
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymers as advanced antibacterial and antibiofilm agents for direct and combination therapies
Zhangyong
Si
a,
Wenbin
Zheng
a,
Dicky
Prananty
 a,
Jianghua
Li
a,
Chong Hui
Koh
a,
En-Tang
Kang
b,
Kevin
Pethe
cd and
Mary B.
Chan-Park
*ace
a,
Jianghua
Li
a,
Chong Hui
Koh
a,
En-Tang
Kang
b,
Kevin
Pethe
cd and
Mary B.
Chan-Park
*ace
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459, Singapore. E-mail: MBEChan@ntu.edu.sg
bDepartment of Chemical & Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Kent Ridge, Singapore 117585, Singapore
cLee Kong Chian School of Medicine, Nanyang Technological University, Singapore 636921, Singapore
dSchool of Biological Sciences, Nanyang Technological University, Singapore 637551, Singapore
eSchool of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
First published on 16th December 2021
Abstract
The growing prevalence of antimicrobial drug resistance in pathogenic bacteria is a critical threat to global health. Conventional antibiotics still play a crucial role in treating bacterial infections, but the emergence and spread of antibiotic-resistant micro-organisms are rapidly eroding their usefulness. Cationic polymers, which target bacterial membranes, are thought to be the last frontier in antibacterial development. This class of molecules possesses several advantages including a low propensity for emergence of resistance and rapid bactericidal effect. This review surveys the structure–activity of advanced antimicrobial cationic polymers, including poly(α-amino acids), β-peptides, polycarbonates, star polymers and main-chain cationic polymers, with low toxicity and high selectivity to potentially become useful for real applications. Their uses as potentiating adjuvants to overcome bacterial membrane-related resistance mechanisms and as antibiofilm agents are also covered. The review is intended to provide valuable information for design and development of cationic polymers as antimicrobial and antibiofilm agents for translational applications.
1. Introduction
Antibiotics underpin most aspects of modern medicine including chemotherapies, surgery, organ transplants and implanted medical devices. However, bacterial infections caused by multi-drug resistant (MDR) organisms are increasingly common. Of particular concern are ESKAPE bacteria which are the Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species that are among the most virulent and resistant nosocomial bacteria commonly associated with antimicrobial resistance.1 The pressing issue of Antimicrobial Resistance (AMR) is aggravated by the slow pace of antibiotic development.2 The discovery of new antibiotic classes has been slow with only a handful of approved drugs active against Gram-positive pathogens, but none against Gram-negative bacteria, discovered in the past 70 years. Classical antibiotics mainly target processes essential for bacterial growth such as syntheses of nucleic acids, proteins, or cell-wall components as well as folate metabolism.3 Emergence of spontaneous resistance to antibiotics acting on specific targets is inevitable.4 Novel concepts are needed to combat AMR through new agents with different mechanisms of action and lower propensity to elicit resistance.Antimicrobial peptides (AMPs) are a class of alternative antimicrobial agents that may potentially replace antibiotics in the therapy of bacterial infections. AMPs such as magainin-2 and β-defensin 3 are membrane-lytic agents and resistance evolution in bacteria against these has been extremely slow.5,6 The pioneering works of Tew,7 Gellman,8 DeGrado,9 and Ikeda et al.10 led to the discovery that polymers with cationic and hydrophobic substituents resemble AMPs in their killing in that these cationic/hydrophobic polymers electrostatically bind to bacterial cytoplasmic membrane or outer membrane of, respectively, Gram-positive and Gram-negative bacteria to form pores or deformations in the membranes resulting in bacterial killing.
Since about two decades ago, many antibacterial polymers have been reported.11,12 It is generally understood that the presence of both cationic and hydrophobic residues, amphiphilicity, charge type, molecular weight, morphology, position of cationic charge (i.e., side-chain versus main-chain) all affect the bactericidal and toxicity properties. Like AMPs, antimicrobial polymers (AMPos) are attractive alternatives to antibiotics because they generally have much lower tendency to evolve resistance in bacteria. Polymers have a rich parameter space in terms of variations in various properties, such as backbone chemistry, biodegradability, morphology, cationic and hydrophobic groups, etc., which have been widely explored as synthetic mimics of antimicrobial peptides to achieve good antibacterial activities coupled with low toxicity.13,14 The main classes of selective antibacterial polymers, reported by the groups of Deming,15 Chan-Park,16 Chen,17 Qiao,18 Gellman,8 Liu,19 Yang,20 Tew,9 DeGrado et al.,9 and others, include poly(alpha-peptides), poly(beta-peptides), polycarbonates, polynorbornenes and polymethacrylates. These works have produced much understanding for tuning the efficacy and safety metrics of (a) antibacterial properties, measured mainly through minimum inhibitory concentrations to eradicate 90% of bacteria (MIC); (b) toxicity towards red blood cells (RBC), measured by hemolytic concentrations, e.g. causing 50% RBC lysis (HC50); (c) inhibitory concentration towards representative mammalian cells (e.g. fibroblasts or kidney cells) (IC50) and (d) figures of merit, termed “selectivity,” typically expressed as HC50/MIC90 or/and IC50/MIC90. To date, the translation of antimicrobial polymers into therapies has been hampered by insufficient selectivity. Only a few groups have reported the translation of these polymers into animal studies. Alternatively, combination therapies with synthetic polymers hold promise to fight bacterial infections by reducing killing dose while limiting emergence of resistance.21 Two other major challenges to translation are the biofilm and persister states of bacteria,22,23 which are important in real clinical settings; these can respond to antimicrobial agents/antibiotics very differently than the planktonic state bacteria employed in early-stage in vitro MIC measurements. Also, real applications in human therapy or in consumer products or agri-/aqua-culture settings invariably must contend with complex environments, such as the presence of proteins or anionic molecules that can impair the efficacy of cationic antimicrobial polymers. Some progress has been made to achieve success in animal studies and in more realistic settings. This review seeks to summarize the potential of polymers in the fight against AMR in more advanced animal studies, but is by no means comprehensive. We shall summarize these advanced antimicrobial polymers in direct monotherapy, as adjuvants in combination therapies and as smart bacteria-responsive systems.
2. Bacterial versus mammalian cell membranes and selectivity
Bacteria are classified into two main categories based on their cell envelope structures: Gram-positive and Gram-negative.24 All bacteria have a cytoplasmic membrane surrounded by a cell wall made of crosslinked peptidoglycan. The peptidoglycan layer provides mechanical support but is highly porous, allowing the passage of molecules as large as 30–57 kDa.25 In Gram-positive bacteria, the cell wall also contains wall teichoic acids (WTA) (Fig. 1a(1)) and lipoteichoic acids (LTA) (Fig. 1a(2)), both of which have a net negative charge under physiological conditions.26 The plasma membrane of Gram-positive bacteria is predominantly constituted of anionic lipids such as phosphatidylglycerol (PG) (Fig. 1b(1)) and cardiolipin (CL) (Fig. 1b(2)), with little content of zwitterionic phospholipids such as phosphatidylethanolamine (PE) (Fig. 1a(3)).27,28 Gram-negative bacteria have an outer membrane encapsulating their thin peptidoglycan layer. The outer leaflet contains lipopolysaccharides (LPS) (Fig. 1a(3)), while the inner leaflet is made of phospholipids similar to those found in the plasma membrane.29,30 LPS are anionic macromolecules with three characteristic sub-structures: lipid A, core oligosaccharide and O-antigen repeats. Lipid A has 2 glucosamine residues linked by a β-1,6 linkage and one phosphate group on each sugar residue, which is conserved amongst Gram-negative bacteria. The phosphate groups in lipid A crosslink with the divalent cations Ca2+ and Mg2+ to overcome their mutual electrostatic repulsion. This forms a penetration barrier to hydrophobic compounds,31 allowing Gram-negative bacteria survival in harsh environments. Gram-negative cytoplasmic membranes are composed mainly of zwitterionic phosphatidylethanolamine (PE) (Fig. 1b(3)), which constitutes more than 60% of the membrane lipids, with substantial fractions of anionic PG (Fig. 1b(1)) and CL (Fig. 1b(2)).27,28 In contrast to the anionic cell envelopes of bacteria, mammalian cells have cytoplasmic membranes that are composed mainly of neutrally charged lipids phosphatidylcholine (PC) (Fig. 1b(4)) and PE (Fig. 1b(3)).32 The charge differential between near neutral mammalian cell cytoplasmic membrane and negatively charged Gram-negative bacteria and Gram-positive bacteria is the basis of the selectivity of AMPs and AMPos, but the differences are small and cationic AMPs and AMPos which are highly effective against bacteria tend to also be excessively toxic to mammalian cells.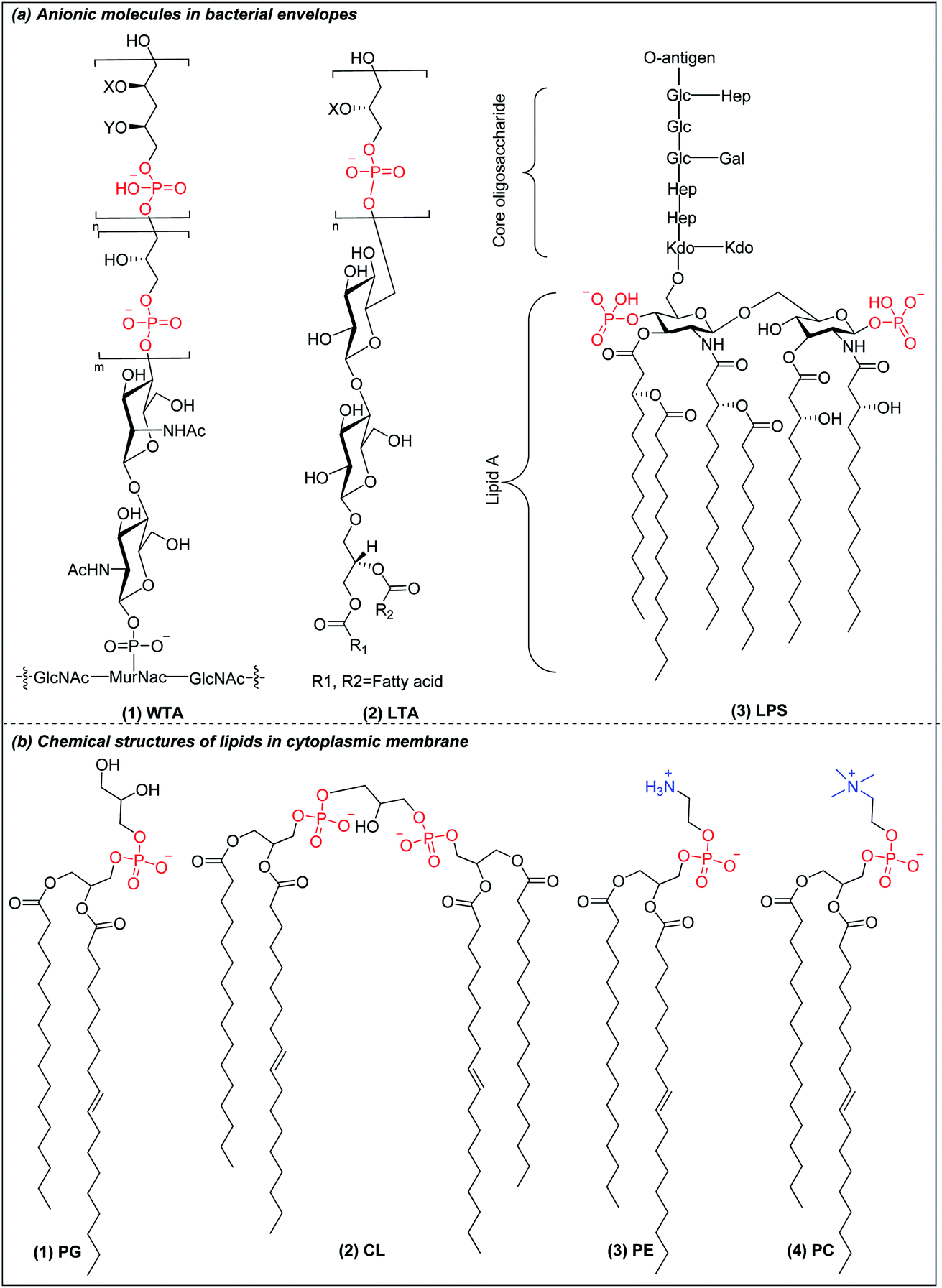 | ||
| Fig. 1 (a) Chemical structure of anionic molecules in bacterial envelopes. (b) Chemical structures of cytoplasmic lipids in bacteria and mammalian cells. | ||
Nearly all multicellular organisms produce cationic antimicrobial peptides (AMPs) that selectively target bacterial membrane based on the charge difference between eukaryotic cells and bacteria.6,33 These molecules are typically 12–50 amino acids in size, amphiphilic, contain cationic residues (i.e., arginine or lysine) and hydrophobic portions. This composition helps AMPs to attach to the bacterial membrane via electrostatic interaction, then fold into an amphiphilic structure that inserts into the membrane, resulting in membrane disruption and bacterial death. The evolution of bacterial resistance to membrane-targeting AMPs is slow as their mechanism of action does not rely on a specific protein target. Nevertheless, the translation of AMPs from research discovery to clinical application has had limited success and very few AMPs have been approved for clinical applications.34 Currently, there are only five AMPs approved in clinical use as alternatives to antibiotics: nisin, gramicidin, polymyxins, daptomycin and melittin. The challenges of AMPs towards clinical application are their limited efficacy, mammalian cell toxicity, peptide instability and high production cost.35,36 To overcome these limitations, more selective antimicrobial polymers with higher constitutional and global compositional diversity need to be discovered and design principles for low MICs and high selectivity need to be uncovered; these specifics will generally depend on the specific class of polymers under investigation. AMPos have high compositional diversity and can be polyamides like AMPs but can also be derived from other functional classes like polycarbonate, polyurethanes, polyimidazoliums etc. and can be linear or have more complex structure, such as star copolymers. A few AMPos have been found with high in vivo selectivity; these include certain molecules in the classes of poly(alpha-peptides), poly(beta-peptides), polycarbonates, star polymers and main-chain cationic polymers.
3. Polymers as direct antibacterial agents
3.1 Poly(α-amino acids)
Deming pioneered α-amino acid N-carboxyanhydrides (NCA) polymerization with transition metal or primary amine as initiator to prepare poly(α-amino acids) with controlled chain length, sequence, composition and polydispersity (Scheme 1a).37–39 His group pioneered peptide synthesis and assembly into vesicles and various novel supramolecular structures.40,41 In 2009, Chan-Park et al. first applied NCA polymerization to prepare a random copolymer, P(K10F15) (Fig. 2a(1)), which effectively eradicated various bacteria. Their optimum peptide consists of 40% polylysine as charged residues together with 60% polyphenylalanine as hydrophobic residues, and showed better antimicrobial potency than naturally occurring AMPs, such as Defensin and Magainin I.42 However, P(K10F15) also caused toxicity to human erythrocytes due to strong non-specific hydrophobic interactions. To improve the biocompatibility, the same group made, via NCA polymerization, a class of antimicrobial peptidopolysaccharide, the graft copolymer chitosan-graft-oligolysine (CS-g-K16) (Fig. 2a(2)). The chitosan in the peptidopolysaccharide mimics bacterial cell wall peptidoglycan and CS-g-K16 displayed high antimicrobial potency against clinically important Gram-negative and Gram-positive bacteria while being non-hemolytic.16 Recently, by reducing the oligolysine chain length from K16 to K5 and the chitosan (CSM) backbone chain length, they prepared a new peptidopolysaccharide in this class, graft copolymer CSM5-K5 (Fig. 2a(3)),43 which eradicated S. aureus in a murine wound study. These graft copolymers of polylysine with chitosan, which have no hydrophobicity, can eradicate Gram-positive bacteria with low toxicity. In spite of having no hydrophobic interactions, CSM-K5 assembles into nanoparticles, due to hydrogen bonding from the chitosan backbones. The nanoparticle assembly concentrates the cationic charge of the oligolysine moiety, leading to stronger membrane perturbation and better bacterial killing. These chitosan-graft-oligolysine complexed and clustered with anionic lipids in bacterial membrane, which lead to membrane phase defects and then membrane lysis via “carpet model” mechanism when a threshold concentration is reached.16,43 Selectivity is improved due to lack of hydrophobicity to avoid insertion into the plasma membrane of mammalian cells.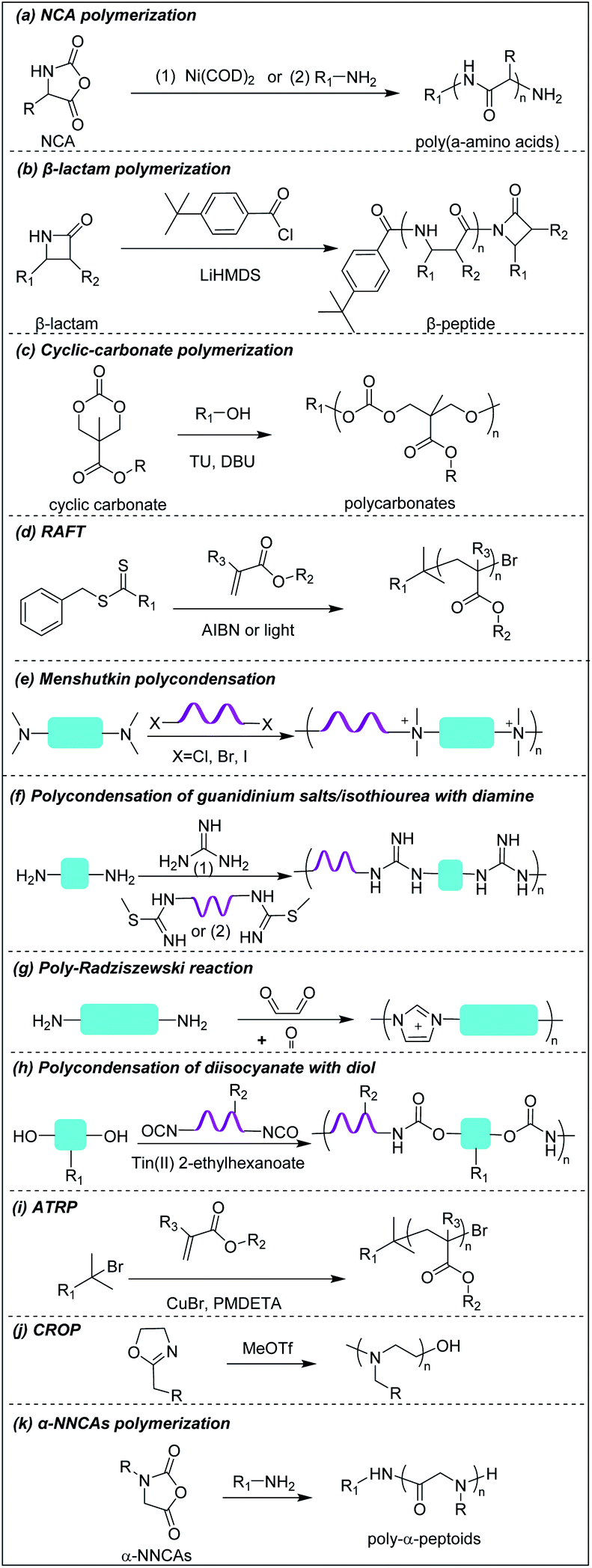 | ||
| Scheme 1 Synthetic schemes to prepare cationic polymers. | ||
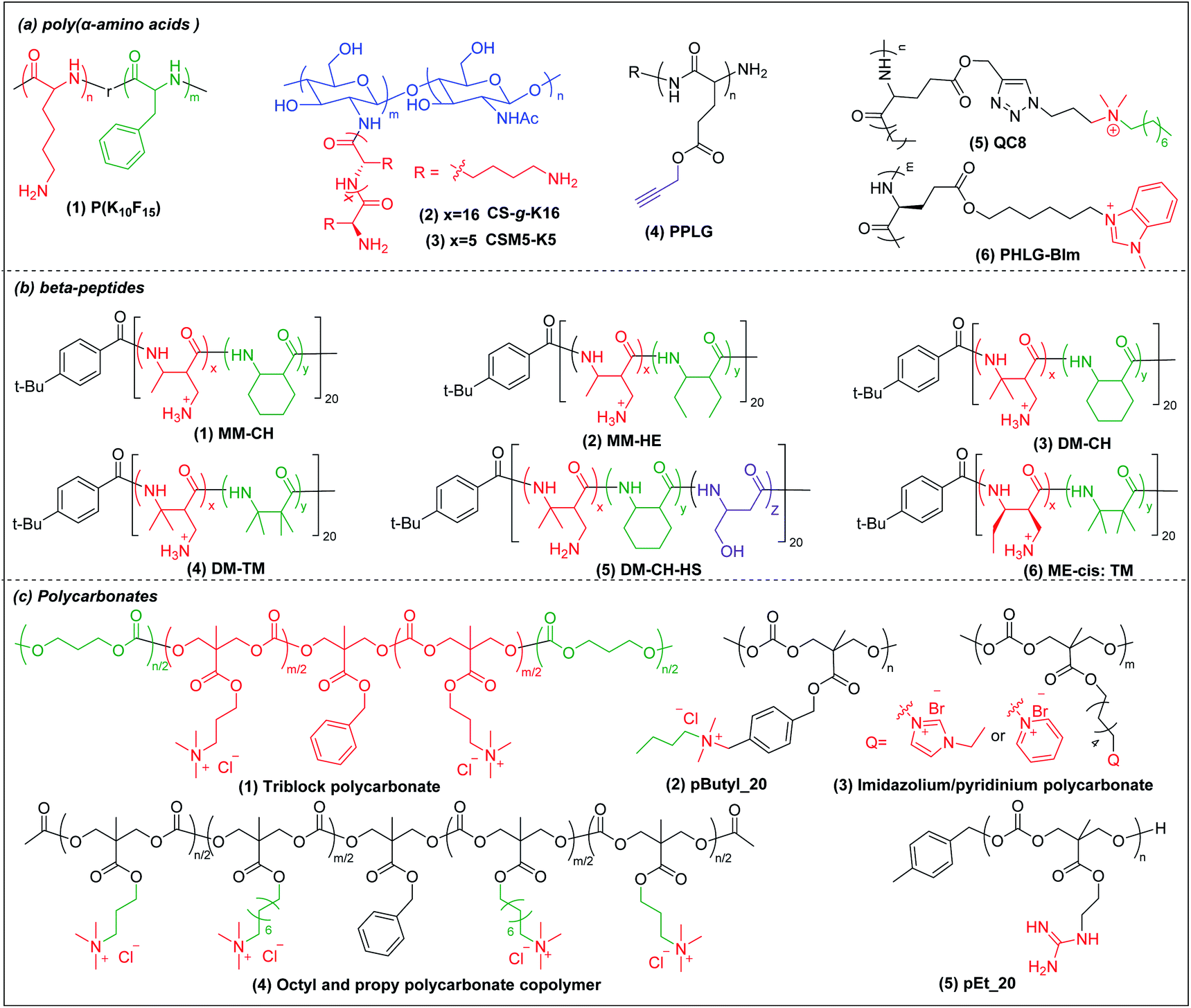 | ||
| Fig. 2 Chemical structures of cationic antimicrobial (a) poly(α-amino acids), (b) β-peptides, and (c) polycarbonates. | ||
Hammond et al. developed, via NCA polymerization, a clickable peptide backbone with a pendant alkyne group (PPLG) (Fig. 2a(4)) and investigated its antimicrobial activity by tailoring the side chains via click chemistry.44,45 The polypeptides started to show bacteria-killing efficacy when the side-chain alkyl length increased to eight carbon (QC8) (Fig. 2a(5)) and exhibited good biocompatibility with no hemolytic effect. Cheng et al. developed, via NCA polymerization, an α-helical polypeptide (PHLG-BIm) (Fig. 2a(6)) with radial amphiphilicity which exhibited lower MIC than the compositionally similar facially amphiphilic peptide.17 The shielding of the hydrophobic helical core minimized the self-aggregation and reduced the interaction with blood proteins, which led to its superior stability in physiological conditions.
3.2 Cationic β-peptides
Compared to α-peptides, β-peptides have an extra methylene group in the backbone. This endows β-peptides with increased flexibility allowing the formation of different types of secondary structures to exhibit amphiphilic conformations, which is one of the key components that contributes to the antimicrobial activity of β-peptides.46–48 Another outstanding feature of β-peptides is their resistance to lysis by proteases, with consequent improved in vivo stability.49,50 In addition, they are usually non-mutagenic.51,52 These properties make β-peptides attractive as candidate antibacterial agents.Inspired by natural AMPs such as magainin,53 Degrado and Gellman et al. first made, via solid-phase synthesis, sequence-defined helical β-peptides with facial amphiphilicity which displayed antimicrobial potency comparable to magainin.54,55 However, the step-by-step coupling reaction by solid-phase synthesis is less efficient, resulting in high-manufacturing cost. Gellman et al. designed and synthesized a new series of antibacterial β-peptide oligomers (also called nylon-3) combining positively charged and hydrophobic residues, via anionic ring-opening polymerization (AROP) of β-lactam monomers (Scheme 1b),8 which formed induced globally amphiphilic conformations when interacting with negatively charged bacterial membranes. The optimized polymer had 63% cationic residues (MM) and 37% hydrophobic residues (CH) with 20 total repeat units in its backbone (MM-CH) (Fig. 2b(1)). It showed antimicrobial activity comparable to the classical AMP magainin II against a panel of bacteria with a significant improvement in biocompatibility compared to magainin II, achieving a high selectivity index of 32.8 Other parameters that might influence the antimicrobial activity and biocompatibility were also investigated, including different cationic and hydrophobic monomers, terminal groups, polymerization degrees and polydispersity.56 Higher molecular weight and higher polydispersity resulted in increased toxicity when the polymerization degree exceeded 30 units. Also, a balance between cationic residues and hydrophobic residues was required to achieve optimum selectivity.
By replacing the cyclic hydrophobic subunits CH with acyclic HE, with the same hydrophobicity (Fig. 2b(2)), both the toxicity and antimicrobial activity decreased, which indicated that the backbone flexibility was important to its biological activity.57 This conclusion was further supported by a study on DM-CH copolymers (Fig. 2b(3)), which showed non-selective toxicity towards both bacteria and erythrocytes.35,37 When the hydrophobic subunits were switched from CH to TM (Fig. 2b(4)), with the same hydrophobicity but with more constrained backbone, the copolymers were found to be less toxic to erythrocytes (HC10 = 400 μg ml−1) without compromising their ability to kill bacteria.58 Conventional AMP mimetics, based on the balance of cationicity and hydrophobicity, show a collective shift between efficacy and toxicity. In order to dissect these two effects, Gellman et al. designed ternary β-peptide copolymers by introducing a third polar, uncharged residue (Fig. 2b(5)).59 Their results showed that replacing a small portion of cationic or hydrophobic residues with neutral-polar residues had a minor influence on antimicrobial potency, but decreased toxicity significantly. Beyond amphiphilicity, the stereochemistry of the monomers significantly influenced toxicity towards eukaryotic cells,60 which may be related to the different conformational propensities of different enantiomeric monomers: ME-cis: TM (Fig. 2b(6)) with extended conformations showed less toxicity to erythrocytes than the corresponding β-peptides with trans-conformation.
Chan-Park and Pethe et al. prepared two glycosylated block co-beta peptides that were the first to demonstrate in vivo eradication of bacteria;61,62 these will be further discussed in Sections 4 and 5 below.
3.3 Cationic polycarbonates
Polycarbonates are attractive antimicrobial polymers due to their intrinsic biocompatibility, biodegradability and tunable chemical structures.63 Yang and Hedrick pioneered antibacterial polycarbonates and developed a versatile synthetic method based on ring-opening polymerization of cyclic carbonate monomers via organocatalysis (Scheme 1c). They first prepared a cationic triblock antimicrobial polycarbonate (Fig. 2c(1)) consisting of one cationic block, PMTC (5-methyl-2-oxo-1,3-dioxane-5-carboxylate, red in the figure), and two hydrophobic PTMC (trimethylene carbonate, green in the figure) blocks, which drove the polymer to self-assemble into nanoparticles.20 The nanoparticles interact with and kill bacteria more effectively than individual polymer chains due to increased local charge concentration, giving low MICs (4.3–10.8 μM) against Bacillus subtilis, S. aureus, methicillin-resistant S. aureus (MRSA), E. faecalis and Cryptococcus neoformans. This triblock antimicrobial polycarbonate did not induce significant toxicity in mice after intravenous injection, which may be due to degradation of the polycarbonate backbone in vivo. However, its in vivo antimicrobial potency was not studied. To increase the structural diversity with degradable polycarbonate backbone, Yang et al. designed a polycarbonate with pendant benzyl chloride group that can be converted into quaternary ammonium or phosphonium group, or azide group, to allow for further post-functionalization using click chemistry.64 Applying this strategy, they prepared antimicrobial polycarbonates with cationic and hydrophobic residues in the same center (pButyl_20) (Fig. 2c(2)).65 Unlike other antimicrobial polymers with separated cationic and hydrophobic residues in different backbone units, incorporation of cationic and hydrophobic residues into the same center allows fine tuning of amphiphilicity, achieving high selectivity. Compared to quaternary ammonium as cationic group, imidazolium and pyridinium (Fig. 2c(3)) grafted polycarbonates have better antimicrobial activity against various bacterial strains.66 The length of hydrophobic linker between the cationic groups to the polymer backbones also affects the antimicrobial activity and biocompatibility of cationic polycarbonates. The longest hydrophobic linker (octyl) gave the best antimicrobial activity but also the most toxicity, whereas the shortest hydrophobic linker (propyl) had the opposite effect. Combining the long and short linkers by copolymerization of two cyclic carbonate monomers with octyl and propyl substitutions produced a non-toxic polycarbonate with broad-spectrum antibacterial activity (Fig. 2c(4)).67 Recently, Yang et al. prepared a polycarbonate (pEt_20, Fig. 2c(5)) with guanidine group as the cationic charge and ethyl as hydrophobic linker, which showed excellent antimicrobial activity in vitro and in vivo with an extremely high in vivo therapeutic index, i.e. ratio of ED50/LD50 (where ED50 is effective dose to cause 50% survival of infected mice and LD50 is lethal dose to cause 50% mice mortality because of polymer toxicity), of 1473.68 The pEt_20 polymer can eradicate MDR ESKAPE bacterial infections in vivo with negligible toxicity. Guanidinium can form stable multidentate hydrogen bonds with membrane phosphate groups,69 which contributes to its high antimicrobial potency. The polycarbonate backbone can be completely degraded into non-toxic products in 3 days after injection into mice, explaining its excellent in vivo biocompatibility. The combination of these two properties makes pEt_20 promising as a potential therapeutic agent.3.4 Star cationic polymer
Star polymers refer to macromolecules with branched architecture, bearing linear “arms” radiating from a core, which may be used to increase the antimicrobial potency of cationic polymers via increasing local concentration of cationic charge.18,70 Star polymers are synthesized following mainly three approaches: core-first, arm-first, and grafting onto.71 Of these, the core-first approach is most frequently used to prepare AMP mimics. Qiao et al. synthesized “structurally nanoengineered antimicrobial peptide polymers” (SNAPPs) (Fig. 3a(1)) via core-first approach with poly(amido amine) (PAMAM) dendrimers as core and lysine and valine copolymer as the arms.18 Interestingly, SNAPPs maintained a nanoparticle form even with infinite dilution, as it is a unimolecular nanoparticle. This endowed SNAPPs with superior activity compared to their linear lysine/valine peptide arms against all Gram-negative strains tested, even colistin- and multi-drug resistant strains. In addition to directly killing bacteria, SNAPPs can also boost host immune response to enhance their in vivo antimicrobial potency, achieving more than 5 log reductions in bacterial cell counts in A. baumannii induced bacteremia. The low toxicity was possibly due to SNAPPs' weak hydrophobicity together with random coil structure in the lysine/valine arm when interacting with mammalian cells.72 Although the antimicrobial activity of SNAPPs was better than classic AMPs, their potency diminished in the presence of divalent cations and serum protein.73 Arm number and length in star-polymer SNAPPs had a significant influence on their antimicrobial activity. Increasing arm number and length would lead to higher localized concentration of peptide arm and increased α-helical content, which is usually correlated with better membrane permeabilization, resulting in enhanced bactericidal efficacy.74 Yang et al. also synthesized star polycarbonate (Fig. 3a(2)) with broad-spectrum antimicrobial activity and low hemolysis, using the core-first approach with β-cyclodextrin as the core.75 Consistent with star polymer SNAPPs, star polycarbonate showed four-fold lower MIC values than its linear analog. In addition to improving the antimicrobial efficacy, star polymers can also be designed to reduce toxicity. Glucosamine-functionalized miktoarm star polymer (Fig. 3a(3)) was synthesized via arm-first approach by crosslinking the polylysine and polyglucosamine macroinitiator with cross-linker, N,N′-methylenebis(acrylamide), via photo-induced reversible addition–fragmentation chain transfer (RAFT) reactions (Scheme 1d).76 This star polymer demonstrated superior biocompatibility over linear analogs due to the shielding effect of the polyglucosamine arms that locked the polylysine arms inside nanoparticles and prevented their interaction with mammalian cells. | ||
| Fig. 3 Chemical structures of cationic antimicrobial (a) star polymers, (b) main-chain cationic polymers and (c) phosphonium polymers. (a(1) adapted from ref. 18 with permission from Springer Nature, copyright (2016); a(3) adapted from ref. 76 with permission from American Chemical Society, copyright (2016)). | ||
3.5 Main-chain cationic polymers (MCCP)
Though less explored, main chain cationic polymers (i.e., polymers with cationic groups located in the backbone as opposed to exclusively on side chains or pendant groups) are attractive due to their good antibacterial efficacy. Similar to other cationic polymers, they usually contain hydrophobic parts, like alkyl chains, and cationic centers consisting of ammonium, guanidine or imidazolium groups, etc. Linear polyethylenimine (LPEI) and polyhexamethylene biguanide (PHMB) are two commercial antimicrobials with the charge on the main chain which are widely used in wound dressing or as antiseptics.77,78 Inspired by these, more main-chain cationic polymers have been developed in recent years. Polyionenes are macromolecules with ammonium in the main chain synthesized via Menshutkin reaction (Scheme 1e).79 Yang et al. prepared polyionenes, via Menshutkin polycondensation of α,ω-tetramethyldiamines and p-xylylene dichlorides. The resulting molecule showed potent antimicrobial activity against a broad spectrum of bacteria (MICs = 3.9–31.3 μg ml−1) with a high hemolytic selectivity (HC50/MIC) of >732 (Fig. 3b(1)).80Main-chain cationic polymers with guanidine group can be synthesized via polycondensation of guanidinium salts/isothiourea and diamine (Scheme 1f).81,82 Bai et al. prepared main-chain cationic alternating amphiphilic oligoguanidine (AAOG) (Fig. 3b(2)), via polycondensation of p-xylenyl diisothiourea and octylenediamine, that had excellent broad-spectrum antimicrobial activity with MICs in the range of 4–8 μg ml−1 against both Gram-negative and Gram-positive bacteria.82 The low MIC of AAOG is related to its main-chain cation placement and its ability to maintain a single chain structure in solution. The optimum AAOG with the incorporation of rigid p-xylenyl groups is important for the formation of single chain alternating amphiphilic structures in solution without self-assembly, as the control oligoguanidine (CTOG) with flexible hexamethylenyl groups self-assembled into folded structures, changing the spatial distribution of amphiphilicity and impairing interaction with bacterial membrane compared with the optimized AAOG. AAOG significantly improved the survival rate of mice with wounds infected with MDR P. aeruginosa.
Main-chain aromatic imidazolium oligomers (IBN-1) (Fig. 3b(3)) were synthesized by Zhang et al. via step-growth method and exhibited strong broad spectrum antimicrobial potency against drug-resistant K. pneumoniae, vancomycin-resistant Enterococcus (VRE), MRSA, and the fluconazole-resistant yeast C. neoformans without causing toxicity to erythrocytes (HC50/MIC > 3000).83 They demonstrated fast killing and in vivo efficacy in a peritonitis model under inflammatory conditions. In order to reduce the accumulation of antimicrobials in the environment, which may aggravate the drug-resistance problem,84 Zhang et al. incorporated carbonate, hemiaminal, ester and urea as biodegradable linkers into the aromatic imidazolium oligomers with excellent bactericidal efficacy.85 The carbonate and ester-linked (Fig. 3b(4)) imidazolium oligomers degraded into inactive and less toxic small molecules within weeks, making them safer for applications in wound dressings or personal care. Recently, Chan-Park, Gründling and Greenberg et al. prepared main-chain cationic alkylated polyimidazoliums (PIM1) (Fig. 3b(5)) which displayed antibiotic-level activities (MICs of 2–8 μg ml−1) even against pan-antibiotic resistant Gram-negative and Gram-positive bacteria86 and a large therapeutic window due to their low mammalian cell toxicity (IC50 against human kidney, human liver, mouse fibroblast and human epithelial cells respectively of >1024, >1024, >1024 and 870 μg ml−1). The PIMs were synthesized via a simple and cost-effective one-pot poly-Radziszewski reaction (Scheme 1g). They also showed that moderate hydrophobicity using a butyl linker between the imidazolium rings, versus the more hydrophilic or hydrophobic linkers, such as ethylene glycol and octyl linkers, achieved the best combination of potent antibacterial activity and good selectivity. The lead compound showed efficacy in treating murine S. aureus and P. aeruginosa infections.
3.6 Phosphonium polymers
Phosphonium polymers are also attractive antimicrobial compounds and some of them have shown enhanced antimicrobial activity and lower cytotoxicity compared to their analogous ammonium polymers. Endo et al. first studied the antimicrobial activities of phosphonium polymers. They found that polytributyl-3-((and 4-)vinylbenzyl) phosphonium (Fig. 3c(1)) exhibited a higher antimicrobial activity by 2 orders of magnitude than its ammonium analogues.87 A similar effect was also observed by Kenawy et al. in poly(glycidyl methacrylate-co-2-hydroxyethyl methacrylate) copolymers (P(GMA-co-HEMA)) (Fig. 3c(2)) and chitosan derivatives (Fig. 3c(3)),88,89 in both of which the phosphonium substituted polymers exhibited higher antimicrobial activity than ammonium substituted polymers. This general effect may be attributed to the difference in phosphorus and nitrogen atoms. Being below nitrogen within the same group in the periodic table, phosphorus has larger atomic radium and is less electronegative than nitrogen,90 which results in a higher positive charge in phosphorus than nitrogen, promoting the absorption of phosphonium polymers on negatively charged bacterial membrane.91Similar to other cationic polymers, both the multivalent interaction and amphiphilic balance play important role in antimicrobial activity of phosphonium polymers. Gillies et al. found that phosphonium polymer micelles (Fig. 3c(4)) with a multivalent display of phosphonium as the corona displayed much higher antimicrobial activity than small-molecule phosphonium cations.92 Eren et al. prepared a series of phosphonium polynorbornenes with different hydrophobic substitutions in phosphine.93 They found that the antimicrobial activity was improved from methyl to tert-butyl substituted phosphonium polynorbornenes without obvious hemolysis. Although more rigid aromatic substituted phosphonium polynorbornenes further enhanced their antimicrobial potency, they also induced remarkable hemolysis. The tert-butyl substituted phosphonium polynorbornene (Fig. 3c(5)) gave the highest selectivity of more than 30 against S. aureus, which is more than 3-fold larger than control AMP magainin. Ragogna et al. prepared a series of hydrophilic and hydrophobic substituted poly(vinylbenzylphosphonium) and investigated their influence on antimicrobial activity and biocompatibility.94 Surprisingly, they found that the hydrophilic hydroxypropyl substituted polymer (HydroxyPP) (Fig. 3c(6)) showed the highest selectivity of more than 392 against both E. coli and S. aureus while the more hydrophobic hexyl substituted polymer (HexylPP) (Fig. 3c(7)) was very hemolytic, resulting in a low selectivity. Although HydroxyPP does not have pendent hydrophobic substitution, it still formed amphiphilic structure, which is derived from its hydrophobic polystyrene backbone and terminal RAFT group.94,95 Consistent with previous findings, they also found that the multivalent polymeric form is important for the high antimicrobial activity and selectivity of HydroxyPP.
Despite being less explored in antimicrobial applications, another potential benefit of phosphonium-containing materials is their relatively lower cytotoxicity compared to their ammonium analogues. Stekar et al. replaced the ammonium group in octadecyl phosphocholine and 2-O-methyl-1-O-octadecyl-rac-glyceryl-3-phosphocholine with phosphonium group,96 which displayed lower acute toxicity in a murine study compared to their parent molecules. Although phosphonium polymers have many advanced properties, they are usually prepared via quaternization by phosphines, which has a propensity to be oxidized.97 This may be the reason for much fewer phosphonium antimicrobial polymers reported than ammonium antimicrobial polymers, and this needs to be carefully considered when researchers design and prepare phosphonium polymers.
4. Polymers as adjuvants: outer membrane permeabilizers and efflux pump inhibitors
The potency of most antibiotics is strictly restricted by the intracellular accumulation of the drugs inside bacteria. Gram-negative bacteria have intrinsic resistance to many classes of antibiotics due to the impermeability of the outer membrane98,99 (Fig. 4a(1)). Antibiotics that are able to cross the outer membrane barrier are typically small and hydrophilic,100 and gain access to the periplasmic space through porin proteins.98,101 However, the small hydrophilic antibiotics that cross the outer membrane barrier may still be prevented from accumulating in the cytoplasm by efflux pumps present in the bacterial membranes,102–104 leading to efflux-mediated antibiotic resistance (Fig. 4a(2)). The efflux pump activities have contributed to multi-drug resistant phenotypes, compromising antibiotic monotherapy. Moreover, outer membrane impermeability and efflux pumps may function simultaneously, leading to a high level of intrinsic resistance to antibiotics.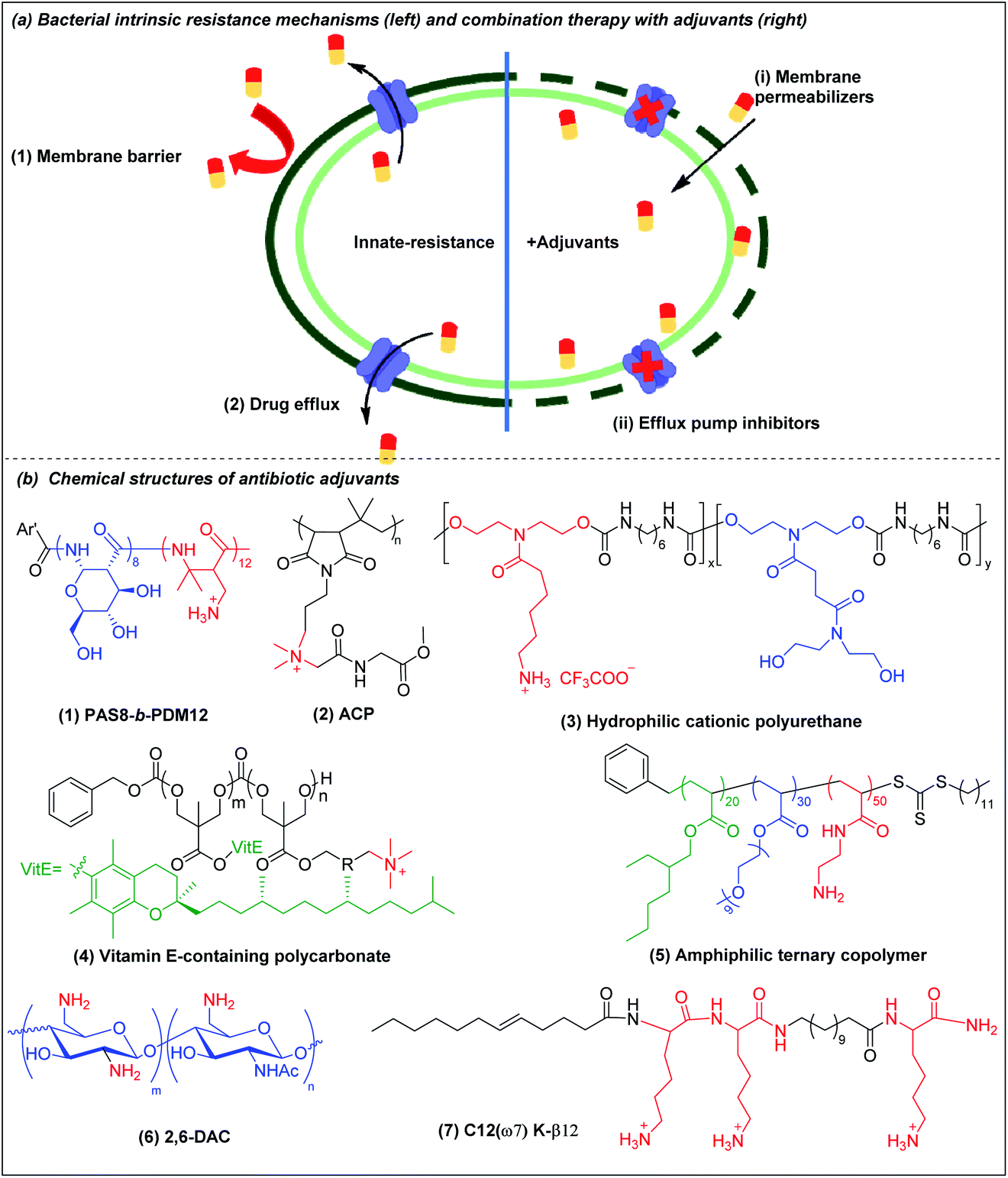 | ||
| Fig. 4 (a) Bacterial membrane-related intrinsic resistance mechanisms (left) and combination therapy with outer membrane permeabilizers and efflux pump inhibitors (right) (adapted from ref. 61 with permission from John Wiley and Sons, copyright (2020)). (b) Chemical structures of outer membrane permeabilizers and efflux pump inhibitors: (1) beta-peptide PAS8-b-PDM12, (2) amino acid conjugated polymer ACP, (3) hydrophilic cationic polyurethane, (4) vitamin E-containing cationic polycarbonate, (5) amphiphilic ternary copolymer, (6) chitosan derivative (2,6-DAC) and (7) cationic lipo-peptide C12(ω7) K-β12. | ||
There are three broad classes of antibiotic adjuvant molecules: (a) outer membrane permeabilizers, (b) efflux pump inhibitors, and (c) beta-lactamase inhibitors. Beta-lactamase inhibitors are usually small molecules and several good reviews are available105–108 and they fall outside the scope of polymers. An attractive and novel strategy is to use a cationic polymer that disrupts the intrinsic antibiotic resistance mechanisms to sensitize bacteria to antibiotics to which they are naturally resistant.106,109 There are relatively few reports on synergistic polymer–antibiotic combinations, especially combinations effective against several multi-drug resistant Gram-negative bacteria, or which potentiate multiple antibiotics. Besides potentiating the antibiotic, the combination strategy allows reduced dosing of the polymer adjuvant, which up to now has a narrower safety window than the antibiotic.
4.1 Outer membrane (OM) permeabilizers
Divalent cations (Ca2+ and Mg2+) crosslink the LPS molecules that are the major component of the Gram-negative outer membrane by the formation of ionic bridges with negatively charged phosphate groups in lipid A. The divalent ions are essential for the outer membrane integrity of Gram-negative bacteria.110 Cationic polymers can target the anionic phosphate groups in lipid A to compromise the physical integrity of the outer membrane by removal of, or competition with, these divalent ions. Chan-Park and Pethe et al. invented a glycosylated block co-beta-peptide (PAS8-b-PDM12) (Fig. 4b(1)) which has a cationic PDM block with a flexible backbone that easily rotates to interact with the anionic phosphate groups in lipid A via both electrostatic and hydrogen bonding.61 As PAS8-b-PDM12 binds to lipid A more strongly than the divalent cations Mg2+ or Ca2+, it competitively replaces these divalent ions, thereby increasing the outer membrane permeability even more effectively than the “gold standard” outer membrane disruptor polymyxin B. Rifampicin is a hydrophobic antibiotic that is approved for several types of bacterial infections such as tuberculosis but that is largely inactive against Gram-negative bacteria due to its inability to cross the outer membrane. PAS8-b-PDM12 enhanced the potency of rifampicin against all the ESKAPE Gram-negative bacteria by increasing the outer membrane permeability in vitro and in vivo. Jayanta et al. also showed that amino acid conjugated polymer (ACP) containing glycine (Fig. 4b(2)) potentiated rifampicin against MDR Gram-negative bacteria by 4–66 fold via a similar mechanism.111Joy et al. prepared a series of hydrophilic cationic polyurethanes without hydrophobic components to avoid toxicity to mammalian cells. The molecules were prepared via polycondensation reaction between diisocyanate and diol monomers using Tin(II) 2-ethylhexanoate as a catalyst (Scheme 1h).112 But the lack of hydrophobic components for membrane insertion also resulted in weak direct antimicrobial potency. Although the series exhibited poor direct antimicrobial activity, the polyurethane (Fig. 4b(3)) with 80% cationic and 20% hydrophilic uncharged residues (from pendant lysine and hydroxyl groups respectively) potentiated the antimicrobial activities of outer membrane impermeable antibiotics, such as rifampicin, oxacillin and erythromycin against E. coli. The cationic polymer forms pores in the outer membrane to allow a higher influx of the previously OM-excluded antibiotic. Further increase in the proportion of the hydrophilic components shielded the cationic groups, impairing their interaction with the anionic membrane, resulting in less membrane permeabilization. In addition to outer membrane, the cytoplasmic membrane may also function as a penetration barrier to hydrophilic antibiotics.113 Yang et al. also explored antibiotic-polymer synergy by incorporating hydrophobic vitamin E into cationic polycarbonates via copolymerization of vitamin E carbonate monomers with chloride carbonate monomers that were further quaternized with triethylamine.114 The resulting vitamin E-containing cationic polymer (Fig. 4b(4)) targets the anionic phospholipid surface and inserts the hydrophobic vitamin E residues into the lipid bilayer to increase the cytoplasmic membrane permeability. This cationic polymer potentiates small hydrophilic antibiotics, such as doxycycline, streptomycin or penicillin G against P. aeruginosa. In addition to working as directly killing agents, Yang et al. also found synergistic killing activity between guanidine-functionalized polycarbonate (pEt_20) (Fig. 2c(5)) with various antibiotics (azithromycin, gentamicin, imipenem and tetracycline) against carbapenem-resistant A. baumannii.69 Boyer et al. also recently demonstrated synergistic killing activity between an amphiphilic ternary copolymer, consisting of low-fouling oligoethylene glycol, hydrophobic ethylhexyl, and cationic primary amine groups (Fig. 4b(5)), and doxycycline and colistin against P. aeruginosa and E. coli.115 Importantly, the rate of resistance development to this combination treatment was also reduced compared to the individual agents.
Chan-Park et al. invented chitosan derivatives, specifically 2,6-diaminochitosan (2,6-DAC) (Fig. 4b(6)) and CSM5-K5 (Fig. 2a(3)), that can potentiate a more diverse array of antibiotics.116,117 2,6-DAC is particularly interesting as it can potentiate small antibiotics (amikacin, tobramycin, novobiocin and tazobactam) against MDR Gram-negative A. baumannii and other antibiotics (carbenicillin, tobramycin, and novobiocin) against MRSA.116 The synergy was also demonstrated in in vivo intraperitoneal and lung infection models. Compared to native chitosan, 2,6-DAC can accept more protons from its surroundings, leading to a stronger proton sponge effect, promoting bacterial membrane permeability to allow the antibiotics to reach their intracellular target. The absence of hydrophobicity in 2,6-DAC contributes to its good in vitro and in vivo biocompatibility. Kline et al. showed that CSM5-K5 synergizes with oxacillin, vancomycin, and streptomycin to clear MRSA, VRE, and MDR E. coli infected wounds in mice.117
4.2 Efflux pump inhibitors
Efflux pumps are transmembrane protein complexes that can “pump out” antimicrobial agents and toxins from the bacterial cytoplasm, thereby reducing their intracellular concentration. Many multidrug-resistant phenotypes have been associated with the presence of efflux pumps.118,119 Some of the most effective antibiotic classes including β-lactams,120 macrolides,121 tetracyclines,122 quinolones,123 trimethoprim,124 sulfonamides124 are efflux pump substrates. Since most efflux pumps are powered by the transmembrane proton motive force (PMF),125 compounds that interfere with membrane integrity have the potential to dissipate the PMF, and thereby inactivate efflux pumps dependent on it.Cationic polymers that target the bacterial cytoplasmic membrane thus have the potential to inhibit efflux pumps, although there are only a few studies of this possibility. One such report was based on a cationic lipo-peptide C12(ω7) K-β12 (Fig. 4b(7)), which potentiates tetracycline and erythromycin against E. coli by targeting PMF.126 Another example is the beta-peptide PAS8-b-PDM12 (Fig. 4b(1)). In addition to potentiating rifampicin against Gram-negative bacteria by increasing outer membrane permeability, PAS8-b-PDM12 also deactivates efflux pump systems by dissipating the PMF.127 This dual mechanism of sensitization is attributed to the formation of H-bonds between PAS8-b-PDM12 and the LPS in the outer membrane and electrostatic/hydrophobic interactions with the cytoplasmic membrane. Novobiocin, which has limited ability to cross the outer membrane and is also an efflux pump substrate in Gram-negative bacteria,128 was highly potentiated by PAS8-b-PDM12 in all the ESKAPE Gram-negative bacteria tested.
5. Antibiofilm agents
Biofilms are communities of microorganisms embedded in a self-produced matrix of extracellular polymeric substances (EPSs).129,130 The US National Institutes of Health has asserted that more than 80% of human infections are biofilm-related.131,132 Infectious biofilms can form on both abiotic and biotic materials.133 Biofilm bacteria are 10 to 1000 times more resistant to conventional antibiotics than are planktonic bacteria.134,135 Once a biofilm is formed, its three-dimensional matrix of EPSs can protect the bacteria from antibacterial agents and immune clearance. In addition, biofilm-embedded bacteria have the ability to switch to a metabolically quiescent phenotype that is less vulnerable to attack by antibiotics.136 The formation of a mature biofilm can be divided into two basic stages: initial bacterial attachment and subsequent three-dimensional biofilm matrix formation (Fig. 5a).136 Biofilm bacteria are phenotypically resistant to antibiotics and there are few reports on the use of antibacterial agents for the dispersion of mature biofilm by targeting the EPSs and/or killing quiescent cells.137 The prevention of biofilm formation essentially involves making a hydrophilic non-fouling surface and this has been extensively reviewed elsewhere.130,138 We shall here discuss only the eradication and dispersion of mature biofilms, which is a treatment rather than a prophylactic measure. The common metrics of antibiofilm efficacy are MBIC (minimum biofilm inhibitory concentration) and MBEC (minimum biofilm eradication concentration) for, respectively, biofilm inhibition and preformed biofilm eradication.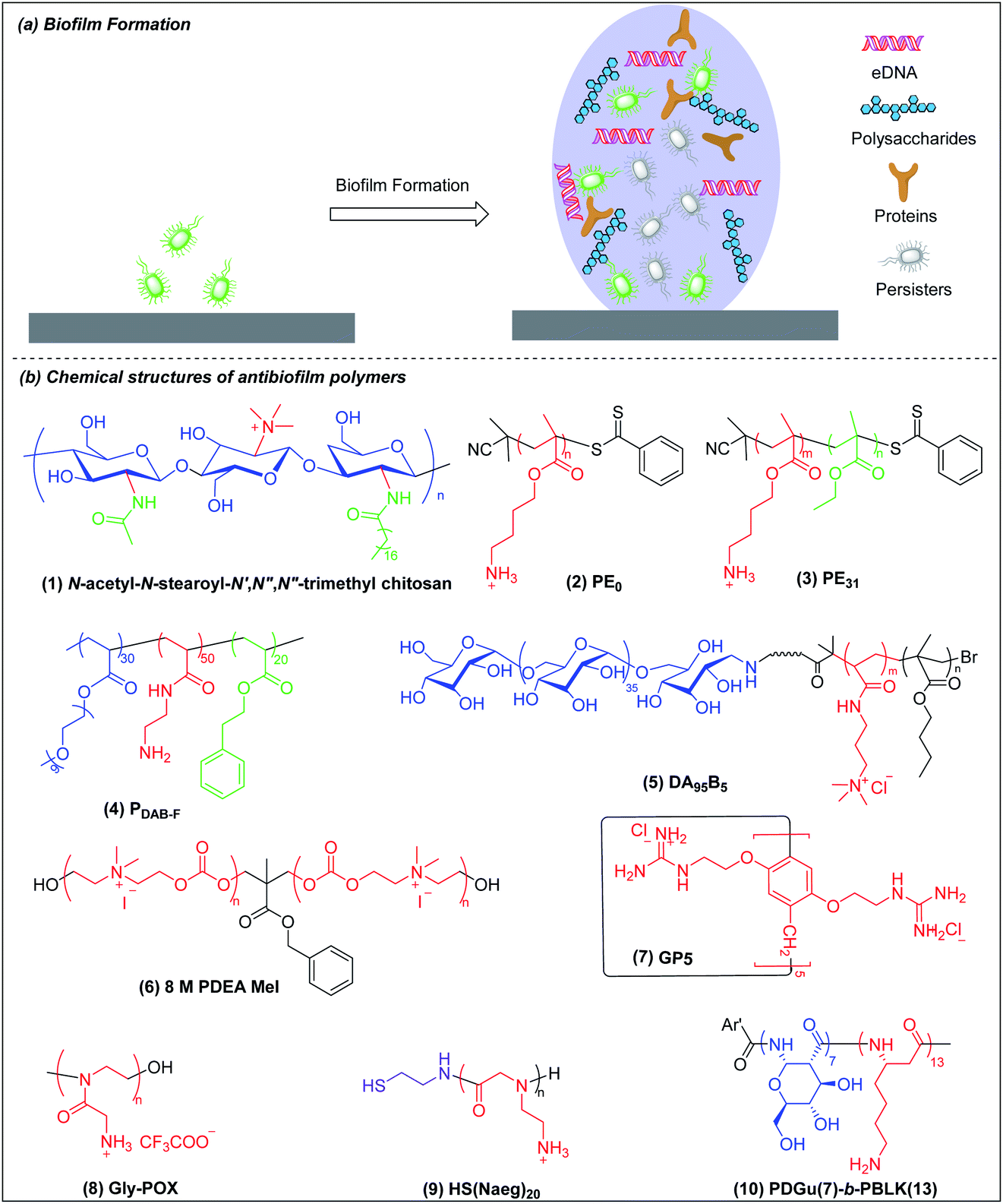 | ||
| Fig. 5 (a) Cartoon representation of biofilm formation and biofilm structure. (b) Chemical structures of antibiofilm polymers: (1) chitosan derivative, (2) and (3) polymethacrylate, (4) amphiphilic ternary copolymer, (5) polysaccharide-based amphiphilic ternary copolymer DA95B5, (6) main-chain cationic polycarbonate 8 M PDEA MeI, (7) guanidinium functionalized pillar[5]arene GP5, (8) cationic peptidomimetic Gly-POX, (9) peptoid HS(Naeg)20 and (10) beta-peptide PDGu(7)-b-PBLK(13). | ||
Biofilm-associated dormant cells include persisters which are a special state of bacteria with very low metabolism and most metabolism-targeting antibiotics are ineffective against them. Persisters are hard to eradicate with conventional antibiotics but there is progress to report. Recently, Mylonakis et al. showed that the new antibiotic candidate retinoid (CD437) kills MRSA persisters and an analog of their retinoid has synergy with gentamicin against MRSA biofilms.139 However, eradication of metabolically inactive cells is challenging and the antibiotic retinoid candidate is also membrane-active.
Chitosan derivatives with chemical modifications such as N-substitution, O-substitution, copolymerization and grafting have been shown to be effective against biofilm bacteria but the mechanisms are not well-studied.140,141 Meyer et al. found that the incorporation of lipophilic moieties into quaternary ammonium-modified chitosan plays a significant role in its antibiofilm performance.142N-Acetyl-N-stearoyl-N′,N′′,N′′-trimethyl chitosan (Fig. 5b(1)), which has two hydrophobic groups, acetyl and stearoyl, displayed the best antibiofilm efficacy against S. aureus. The MBEC of N-acetyl-N-stearoyl-N′,N′′,N′′-trimethyl chitosan decreases up to 4-fold compared to a control without lipophilic moieties, which may be attributed to its enhanced penetration into biofilm.
Kuroda et al. synthesized homocationic PE0 (Fig. 5b(2)) and amphiphilic PE31 (Fig. 5b(3)) copolymer containing 69% cationic residues and 31% hydrophobic ethyl groups by RAFT polymerization with similar molecular weights (2400–2500 g mol−1) and compared their antibiofilm activity.143 These two polymers can both reduce at least 80% of biofilm biomass at a concentration of 1000 μg ml−1 in 2 h, which appears superior to chlorhexidine and the commercially available surfactant cetrimonium bromide (CTAB). However, at a lower concentration of 125 and 250 μg ml−1, homocationic polymer reduced more biofilm biomass than copolymer, which suggests that cationic groups rather than hydrophobicity play the dominant role in their removal of biofilm biomass. This may be attributed to binding of cationic residues to the negatively charged biopolymers in the EPSs, causing physical disruption of the biofilm matrix. However, PE0 cannot kill biofilm bacteria although it can remove the biomass while PE31 can both disperse and eradicate biofilm bacteria. Hence, the capabilities for dispersal and eradication of the biofilm bacteria depend on the cationic and hydrophobic properties of this amphiphilic copolymer.
Boyer and Wong et al. prepared, via RAFT polymerization, another amphiphilic ternary copolymers containing anti-fouling poly(ethylene glycol), primary amine and hydrophobic residue, which can self-assembled into single-chain polymeric nanoparticles (SCPNs) driven by intramolecular hydrophobic interactions.144 The optimized copolymer (denoted PDAB-F, Fig. 5b(4), where DAB is diaminobutyric acid and F is phenylalanine) showed good biofilm killing efficacy of >99.99% within 1 hour towards the Gram-negative pathogen P. aeruginosa.
Chan-Park and Yang et al. also developed a polysaccharide-based amphiphilic ternary copolymer, dextran-block-(poly(3-acrylamidopropyl) trimethylammonium chloride (AMPTMA)-co-butyl methacrylate (BMA), DA95B5 (Fig. 5b(5)), via ATPR polymerization (Scheme 1i). DA95B5 dispersed mature biofilms of multi-drug resistant and clinically relevant Gram-positive bacteria strains, with efficacy much higher than and/or similar to conventional antibiotics.145 DA95B5 assembles into a nanoparticle with hydrophilic dextran as the corona, which diffuses deep into mature biofilm of Gram-positive bacteria and detaches the bacteria from biofilm, leading to biofilm dispersal via a mechanism termed nanoscale bacterial debridement (i.e. the nanoparticles aggregate at the bacteria–matrix interface and cause the slow “dissolution” and dispersal of the bacteria from the biofilm).145 Although DA95B5 effectively disperses mature biofilm, it does not kill the dispersed bacteria, which is not ideal since dispersed bacteria may colonize other sites, resulting in disseminated infection.
Hedrick et al. prepared a main-chain cationic polycarbonate (8 M PDEA MeI) (Fig. 5b(6)) via organocatalytic ring-opening polymerization of eight-member heterocyclic carbonate monomers and quaternization with methyl iodide, which not only dispersed biofilm biomass, but also killed the embedded bacteria.146 8 M PDEA MeI reduced Gram-positive S. aureus and Gram-negative E. coli biofilm biomass to around 25% and killed around 90% embedded bacteria in biofilms at 8-fold MIC. Although 8 M PDEA MeI killed around 90% of biofilm bacteria, the surviving 10% could regrow to re-establish a biofilm infection. Wang et al. prepared guanidinium-functionalized pillar[5]arene (GP5) (Fig. 5b(7)), which not only dispersed and killed biofilm bacteria, but also formed host–guest complexes with clofazimine (CFZ).147 The GP5 ⊃ CFZ complex showed synergistic killing activity against biofilm bacteria, resulting in near complete eradication of the biofilm bacteria. The survival of small numbers of biofilm bacteria after attack by GP5 ⊃ CFZ complex may be due to the presence of persister cells in the biofilm.
Liu et al. prepared a cationic peptidomimetic Gly-POX (Fig. 5b(8)), via cationic ring-opening polymerization (CROP) of N-Boc-aminomethyl-2-oxazoline monomers with trifluoromethanesulfonate (MeOTf) as the initiator (Scheme 1j), which effectively killed MRSA persisters at 4× MIC due to its capacity to damage membrane.148 Recently, Liu et al. also prepared a series of peptoids via ring-opening polymerization (ROP) of α-amino acid N-substituted N-carboxyanhydrides (α-NNCAs) (Scheme 1k) and the optimized polymer (HS(Naeg)20) (Fig. 5b(9)) displayed superior antimicrobial activity against MRSA persister cells and biofilms at 8× MIC, while the conventional antibiotic vancomycin and norfloxacin did not show potency even at concentrations up to 1024–2048× MIC.149 The superior antimicrobial and antibiofilm activities of HS(Naeg)20 was also validated in mouse wound model, mouse keratitis model and mouse peritonitis model induced by MRSA.
Chan-Park and Pethe et al. developed another glycosylated cationic co-beta-peptide poly(D-glucose)-block-poly(beta-L-lysine) (PDGu(7)-b-PBLK(13)) (Fig. 5b(10)), which can completely eradicate biofilms of the Gram-positive bacterium MRSA through its combination of bactericidal, anti-persister and biofilm dispersal abilities.150 PDGu(7)-b-PBLK(13) electrostatically binds to the anionic cell membrane and WTA, leading to defects of cell envelope integrity and eventually cell death. The membrane-targeting mechanism is not related to metabolism so that PDGu(7)-b-PBLK(13) can act on persister bacteria. Upon surface contact with bacterial membrane, the cationic beta-peptide block undergoes transition from random-coil to helical conformation to expose the cationic charges, leading to strong bacteria-killing effect. PDGu(7)-b-PBLK(13) possesses a unique bacteria-triggered surfactant effect: the cationic block adsorbs onto the negatively charged bacterial envelope while the hydrophilic sugar block promotes dissolution, resulting in “surfactant-like” solvation of bacteria from biofilm. The co-beta-peptide not only disperses established biofilms but also totally eradicates the biofilm bacteria, which also has a good safety window and demonstrates no acute in vivo toxicity in repeated dosing studies at levels above those required for therapeutic efficacy.
Further, various groups have formulated nanoparticles from polymers to contain other active ingredients like nitric oxide (NO),151D-amino acid,152 essential oils153 and antibiotics154 for eradicating biofilms, and excellent reviews have been written on them,136,155,156 and they are not covered here.
6. Smart responsive antibacterial systems
To reduce the non-specific toxicity of typical cationic polymers, smart antimicrobial systems have been designed with “on-demand” antimicrobial ability. These agents are responsive to pH, enzymes or bacterial membrane charge (Fig. 6).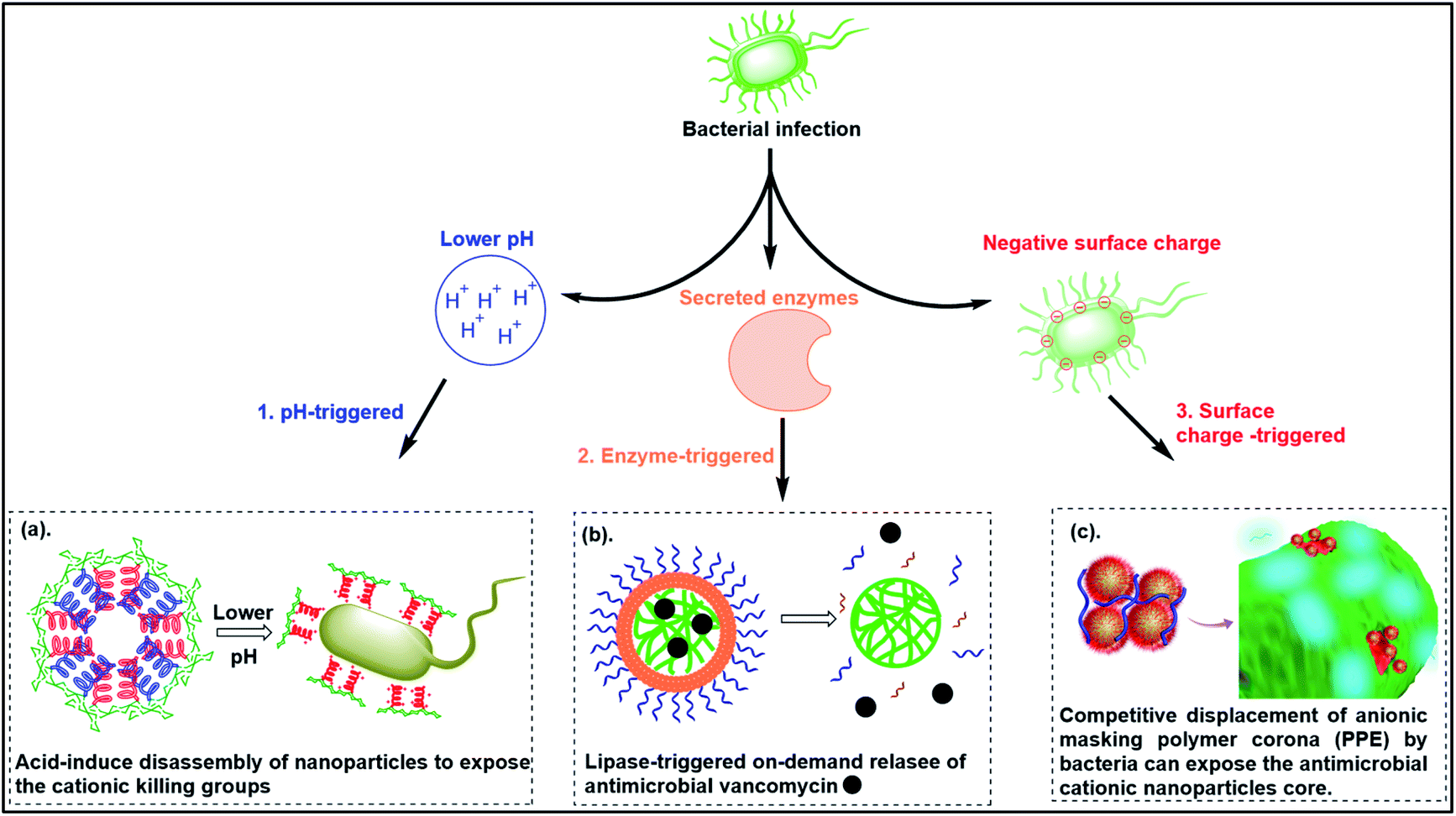 | ||
| Fig. 6 Smart responsive antibacterial systems triggered by (a) pH, (b) enzymes and (c) bacterial surface anionic charge. ((a) adapted from ref. 161 with permission from the Royal Society of Chemistry under the terms and conditions of the Creative Commons Attribution License; (b) adapted from ref. 157 with permission from American Chemical Society, copyright (2012); (c) adapted from ref. 158 with permission from American Chemical Society, copyright (2021)). | ||
6.1 pH-triggered antimicrobial polymers
Bacterial infections usually induce an acidic microenvironment that results from bacterial anaerobic fermentation and subsequent inflammation.159 Therefore, pH-sensitive polymers have been extensively exploited for “on-demand” antimicrobial therapy. Cheng et al. prepared a pH-sensitive, helix-coil conformation transitionable antimicrobial polypeptide ((PGA)m-r-(PHLG-MHH)n) consisting of poly(glutamic acid) and poly(γ-6-N-(methyldihexylammonium)hexyl-L-glutamate) to combat Helicobacter pylori infection (Fig. 7a(1)).160 The negatively charged glutamic acid and positively charged ammonium groups in the polymer side chains have strong electrostatic attractions, which drive the peptides to adopt a random-coil conformation without obvious toxicity at physiological pH. Under acidic conditions of H. pylori infected stomach, the protonation of glutamic acid breaks the electrostatic attraction, and the polymer transforms to helical structure with exposed cationic killing groups, which exhibits high antibacterial activity against H. pylori, including antibiotic-resistant clinical isolates.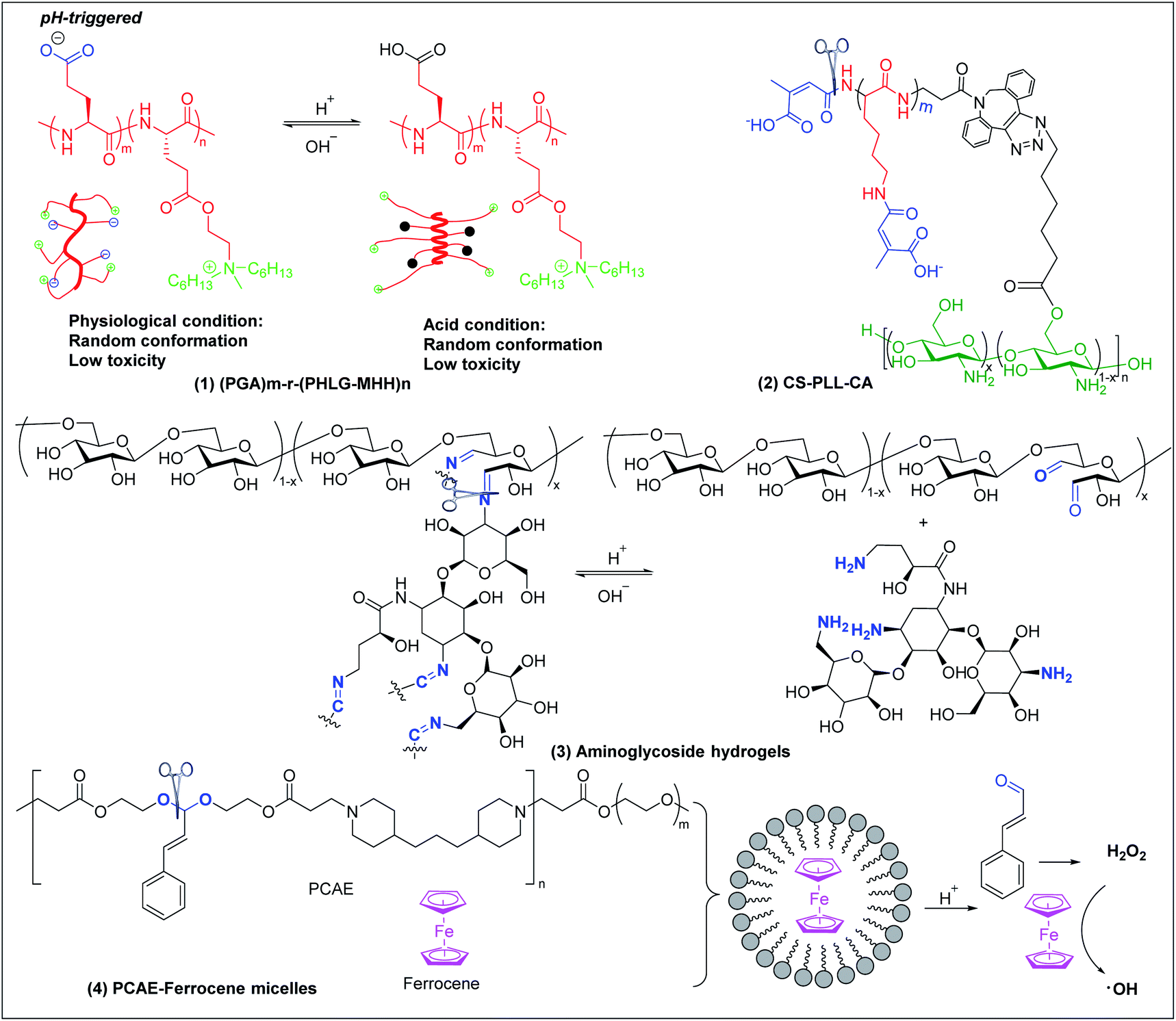 | ||
| Fig. 7 Chemical structures and cartoon schemes illustrating pH-triggered antimicrobial polymers ((1) adapted from ref. 160 with permission from National Academy of Sciences, copyright (2017)). | ||
Citraconyl amide is acid cleavable at pH 5 and below, which provides another strategy to rationally switch ON-or-OFF the cationic motif of cationic antimicrobial polymers via pH to specifically activate bactericidal activity at acidic but not physiological pH. Chan-Park and Kang et al. added this pH-cleavable citraconyl amide bond onto highly potent but toxic high molecular weight chitosan (20)-graft-polylysine (40) (CS-PLL) via coupling with citraconic anhydride.161 The obtained anionic citraconyl-functionalized CS-PLL (CS-PLL-CA) (Fig. 7a(2)) self-assembled with cationic CS-PLL into nanomicelles via anionic complex coacervation.161 The cationic PLL was neutralized by the anionic PLL-CA within the core and protected by the chitosan shell, resulting in excellent hemo- and cytocompatibility of the nanomicelles (Fig. 6a). At pH 5 and below, the citraconyl amide caps were hydrolyzed, causing destabilization of the cationic polylysine core and disassembly of the nanomicelles due to strong electrostatic repulsion. The disassembly released the cationic CS-PLL, which killed bacteria upon contact.
pH-sensitive systems have also been investigated for controlled release of antimicrobial agents. Imine bonds formed by Schiff-base reaction between aldehyde and amine groups are stable at alkaline and cleaved at acidic conditions. This has been extensively utilized to fabricate pH-sensitive systems for acidity-triggered release of antimicrobial agents.162,163 Cheng et al. developed a series of smart aminoglycoside hydrogels with tunable gel degradation and “on-demand” drug release via an imine linkage between aminoglycoside and oxidized polysaccharides (Fig. 7a(3)) such as dextran, carboxymethyl cellulose, alginate, and chondroitin.164 This hydrogel system showed fast gelation, self-healing and “on-demand” aminoglycoside release with tunable release kinetics.164 Potent antibacterial activities in vitro and in vivo against both Gram-positive and Gram-negative bacteria were observed.
Acetal linkage is another acid-labile chemical bond that can be used to fabricate antimicrobial delivery systems. It can be prepared by reaction between aldehyde and hydroxyl groups.165 Cinnamaldehyde is a type of antibiotic with aldehyde groups. Lee et al. developed a cinnamaldehyde-based pH-sensitive system for antibacterial therapy through reactive oxygen species (ROS) generated from the Fenton reaction (Fig. 7a(4)).166 Cinnamaldehyde was covalently incorporated, through an acid cleavable acetal linkage, in the backbone of the amphiphilic polymer poly[(3-phenylprop-2-ene-1,1-diyl)bis(oxy)bis(ethane-2,1-diyl)diacrylate]-co-4,4′(trimethylene dipiperidine)-co-poly (ethylene glycol) (PCAE), which can self-assemble with ferrocene into micelles. At acidic conditions, cinnamaldehyde is released from the micelles by cleavage of the acetal linkage and generates hydrogen peroxide (H2O2). Ferrocene converts mildly oxidizing H2O2 into highly reactive and toxic hydroxyl radicals that can effectively kill bacteria through membrane damage.
6.2 Enzyme-triggered antimicrobial polymers
Lipase is one of the most important enzymes secreted by bacteria167 that can rapidly degrade ester bonds. Antibiotic delivery systems fabricated from polymers with ester bonds can be degraded in the presence of lipase, releasing the antibiotic rapidly.168 Wang et al. prepared lipase-sensitive polymeric triple-layered nanogels (TLN) for “on-demand” antibiotic release (Fig. 6b and 8a(1)).157 They used monomethoxy poly(ethylene glycol)-b-poly(ε-caprolactone) to initiate the ring-opening polymerization of difunctional monomer 3-oxapentane-1,5-diyl bis(ethylene phosphate), which resulted in TLN with polyphosphoester as the inner core, poly(ε-caprolactone) (PCL) as the middle layer and poly(ethylene glycol) as the shell. Vancomycin was loaded in the polyphosphoester core, and the middle hydrophobic PCL layer formed a compact molecular fence to inhibit leakage of vancomycin from the core. The PCL layer is degraded by bacteria-secreted lipase, resulting in rapid vancomycin release at the site of infection.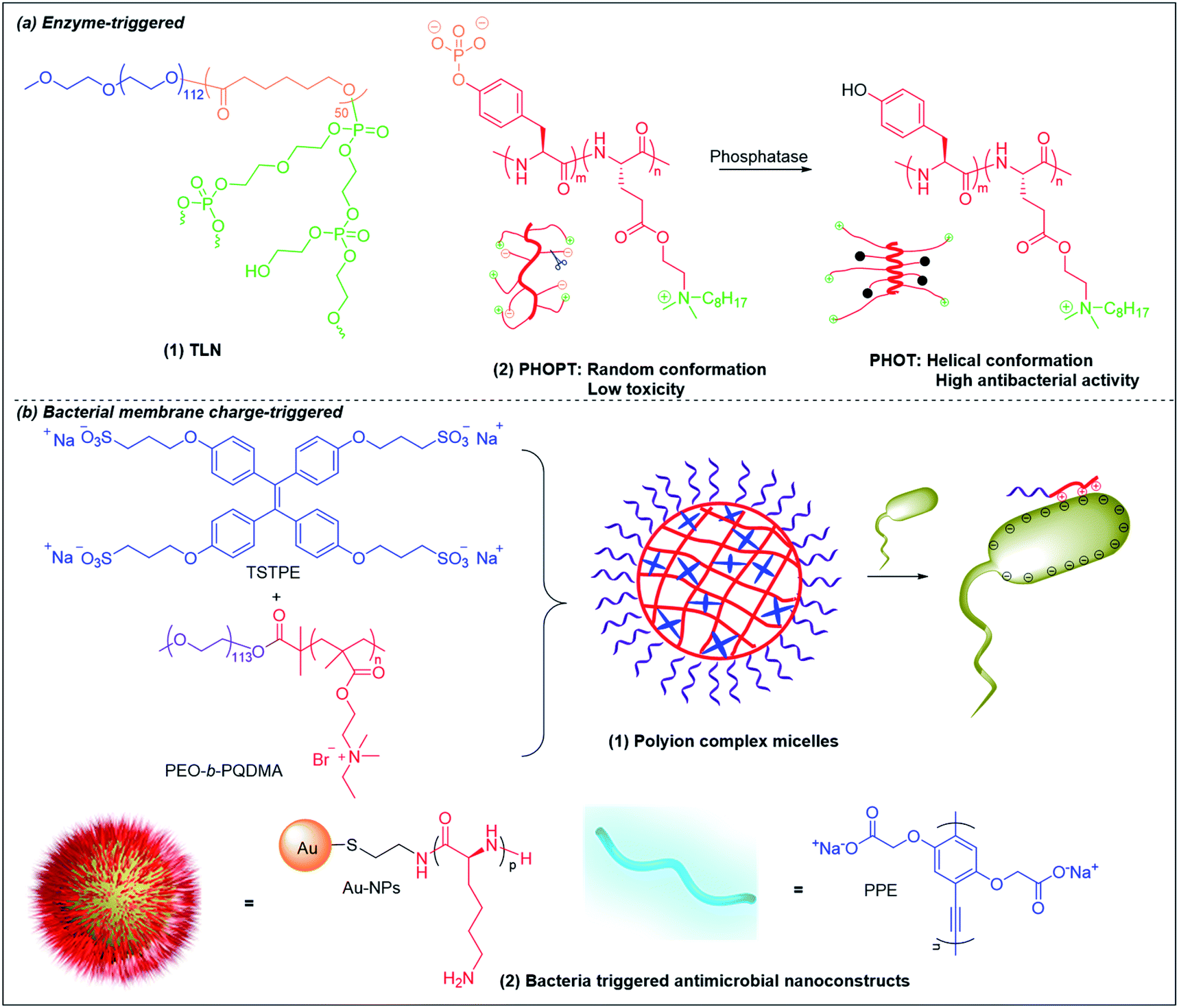 | ||
| Fig. 8 Chemical structures and cartoon schemes illustrating (a) enzyme-triggered antimicrobial polymers and (b) bacterial membrane charge-triggered antimicrobial polymers. (a(2) adapted from ref. 170 with permission from John Wiley and Sons, copyright (2017); b(2) adapted from ref. 158 with permission from American Chemical Society, copyright (2021)). | ||
Bacterial phosphatase is another key virulence factor that is over-excreted in infection sites and has been used to activate the antimicrobial activity of cationic polymers.169,170 Similar to the pH-triggered (PGA)m-r-(PHLG-MHH)n discussed above, Chen et al. also incorporated anionic phosphorylated tyrosine into a helical non-selective cationic polypeptide (Fig. 8a(2)).170 The electrostatic interaction between anionic phosphate and cationic ammonium breaks the helical structure and results in a random-coli conformation (PHOPT), which does not exhibit toxicity. However, when PHOPT reaches the infection site, bacterial phosphatase removes the anionic phosphate from the polymers, which switch back to a helical conformation (PHOT) with high membrane toxicity towards the bacteria.
In addition to the two enzymes discussed above, bacterial proteases,171,172 nitroreductases,173 hyaluronidases174 and esterases,175etc. can also be used as triggers to design smart antimicrobial prodrugs with on-demand antimicrobial activity.
6.3 Bacterial membrane charge-triggered antimicrobial polymers
As bacterial membranes are more negatively charged than eukaryotic membranes, it is possible to design smart systems that are responsive upon contact with bacterial membrane but nonresponsive towards eukaryotic cells based on the difference in membrane charge state. Liu et al. fabricated polyion complex micelles by mixing neutral-cationic diblock poly(ethylene oxide)-b-quaternary poly(2-(dimethylamino)ethyl methacrylate) (PEO-b-PQDMA) with anionic tetraphenylethylene (TPE) sulfonate derivatives (Fig. 8b(1)).176 The cationic PQDMA block was neutralized by the anionic TPE in the core and protected by the neutral PEO block that formed the shell, rendering the micelles nonhemolytic towards human red blood cells. Upon contact with bacteria, the cationic PQDMA block detached from the anionic TPE molecules for preferential interaction with bacterial membrane, leading to membrane disruption and bacterial killing. A similar concept with higher cationic exposure at triggered release was developed by decorating cationic antimicrobial peptides-functionalized Au nanoparticles (NPs) with anionic poly(p-phenyleneethynylene) (PPE) derivatives to form nanoconstructs (Fig. 6c and 8b(2)).158 Due to electrostatic neutralization of the cationic NP surface by the anionic PPE polymer, the nanoconstructs showed ultralow hemolytic activity and cytotoxicity in vitro, as well as nontoxicity towards mice in vivo. Upon contact with bacteria, the negatively charged bacterial membrane competitively displaced and drove away the anionic PPE polymers to interact with the highly cationic NP surface. These nanoconstructs were able to effectively eradicate Gram-positive and Gram-negative bacteria.1587. Conclusion and outlook
The structural difference between Gram-positive, Gram-negative bacterial and mammalian cell envelopes makes it possible to design cationic polymers that selectively combat bacteria by finely tuning their amphiphilicity with choice of cationic and hydrophobic monomers and their ratios. Instead of targeting specific molecules or essential metabolic pathways, cationic polymers kill bacteria via a more general membrane-disruption mechanism, which has a lower propensity to elicit resistance. The antimicrobial activity, biocompatibility and stability of various cationic polymers can be tuned via their backbone chemistry, cationic and hydrophobic component structures, and morphology. Although less explored, main-chain cationic polymers show the highest antimicrobial potency with potency comparable to conventional antibiotics, which may be due to the reduced freedom of motion of the charges, and should be the next research focus of cationic polymers as antimicrobial compounds. Other cationic polymers, like poly(α-amino acids), β-peptides, polycarbonates and even their assembled structures, usually have MIC values much higher than conventional antibiotics and may not be suitable for use as direct antimicrobial agents. Instead, as they target the bacterial membrane, they may make excellent antibiotic adjuvants to overcome membrane-related antibiotic resistances, including both the outer membrane barrier and efflux pumps. Another possible application for cationic polymers is to treat biofilms, toward which conventional antibiotics are typically ineffective due to the multiple resistance mechanisms possessed by biofilms,177e.g. the EPS barrier, the persister phenotype, overexpressed antibiotic resistance enzymes and efflux pumps, etc. Multifunctional cationic polymers combining biofilm dispersal, bactericidal and anti-persister activity can be designed to be highly effective antibiofilm agents, which fills a significant gap in the current therapeutic armamentarium. Another noteworthy point is that many cationic polymers have exhibited nephrotoxicity and hepatotoxicity in vivo or in clinical studies due to their cationicity,178–181 even though they do not show any toxicity in vitro. These in vivo toxicities may be addressable with cationic polymers with “on-demand” antimicrobial ability triggered by bacterial infectious environment. Further, for biomedical and environmental applications, the cationic polymers should preferably be designed with biodegradability, so that only the polymeric form is antibacterial, while the degraded monomeric forms have no toxicity to mammalian cells and the environment.In summary, antimicrobial polymers have been a focus of intensive research for decades. The urgency of this research is heightened by the intensifying problem of antimicrobial resistance. Although numerous antimicrobial polymers have been designed and prepared, none of them have been shown efficacy in the real sepsis infectious model and thigh infectious model, which are more translational animal models for evaluating antimicrobial drugs. In view of the rapid advances and innovations in diverse chemistries achieved in recent years, it appears likely that biodegradable cationic polymers will be able to achieve efficacy and selectivity sufficient for their employment as direct antibacterial or antibiofilm agents, or as adjuvants in combination therapies, or as smart systems. This review covers the structure–activity of antimicrobial polymers and their potential applications, which shall contribute to the future development of polymers as antimicrobial and antibiofilm agents with translational applications.
Data availability
Data supporting the findings of this study are available in this article.Author contributions
Z. S. and M. B. C. conceived the project. Z. S. and C. H. K. wrote the membrane structures of bacteria and mammalian cells. Z. S. and W. Z. wrote the polymers as direct antimicrobial agents. Z. S. and J. L. wrote the antibiofilm polymers. Z. S. and D. P. wrote the smart responsive antimicrobial systems. Z. S., E.-T. K., K. P., and M. B. C. wrote the manuscript with contribution from the other authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research is supported by the Ministry of Education, Singapore, under its Academic Research Fund Tier 3 (AcRF Tier 3): MOE2018-T3-1-003.References
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microb., 2019, 10, 539 CrossRef PubMed.
- K. Lewis, Biochemistry, 2020, 85, 1469–1483 CAS.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- J. Portelinha, S. S. Duay, S. I. Yu, K. Heilemann, M. D. J. Libardo, S. A. Juliano, J. L. Klassen and A. M. Angeles-Boza, Chem. Rev., 2021, 121, 2648–2712 CrossRef CAS PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- M. F. Ilker, K. Nüsslein, G. N. Tew and E. B. Coughlin, J. Am. Chem. Soc., 2004, 126, 15870–15875 CrossRef CAS PubMed.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS PubMed.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed.
- T. Ikeda, S. Tazuke and Y. Suzuki, Die Makromolekulare Chemie: Macromol. Chem. Phys., 1984, 185, 869–876 CrossRef CAS.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- K. Jung, N. Corrigan, E. H. Wong and C. Boyer, Adv. Mater., 2021, 2105063 CrossRef PubMed.
- M. S. Ganewatta and C. Tang, Polymer, 2015, 63, A1–A29 CrossRef CAS.
- A. Muñoz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef.
- M. P. Bevilacqua, D. J. Huang, B. D. Wall, S. J. Lane, C. K. Edwards III, J. A. Hanson, D. Benitez, J. S. Solomkin and T. J. Deming, Macromol. Biosci., 2017, 17, 1600492 CrossRef PubMed.
- P. Li, C. Zhou, S. Rayatpisheh, K. Ye, Y. F. Poon, P. T. Hammond, H. Duan and M. B. Chan-Park, Adv. Mater., 2012, 24, 4130–4137 CrossRef CAS PubMed.
- M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek, C. Yao, L.-F. Chen, A. L. Ferguson and G. C. Wong, Proc. Natl. Acad. Sci., 2015, 201507893 Search PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, S. Adrian, E. H. H. Wong, Yu-Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 1–11 Search PubMed.
- M. Zhou, X. Xiao, Z. Cong, Y. Wu, W. Zhang, P. Ma, S. Chen, H. Zhang, D. Zhang and D. Zhang, Angew. Chem., Int. Ed., 2020, 59, 7240–7244 CrossRef CAS PubMed.
- F. Nederberg, Y. Zhang, J. P. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima and L. Li, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- R. Namivandi-Zangeneh, E. H. Wong and C. Boyer, ACS Infect. Dis., 2021, 7, 215–253 CrossRef CAS PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- R. A. Fisher, B. Gollan and S. Helaine, Nat. Rev. Microbiol., 2017, 15, 453 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- G. Cox and G. D. Wright, Int. J. Med. Microbiol., 2013, 303, 287–292 CrossRef CAS PubMed.
- M. Gross, S. E. Cramton, F. Götz and A. Peschel, Infect. Immun., 2001, 69, 3423–3426 CrossRef CAS PubMed.
- R. M. Epand and R. F. Epand, J. Pept. Sci., 2011, 17, 298–305 CrossRef CAS PubMed.
- K. L. Hilton, C. Manwani, J. E. Boles, L. J. White, S. Ozturk, M. D. Garrett and J. R. Hiscock, Chem. Sci., 2021, 12, 13273–13282 RSC.
- P. Sperandeo, A. M. Martorana and A. Polissi, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1451–1460 CrossRef CAS PubMed.
- H. Dong, X. Tang, Z. Zhang and C. Dong, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1461–1467 CrossRef CAS PubMed.
- J. M. Stokes, C. R. MacNair, B. Ilyas, S. French, J.-P. Côté, C. Bouwman, M. A. Farha, A. O. Sieron, C. Whitfield and B. K. Coombes, Nat. Microbiol., 2017, 2, 1–8 Search PubMed.
- G. J. Gabriel, A. Som, A. E. Madkour, T. Eren and G. N. Tew, Mater. Sci. Eng., R, 2007, 57, 28–64 CrossRef PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111, e24122 Search PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- G. S. Dijksteel, M. M. Ulrich, E. Middelkoop and B. K. Boekema, Front. Microbiol., 2021, 12, 287 Search PubMed.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS PubMed.
- T. J. Deming and S. A. Curtin, J. Am. Chem. Soc., 2000, 122, 5710–5717 CrossRef CAS.
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- J. N. Cha, G. D. Stucky, D. E. Morse and T. J. Deming, Nature, 2000, 403, 289–292 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- C. Zhou, X. Qi, P. Li, W. N. Chen, L. Mouad, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomacromolecules, 2009, 11, 60–67 CrossRef PubMed.
- Z. Hou, Y. V. Shankar, Y. Liu, F. Ding, J. L. Subramanion, V. Ravikumar, R. Zamudio-Vazquez, D. Keogh, H. Lim, M. Y. F. Tay, S. Bhattacharjya, S. A. Rice, J. Shi, H. Duan, X. W. Liu, Y. Mu, N. S. Tan, K. C. Tam, K. Pethe and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2017, 9, 38288–38303 CrossRef CAS PubMed.
- A. C. Engler, H. i. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS PubMed.
- A. C. Engler, A. Shukla, S. Puranam, H. G. Buss, N. Jreige and P. T. Hammond, Biomacromolecules, 2011, 12, 1666–1674 CrossRef CAS PubMed.
- C. Cabrele, T. s. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- M.-R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, ACS Chem. Biol., 2017, 12, 2975–2980 CrossRef CAS PubMed.
- O. G. Travkova, H. Moehwald and G. Brezesinski, Adv. Colloid Interface Sci., 2017, 247, 521–532 CrossRef CAS PubMed.
- H. M. Werner, C. C. Cabalteja and W. S. Horne, ChemBioChem, 2016, 17, 712–718 CrossRef CAS PubMed.
- Q. Chen, D. Zhang, W. Zhang, H. Zhang, J. Zou, M. Chen, J. Li, Y. Yuan and R. Liu, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- M. Zasloff, Proc. Natl. Acad. Sci., 1987, 84, 5449–5453 CrossRef CAS PubMed.
- E. A. Porter, X. Wang, H.-S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS PubMed.
- Y. Hamuro, J. P. Schneider and W. F. DeGrado, J. Am. Chem. Soc., 1999, 121, 12200–12201 CrossRef CAS.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS PubMed.
- S. Chakraborty, R. Liu, J. J. Lemke, Z. Hayouka, R. A. Welch, B. Weisblum, K. S. Masters and S. H. Gellman, ACS Macro Lett., 2013, 2, 753–756 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. Chakraborty, J. J. Lemke, Z. Hayouka, C. Chow, R. A. Welch, B. Weisblum, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4410–4418 CrossRef CAS PubMed.
- S. Chakraborty, R. Liu, Z. Hayouka, X. Chen, J. Ehrhardt, Q. Lu, E. Burke, Y. Yang, B. Weisblum, G. C. L. Wong, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 14530–14535 CrossRef CAS PubMed.
- L. Liu, K. C. Courtney, S. W. Huth, L. A. Rank, B. Weisblum, E. R. Chapman and S. H. Gellman, J. Am. Chem. Soc., 2021, 143, 3219–3230 CrossRef CAS PubMed.
- Z. Si, H. W. Lim, M. Y. Tay, Y. Du, L. Ruan, H. Qiu, R. Zamudio-Vazquez, S. Reghu, Y. Chen and W. S. Tiong, Angew. Chem., 2020, 132, 6886–6893 CrossRef.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy, S. Reghu, K. Marimuthu, P. P. De, O. T. Ng, J. R. Mediavilla, B. N. Kreiswirth, Y. R. Chi, J. Ren, K. C. Tam, X. W. Liu, H. Duan, Y. Zhu, Y. Mu, P. T. Hammond, G. C. Bazan, K. Pethe and M. B. Chan-Park, Nat. Commun., 2019, 10, 4792 CrossRef PubMed.
- J. Tan, J. Tay, J. Hedrick and Y. Y. Yang, Biomaterials, 2020, 252, 120078 CrossRef CAS PubMed.
- R. J. Ono, Q. L. Shao, S. Venkataraman, W. Chin, Y. Y. Yi and J. L. Hedrick, Macromolecules, 2014, 47, 7725–7731 CrossRef CAS.
- W. Chin, C. Yang, V. W. L. Ng, H. Yuan, J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yi, Macromolecules, 2013, 46, 8797–8807 CrossRef CAS.
- V. W. L. Ng, J. P. K. Tan, J. Leong, X. V. Zhi, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS.
- A. C. Engler, J. P. K. Tan, O. Z. Yuin, D. J. Coady, V. W. L. Ng, Y. Y. Yan and J. L. Hedrick, Biomacromolecules, 2013, 14, 4331–4339 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. D. Sessions, B. Periaswamy, A. Lee, Z. C. Liang and X. Ding, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- X. Ding, C. Yang, W. Moreira, P. Yuan, B. Periaswamy, P. F. de Sessions, H. Zhao, J. Tan, A. Lee and K. X. Ong, Adv. Sci., 2020, 7, 2001374 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. J. Tan, W. Fan, S. S. Venkatraman, L. Li and Y.-Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- Y. Zhang, T. Chen, Z. Pan, X. Sun, X. Yin, M. He, S. Xiao and H. Liang, Langmuir, 2018, 34, 13438–13448 CrossRef CAS PubMed.
- S. J. Lam, E. H. Wong, N. M. O'Brien-Simpson, N. Pantarat, A. Blencowe, E. C. Reynolds and G. G. Qiao, ACS Appl. Mater. Interfaces, 2016, 8, 33446–33456 CrossRef CAS PubMed.
- S. J. Shirbin, I. Insua, J. A. Holden, J. C. Lenzo, E. C. Reynolds, N. M. O'Brien-Simpson and G. G. Qiao, Adv. Healthcare Mater., 2018, 7, e1800627 CrossRef PubMed.
- C. Yang, S. Krishnamurthy, J. Liu, S. Liu, X. Lu, D. J. Coady, W. Cheng, G. De Libero, A. Singhal, J. L. Hedrick and Y. Y. Yang, Adv. Healthcare Mater., 2016, 5, 1272–1281 CrossRef CAS PubMed.
- E. H. Wong, M. M. Khin, V. Ravikumar, Z. Si, S. A. Rice and M. B. Chan-Park, Biomacromolecules, 2016, 17, 1170–1178 CrossRef CAS PubMed.
- H. Fjeld and E. Lingaas, Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke, 2016, vol. 136, pp. 707–711 Search PubMed.
- S. J. Fox, M. H. Fazil, C. Dhand, M. Venkatesh, E. T. Goh, S. Harini, C. Eugene, R. R. Lim, S. Ramakrishna, S. S. Chaurasia, R. W. Beuerman, C. S. Verma, N. K. Verma, X. J. Loh and R. Lakshminarayanan, Acta Biomater., 2016, 37, 155–164 CrossRef CAS PubMed.
- S. R. Williams and T. E. Long, Prog. Polym. Sci., 2009, 34, 762–782 CrossRef CAS.
- S. Liu, R. J. Ono, H. Wu, J. Y. Teo, Z. C. Liang, K. Xu, M. Zhang, G. Zhong, J. P. Tan and M. Ng, Biomaterials, 2017, 127, 36–48 CrossRef CAS PubMed.
- Y. Zhang, J. Jiang and Y. Chen, Polymer, 1999, 40, 6189–6198 CrossRef CAS.
- Z. Chen, C. Zhou, Y. Xu, K. Wen, J. Song, S. Bai, C. Wu, W. Huang, Q. Cai and K. Zhou, Biomaterials, 2021, 275, 120858 CrossRef CAS PubMed.
- L. Liu, Y. Huang, S. N. Riduan, S. Gao, Y. Yang, W. Fan and Y. Zhang, Biomaterials, 2012, 33, 8625–8631 CrossRef CAS PubMed.
- J. Lu and J. Guo, Science, 2021, 371, 474 CrossRef PubMed.
- Y. Yuan, D. S. W. Lim, H. Wu, H. Lu, Y. Zheng, A. C. A. Wan, J. Y. Ying and Y. Zhang, Biomater. Sci., 2019, 7, 2317–2325 RSC.
- W. Zhong, Z. Shi, S. H. Mahadevegowda, B. Liu, K. Zhang, C. H. Koh, L. Ruan, Y. Chen, M. S. Zeden and C. J. Pee, Proc. Natl. Acad. Sci., 2020, 117, 31376–31385 CrossRef CAS PubMed.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 335–343 CrossRef CAS.
- E. R. Kenawy, F. I. Abdel-Hay, A. E. R. R. El-Shanshoury and M. H. El-Newehy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2384–2393 CrossRef CAS.
- M. H. El-Newehy, E.-R. Kenawy and S. S. Al-Deyab, Int. J. Polym. Mater. Polym. Biomater., 2014, 63, 758–766 CrossRef CAS.
- V. L. Rose, F. Mastrotto and G. Mantovani, Polym. Chem., 2017, 8, 353–360 RSC.
- Y. Xue, Y. Pan, H. Xiao and Y. Zhao, RSC Adv., 2014, 4, 46887–46895 RSC.
- B. Hisey, P. J. Ragogna and E. R. Gillies, Biomacromolecules, 2017, 18, 914–923 CrossRef CAS PubMed.
- N. C. Süer, C. Demir, N. A. Ünübol, Ö. Yalçın, T. Kocagöz and T. Eren, RSC Adv., 2016, 6, 86151–86157 RSC.
- T. J. Cuthbert, B. Hisey, T. D. Harrison, J. F. Trant, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2018, 57, 12707–12710 CrossRef CAS PubMed.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., 2019, 131, 3728–3731 CrossRef.
- J. Stekar, G. Nössner, B. Kutscher, J. Engel and P. Hilgard, Angew. Chem., Int. Ed. Engl., 1995, 34, 238–240 CrossRef CAS.
- Y. Xue, H. Xiao and Y. Zhang, Int. J. Mol. Sci., 2015, 16, 3626–3655 CrossRef CAS PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808–816 CrossRef CAS PubMed.
- H. I. Zgurskaya, C. A. López and S. Gnanakaran, ACS Infect. Dis., 2015, 1, 512–522 CrossRef CAS PubMed.
- J.-M. Pages, C. E. James and M. Winterhalter, Nat. Rev. Microbiol., 2008, 6, 893 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299 CrossRef CAS PubMed.
- X.-Z. Li, P. Plésiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef PubMed.
- B. D. Schindler and G. W. Kaatz, Drug Resist. Updates, 2016, 27, 1–13 CrossRef PubMed.
- X. Z. Li, P. Plesiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef PubMed.
- K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS PubMed.
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS PubMed.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
- J.-D. Docquier and S. Mangani, Drug Resist. Updates, 2018, 36, 13–29 CrossRef PubMed.
- M. Chang, K. V. Mahasenan, J. A. Hermoso and S. Mobashery, Acc. Chem. Res., 2021, 1738–1741 Search PubMed.
- R. Domalaon, T. Idowu, G. G. Zhanel and F. Schweizer, Clin. Microbiol. Rev., 2018, 31, e00077-17 CrossRef PubMed.
- S. Barman, S. Mukherjee, S. Ghosh and J. Haldar, ACS Appl. Bio Mater., 2019, 2, 5404–5414 CrossRef CAS.
- C. Tantisuwanno, F. Dang, K. Bender, J. D. Spencer, M. E. Jennings, H. A. Barton and A. Joy, Biomacromolecules, 2021, 2910–2920 CrossRef CAS PubMed.
- K. Lewis, Cell, 2020, 181, 29–45 CrossRef CAS PubMed.
- V. W. L. Ng, X. Ke, A. L. Lee, J. L. Hedrick and Y. Y. Yang, Adv. Mater., 2013, 25, 6730–6736 CrossRef CAS PubMed.
- R. Namivandi-Zangeneh, Z. Sadrearhami, D. Dutta, M. Willcox, E. H. Wong and C. Boyer, ACS Infect. Dis., 2019, 5, 1357–1365 CrossRef CAS PubMed.
- Z. Si, Z. Hou, Y. S. Vikhe, K. R. V. Thappeta, K. Marimuthu, P. P. De, O. T. Ng, P. Li, Y. Zhu and K. Pethe, ACS Appl. Mater. Interfaces, 2021, 13, 3237–3245 CrossRef CAS PubMed.
- K. R. Thappeta, Y. S. Vikhe, A. M. Yong, M. B. Chan-Park and K. A. Kline, ACS Infect. Dis., 2020, 6, 1228–1237 CrossRef CAS PubMed.
- M. Webber and L. Piddock, J. Antimicrob. Chemother., 2003, 51, 9–11 CrossRef CAS PubMed.
- J. Sun, Z. Deng and A. Yan, Biochem. Biophys. Res. Commun., 2014, 453, 254–267 CrossRef CAS PubMed.
- T. Nakae, A. Nakajima, T. Ono, K. Saito and H. Yoneyama, Antimicrob. Agents Chemother., 1999, 43, 1301–1303 CrossRef CAS PubMed.
- P. Zhong and V. D. Shortridge, Drug Resist. Updates, 2000, 3, 325–329 CrossRef CAS PubMed.
- X.-Z. Li, D. M. Livermore and H. Nikaido, Antimicrob. Agents Chemother., 1994, 38, 1732–1741 CrossRef CAS PubMed.
- A. Mahamoud, J. Chevalier, S. Alibert-Franco, W. V. Kern and J.-M. Pages, J. Antimicrob. Chemother., 2007, 59, 1223–1229 CrossRef CAS PubMed.
- T. Köhler, M. Kok, M. Michea-Hamzehpour, P. Plesiat, N. Gotoh, T. Nishino, L. K. Curty and J.-C. Pechere, Antimicrob. Agents Chemother., 1996, 40, 2288–2290 CrossRef PubMed.
- H. Nikaido, Science, 1994, 264, 382–388 CrossRef CAS PubMed.
- K. Goldberg, H. Sarig, F. Zaknoon, R. F. Epand, R. M. Epand and A. Mor, FASEB J., 2013, 27, 3818–3826 CrossRef CAS PubMed.
- Z. Si, H. W. Lim, M. Y. Tay, Y. Du, L. Ruan, H. Qiu, S. Reghu, Y. Chen, W. S. Tiong and K. Marimuthu, Angew. Chem., Int. Ed., 2020, 6819–6826 CrossRef CAS PubMed.
- G. Krishnamoorthy, I. V. Leus, J. W. Weeks, D. Wolloscheck, V. V. Rybenkov and H. I. Zgurskaya, mBio, 2017, 8, e01172-17 CrossRef PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563 CrossRef CAS PubMed.
- L. D. Blackman, Y. Qu, P. Cass and K. E. Locock, Chem. Soc. Rev., 2021, 50, 1587–1616 RSC.
- A. Houry, M. Gohar, J. Deschamps, E. Tischenko, S. Aymerich, A. Gruss and R. Briandet, Proc. Natl. Acad. Sci., 2012, 109, 13088–13093 CrossRef CAS PubMed.
- https://grants.nih.gov/grants/guide/pa-files/PA-03-047.html .
- D. Lebeaux, J.-M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543 CrossRef PubMed.
- D. Sharma, L. Misba and A. U. Khan, Antimicrobial Resistance & Infection Control, 2019, 8, 1–10 Search PubMed.
- Y. Bi, G. Xia, C. Shi, J. Wan, L. Liu, Y. Chen, Y. Wu, W. Zhang, M. Zhou and H. He, Fundamental Research, 2021 Search PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740 CrossRef CAS PubMed.
- R. Srinivasan, S. Santhakumari, P. Poonguzhali, M. Geetha, M. Dyavaiah and L. Xiangmin, Front. Microb., 2021, 12, 1106 Search PubMed.
- W. Kim, W. Zhu, G. L. Hendricks, D. Van Tyne, A. D. Steele, C. E. Keohane, N. Fricke, A. L. Conery, S. Shen and W. Pan, Nature, 2018, 556, 103–107 CrossRef CAS PubMed.
- F. Khan, D. T. N. Pham, S. F. Oloketuyi, P. Manivasagan, J. Oh and Y.-M. Kim, Colloids Surf., B, 2020, 185, 110627 CrossRef CAS PubMed.
- P. Sahariah and M. Masson, Biomacromolecules, 2017, 18, 3846–3868 CrossRef CAS PubMed.
- P. Sahariah, M. Masson and R. L. Meyer, Biomacromolecules, 2018, 19, 3649–3658 CrossRef CAS PubMed.
- H. Takahashi, E. T. Nadres and K. Kuroda, Biomacromolecules, 2016, 18, 257–265 CrossRef PubMed.
- T.-K. Nguyen, S. J. Lam, K. K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed.
- J. Li, K. Zhang, L. Ruan, S. F. Chin, N. Wickramasinghe, H. Liu, V. Ravikumar, J. Ren, H. Duan, L. Yang and M. B. Chan-Park, Nano Lett., 2018, 18, 4180–4187 CrossRef CAS PubMed.
- J. P. Tan, D. J. Coady, H. Sardon, A. Yuen, S. Gao, S. W. Lim, Z. C. Liang, E. W. Tan, S. Venkataraman and A. C. Engler, Adv. Healthcare Mater., 2017, 6, 1601420 CrossRef PubMed.
- S. Guo, Q. Huang, Y. Chen, J. Wei, J. Zheng, L. Wang, Y. Wang and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 618–623 CrossRef CAS PubMed.
- M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong and Z. Ji, Angew. Chem., Int. Ed., 2020, 59, 6412–6419 CrossRef CAS PubMed.
- J. Xie, M. Zhou, Y. Qian, Z. Cong, S. Chen, W. Zhang, W. Jiang, C. Dai, N. Shao and Z. Ji, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy and S. Reghu, Nat. Commun., 2019, 10, 1–14 CrossRef PubMed.
- Z. Sadrearhami, T.-K. Nguyen, R. Namivandi-Zangeneh, K. Jung, E. H. Wong and C. Boyer, J. Mater. Chem. B, 2018, 6, 2945–2959 RSC.
- M. Chen, J. Wei, S. Xie, X. Tao, Z. Zhang, P. Ran and X. Li, Nanoscale, 2019, 11, 1410–1422 RSC.
- R. F. Landis, C.-H. Li, A. Gupta, Y.-W. Lee, M. Yazdani, N. Ngernyuang, I. Altinbasak, S. Mansoor, M. A. Khichi and A. Sanyal, J. Am. Chem. Soc., 2018, 140, 6176–6182 CrossRef CAS PubMed.
- S. Tian, L. Su, Y. Liu, J. Cao, G. Yang, Y. Ren, F. Huang, J. Liu, Y. An, H. C. van der Mei, H. J. Busscher and L. Shi, Sci. Adv., 2020, 6, eabb1112 CrossRef CAS PubMed.
- J. M. V. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23–36 CrossRef CAS PubMed.
- Y. Liu, L. Shi, L. Su, H. C. van der Mei, P. C. Jutte, Y. Ren and H. J. Busscher, Chem. Soc. Rev., 2019, 48, 428–446 RSC.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-C. Wang, B. Sun and J. Wang, J. Am. Chem. Soc., 2012, 134, 4355–4362 CrossRef CAS PubMed.
- D. Pranantyo, C. Raju, Z. Si, X. Xu, K. Pethe, E.-T. Kang and M. B. Chan-Park, Nano Lett., 2021, 21, 899–906 CrossRef CAS PubMed.
- M. Ye, Y. Zhao, Y. Wang, N. Yodsanit, R. Xie and S. Gong, Adv. Funct. Mater., 2020, 30, 2002655 CrossRef CAS.
- M. Xiong, Y. Bao, X. Xu, H. Wang, Z. Han, Z. Wang, Y. Liu, S. Huang, Z. Song, J. Chen, R. M. Peek, L. Yin, L.-F. Chen and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12675–12680 CrossRef CAS PubMed.
- D. Pranantyo, E.-T. Kang and M. B. Chan-Park, Biomater. Sci., 2021, 9, 1627–1638 RSC.
- J. Hoque, B. Bhattacharjee, R. G. Prakash, K. Paramanandham and J. Haldar, Biomacromolecules, 2018, 19, 267–278 CrossRef CAS PubMed.
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Biomaterials, 2010, 31, 1444–1452 CrossRef CAS PubMed.
- J. Hu, Y. Quan, Y. Lai, Z. Zheng, Z. Hu, X. Wang, T. Dai, Q. Zhang and Y. Cheng, J. Controlled Release, 2017, 247, 145–152 CrossRef CAS PubMed.
- D. V. Amato, D. N. Amato, L. T. Blancett, O. V. Mavrodi, W. B. Martin, S. N. Swilley, M. J. Sandoz, G. Shearer, D. V. Mavrodi and D. L. Patton, Acta Biomater., 2018, 67, 196–205 CrossRef CAS PubMed.
- S.-C. Park, N.-H. Kim, W. Yang, J.-W. Nah, M.-K. Jang and D. Lee, J. Controlled Release, 2016, 221, 37–47 CrossRef CAS PubMed.
- V. V. Komnatnyy, W. C. Chiang, T. Tolker-Nielsen, M. Givskov and T. E. Nielsen, Angew. Chem., Int. Ed., 2014, 53, 439–441 CrossRef CAS PubMed.
- B. Wang, H. Liu, Z. Wang, S. Shi, K. Nan, Q. Xu, Z. Ye and H. Chen, J. Mater. Chem. B, 2017, 5, 1498–1506 RSC.
- M. H. Xiong, Y. J. Li, Y. Bao, X. Z. Yang, B. Hu and J. Wang, Adv. Mater., 2012, 24, 6175–6180 CrossRef CAS PubMed.
- M. Xiong, Z. Han, Z. Song, J. Yu, H. Ying, L. Yin and J. Cheng, Angew. Chem., Int. Ed., 2017, 56, 10826–10829 CrossRef CAS PubMed.
- S. Mizukami, M. Kashibe, K. Matsumoto, Y. Hori and K. Kikuchi, Chem. Sci., 2017, 8, 3047–3053 RSC.
- J. H. Boyce, B. Dang, B. Ary, Q. Edmondson, C. S. Craik, W. F. DeGrado and I. B. Seiple, J. Am. Chem. Soc., 2020, 142, 21310–21321 CrossRef CAS PubMed.
- H. A. Hibbard and M. M. Reynolds, Bioorg. Chem., 2019, 93, 103318 CrossRef CAS PubMed.
- H. Ji, K. Dong, Z. Yan, C. Ding, Z. Chen, J. Ren and X. Qu, Small, 2016, 12, 6200–6206 CrossRef CAS PubMed.
- W. Neumann, M. Sassone-Corsi, M. Raffatellu and E. M. Nolan, J. Am. Chem. Soc., 2018, 140, 5193–5201 CrossRef CAS PubMed.
- Y. Li, X. Hu, S. Tian, Y. Li, G. Zhang, G. Zhang and S. Liu, Biomaterials, 2014, 35, 1618–1626 CrossRef CAS PubMed.
- T. Sen, S. Karmakar and R. Sarkar, in Evidence-Based Validation of Herbal Medicine, Elsevier, 2015, pp. 321–338 Search PubMed.
- S. K. Samal, M. Dash, S. Van Vlierberghe, D. L. Kaplan, E. Chiellini, C. Van Blitterswijk, L. Moroni and P. Dubruel, Chem. Soc. Rev., 2012, 41, 7147–7194 RSC.
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. Satchi-Fainaro, J. Controlled Release, 2012, 161, 446–460 CrossRef CAS PubMed.
- R. Toy, P. Pradhan, V. Ramesh, N. C. Di Paolo, B. Lash, J. Liu, E. L. Blanchard, C. J. Pinelli, P. J. Santangelo and D. M. Shayakhmetov, Biomaterials, 2019, 225, 119512 CrossRef CAS PubMed.
- M. E. Falagas and S. K. Kasiakou, Crit. Care, 2006, 10, 1–13 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |