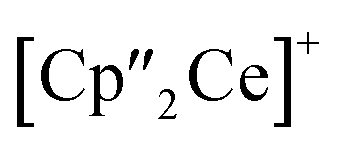 cations also coordinate to the two respective charged equatorial arsenic atoms (Ce–As2 3.0598(8) Å, Ce–As4 3.0417(7) Å), with bond lengths comparable to the corresponding bonds in 2. Analogous to 3, the [K(18-crown-6)]+ fragments are disordered over two positions (Fig. S11‡).
cations also coordinate to the two respective charged equatorial arsenic atoms (Ce–As2 3.0598(8) Å, Ce–As4 3.0417(7) Å), with bond lengths comparable to the corresponding bonds in 2. Analogous to 3, the [K(18-crown-6)]+ fragments are disordered over two positions (Fig. S11‡).
In contrast to B(Ce), the use of the 4-electron Nd reducing agent B(Nd) at room temperature resulted in mixture of even more products upon crystallization (Scheme 4). As result, four different species were formed under these conditions, indicating an influence of the different used lanthanides and their reactivity on the reaction. The obtained products are the As33− species [K(18-crown-6)][(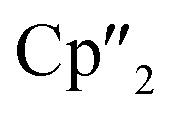 Nd)2(μ-η3:η3-As3)] (5), two compounds with an As73− motif [{K(18-crown-6)}(
Nd)2(μ-η3:η3-As3)] (5), two compounds with an As73− motif [{K(18-crown-6)}(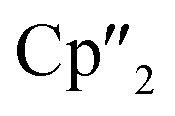 Nd)2(μ3-η2:η2:η2-As7)] (6) and [{K(18-crown-6)}2(
Nd)2(μ3-η2:η2:η2-As7)] (6) and [{K(18-crown-6)}2(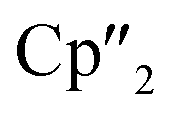 Nd)(μ3-η2:η2:η2-As7)] (7) as well as the isolable As144−-compound (3).
Nd)(μ3-η2:η2:η2-As7)] (7) as well as the isolable As144−-compound (3).
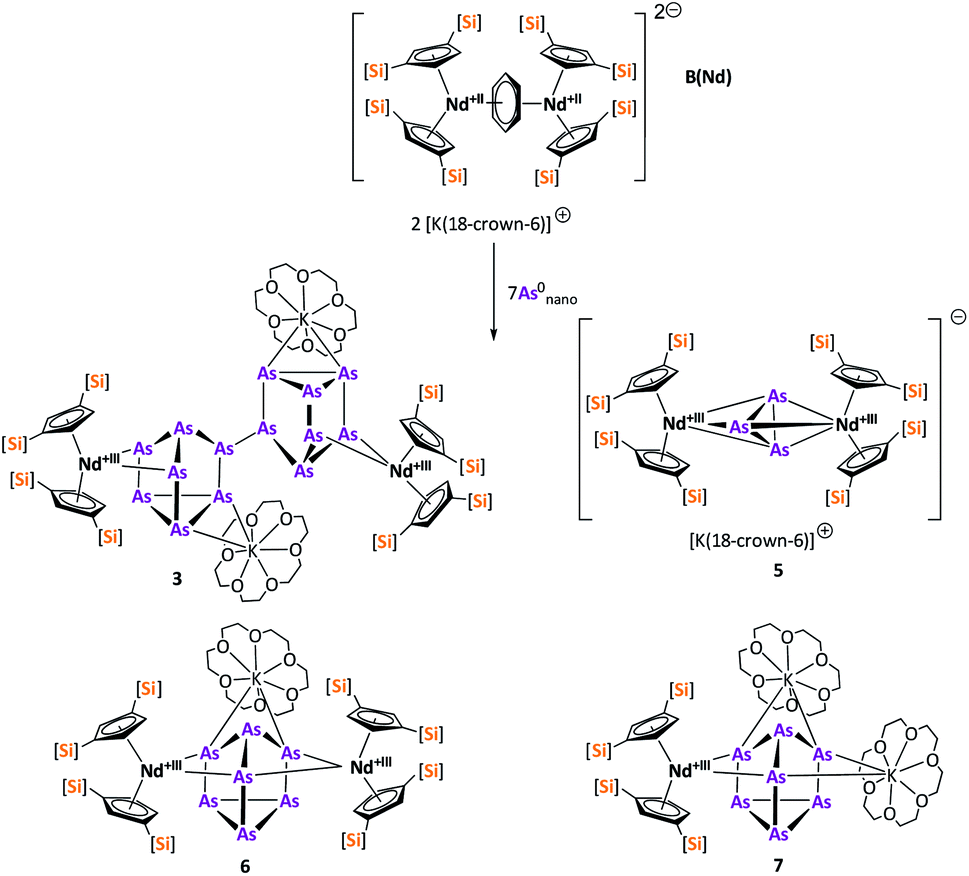 | ||
| Scheme 4 Synthesis of compounds 3, 5, 6 and 7. | ||
Within the mixture obtained at room temperature, [K(18-crown-6)][( Nd)2(μ-η3:η3-As3)] (5) (Fig. 5) features the smallest of the organo-lanthanide-polyarsenides obtained here (As33− unit) and – similar to [(
Nd)2(μ-η3:η3-As3)] (5) (Fig. 5) features the smallest of the organo-lanthanide-polyarsenides obtained here (As33− unit) and – similar to [( Sm)2(μ-η2:η2-As2)] from the activation of As0nano by SET – may be regarded as an intermediate in the formation of the larger polyarsenides.26 The
Sm)2(μ-η2:η2-As2)] from the activation of As0nano by SET – may be regarded as an intermediate in the formation of the larger polyarsenides.26 The  fragments are slightly offset from each other and both η3-coordinated by the central As33− moiety, which in turn forms an equilateral triangle. The Nd–As bond lengths range from 2.9311(7) to 3.0481(7) Å and are thus comparable to the shortest Nd–As distance in [K(18-crown-6)][
