
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unified total synthesis of the brevianamide alkaloids enabled by chemical investigations into their biosynthesis†
Robert C.
Godfrey‡
,
Helen E.
Jones‡
 ,
Nicholas J.
Green§
and
Andrew L.
Lawrence
*
,
Nicholas J.
Green§
and
Andrew L.
Lawrence
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: a.lawrence@ed.ac.uk
First published on 29th December 2021
Abstract
The bicyclo[2.2.2]diazaoctane alkaloids are a vast group of natural products which have been the focus of attention from the scientific community for several decades. This interest stems from their broad range of biological activities, their diverse biosynthetic origins, and their topologically complex structures, which combined make them enticing targets for chemical synthesis. In this article, full details of our synthetic studies into the chemical feasibility of a proposed network of biosynthetic pathways towards the brevianamide family of bicyclo[2.2.2]diazaoctane alkaloids are disclosed. Insights into issues of reactivity and selectivity in the biosynthesis of these structures have aided the development of a unified biomimetic synthetic strategy, which has resulted in the total synthesis of all known bicyclo[2.2.2]diazaoctane brevianamides and the anticipation of an as-yet-undiscovered congener.
Introduction
The fungal-derived bicyclo[2.2.2]diazaoctane alkaloids have been the focus of attention from the scientific community for several decades.1 They are of significant interest to synthetic chemists as topologically complex targets, which have diverse biosynthetic origins and exhibit a broad range of biological activities. There are two types of biosynthetically distinct bicyclo[2.2.2]diazaoctane alkaloid, the monooxopiperazine structures, which include the paraherquamides2 and malbrancheamides;3 and the dioxopiperazine structures, which include the stephacidins4 and notoamides (Fig. 1).5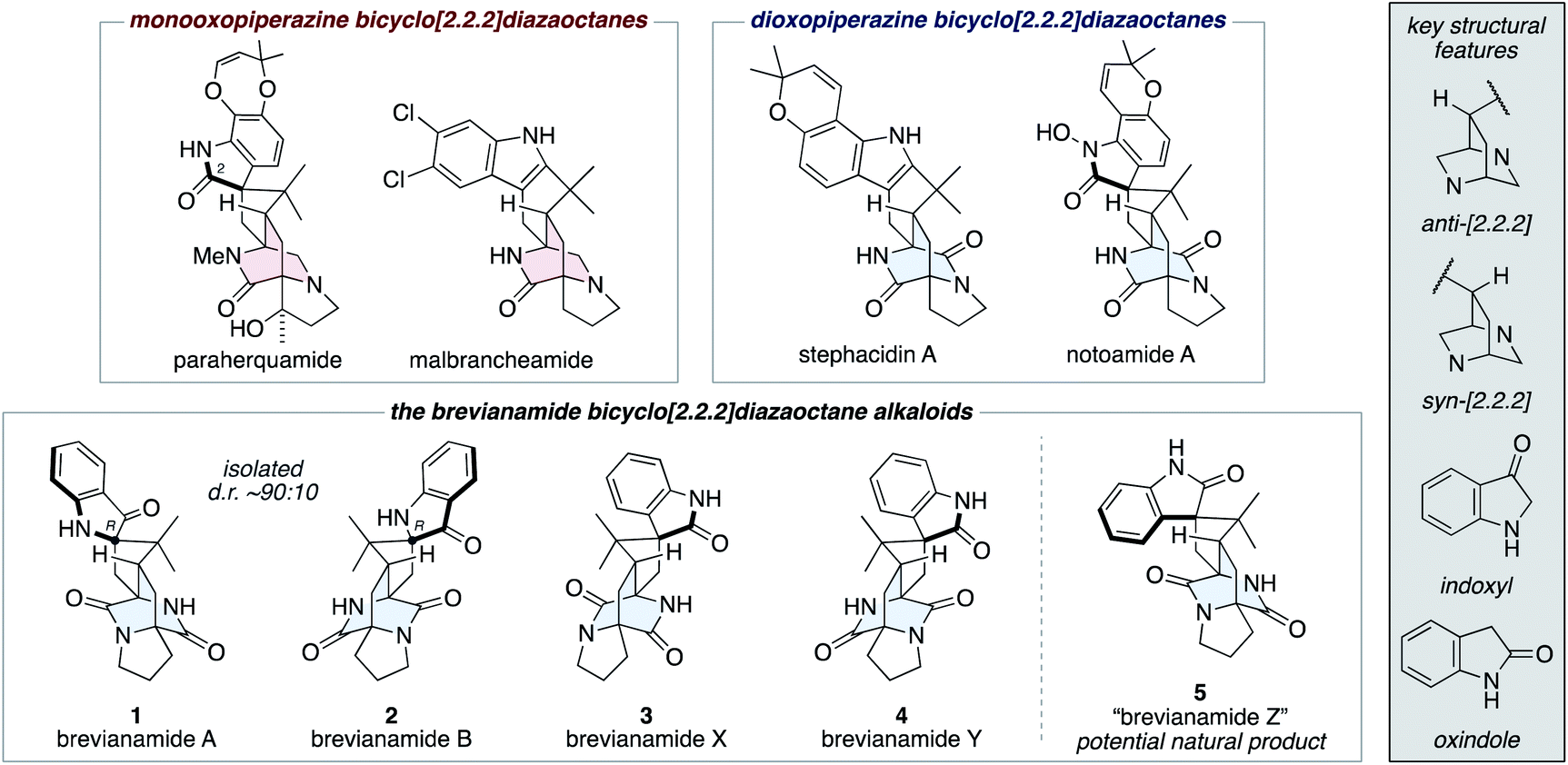 | ||
| Fig. 1 Representative bicyclo[2.2.2]diazaoctane alkaloids, including brevianamides A (1), B (2), X (3), Y (4), and Z (5). | ||
The brevianamide alkaloids
The first reported bicyclo[2.2.2]diazaoctane alkaloids were brevianamide A (1) and B (2), which are diastereomeric dioxopiperazine-type structures, isolated by Birch and Wright from the fungus Penicillium brevicompactum in 1969 (Fig. 1).6 Structurally, they share a common (R)-configured spiro-indoxyl stereogenic centre and they possess ‘enantiomeric’ anti-configured bicyclo[2.2.2]diazaoctane cores (anti refers to the relative configuration of the methine C–H and proximal piperazine nitrogen, see grey box in Fig. 1). They are isolated from the fungus in a consistent diastereomeric ratio of approximately 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.7 In 2017, the closely related natural products, brevianamides X (3) and Y (4) were isolated by Qi and co-workers from the deep-sea-derived P. brevicompactum DFFSCS025.8 In contrast to brevianamides A (1) and B (2), they contain spiro-oxindole rings and brevianamide X (3) is further differentiated by having a syn-configured bicyclo[2.2.2]-diazaoctane core.
:10.7 In 2017, the closely related natural products, brevianamides X (3) and Y (4) were isolated by Qi and co-workers from the deep-sea-derived P. brevicompactum DFFSCS025.8 In contrast to brevianamides A (1) and B (2), they contain spiro-oxindole rings and brevianamide X (3) is further differentiated by having a syn-configured bicyclo[2.2.2]-diazaoctane core.
We recently reported the first chemical synthesis of brevianamide A (1),9 the archetypal bicyclo[2.2.2]diazaoctane alkaloid.1 Our synthetic strategy was modelled on a proposed biosynthetic pathway that built upon decades of biosynthetic speculation and investigation. In this article we provide full details of our synthetic endeavors, which have resulted in the development of a unified total synthesis of all known bicyclo[2.2.2]diazaoctane brevianamide alkaloids (1–4) and the anticipation of an as-yet-undiscovered congener (5) (Fig. 1). We also outline experimental investigations into complexity-generating biomimetic cascade reactions and discuss how these relate to recent biosynthetic studies.10 Our exploration of the (bio)synthetic relationships between these complex alkaloids has allowed us to identify conditions that favor formation of specific scaffolds from a common intermediate.
Biological activity
The bicyclo[2.2.2]diazaoctane alkaloids have diverse and often potent biological activities. For example, the stephacidins and notoamides are cytotoxic,4,5 the malbrancheamides are calmodulin inhibitors,3 and the paraherquamides are anthelmintics.2 A modified paraherquamide, derquantel (2-deoxy-paraherquamide), is currently used in combination with the macrocyclic lactone, abamectin, under the trade name STARTECT® to treat sheep with gastrointestinal nematodes.2 With regards to the brevianamides, brevianamide A (1) is reported to have potent antifeedant activity against the larvae of the insect pests Spodoptera frugiperda (fall armyworm) and Heliothis virescens (tobacco budworm),11 but no biological activity has yet been reported for brevianamides B (2), X (3) and Y (4). Our synthetic access to practical quantities of brevianamide A (1) is facilitating ongoing studies into its antifeedant properties and we envisage that access to the other brevianamide alkaloids, and synthetic analogues, will similarly enable investigations into their biological profiles.Previous synthetic studies
The historical failure to develop a viable synthetic route to brevianamide A (1) is attributable to all previously explored approaches featuring a common and ultimately doomed end-game strategy (Scheme 1a). This strategy hinges on a polycyclic indole intermediate (e.g., 6) undergoing a selective indole-oxidation and [1,2]-shift ring contraction to give brevianamide A (1) (Scheme 1a). Unfortunately, all examples of this type of late-stage indole-oxidation exhibit complete selectivity for oxidation occurring on the sterically more accessible convex face. Following the stereospecific [1,2]-shift this then leads to the unnatural enantiomeric framework of brevianamide B (Scheme 1a).12 Thus, although this strategy hasn't provided a route to brevianamide A (1), several syntheses of brevianamide B (2) have been achieved that proceed via intermediate 6, or protected analogues thereof (see structures in Scheme 1b).12 Most notably, Williams and co-workers have achieved four syntheses of brevianamide B (2). In 1988, they reported the first total synthesis of ent-brevianamide B (ent-2) using a late-stage cationic cyclization, which resulted in an 18-step total synthesis.12a–c A decade later they reported a 12-step total synthesis of (±)-brevianamide B (2), in which they utilized a bio-inspired intramolecular Diels–Alder reaction to forge the bicyclo[2.2.2]diazaoctane core, albeit with diastereoselectivity not favoring the desired diastereomer (d.r. 2:1, vide infra).12d,e In their third synthesis, Williams and co-workers utilized a late-stage Fischer indole-synthesis to gain access to (±)-brevianamide B (2) in 12 steps.12f Finally, in 2007 they reappraised their biomimetic intramolecular Diels–Alder strategy, this time avoiding protection of the lactam functionality, and achieved a 14-step synthesis of (±)-brevianamide B (2).12g,h The shortest total synthesis of ent-brevianamide B (ent-2), prior to this present study, was achieved by Simpkins and co-workers in 2009.12i,j The brevity of their 9-step synthesis can be traced back to an elegant cationic double-cyclization strategy, wherein two new C–C bonds and three new rings are formed in one step. Unfortunately, the diastereoselectivity of this cascade reaction did not favor the required diastereomer (d.r. 4:1). In 2016 and 2017, Scheerer and co-workers reported two formal syntheses of (±)-brevianamide B (2), which rely on early intermolecular Diels–Alder reactions to forge the bicyclo[2.2.2]diazaoctane ring system.12k,l A series of functional group interconversions and a late-stage cationic cyclization, as pioneered by Williams and co-workers in 1988,12a result in 13 and 15 step formal syntheses of (±)-brevianamide B (2). In summary, the confluence of previous synthetic approaches on one common end-game strategy (Scheme 1a) has precluded access to brevianamide A (1) but resulted in several syntheses of brevianamide B (2), either in its unnatural enantiomeric form or as a racemate (Scheme 1b).
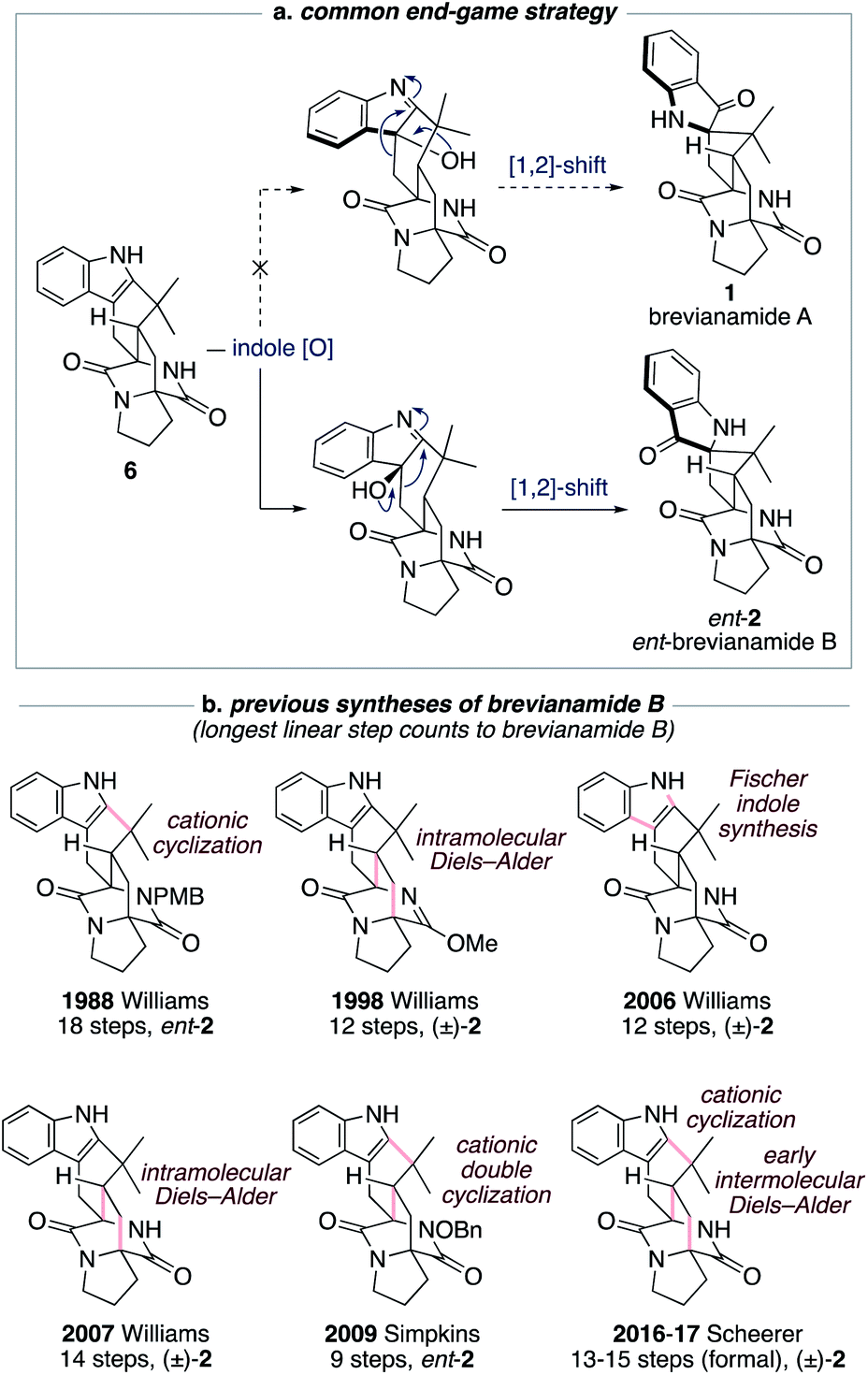 | ||
| Scheme 1 Previous synthetic approaches towards brevianamides A (1) and B (2). | ||
Biosynthesis of the brevianamides
The most notable transformation in the biosynthesis of the brevianamides (1-4) is an intramolecular Diels–Alder reaction, which was originally proposed by Sammes and Porter in 1970 (see grey box in Scheme 2).13 This insightful proposal has since been extended to other bicyclo[2.2.2]diazaoctane alkaloid biosyntheses.1 Diels–Alderase enzymes have been identified in the biosynthetic gene clusters responsible for the malbrancheamide and paraherquamide monooxopiperazine alkaloids.14 However, the involvement of Diels–Alderase enzymes in the biosynthesis of the dioxopiperazine alkaloids has been a topic of debate for decades, particularly with regards to the brevianamides.1,15 DFT calculations by Domingo and co-workers in 1997 indicated that for precursor 7, where the azadiene and dienophile are tethered via three atoms through an indoxyl ring system (i.e., post indole oxidation), the Diels–Alder reaction is expected to be inherently anti-diastereoselective (Scheme 2a).16 This was further supported last year in computational studies reported by Williams, Sherman, Li and co-workers (Scheme 2a).10 Both studies also revealed that an intramolecular hydrogen bond, between the indoxyl N–H and proximal lactam C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, can stabilize the transition state for brevianamide A (1), relative to B (2), thus potentially accounting for the greater quantity of brevianamide A (1) isolated from the fungus (d.r. A:B ∼90:10).10,16 On the other hand, in 1998 Williams and co-workers discovered that the Sammes-type intramolecular Diels–Alder reaction on an unoxidized indole intermediate, 8, exhibits moderate syn-diastereoselectivity (Scheme 2b).12d,e Thus, the necessity of a Diels–Alderase enzyme to control stereoselectivity in the biogenesis of the brevianamides appeared to be dependent on the relative order of the indole-oxidation and Diels–Alder reaction.17 Indeed, this point highlights the fact that without experimental data (synthetic, biochemical, or computational) there are numerous plausible permutations of biosynthetic transformations towards these alkaloids which could be, and indeed have been, proposed.1
O, can stabilize the transition state for brevianamide A (1), relative to B (2), thus potentially accounting for the greater quantity of brevianamide A (1) isolated from the fungus (d.r. A:B ∼90:10).10,16 On the other hand, in 1998 Williams and co-workers discovered that the Sammes-type intramolecular Diels–Alder reaction on an unoxidized indole intermediate, 8, exhibits moderate syn-diastereoselectivity (Scheme 2b).12d,e Thus, the necessity of a Diels–Alderase enzyme to control stereoselectivity in the biogenesis of the brevianamides appeared to be dependent on the relative order of the indole-oxidation and Diels–Alder reaction.17 Indeed, this point highlights the fact that without experimental data (synthetic, biochemical, or computational) there are numerous plausible permutations of biosynthetic transformations towards these alkaloids which could be, and indeed have been, proposed.1
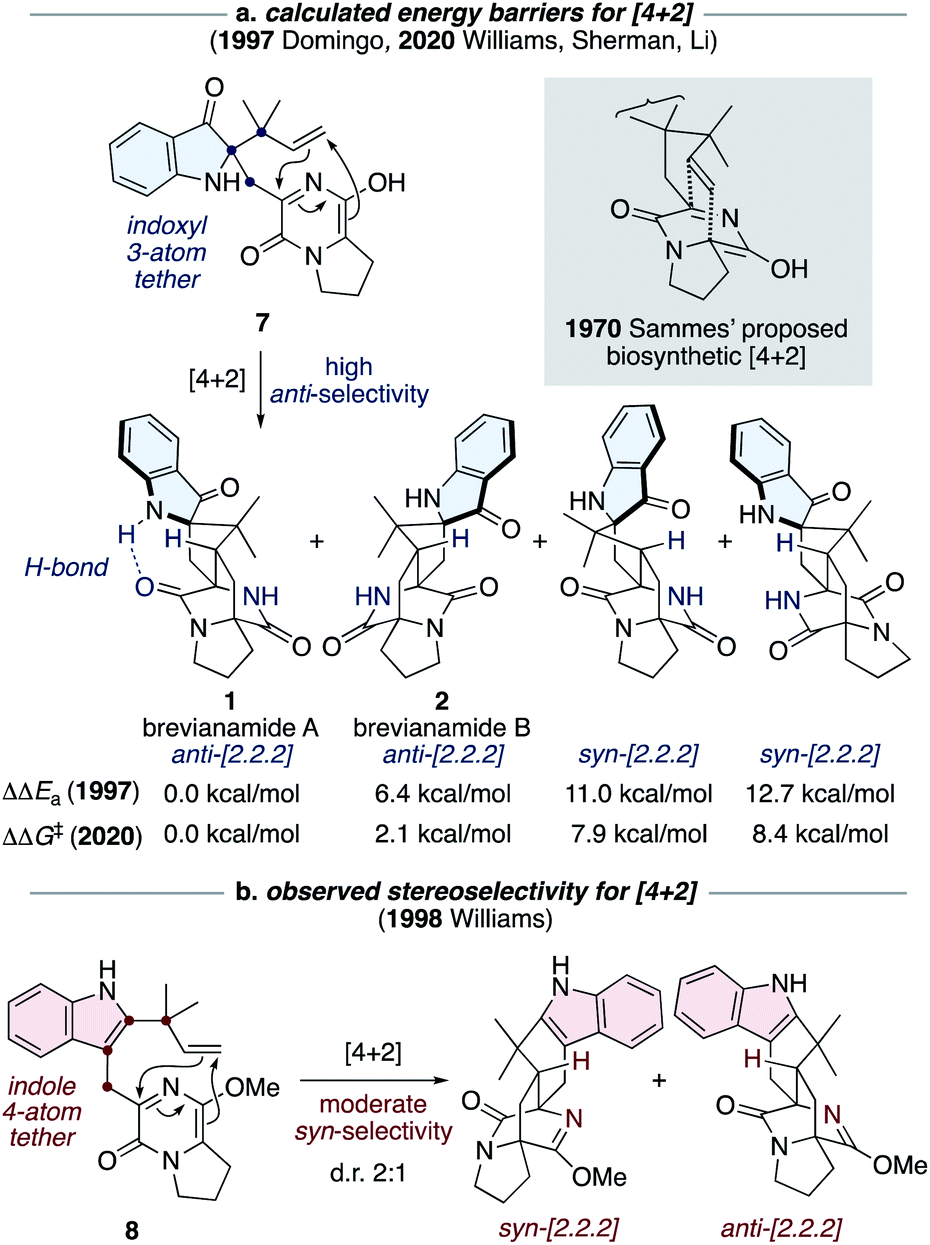 | ||
| Scheme 2 Diastereoselectivity of intramolecular Diels–Alder reactions related to Sammes' biosynthetic proposal.10,12d,e,16 | ||
We proposed a modified biosynthetic pathway for brevianamide A (1) in 2020.9 Our modified pathway relies on an early indole oxidation followed by a late-stage, substrate controlled, anti-diastereoselective Diels–Alder reaction (Scheme 3a).16 A key modification in our proposed pathway was the involvement of dehydrodeoxybrevianamide E (9), a natural product isolated from various Penicillium and Aspergillus fungi.18 We invoked this known natural product, 9, as a biosynthetic intermediate because its diketopiperazine ring is already at the correct oxidation level for the late-stage Diels–Alder reaction.19 Our pathway begins with diastereoselective indole-oxidation of 9 to give dehydrobrevianamide E (10), which we propose may be an as-yet-undiscovered natural product. Retro-5-exo-trig ring opening, followed by stereospecific [1,2]-shift and tautomerization then gives enantiopure azadiene 7 (Scheme 3a). A final Sammes-type Diels–Alder cycloaddition then gives brevianamides A (1) and B (2), which we were confident would proceed with the required anti-diastereoselectivity thanks to the DFT calculations reported by Domingo and co-workers.16 Our recent success in mimicking this proposed pathway in vitro (vide infra) led us to expand this biosynthetic speculation to account for the formation of all known bicyclo[2.2.2]diazaoctane brevianamides (1–4).
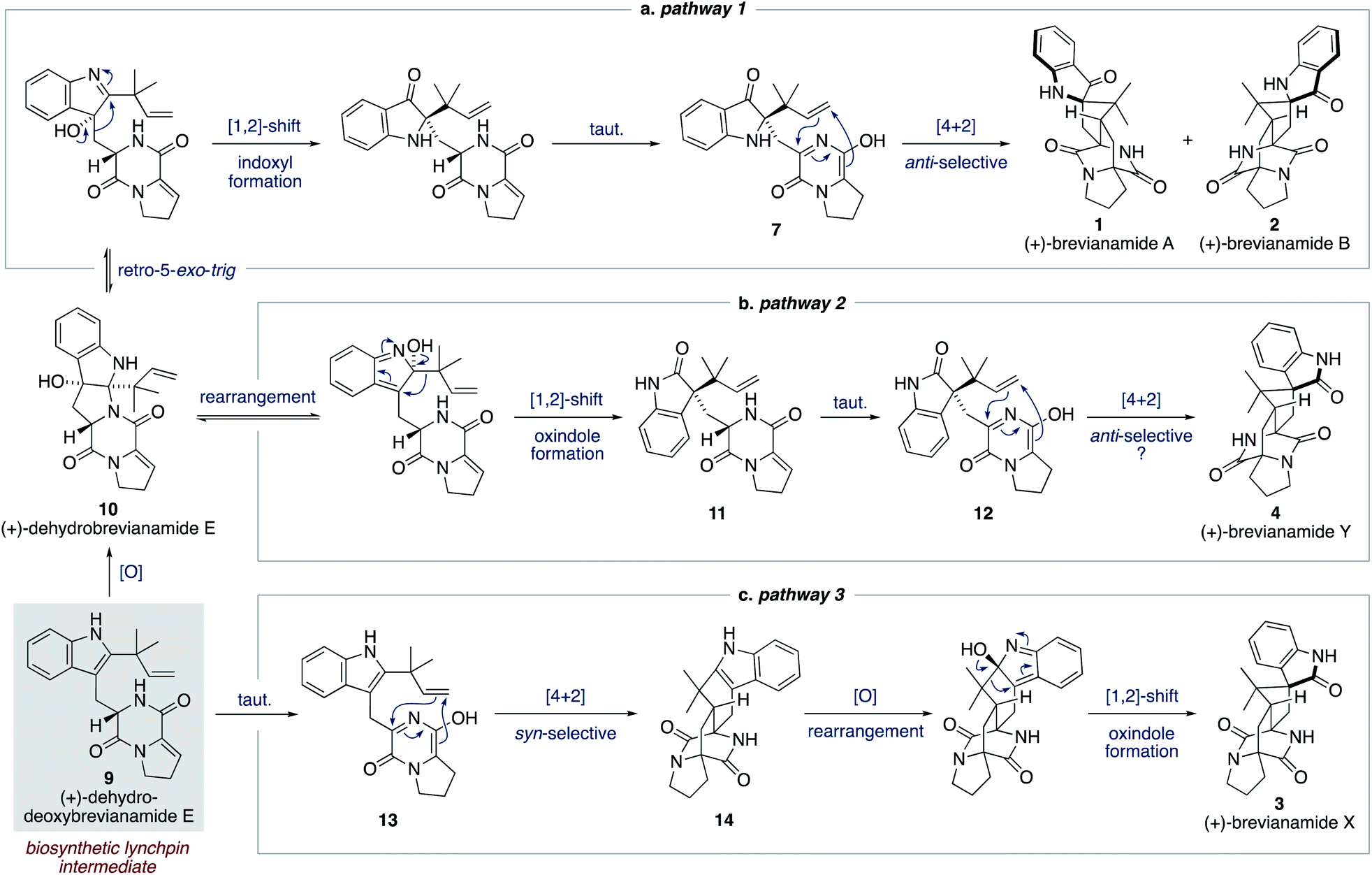 | ||
| Scheme 3 Our proposed biosynthesis of the bicyclo[2.2.2]diazaoctane brevianamides A, B, X and Y (1–4) from dehydrodeoxybrevianamide E (9). | ||
For brevianamide Y (4), we propose that dehydrobrevianamide E (10) undergoes a different rearrangement to give a thermodynamically more favorable oxindole intermediate 11 (Scheme 3b). This type of rearrangement (hydroxyindolenine to oxindole) is well known and is believed to proceed via transient epoxide intermediates.20 Subsequent tautomerization of oxindole 11 then gives an enantiopure azadiene intermediate 12, which undergoes a Sammes-type Diels–Alder cycloaddition to give brevianamide Y (4). We set-out to demonstrate the feasibility of this new Diels–Alder reaction and to probe its inherent diastereoselectivity through chemical synthesis.
To account for the formation of the atypical syn-configured bicyclo[2.2.2]diazaoctane core of brevianamide X (3) we propose an early Diels–Alder cycloaddition of dehydrodeoxybrevianamide E (9) (Scheme 3c). This is predicated on Williams' observation of inherent syn-diastereoselectivity in Diels–Alder reactions with indole oxidation-level substrates (e.g., 8) (Scheme 2b).12d,e It is reasonable to propose that a Diels–Alderase enzyme might be involved in this pathway as it involves an achiral intermediate, 13.21 There are, however, alternative explanations for the isolation of brevianamide X (3) in enantioenriched form. A spontaneous (i.e., non-enzyme mediated) biosynthetic Diels–Alder reaction might be followed by a kinetic resolution of racemic 14 in the subsequent enzymatic oxidation, or semi-pinacol [1,2]-shift.22 Alternatively, selective degradation of the unobserved enantiomer might be occurring. Detailed biosynthetic investigations will be required to unequivocally explain the isolation of brevianamide X (3) in enantioenriched form. Nevertheless, we were hopeful that we could establish the chemical feasibility of this pathway through chemical synthesis.
Results and discussion
Total synthesis of dehydrodeoxybrevianamide E
To fully investigate the chemical feasibility of our proposed network of biosynthetic pathways (Scheme 3) we first needed access to our lynchpin (bio)synthetic intermediate, (+)-dehydrodeoxybrevianamide E (9). Phthaloyl protection of L-tryptophan methyl ester 15 was followed by reverse prenylation of the indole-C2 position using Danishefsky's conditions to give intermediate 17 in 71% yield over the two steps (Scheme 4).23,24 Hydrolysis of methyl ester 17 resulted in unwanted ring opening of the phthaloyl group to give diacid 18. This presumably explains why a deprotection/reprotection sequence is undertaken in related synthetic endeavours.24,25 To avoid these extraneous two steps, we explored the possibility of a Krapcho-type demethylation of methyl ester 17. Use of LiI in EtOAc, as reported by Fisher and Trinkle,26 successfully gave the desired product 19. However, we found using LiCl in DMF resulted in a cleaner reaction. Lithium carboxylate 19 was then converted to N-acyl enamine 21 through a one-pot acyl chloride formation and imine acylation reaction using dehydroproline 20,27 which can be easily accessed in practical quantities from proline methyl ester using N-chlorosuccinimide as the oxidant (Scheme 4). Standard phthaloyl deprotection reagents, such as hydrazine, ethylene diamine, methylamine, hydroxylamine, ethanolamine and phenylhydrazine, resulted in unwanted cleavage of the amide bond in intermediate 21. However, the less nucleophilic ammonia in methanol not only deprotected the primary amine but also resulted in spontaneous cyclization to give (+)-dehydrodeoxybrevianamide E (9) in 49% yield over the three steps from methyl ester 17.28 Thus, the total synthesis of (+)-dehydrodeoxybrevianamide E (9) has been achieved in a longest linear sequence of 5 steps, in 35% overall yield, and requires just 2 chromatographic purifications (cf. previous synthesis: 12 steps, 8% overall yield).12g,h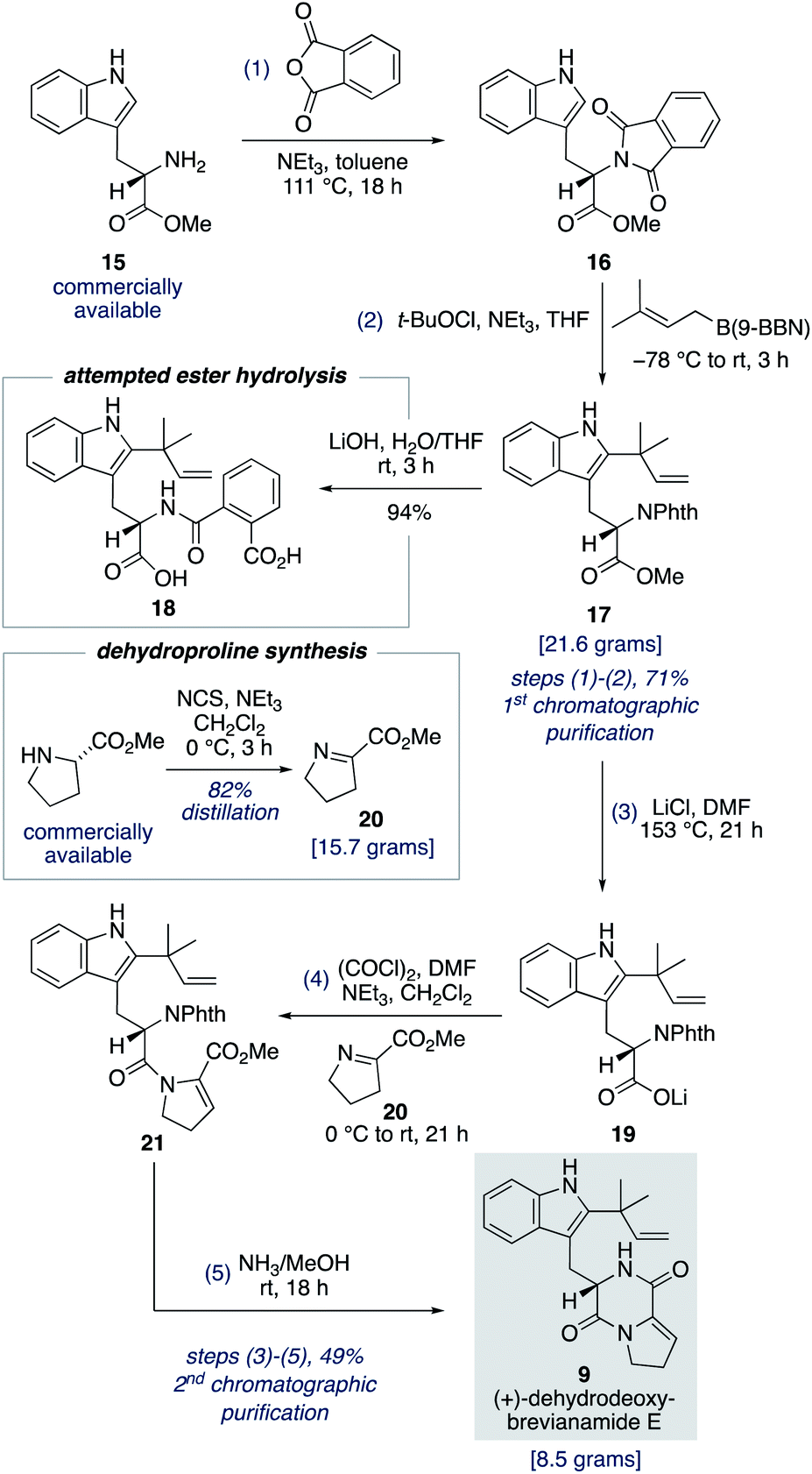 | ||
| Scheme 4 Five-step, gram-scale total synthesis of (+)-dehydrodeoxybrevianamide E (9). | ||
Total synthesis of brevianamides A and B
Oxidation of (+)-dehydrodeoxybrevianamide E (9) using a variety of oxidants (for example, peroxy acids, singlet oxygen,29 dioxiranes,24 and oxaziridines15a) gave dehydrobrevianamide E (10), alongside diastereomer 22 (Scheme 5). The stereoselectivity of the corresponding biosynthetic indole oxidation is controlled by flavin-dependent monooxygenase enzymes.1b Achieving diastereoselectivity using chemical oxidants proved to be very challenging. For example, use of Davis' oxaziridine resulted in a relatively clean reaction but with no diastereoselectivity. The best we could achieve was a d.r. of 63:37 (10:22) using m-CPBA at room temperature, with no appreciable improvement observed at lower temperatures or when using other peroxy acids. Use of an excess of m-CPBA, to improve conversion, resulted in the formation of an unwanted side-product, dehydro-depyranoamoenamide A (Scheme 5). This unexpected double oxidation/fragmentation reaction could be of future utility for the total synthesis of amoenamide A, a natural product isolated from Aspergillus amoenus NRRL 35600 by Tsukamoto and co-workers in 2017 (Scheme 5).30 To suppress this unwanted reaction an equimolar quantity of m-CPBA was added slowly to (+)-dehydrodeoxybrevianamide E (9) at room temperature to give dehydrobrevianamide E (10) and diastereomer 22 in 57% combined yield (d.r. 63:37). Under these conditions, conversion was satisfactorily high with minimal formation of unwanted side-products; 5% dehydro-depyranoamoenamide A, 5% oxindole 11, and 7% recovered starting material. Synthetic access to both diastereomers 10 and 22 is valuable as it enables the preparation of both the natural-(+) and unnatural-(−) enantiomers of brevianamides A (1) and B (2) (vide infra). Exposure of dehydrobrevianamide E (10) to LiOH in water at ambient temperature for 30 min successfully gave the natural (+)-enantiomers of brevianamide A (1) and B (2) in a combined 63% yield. Keeping the reaction time to a minimum and avoiding elevated temperatures was essential to prevent alkaline hydrolysis of the products, as previously observed by Birch and co-workers.31 Following only the fourth chromatographic purification in the entire seven-step synthesis, 750 mg of (+)-brevianamide A (1) and 60 mg of (+)-brevianamide B (2) were isolated. Chiral-HPLC analysis revealed both products (1 and 2) were isolated in a 93:7 enantiomeric ratio, which is not surprising given many of our synthetic intermediates may have undergone partial racemization. Recrystallization gave (+)-brevianamide A (1) in an enantiomeric ratio of 99:1. The unnatural (−)-enantiomers of brevianamide A (ent-1) and B (ent-2) were similarly accessed by subjecting the minor diastereomer, compound 22, to the same reaction conditions (Scheme 5).
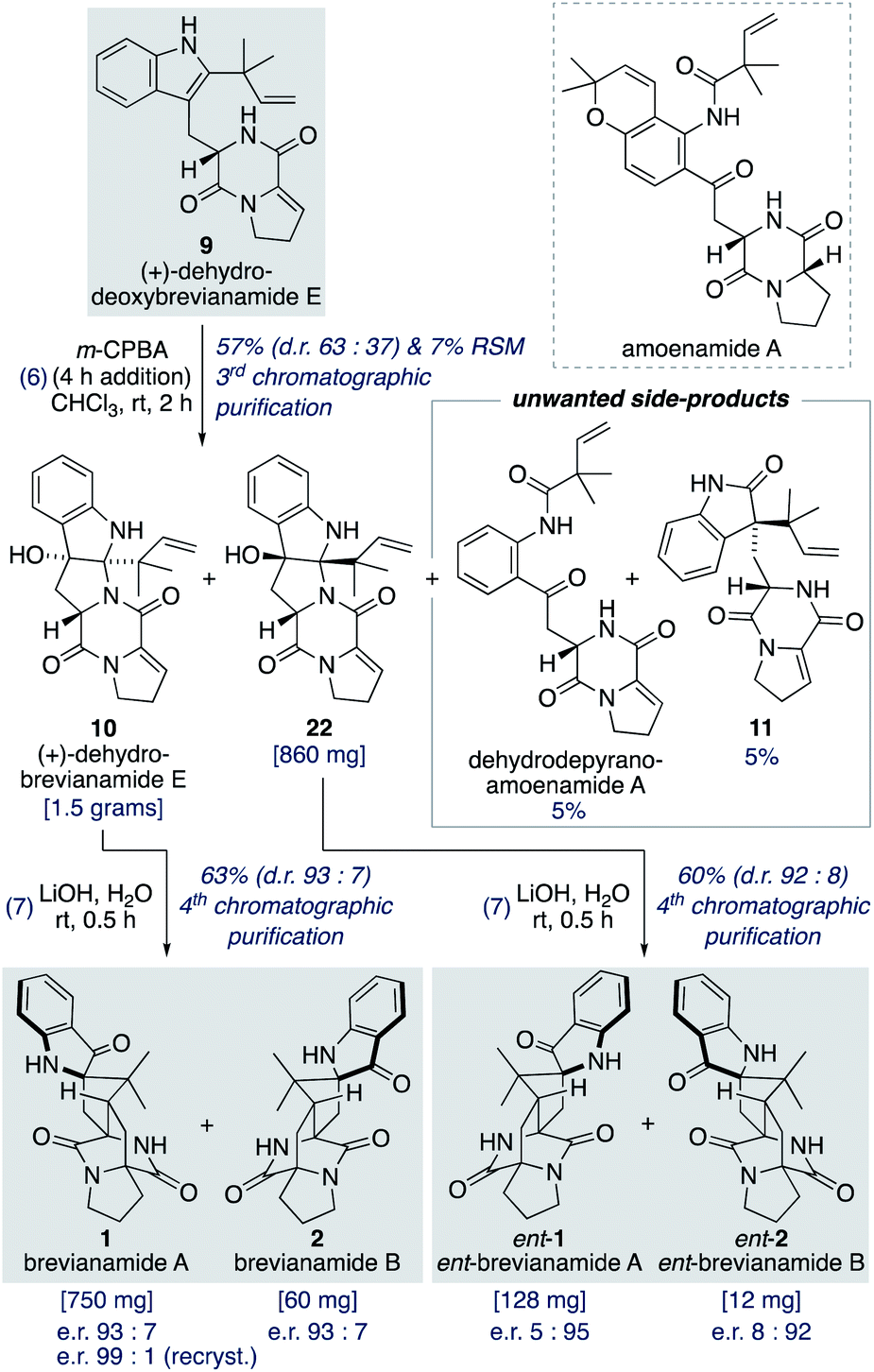 | ||
| Scheme 5 Total synthesis of (+) and (−)-brevianamides A (1) and B (2). | ||
Williams and co-workers have reported the use of aqueous KOH in methanol for closely related biomimetic reactions.12d,g For our reaction, however, we found that aqueous KOH in methanol resulted in a lower yield (44%) with the formation of a minor hydroxy-indolenine side-product 23 (11%) (vide infra). Use of KOH or NaOH in water also resulted in slightly lower yields. Mechanistic insights into the importance of the reaction conditions for the final cascade reaction (i.e., 10 to 1/2 and 22 to ent-1/ent-2) were acquired through further investigations using the minor diastereomer 22. It was found that when using a weaker base (aqueous K2CO3) in methanol two diastereomeric hydroxy-indolenine structures, 23 and 24, could be formed as the major products (Scheme 6). This presumably occurs via a domino retro-5-exo-trig/tautomerization/Diels–Alder reaction sequence, with the Diels–Alder reaction exhibiting anti-diastereoselectivity. These hydroxy-indolenines, 23 and 24, could conceivably serve as intermediates in the synthesis of ent-brevianamides A (ent-1) and B (ent-2) through a final semi-pinacol [1,2]-shift (Scheme 6). We conducted a series of experiments to further probe this mechanistic possibility. Hydroxy-indolenine 23 was subjected to aqueous LiOH conditions and no conversion to ent-brevianamide A (ent-1) was observed. In contrast, ent-brevianamide B (ent-2) was formed when hydroxy-indolenine 24 was subjected to aqueous LiOH conditions. We suspect that this stark difference in reactivity might be due to a stabilizing intramolecular ionic hydrogen bond in alkoxy-indolenine 23 (Scheme 6).32 Overall, these results indicate that our successful synthesis of brevianamides A (1) and B (2) does not involve hydroxy-indolenine intermediates (23/24). It appears that use of a strong base in water ensures the semi-pinacol [1,2]-shift occurs prior to the Diels–Alder reaction. This reactivity issue is reflected in the recent biosynthetic studies reported by Williams, Sherman, Li and co-workers,10 which revealed that the final enzyme in the biogenesis of brevianamides A (1) and B (2) is a semi-pinacolase. Thus, our use of LiOH in the chemical synthesis of brevianamides A (1) and B (2) appears to be serving the same role as the semi-pinacolase enzyme in the biosynthesis.
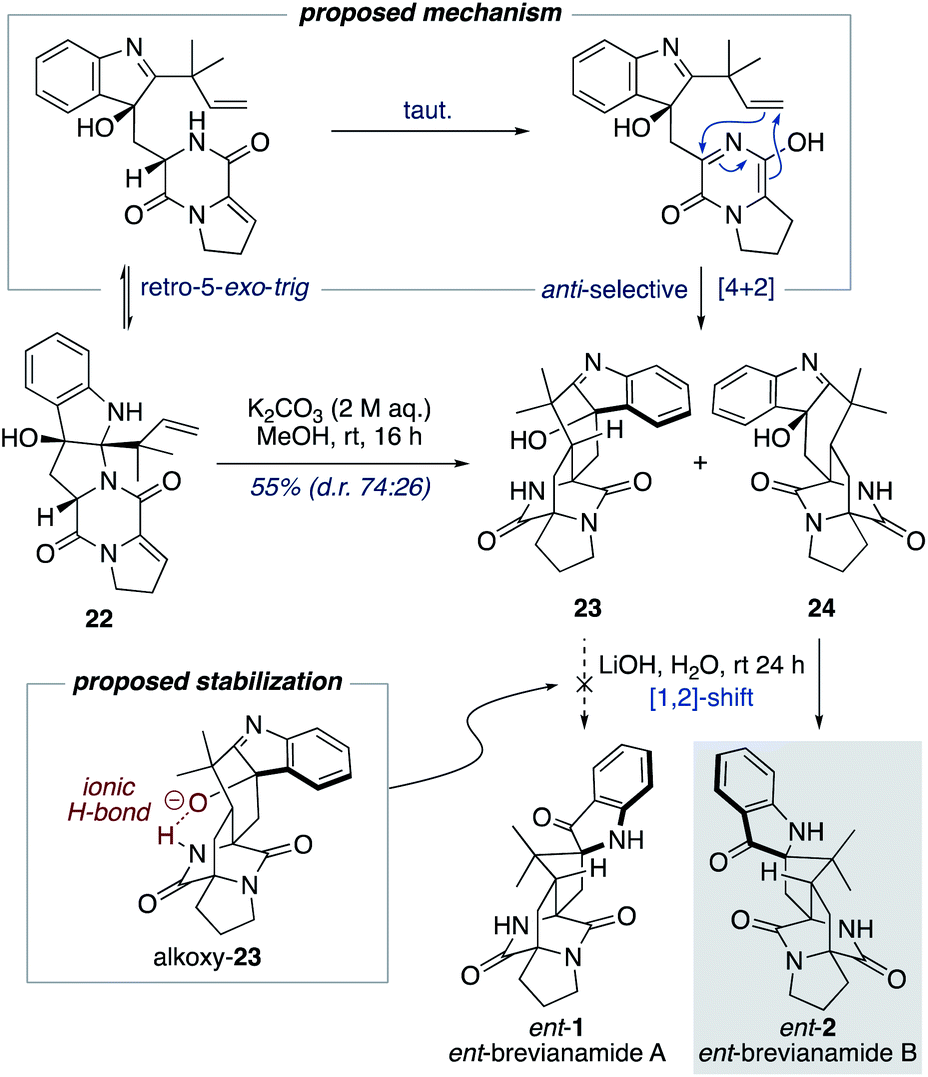 | ||
| Scheme 6 Mechanistic investigations into the final biomimetic cascade reaction. | ||
The close agreement between the diastereoselectivity observed in our biomimetic Diels–Alder reaction (d.r. 93:7) and the ratio in which brevianamides A (1) and B (2) are isolated from P. brevicompactum (appx. 90:10) prompted us to speculate that “this Diels–Alder reaction might be a spontaneous process that occurs naturally without direct enzyme participation”.9 Our proposal of a Diels–Alderase-free biosynthesis, based purely on synthetic observations, aligns with the subsequent biosynthetic studies reported by Williams, Sherman, Li and co-workers.10 This result underlines the important role synthetic studies can have for our understanding of reactivity and selectivity in biosynthetic pathways.
During the preparation of this manuscript, a second total synthesis of brevianamide A (1) was reported by Smith and Xu.33 In their synthesis, they also relied on an inherently anti-diastereoselective final Diels–Alder reaction to complete an impressive seven-step synthesis of brevianamide A (1).
Total synthesis of brevianamide Y
Although brevianamide Y (4) was only identified as a natural product in 2017,8 it was prepared in racemic-form a decade earlier by Williams and co-workers in a 14-step, non-targeted synthesis (Scheme 7a).12g,h During a screen of conditions to oxidize indole 6 to give (±)-brevianamide B (2), Williams found that using oxaziridine 25 followed by treatment with 2 M HCl, to facilitate the hydroxyindolenine to oxindole rearrangement, gave exclusive formation of oxindole 4 (i.e., (±)-brevianamide Y). In 2015, Qin and co-workers reported a targeted 18-step synthesis of the unnatural (−)-enantiomer of brevianamide Y (ent-4), as a simplified analogue of the related natural product, (−)-versicolamide B (Scheme 7b).34 An indoline intermediate 26, which was accessed through Diels–Alder chemistry, was oxidized over two steps to give ent-4. In 2020, as part of their detailed biosynthetic studies, Williams, Sherman, Li and co-workers achieved a 12-step synthesis of brevianamide Y (4) (Scheme 7c).10 This targeted synthesis involved a biomimetic Diels–Alder reaction of oxindole 11, which aligns with our biosynthetic speculation (Scheme 3b). We envisaged that an extension of our synthetic strategy could provide an even shorter route to brevianamide Y (4).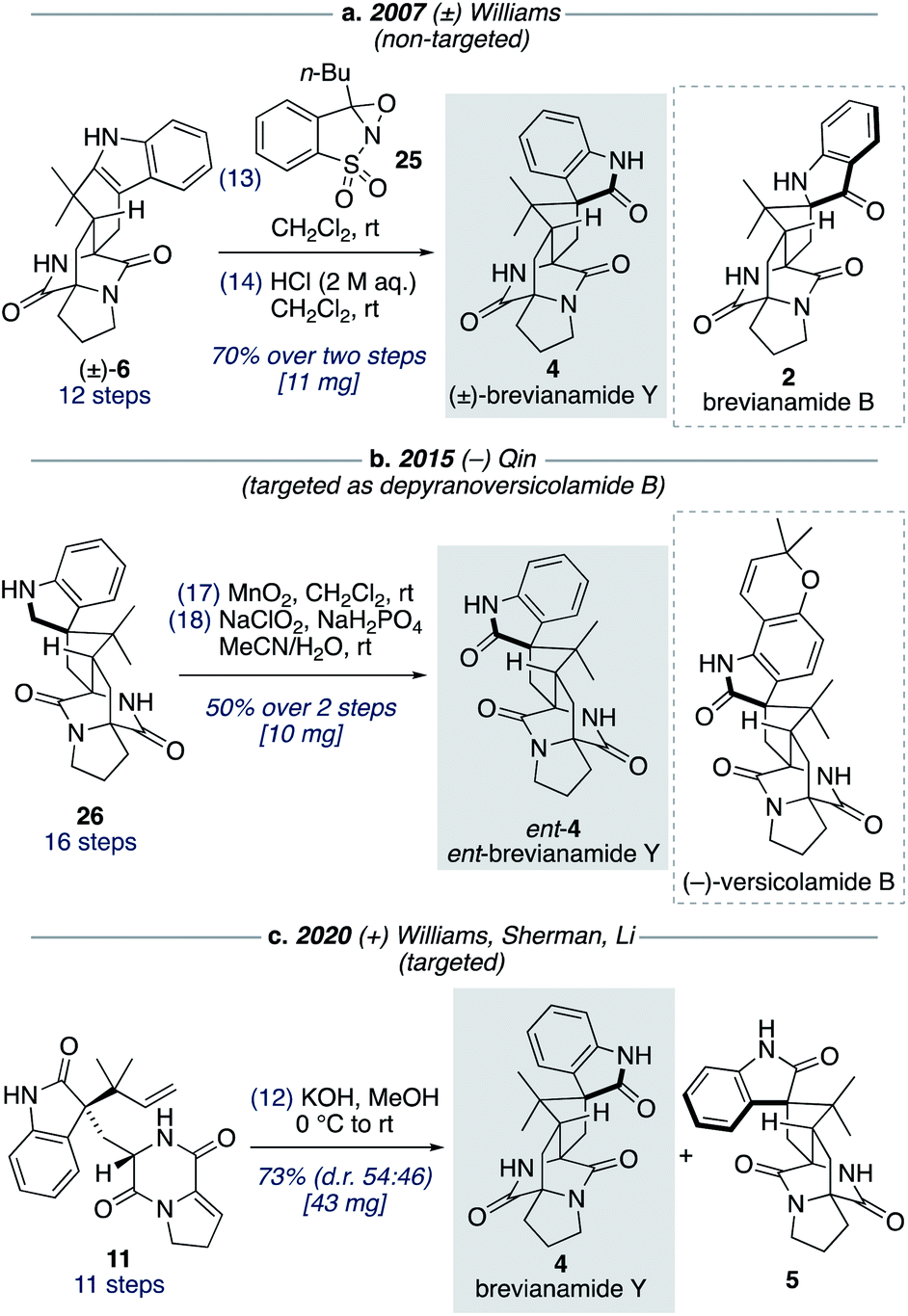 | ||
| Scheme 7 Previous synthetic approaches towards brevianamide Y (4). | ||
Our studies commenced with investigating the rearrangement of (+)-dehydrobrevianamide E (10) to oxindole 11 (Scheme 8, preliminary experiments). Under acidic conditions we were pleased to observe formation of the desired oxindole 11, albeit in low yield. Similarly, we were able to transform the minor diastereomer 22 into the corresponding oxindole 27. However, we had previously observed direct formation of oxindole 11 as a minor side-product when oxidizing (+)-dehydrodeoxybrevianamide E (9) with m-CPBA (Scheme 5). Through further experimentation with different oxidants and acidic additives, we found that use of N-chlorosuccinimide and aqueous trifluoroacetic acid in methanol converted (+)-dehydrodeoxybrevianamide E (9) directly into oxindoles 11 and 27 in excellent yield and with slightly improved diastereoselectivity (d.r. 69:31). Reactions conducted in chloroform gave less clean reactions, and the use of N-iodosuccinimide gave unreliable results. Access to both diastereomers, 11 and 27, was again fortuitous as it enables access to both enantiomers of the target molecule, brevianamide Y (4). When the major oxindole 11 was subjected to LiOH in water at ambient temperature for 24 h (+)-brevianamide Y (4) was formed in 32% combined yield alongside a minor diastereomer 5, which we herein name (+)-brevianamide Z (Scheme 8). Thus, our results alongside the synthetic results obtained by Williams, Sherman, Li and co-workers (Scheme 7c),10 demonstrate that the Sammes-type Diels–Alder reaction of oxindole 12 proceeds with the same inherent anti-diastereoselectivity as indoxyl 7 (Scheme 3a/b). The diastereomeric relationship between brevianamide Y (4) and Z (5) is similar to that which exists between brevianamide A (1) and B (2). That is, they share a common spiro-stereogenic centre and have ‘enantiomeric’ bicyclo[2.2.2]diazaoctane cores. Thus, we propose that (+)-brevianamide Z (5) is an as-yet-undiscovered natural product, worthy of directed isolation efforts. Indeed, if (+)-brevianamide Z (5) is later identified as a minor natural product isolated alongside (+)-brevianamide Y (4), akin to the isolation of B (2) alongside A (1), it would lend support to our biosynthetic proposal and suggest that the biosynthetic Diels–Alder reaction might also be a spontaneous process that occurs without direct enzyme participation. The comparatively lower yield for the final biomimetic transformation towards brevianamides Y (4) and Z (5) (32% yield; Scheme 8) compared to that for brevianamides A (1) and B (2) (63% yield; Scheme 5) is attributable to nonspecific decomposition and associated difficulties encountered with chromatographic purification. The unnatural (−)-enantiomers of brevianamide Y (ent-4) and Z (ent-5) were accessed under identical conditions from the minor oxindole 27. Again, we were not surprised to discover that some erosion of enatiopurity had occurred during these syntheses.35 We discovered that the enantiopurity of (+)-brevianamide Y (4) could be improved through selective crystallization and removal of the racemate. In summary, through our biomimetic studies we have developed a 7-step total synthesis of (+)-brevianamide Y (4), proceeding in 4.1% overall yield and requiring just 4 chromatographic purifications.
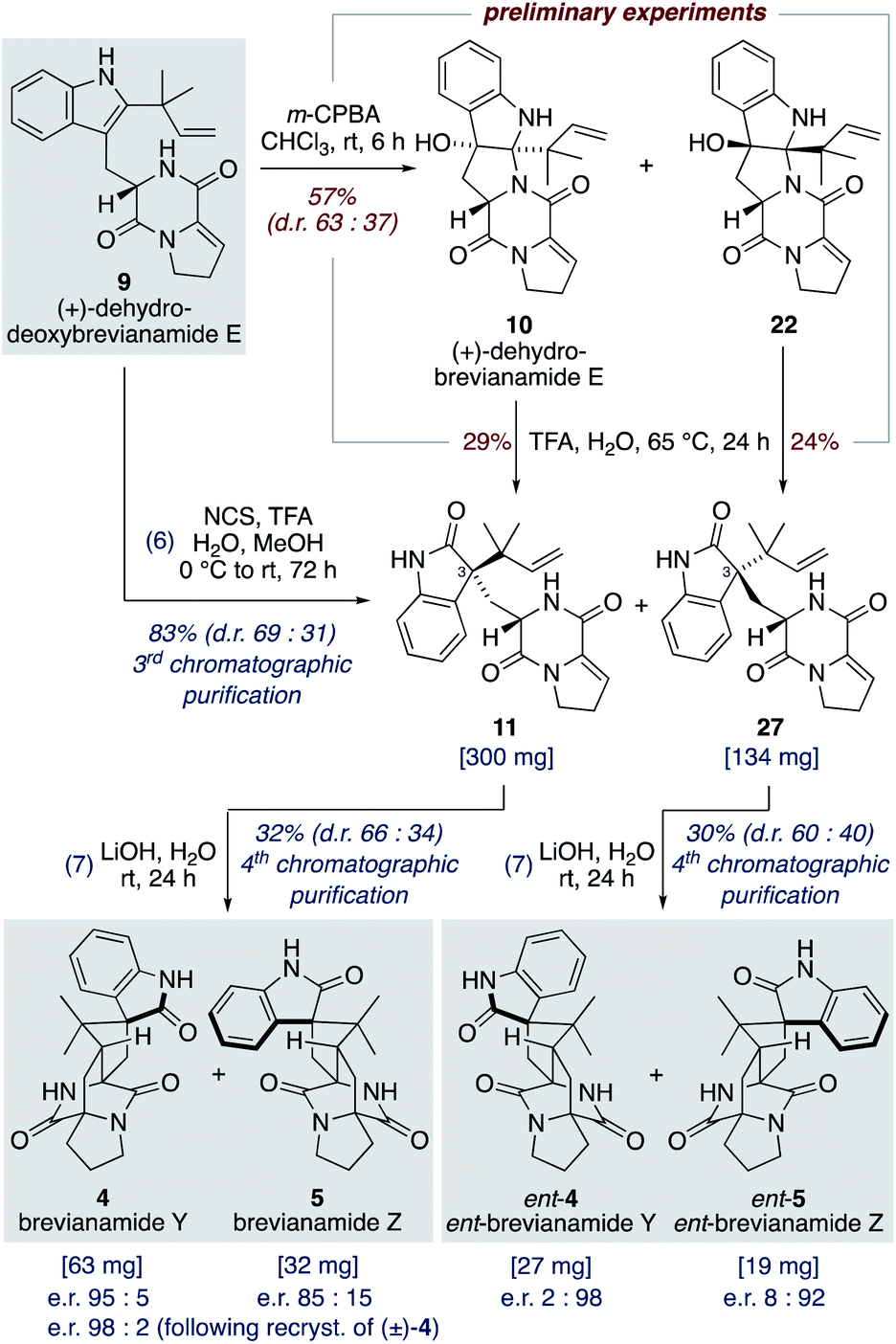 | ||
| Scheme 8 Total synthesis of (+) and (−)-brevianamides Y (4) and Z (5). | ||
Total synthesis of (±)-brevianamide X
We found that for the Diels–Alder cyclization of (+)-dehydrodeoxybrevianamide E (9) Williams' conditions of methanolic KOH gave a better result than our aqueous LiOH conditions (Scheme 9).12 Racemic syn- and anti-configured bicyclo[2.2.2]diazaoctane products, 14 and 6, were isolated in 53% yield with modest syn-diastereoselectivity (d.r. 57:43). The major diastereomer 14 was then oxidized with m-CPBA to give exclusive formation of hydroxyindolenine 28. This crude material, 28, was then immediately treated to acidic conditions to promote the hydroxyindolenine-to-oxindole rearrangement to give (±)-brevianamide X (3) in 50% yield over the two steps. Thus, we have achieved the total synthesis of (±)-brevianamide X (3) in 8 steps and 5.2% overall yield, with just 4 chromatographic purifications required. In 2020, Williams, Sherman, Li and co-workers reported a 12-step synthesis of brevianamide X (3),10 which utilized the same end-game strategy. Our attempts to access brevianamide X (3) in enantioenriched form via kinetic resolution of indole 14, using chiral enantiopure oxidants, have so far failed. Investigations into how (+)-brevianamide X (3) is biosynthesized in enantioenriched form will hopefully provide further inspiration for the development of a chemical synthesis of enantioenriched material.
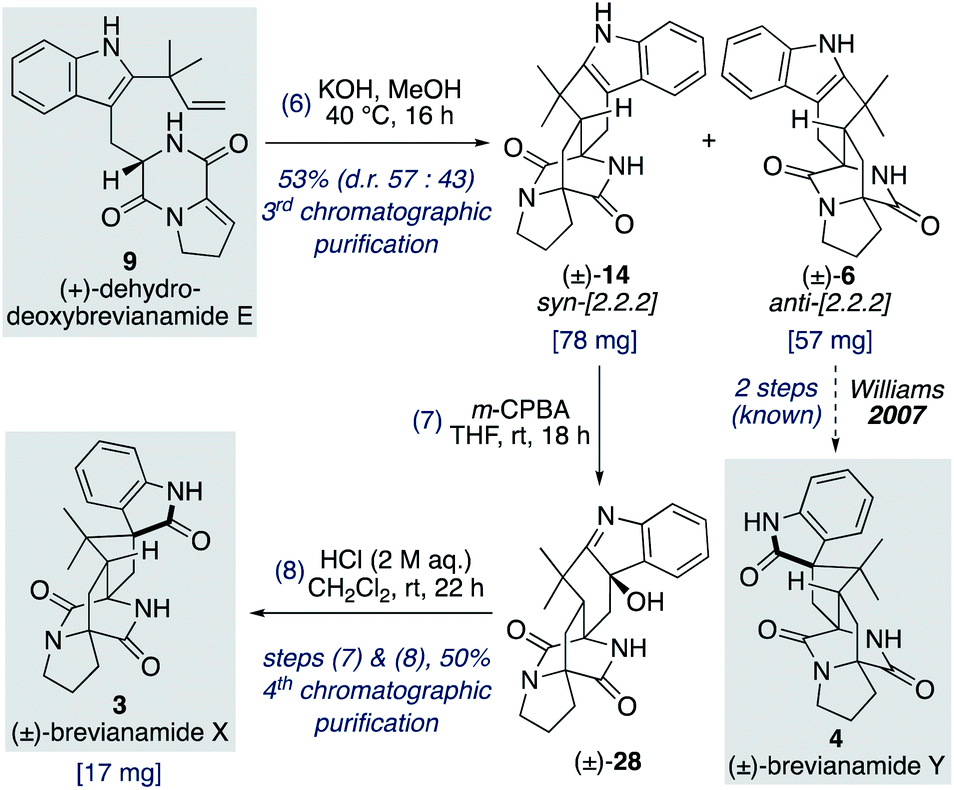 | ||
| Scheme 9 Total synthesis of (±)-brevianamide X (3). | ||
Conclusions
By undertaking a curiosity-driven study to probe the chemical feasibility of several proposed biosynthetic pathways, we have developed a unified synthetic strategy to the bicyclo[2.2.2]diazaoctane brevianamide alkaloids. All our syntheses are either the first and/or shortest reported total syntheses of the target structures, thus demonstrating the enabling power of the biomimetic approach in natural product synthesis. We have also synthesized an as-yet-undiscovered congener, (+)-brevianamide Z (5), which we hope will help direct future isolation studies. Work is ongoing in our laboratories to now further expand this biomimetic strategy to access other bicyclo[2.2.2]diazaoctane alkaloids in similarly step-economical syntheses.Data availability
All experimental procedures and spectral data are available in the ESI.†Author contributions
The manuscript was written through contributions of all authors. R. C. G. and H. E. J. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 759552), a Marie Curie Career Integration Grant (631132, POSIN), and by an EPSRC First Grant (EP/N029542/1). H. E. J. thanks the University of Edinburgh for the provision of a studentship. We thank T. Herlt for assistance and advice regarding chromatography, and acknowledge SIRCAMS at the University of Edinburgh for mass spectrometry. Finally, the authors dedicate this paper to Professor Robert M. Williams (1953–2020), pioneer of the biomimetic approach towards bicyclo[2.2.2]diazaoctane alkaloids.Notes and references
- (a) J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812 CrossRef CAS PubMed; (b) K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Nat. Prod. Rep., 2018, 35, 532 RSC.
- (a) A. P. Robertson, C. L. Clark, T. A. Burns, D. P. Thompson, T. G. Geary, S. M. Trailovic and R. J. Martin, J. Pharmacol. Exp. Ther., 2002, 303, 853 CrossRef PubMed; (b) P. R. Little, A. Hodge, S. J. Maeder, N. C. Wirtherle, D. R. Nicholas, G. C. Cox and G. A. Conder, Vet. Parasitol., 2011, 181, 180 CrossRef CAS PubMed; (c) S. K. Buxton, C. L. Charvet, C. Neveu, J. Cabaret, J. Cortet, N. Peineau, M. Abongwa, E. Courtot, A. P. Robertson and R. J. Martin, PLoS Pathog., 2014, 10, e1003870 CrossRef PubMed.
- S. Martínez-Luisa, R. Rodríguez, L. Acevedo, M. C. González, A. Lira-Rocha and R. Mata, Tetrahedron, 2006, 62, 1817 CrossRef.
- J. Qian-Cutrone, S. Huang, Y.-Z. Shu, D. Vyas, C. Fairchild, A. Menendez, K. Krampitz, R. Dalterio, S. E. Klohr and Q. Gao, J. Am. Chem. Soc., 2002, 124, 14556 CrossRef CAS PubMed.
- H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Angew. Chem., Int. Ed., 2007, 46, 2254 CrossRef CAS PubMed.
- A. J. Birch and J. J. Wright, J. Chem. Soc. D, 1969, 644 RSC.
- (a) B. A. Bird and I. M. Campbell, Appl. Environ. Microbiol., 1981, 42, 521 CrossRef CAS PubMed; (b) B. A. Bird, A. T. Remaley and I. M. Campbell, Appl. Environ. Microbiol., 1982, 43, 345 CrossRef CAS PubMed.
- X. Xu, X. Zhang, X. Nong, J. Wang and S. Qi, Mar. Drugs, 2017, 15, 43 CrossRef PubMed.
- R. C. Godfrey, N. J. Green, G. S. Nichol and A. L. Lawrence, Nat. Chem., 2020, 12, 615 CrossRef CAS PubMed.
- Y. Ye, L. Du, X. Zhang, S. A. Newmister, M. McCauley, J. V. Alegre-Requena, W. Zhang, S. Mu, A. Minami, A. E. Fraley, M. L. Adrover-Castellano, N. A. Carney, V. V. Shende, F. Qi, H. Oikawa, H. Kato, S. Tsukamoto, R. S. Paton, R. M. Williams, D. H. Sherman and S. Li, Nat. Catal., 2020, 3, 497 CrossRef CAS PubMed.
- R. R. M. Paterson, M. J. S. Simmonds, C. Kemmelmeier and W. M. Blaney, Mycol. Res., 1990, 94, 538 CrossRef CAS.
- (a) R. M. Williams, T. Glinka and E. Kwast, J. Am. Chem. Soc., 1988, 110, 5927 CrossRef CAS; (b) R. M. Williams and E. Kwast, Tetrahedron Lett., 1989, 30, 451 CrossRef CAS; (c) R. M. Williams, T. Glinka, E. Kwast, H. Coffman and J. K. Stille, J. Am. Chem. Soc., 1990, 112, 808 CrossRef CAS; (d) R. M. Williams, J. F. Sanz-Cervera, F. Sancenón, J. A. Marco and K. Halligan, J. Am. Chem. Soc., 1998, 120, 1090 CrossRef CAS; (e) R. M. Williams, J. F. Sanz-Cervera, F. Sancenón, J. A. Marco and K. Halligan, Bioorg. Med. Chem., 1998, 6, 1233 CrossRef CAS PubMed; (f) L. A. Adams, M. W. N. Valente and R. M. Williams, Tetrahedron, 2006, 62, 5195 CrossRef CAS; (g) T. J. Greshock and R. M. Williams, Org. Lett., 2007, 9, 4255 CrossRef CAS PubMed; (h) T. J. Greshock and R. M. Williams, Org. Lett., 2012, 14(24), 6377 CrossRef CAS; (i) F. C. Frebault, N. S. Simpkins and A. Fenwick, J. Am. Chem. Soc., 2009, 131, 4214 CrossRef CAS PubMed; (j) F. C. Frebault and N. S. Simpkins, Tetrahedron, 2010, 66, 6585 CrossRef CAS; (k) J. G. Robins, K. J. Kim, A. J. Chinn, J. S. Woo and J. R. Scheerer, J. Org. Chem., 2016, 81, 2293 CrossRef CAS PubMed; (l) J. C. Perkins, X. Wang, R. D. Pike and J. R. Scheerer, J. Org. Chem., 2017, 82, 13656 CrossRef CAS PubMed.
- A. E. A. Porter and P. G. Sammes, J. Chem. Soc. D, 1970, 1103a RSC.
- Q. Dan, S. A. Newmister, K. R. Klas, A. E. Fraley, T. J. McAfoos, A. D. Somoza, J. D. Sunderhaus, Y. Ye, V. V. Shende, F. Yu, J. N. Sanders, W. C. Brown, L. Zhao, R. S. Paton, K. N. Houk, J. L. Smith, D. H. Sherman and R. M. Williams, Nat. Chem., 2019, 11, 972 CrossRef CAS PubMed.
- (a) K. A. Miller, S. Tsukamoto and R. M. Williams, Nat. Chem., 2009, 1, 68 Search PubMed; (b) S. Li, K. Srinivasan, H. Tran, F. Yu, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, S. Tsukamoto, R. M. Williams and D. H. Sherman, Med. Chem. Commun., 2012, 3, 987 RSC.
- L. R. Domingo, J. F. Sanz-Cervera, R. M. Williams, M. T. Picher and J. A. Marco, J. Org. Chem., 1997, 62, 1662 CrossRef CAS.
- Sammes' proposal was originally presented in a manner as depicted in the grey box in Scheme 2 (see ref. 13), which doesn't make explicitly clear if Sammes intended one particular substrate.
- (a) P. S. Steyn, Tetrahedron, 1973, 29, 107 CrossRef CAS; (b) P. M. Scott, B. P. C. Kennedy, J. Harwig and Y.-K. Chen, Appl. Microbiol., 1974, 28, 892 CrossRef CAS PubMed; (c) A. Kaur, H. A. Raja, G. Deep, R. Agarwal and N. H. Oberlies, Magn. Reson. Chem., 2016, 54, 164 CrossRef CAS PubMed.
- The previous identification of 9 as a natural product lends credence to it being a biosynthetic intermediate. However, other related structures, at the same overall oxidation state, are also potentially viable biosynthetic intermediates.
- For recent examples of the rearrangement of hydroxyindolenines to oxindoles, see; (a) S. Han, K. C. Morrison, P. J. Hergenrother and M. Movassaghi, J. Org. Chem., 2014, 79, 473 CrossRef CAS PubMed; (b) A. Umehara, H. Ueda and H. Tokuyama, Org. Lett., 2014, 16, 2526 CrossRef CAS PubMed; (c) E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318 CrossRef CAS PubMed; (d) N. David, R. Pasceri, R. R. A. Kitson, A. Pradal and C. J. Moody, Chem. - Eur. J., 2016, 22, 10867 CrossRef CAS PubMed; (e) E. Schendera, S. Lerch, T. von Drathen, L.-N. Unkel and M. Brasholz, Eur. J. Org. Chem., 2017, 22, 3134 CrossRef; (f) B. Alcaide, P. Almendros, E. Busto, F. Herrera, C. Lázaro-Milla and A. Luna, Adv. Synth. Catal., 2017, 359, 2640 CrossRef CAS; (g) S. Jia, G. Dong, C. Ao, X. Jiang and W. Hu, Org. Lett., 2019, 21, 4322 CrossRef CAS PubMed.
- For reviews on enzymatic catalysis of Diels–Alder reactions, see; (a) E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078 CrossRef CAS PubMed; (b) H. Oikawa and T. Tokiwano, Nat. Prod. Rep., 2004, 21, 321 RSC; (c) K. Klas, S. Tsukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672 CrossRef CAS PubMed; (d) A. Minami and H. Oikawa, J. Antibiot., 2016, 69, 500 CrossRef CAS PubMed; (e) C. S. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. N. Houk, Nat. Prod. Rep., 2019, 36, 698 RSC.
- For an example of a previously proposed biosynthetic kinetic resolution, see: N. J. Lajkiewicz, S. P. Roche, B. Gerard and J. A. Porco Jr, J. Am. Chem. Soc., 2012, 134, 13108 CrossRef CAS PubMed.
- H. Liu, V. R. Pattabiraman and J. C. Vederas, Org. Lett., 2007, 9, 4211 CrossRef CAS PubMed.
- J. M. Schkeryantz, J. C. G. Woo, P. Siliphaivanh, K. M. Depew and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11964 CrossRef CAS.
- L. Zhao, J. P. May, J. Huang and D. M. Perrin, Org. Lett., 2012, 14, 90 CrossRef CAS PubMed.
- J. W. Fisher and K. L. Trinkle, Tetrahedron Lett., 1994, 35, 2505 CrossRef CAS.
- P. Huy, J.-M. Neudörfl and H.-G. Schmalz, Org. Lett., 2011, 13, 216 CrossRef CAS PubMed.
- T. A. Nigst, A. Antipova and H. Mayr, J. Org. Chem., 2012, 77, 8142 CrossRef CAS PubMed.
- T. Kametani, N. Kanaya and M. Ihara, J. Am. Chem. Soc., 1980, 102, 3974 CrossRef CAS.
- K. Sugimoto, Y. Sadahiro, I. Kagiyama, H. Kato, D. H. Sherman, R. M. Williams and S. Tsukamoto, Tetrahedron Lett., 2017, 58, 2797 CrossRef CAS PubMed.
- A. J. Birch and J. J. Wright, Tetrahedron, 1970, 26, 2329 CrossRef CAS PubMed.
- (a) M. Meot-Ner, Chem. Rev., 2005, 105, 213 CrossRef CAS PubMed; (b) M. Meot-Ner, Chem. Rev., 2012, 112, PR22 CrossRef PubMed.
- F. Xu and M. W. Smith, Chem. Sci., 2021, 12, 13756 RSC.
- W.-F. Qin, T. Xiao, L.-F. Deng, Y. Wang and Y. Qin, Chem. Commun., 2015, 51, 16143 RSC.
- The significant difference in enantiopurity for isolated brevianamide Y (4/ent-4) and Z (5/ent-5) is potentially a result of either self-disproportionation of enantiomers occurring during the chromatographic purifications, or an unexpected non-linear effect in the reaction, see; (a) J. Han, O. Kitagawa, A. Wzorek, K. D. Klika and V. A. Soloshonok, Chem. Sci., 2018, 9, 1718 RSC; (b) H. B. Kagan, Nonlinear Effects in Asymmetric Catalysis, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, Hoboken, NJ, 2007, pp. 207–219 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data for all compounds, and mechanistic studies. See DOI: 10.1039/d1sc05801k |
| ‡ These authors contributed equally. |
| § Present address: MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2022 |