fragments are slightly offset from each other and both η3-coordinated by the central As33− moiety, which in turn forms an equilateral triangle. The Nd–As bond lengths range from 2.9311(7) to 3.0481(7) Å and are thus comparable to the shortest Nd–As distance in [K(18-crown-6)][ Nd(μ-η4:η4-As5)FeCp*] (2.9258(4) Å).33 The As–As distances (2.4198(7)–2.4388(8) Å), all of approximately equal length, are in the range of single bonds.42 Additionally, they are comparable to As33− units in solid state compounds (2.43–2.47 Å in CsAs).38 While an example of an uranium complex with such an As33− unit is reported for the actinides,45 this has not been accessible for the lanthanides so far.
Nd(μ-η4:η4-As5)FeCp*] (2.9258(4) Å).33 The As–As distances (2.4198(7)–2.4388(8) Å), all of approximately equal length, are in the range of single bonds.42 Additionally, they are comparable to As33− units in solid state compounds (2.43–2.47 Å in CsAs).38 While an example of an uranium complex with such an As33− unit is reported for the actinides,45 this has not been accessible for the lanthanides so far.
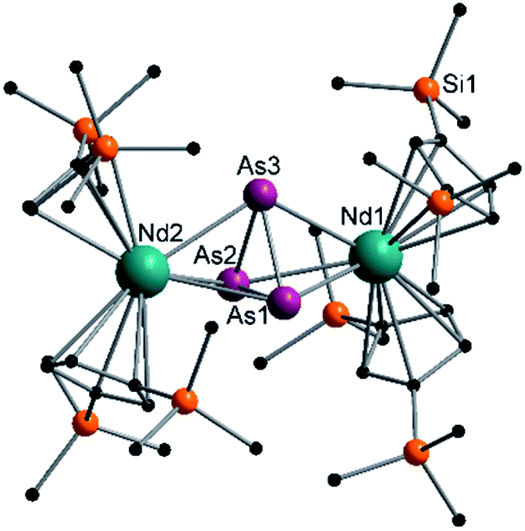 | ||
| Fig. 5 Molecular structure of 5 in the solid state. Solvent molecules, counter cation [K(18-crown-6)]+ and hydrogen atoms are omitted for clarity. For bond lengths and angles see ESI (Fig. S12‡). | ||
In addition, two different compounds with an As73− structural motif were also found in the mixture (Fig. 6). Here, [{K(18-crown-6)}(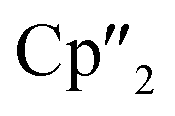 Nd)2(μ3-η2:η2:η2-As7)] (6) represents the Nd analogue to 2. The observed bond lengths are all comparable to the latter. In contrast, [{K(18-crown-6)}2(
Nd)2(μ3-η2:η2:η2-As7)] (6) represents the Nd analogue to 2. The observed bond lengths are all comparable to the latter. In contrast, [{K(18-crown-6)}2(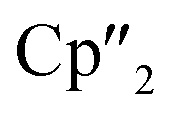 Nd)(μ3-η2:η2:η2-As7)] (7) shows an altered composition, since the charge balance takes place by means of a
Nd)(μ3-η2:η2:η2-As7)] (7) shows an altered composition, since the charge balance takes place by means of a 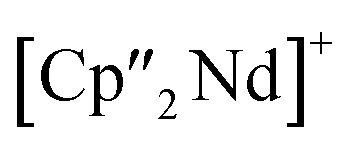 fragment and two [K(18-crown-6)]+ units (vice versa for 1, 2, and 6). All bond lengths are within the previously observed ranges.
fragment and two [K(18-crown-6)]+ units (vice versa for 1, 2, and 6). All bond lengths are within the previously observed ranges.
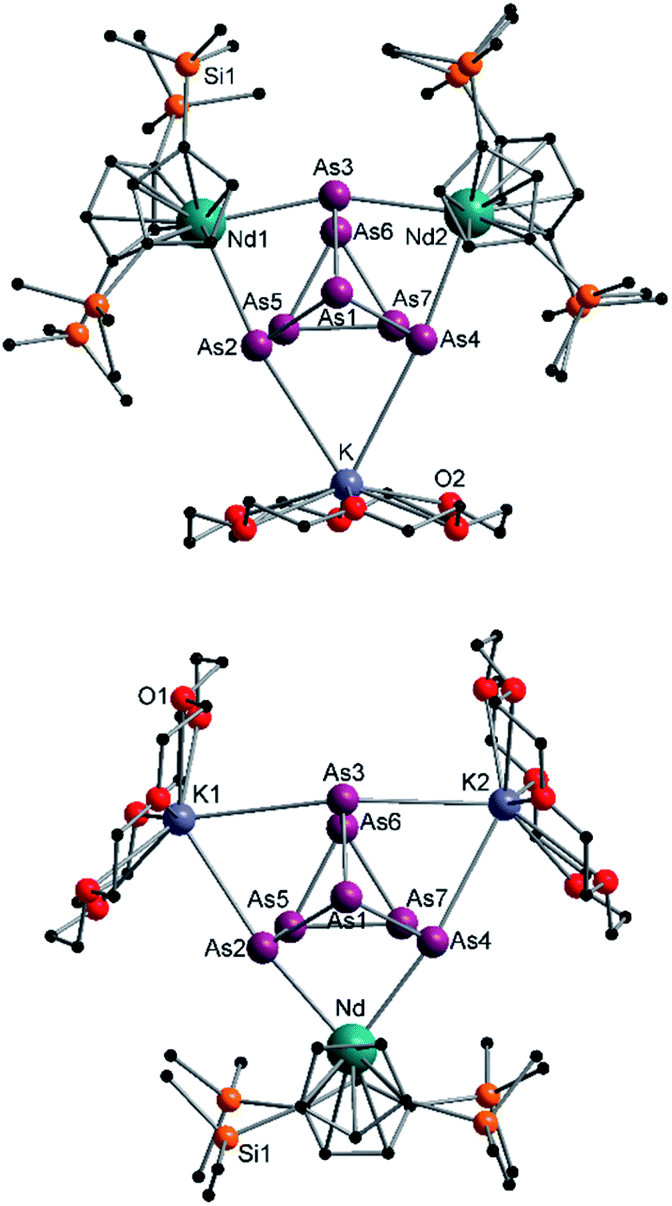 | ||
| Fig. 6 Molecular structures of 6 (top) and 7 (bottom) in the solid state. Solvent molecules and hydrogen atoms are omitted for clarity. For bond lengths and angles see ESI (Fig. S13 and S14‡). | ||
Finally, the last compound that can be obtained in the mixture is the already described As144− compound 3. As mentioned above, this compound could also be obtained in pure form in the presence of an excess of As0nano and by prolonged heating (Scheme 2).
Conclusions
In summary, we have demonstrated that the solid state material As0nano can be activated via 3- and 4-electron reducing agents of the early non-classical divalent lanthanides to obtain a variety of new molecular organo-lanthanide-polyarsenides. This significantly extends the bridge from solid-state arsenic to molecular f-element polyarsenides, contributing to a better understanding of the formation and properties of such polyarsenide materials.On the one hand, the clean formation of an As73− Zintl anion with a nortricyclic structure in 1 and 2 is observed by using the 3-electron reducing agents A(La) and A(Ce). Although lanthanide compounds with As73− Zintl anions have already been reported, the nortricyclane structural motif was previously unknown in this chemistry. On the other hand, the formation the As144− Zintl species 3 as sole isolable product is seen by applying the 4-electon reducing agents B(Nd) at elevated temperature. Compound 3 represents the largest known organo-lanthanide-polyarsenides to date. In between these boundaries, mixtures of various compounds with a polyarsenide as central motif were obtained. These results show that the formation of sophisticated structures directly out of nanoscale gray arsenic, which is a kind of polymer, is a complex process with various intermediates. Only careful tuning of the reaction conditions and the use of an optimized reducing reagent leads to isolable and unprecedented products.
Data availability
All synthetic protocols, spectroscopic data, supplementary figures and tables, and detailed crystallographic information can be found in the ESI.‡ Crystallographic data are available via the Cambridge Crystallographic Data Centre (CCDC): 2094692–2094698.Author contributions
All authors have given approval to the final version of the manuscript. N. R. and A. H. synthesized and analyzed all compounds. L. M conducted X-ray experiments. PWR originated the idea, supervised the work, and interpreted the results. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft (DFG) (No. 266153560, Ro 2008/17–2) for financial support. N. R.'s PhD study is additionally funded by the Fonds der Chemischen Industrie (102431).References
- Zintl Ions Principles and Recent Developments, ed. T. F. Faessler, Springer Verlag, Berlin, 2011 Search PubMed.
- R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC.
- J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142–11151 CrossRef PubMed.
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS PubMed.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed.
- L. Giusti, V. R. Landaeta, M. Vanni, J. A. Kelly, R. Wolf and M. Caporali, Coord. Chem. Rev., 2021, 441, 213927 CrossRef CAS.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- A. Wiesner, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Chem. Sci., 2018, 9, 7169–7173 RSC.
- C. Schoo, S. Bestgen, R. Köppe, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2018, 54, 4770–4773 RSC.
- O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110–4112 CrossRef CAS.
- S. Pelties, D. Herrmann, B. de Bruin, F. Hartl and R. Wolf, Chem. Commun., 2014, 50, 7014–7016 RSC.
- S. Heinl and M. Scheer, Chem. Sci., 2014, 5, 3221–3225 RSC.
- C. Schwarzmaier, A. Y. Timoshkin, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 9077–9081 CrossRef CAS PubMed.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS PubMed.
- T. Li, S. Kaercher and P. W. Roesky, Chem. Soc. Rev., 2014, 43, 42–57 RSC.
- N. Arleth, M. T. Gamer, R. Köppe, N. A. Pushkarevsky, S. N. Konchenko, M. Fleischmann, M. Bodensteiner, M. Scheer and P. W. Roesky, Chem. Sci., 2015, 6, 7179–7184 RSC.
- F. Jaroschik, A. Momin, F. Nief, X.-F. LeGoff, G. B. Deacon and P. C. Junk, Angew. Chem., Int. Ed., 2009, 48, 1117–1121 CrossRef CAS PubMed.
- P. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2021, 60, 473–479 CrossRef CAS PubMed.
- A. F. Hollemann and E. Wiberg, Lehrbuch der Anorganischen Chemie, Walter deGruyter, Berlin, 102nd edn, 2007 Search PubMed.
- H. A. Spinney, N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 16233–16243 CrossRef CAS PubMed.
- F. Spitzer, M. Sierka, M. Latronico, P. Mastrorilli, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 4392–4396 CrossRef CAS PubMed.
- C. Schwarzmaier, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 858–861 CrossRef CAS PubMed.
- C. Schwarzmaier, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 7600–7603 CrossRef CAS PubMed.
- A. E. Seitz, F. Hippauf, W. Kremer, S. Kaskel and M. Scheer, Nat. Commun., 2018, 9, 361 CrossRef PubMed.
- C. Schoo, S. Bestgen, A. Egeberg, J. Seibert, S. N. Konchenko, C. Feldmann and P. W. Roesky, Angew. Chem., Int. Ed., 2019, 58, 4386–4389 CrossRef CAS PubMed.
- A. Hauser, L. Münzfeld and P. W. Roesky, Chem. Commun., 2021, 57, 5503–5506 RSC.
- W. Fang, I. Douair, A. Hauser, K. Li, Y. Zhao, P. W. Roesky, S. Wang, L. Maron and C. Zhu, CCS Chem, 2021, 3, 3268–3276 CrossRef.
- C. E. Kefalidis, S. Essafi, L. Perrin and L. Maron, Inorg. Chem., 2014, 53, 3427–3433 CrossRef CAS PubMed.
- W. Kim, J. Zide, A. Gossard, D. Klenov, S. Stemmer, A. Shakouri and A. Majumdar, Phys. Rev. Lett., 2006, 96, 045901 CrossRef PubMed.
- M. P. Hanson, A. C. Gossard and E. R. Brown, Appl. Phys. Lett., 2006, 89, 111908 CrossRef.
- J. M. O. Zide, A. Kleiman-Shwarsctein, N. C. Strandwitz, J. D. Zimmerman, T. Steenblock-Smith, A. C. Gossard, A. Forman, A. Ivanovskaya and G. D. Stucky, Appl. Phys. Lett., 2006, 88, 162103 CrossRef.
- N. Reinfandt, N. Michenfelder, C. Schoo, R. Yadav, S. Reichl, S. N. Konchenko, A. N. Unterreiner, M. Scheer and P. W. Roesky, Chem. -Eur. J., 2021, 27, 7862–7871 CrossRef CAS PubMed.
- C. T. Palumbo, L. E. Darago, M. T. Dumas, J. W. Ziller, J. R. Long and W. J. Evans, Organometallics, 2018, 37, 3322–3331 CrossRef CAS.
- N. Reinfandt, Untersuchungen zur Reaktivität klassischer und nicht-klassischer divalenter Lanthanoidverbindungen gegenüber Pnictogenen und deren Verbindungen sowie Darstellung heterobimetallischer Lanthanoid-Münzmetallkomplexe, Cuvillier Verlag, Göttingen, 2021 Search PubMed.
- N. Arleth, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Fleischmann, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2016, 55, 1557–1560 CrossRef CAS PubMed.
- K. Hübler and G. Becker, Z. Anorg. Allg. Chem., 1998, 624, 483–496 CrossRef.
- F. Emmerling and C. Röhr, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 963–975 CrossRef CAS.
- A. W. Castleman, S. N. Khanna, A. Sen, A. C. Reber, M. Qian, K. M. Davis, S. J. Peppernick, A. Ugrinov and M. D. Merritt, Nano Lett., 2007, 7, 2734–2741 CrossRef CAS PubMed.
- S. Mitzinger, L. Guggolz, W. Massa and S. Dehnen, Z. Anorg. Allg. Chem., 2019, 645, 153–157 CrossRef CAS.
- S. Mandal, R. Liu, A. C. Reber, M. Qian, H. M. Saavedra, X. Ke, P. Schiffer, S. Sen, P. S. Weiss, S. N. Khanna and A. Sen, Chem. Commun., 2011, 47, 3126–3128 RSC.
- M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Chem. -Eur. J., 2010, 16, 432–435 CrossRef CAS PubMed.
- M. Schmidt, D. Konieczny, E. V. Peresypkina, A. V. Virovets, G. Balázs, M. Bodensteiner, F. Riedlberger, H. Krauss and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 7307–7311 CrossRef CAS PubMed.
- T. Hanauer, J. C. Aschenbrenner and N. Korber, Inorg. Chem., 2006, 45, 6723–6727 CrossRef CAS PubMed.
- B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582–590 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Peter C. Junk on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Full experimental procedures, spectra, and analytical data is provided. CCDC 2094692–2094698. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05797a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |