
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIr/f-Ampha complex catalyzed asymmetric sequential hydrogenation of enones: a general access to chiral alcohols with two contiguous chiral centers†
Wendian
Li‡
ad,
Tilong
Yang‡
 c,
Nan
Song
a,
Ruihao
Li
a,
Jiao
Long
a,
Lin
He
b,
Xumu
Zhang
c and
Hui
Lv
*a
c,
Nan
Song
a,
Ruihao
Li
a,
Jiao
Long
a,
Lin
He
b,
Xumu
Zhang
c and
Hui
Lv
*a
aSauvage Center for Molecular Sciences, Key Laboratory of Biomedical Polymers of Ministry of Education & College of Chemistry and Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, Wuhan University, Wuhan, Hubei 430072, China. E-mail: huilv@whu.edu.cn
bKey Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, School of Chemistry and Chemical Engineering, Shihezi University, Xinjiang Uygur Autonomous Region, 832000, China
cDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China
dChina Tobacco Sichuan Industrial Company, Ltd., Chengdu, Sichuan 610065, China
First published on 18th January 2022
Abstract
A general and highly efficient method for asymmetric sequential hydrogenation of α,β-unsaturated ketones has been developed by using an iridium/f-Ampha complex as the catalyst, furnishing corresponding chiral alcohols with two contiguous stereocenters in high yields with excellent diastereo- and enantioselectivities (up to 99% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and >99% ee). Control experiments indicated that the C
:1 dr and >99% ee). Control experiments indicated that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds of the enones were hydrogenated sequentially, and the final stereoselectivities were determined by the dynamic kinetic resolution of ketones. Moreover, DFT calculations revealed that an outer sphere pathway was involved in both reduction of CC and CO bonds of enones. The synthetic utility of this method was demonstrated by a gram-scale reaction with very low catalyst loading (S/C = 20000) and a concise synthetic route to key chiral intermediates of the antiasthmatic drug CP-199,330.
C and CO bonds of the enones were hydrogenated sequentially, and the final stereoselectivities were determined by the dynamic kinetic resolution of ketones. Moreover, DFT calculations revealed that an outer sphere pathway was involved in both reduction of CC and CO bonds of enones. The synthetic utility of this method was demonstrated by a gram-scale reaction with very low catalyst loading (S/C = 20000) and a concise synthetic route to key chiral intermediates of the antiasthmatic drug CP-199,330.
Introduction
Owing to the perfect atom-economy, environmental benignancy and operational simplicity, asymmetric hydrogenation has been widely investigated both in academia and in industry, and it has emerged as one of the most important approaches to prepare chiral molecules.1 In this context, the selective asymmetric hydrogenation of CO2 or CC3 bonds of α,β-unsaturated ketones has been well established by choosing a well-designed ligand combined with optimization of reaction conditions. In comparison, asymmetric sequential hydrogenation of multiply substituted α,β-unsaturated ketones in one step is rarely exploited although it provides a powerful strategy for the construction of molecules with multi-chiral centers.4 The possible reasons may be attributed to the following challenges: first, the reaction mechanism of asymmetric hydrogenation of ketones is different from that of alkenes, it usually needs different catalysts and different reaction conditions to obtain good results in the asymmetric hydrogenation of ketones or olefins, thus it's very difficult to achieve the asymmetric sequential hydrogenation of multisubstituted enones with high yields and excellent stereoselectivities by using a single catalyst in one step. In addition, the first formed chiral center usually has an impact on the stereocontrol of the next chiral center generated, which makes it difficult to obtain high diastereoselectivity. Moreover, due to the different reactivities of ketones and olefins in asymmetric hydrogenation and different coordination models between the metal catalyst and substrate (inner sphere vs. outer sphere), it's a challenge to obtain high turnover numbers (TONs) for the sequential reduction of α,β-unsaturated ketones. For example, the Ru–PHOX–Ru catalytic system developed by the Zhang group exhibited excellent performance in the selective asymmetric hydrogenation of the CO bonds of exocyclic α,β-unsaturated pentenones (S/C is up to 10000), but failed to complete both reduction of CO and CC bonds even by increasing the catalyst loading (S/C = 100), hydrogen pressure and reaction temperature, only less than 50% sequential reduction product was obtained with poor diastereoselectivity (2:1) (Scheme 1-2).2a As a result, the asymmetric sequential hydrogenation of enones could be achieved only in a few cases, and most of them featured moderate diastereoselectivity, low turnover number and limited substrate scope.4b–f To date, multistep reduction is still the main method to achieve both asymmetric reduction of CO and CC bonds of enones.2a–c,3j Thus, it's highly desirable to develop new protocols to achieve asymmetric sequential hydrogenation of multiply substituted α,β-unsaturated ketones in a simple and efficient manner.
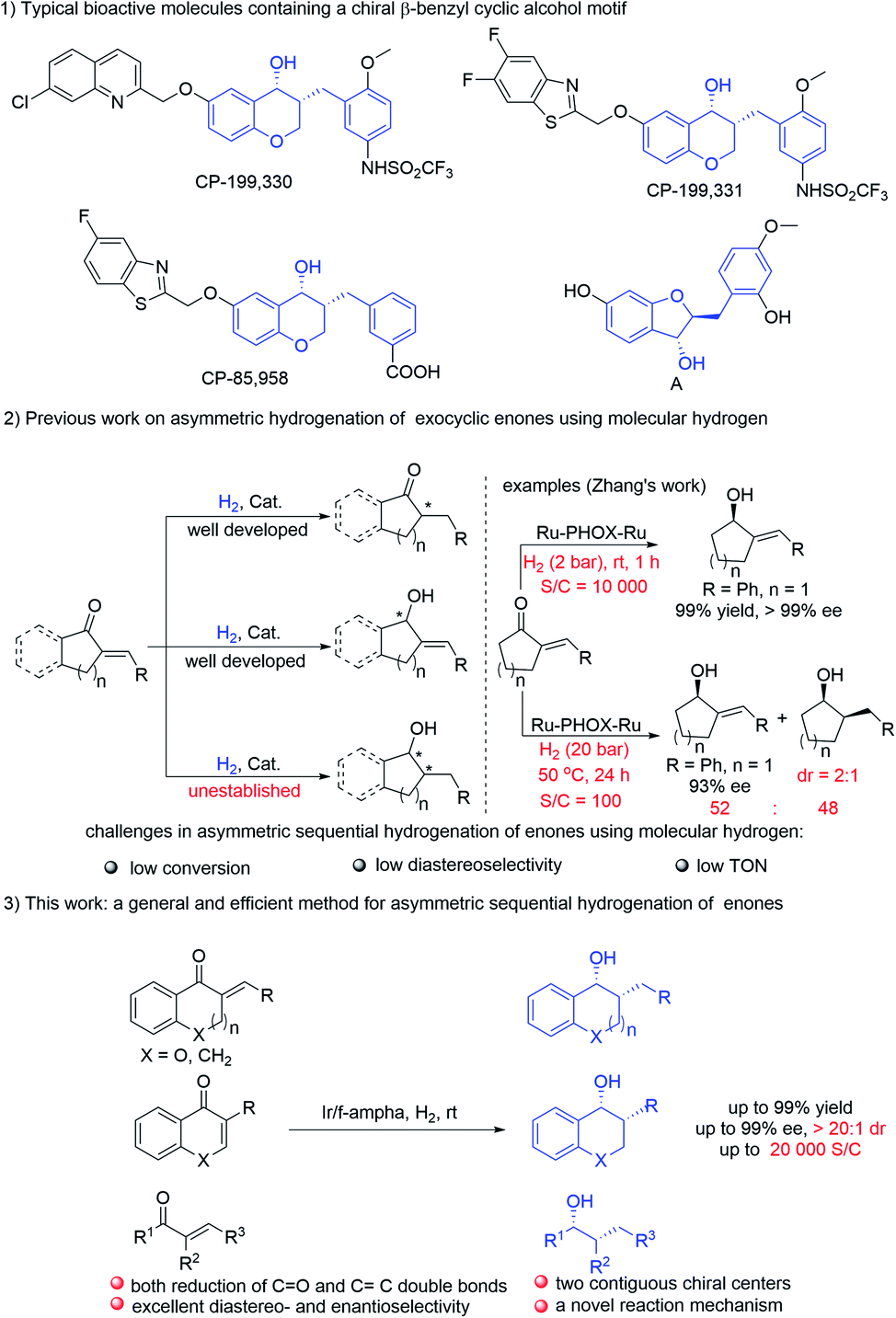 | ||
| Scheme 1 Asymmetric hydrogenation of exocyclic enones using H2 and typical bioactive molecules containing a chiral β-benzyl cyclic alcohol unit. | ||
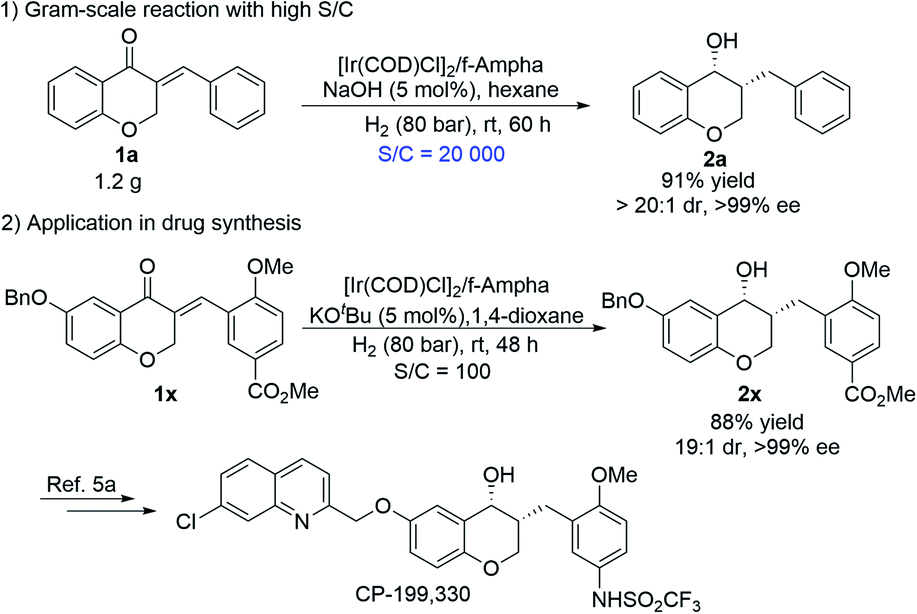 | ||
| Scheme 2 Gram-scale experiment with high TON and asymmetric synthesis of the antiasthmatic drug CP-199,330. | ||
Chiral 4-chromanols and their derivatives are privileged structural motifs widely occurring in natural products and drug lead compounds (Scheme 1-1).5 Particularly, chiral 3-alkyl-chroman-4-ols are known to be medicinally important motifs with a wide range of biological activities. For instance, CP-199,330 and CP-199,331 are potent cysteinyl leukotriene-1 (LT1) receptor antagonists, targeted for the treatment of asthma.5a,i CP-85,958 is useful in the treatment of arthritis and asthma.5f,g Compound A, which was isolated from dehulled adlay seeds, displays an anti-inflammatory effect.6 Considerable efforts have been directed toward the synthesis of chiral chromanols.5a,f,g However, protocols for their enantioselective preparation are still very limited. To our surprise, asymmetric sequential reduction of exocyclic α,β-unsaturated ketones using molecular hydrogen, one of the most straightforward methods to synthesize chiral 3-alkylchroman-4-ols, has not been established.7 Devising new methods for the asymmetric synthesis of 3-alkylchroman-4-ols is urgently needed.
Recently, our group developed a series of ferrocene-based tridentate amino-phosphine ligands, such as f-Amphox,8 f-Ampha9 and f-Amphol,10 which showed excellent performance in the Ir-catalyzed asymmetric hydrogenation of prochiral ketones. We envision that the active Ir–H species may react with multiply substituted enones through 1,4-addition to form α-substituted ketones, followed by dynamic kinetic resolution (DKR) of ketones to achieve the asymmetric sequential hydrogenation of enones with high diastereo- and enantioselectivity. Herein, we report a general and efficient approach for the synthesis of chiral alcohols with two contiguous chiral centers by the Ir/f-Ampha complex enabled asymmetric sequential hydrogenation of α,β-unsaturated ketones.
Results and discussion
Our initial studies began with the optimization of reaction conditions by using the asymmetric hydrogenation of (E)-3-benzylidene chroman-4-one 1a as a model reaction. With 0.1 mol% [Ir(COD)Cl]2 and 5 mol% KOtBu in iPrOH at room temperature, a set of chiral tridentate ligands developed by our group were evaluated. When f-Amphox L1 was used, only 13% syn 3-benzylchroman-4-ol 2a was obtained along with a large amount of the ketone reduction product 2a′′ (Table 1, entry 1). f-Amphol L2 and f-Ampha L3 exhibited good reactivity for sequential reduction of enone 1a, both of them only afforded target product 2a with excellent yields and good ee values (Table 1, entries 2–3). Considering the product distribution and enantioselectivity, L3 was chosen as the best ligand. Then, solvent screening was conducted and the results disclosed that solvents have a significant impact on the yield and the enantioselectivity of 2a. Most of the solvents tested here, such as dioxane, dichloromethane, dichloroethane, tetrahydrofuran, ethanol, and trifluoroethanol, are mainly beneficial for the reduction of CC double bonds, delivering the α-chiral ketone 2a′ as the main product (Table 1, entry 4 and entries 7–11). Fortunately, the desired product 2a was obtained in a quantitative yield with excellent enantioselectivity by using hexane as the solvent (Table 1, entry 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Conv.b (%) | 2a/2a′/2a′′b | drb (2a) | eec (%) |
|
a [Ir(COD)Cl]2/ligand/1a (0.1 mmol) ratio of 0.5:1.1:500 in 1.0 mL solvent. In all cases described in this manuscript, little iPrOH as the solvent of the catalyst was introduced into the reaction mixture.
b Determined by 1H NMR analysis.
c Determined by HPLC analysis using a chiral stationary phase. The configuration of 2a was determined by comparing the specific rotation of 2a with the data in the literature.7a
|
||||||
| 1 | L1 | i PrOH | >99 | 13/0/87 | >20:1 |
>99 |
| 2 | L2 | i PrOH | >99 | 100/0/0 | >20:1 |
87 |
| 3 | L3 | i PrOH | >99 | 100/0/0 | >20:1 |
90 |
| 4 | L3 | Dioxane | >99 | 14/86/0 | >20:1 |
90 |
| 5 | L3 | Toluene | >99 | 65/35/0 | >20:1 |
97 |
| 6 | L3 | Hexane | >99 | 100/0/0 | >20:1 |
98 |
| 7 | L3 | THF | >99 | 36/64/0 | >20:1 |
92 |
| 8 | L3 | DCM | >99 | 21/79/0 | >20:1 |
83 |
| 9 | L3 | DCE | >99 | 5/95/0 | >20:1 |
11 |
| 10 | L3 | EtOH | >99 | 31/69/0 | >20:1 |
19 |
| 11 | L3 | TFE | >99 | 3/97/0 | >20:1 |
— |
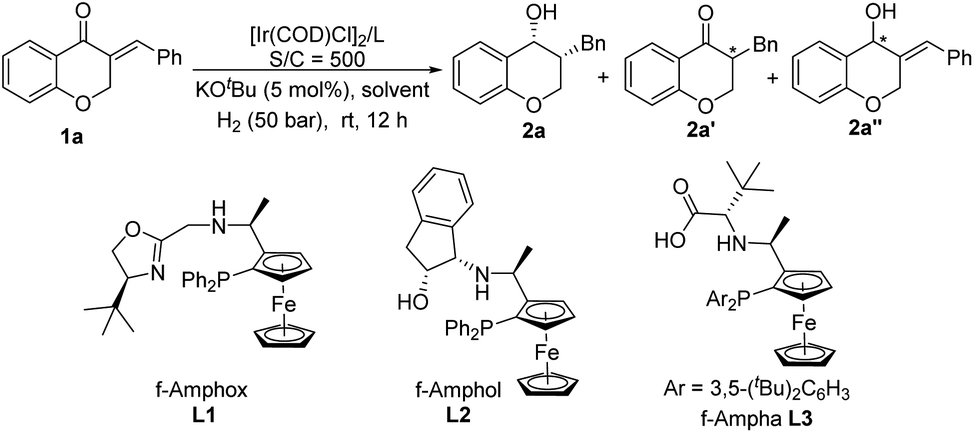
Subsequently, the effect of bases was investigated and the results are summarized in Table 2. The reaction did not occur in the absence of a base (Table 2, entry 1). Strong bases and moderately strong bases, such as NaOH, NaOMe, NaOtBu, KOtBu, and Cs2CO3, were good choices for this transformation, giving 2a with high yields and excellent stereoselectivities (Table 2, entries 2–6). When Na2CO3 was used, only 65% conversion was obtained and more than half of the converted starting material was only transformed to the product hydrogenated at the CC double bond, which indicated that weak bases had a detrimental impact on the sequential hydrogenation of 1a (Table 2, entry 7). To screen out the best base, a series of bases were re-evaluated under 0.05 mol% catalyst loading (S/C = 2000), and the results show that NaOH was superior to others (Table 2, entries 8–11). In addition, the effect of catalyst loading was also investigated. To our delight, on decreasing the catalyst loading to 0.01 mol% (S/C = 10000), there was no loss of yield or diastereo- and enantioselectivity (Table 2, entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Conv.b (%) |
2a:2a′b |
drb (2a) | eec (%) |
|
a Unless otherwise mentioned, all reactions were carried out with a [Ir(COD)Cl]2/f-Ampha/1a (0.1 mmol) ratio of 0.5:1.1:500 in 1.0 mL of hexane.
b Determined by 1H NMR analysis.
c Determined by HPLC analysis using a chiral stationary phase.
d S/C = 2000.
e S/C = 10000. NR = no reaction. NA = not available.
|
|||||
| 1 | — | NR | NA | NA | NA |
| 2 | NaOH | 99 | >20:1 |
>20:1 |
98 |
| 3 | NaOMe | 99 | 15:1 |
>20:1 |
97 |
| 4 | NaOtBu | 99 | >20:1 |
15:1 |
98 |
| 5 | KOtBu | 99 | >20:1 |
>20:1 |
98 |
| 6 | Cs2CO3 | 99 | >20:1 |
>20:1 |
98 |
| 7 | Na2CO3 | 65 | 0.9:1 |
15:1 |
94 |
| 8d | NaOH | 99 | >20:1 |
>20:1 |
99 |
| 9d | Cs2CO3 | 99 | 2.3:1 |
17:1 |
97 |
| 10d | KOtBu | 99 | 0.1:1 |
>20:1 |
87 |
| 11d | NaOtBu | 99 | >20:1 |
15:1 |
99 |
| 12e | NaOH | 99 | >20:1 |
>20:1 |
99 |
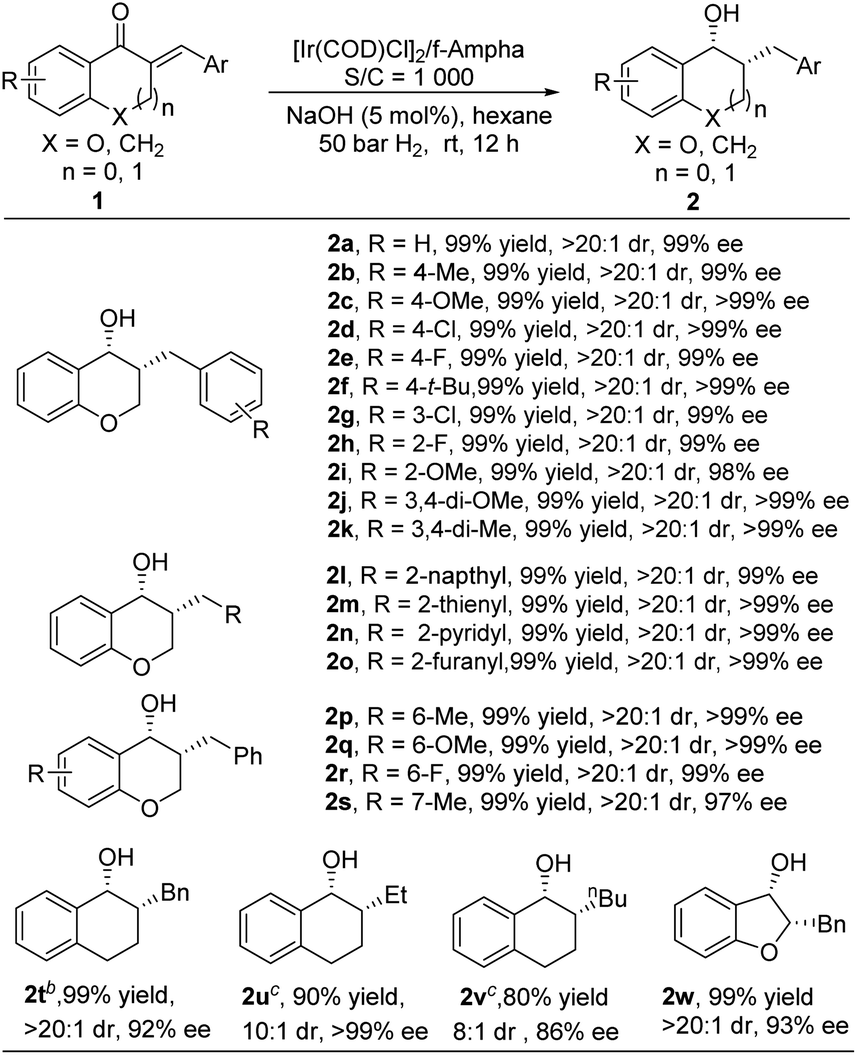
With the optimal conditions in hand, the substrate scope of this reaction was explored with 0.1 mol% catalyst and the results are summarized in Table 3. Delightfully, the reaction exhibited good tolerance to a variety of exocyclic α,β-unsaturated ketones, furnishing the corresponding products in quantitative yields with excellent diastereo- and enantioselectivities. Generally, the reaction was not affected by the substitution pattern and the electronic properties of the aryl group, giving target products with 99% yield and 99% ee (2a–2l). Heteroaryl substituted substrates (2m–2o) were also well tolerated. Changing the position and the electronic properties of the R group didn't cause any changes in reactivity and in stereocontrol, affording target products with high yields and excellent diastereo- and enantioselectivities (2p–2s). When the O atom on the ring was replaced by a methylene group, the reactivity dropped, it required 0.5–1 mol% catalyst loading to achieve this transformation, delivering 2t–2v with good yields and high stereoselectivities. Interestingly, when the ring size was changed from a 6-membered to a 5-membered ring, the reaction proceeded smoothly, offering chiral 3-hydroxyl-2,3-dihydrobenzofurans, the core structure of several natural products,11 in 99% yield with 93% ee (2w).
|
a Unless otherwise mentioned, all reactions were carried out with a [Ir(COD)Cl]2/f-Ampha/1 (0.5 mmol) ratio of 0.5:1.1:1000 in 2.0 mL of hexane. The dr was determined by 1H NMR. The yield was an isolated yield. The ee was determined by HPLC analysis using a chiral stationary phase.
b 24 h, S/C = 200.
c 36 h, S/C = 100, NaOMe was used instead of NaOH.
|
|---|
|
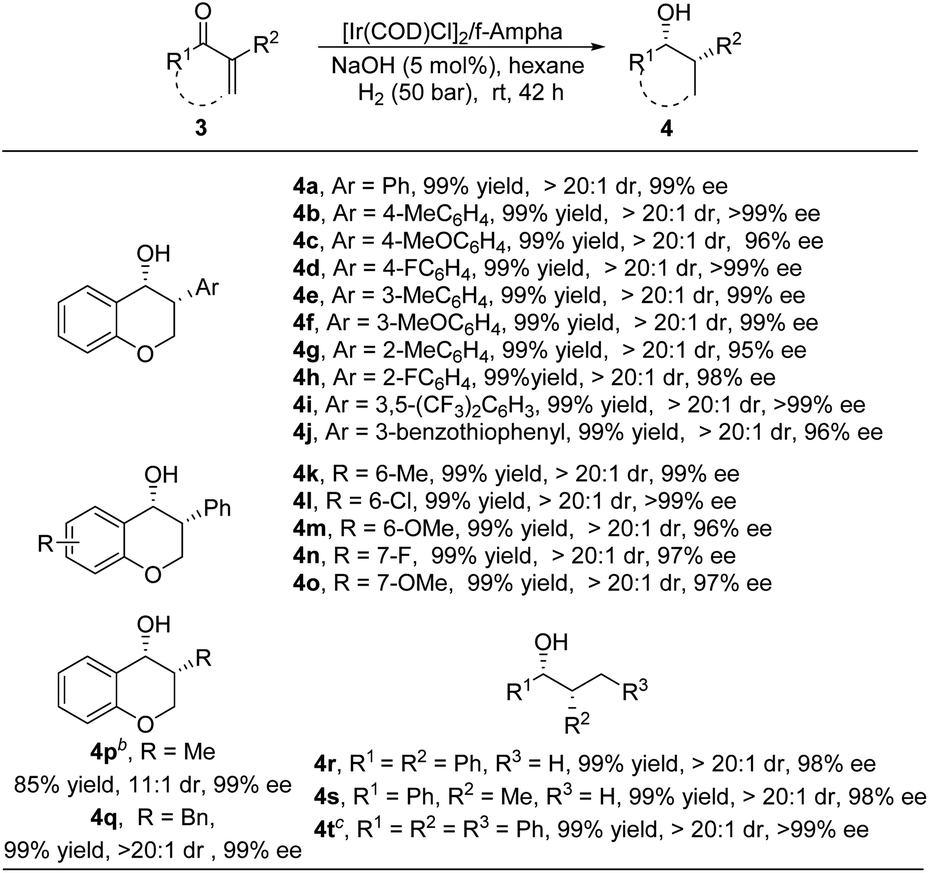
To further explore the substrate scope of this methodology, other types of enones, such as endocyclic enones and acyclic enones, were also examined under standard conditions. As shown in Table 4, various 3-aryl substituted endocyclic enones, regardless of the electronic properties and the position of the substituents on the benzene ring, were well tolerated in this transformation, furnishing 3-arylchroman-4-ones 4a–4o with high yields and excellent enantioselectivities. Alkyl substituted endocyclic enones were also compatible in this reaction, delivering target products 4p–4q in high yields with good diastereo- and enantioselectivities. Moreover, acyclic enones also proved to be good substrates for this transformation, affording corresponding alcohols 4r–4t in 99% yield with 98–99% ee.
|
a Unless otherwise mentioned, all reactions were carried out with an [Ir(COD)Cl]2/f-Ampha/3 (0.2 mmol) ratio of 0.5:1.1:1000 in 2.0 mL of hexane. The dr was determined by 1H NMR. The yield was an isolated yield. The ee was determined by HPLC analysis using a chiral stationary phase.
b 5 mol% KOtBu, 16% TBAOH (tetrabutylammonium hydroxide).
c S/C = 100, 4 days.
|
|---|
|
To demonstrate the potential utility of this catalytic system, the gram-scale reaction with 0.005 mol% catalyst loading (S/C = 20000) was conducted, and it proceeded very smoothly, affording 2a in high yield and excellent diastereo- and enantioselectivity (Scheme 2-1, 91% yield, >99% ee, >20:1 dr), which indicated that the Ir/f-Ampha complex is very efficient for the sequential asymmetric hydrogenation of enones, and has a practical application prospect. In addition, a highly efficient and concise synthetic route to the antiasthmatic drug CP-199,330 was developed. As shown in Scheme 2-2, 1x can be sequentially hydrogenated efficiently to afford 2x in 88% yield and 99% ee, which can be transformed into the antiasthmatic drug CP-199,330 according to the reported procedure.5a Compared with the previous method through racemic resolution,12 the sequential asymmetric hydrogenation strategy is more cost-effective and more environmentally friendly.
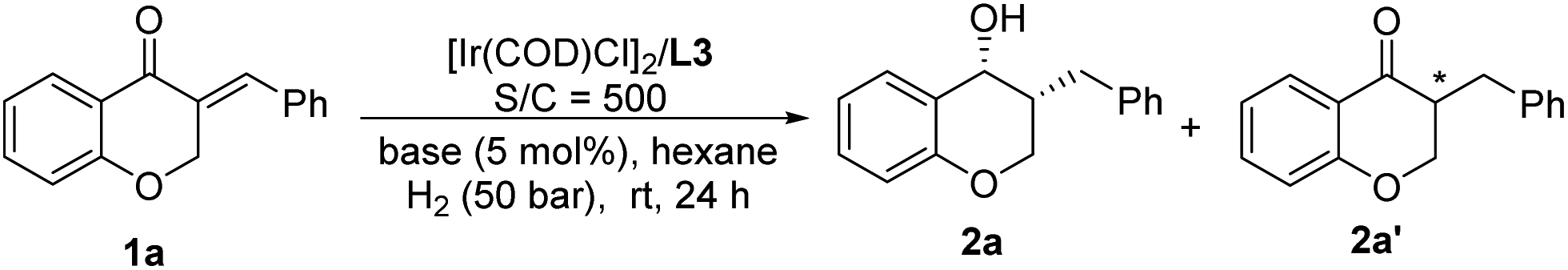
The change of product distribution with reaction time was investigated to shed light on the reaction mechanism. As shown in Fig. 1, most of 1a was quickly transformed into 2a′ within 0.17 hours by selective reduction of CC bonds, without any 2a′′ and 2a being detected. Prolonging the reaction time to one hour, the starting material 1a was completely consumed, affording target product 2a in 82% yield along with 2a′ in 18% yield. On further increasing the reaction time, the amount of 2a′ decreased gradually until it was totally transformed into 2a. It was noteworthy that no 2a′′ was detected in these experiments. In order to further identify the reaction pathway, the possible intermediates 2a′ and 2a′′ were synthesized and then hydrogenated under the standard reaction conditions. The results show that the intermediate 2a′ was hydrogenated smoothly, affording chiral 3-benzylchroman-4-ol 2a in a quantitative yield with excellent enantioselectivity and diastereoselectivity (Scheme 3-1), which demonstrated that a dynamic kinetic resolution (DKR) was involved in the reaction process. 2a′′ can't be hydrogenated under standard conditions, which indicated that the reaction pathway involving the asymmetric hydrogenation of a ketone followed by the reduction of CC double bonds was excluded (Scheme 3-2). Taking together, these results clearly demonstrated that the hydrogenation of 1a proceeded through the hydrogenation of the CC double bonds of 2a to form 2a′, then asymmetric hydrogenation of 2a′via DKR to afford the desired product 2a.
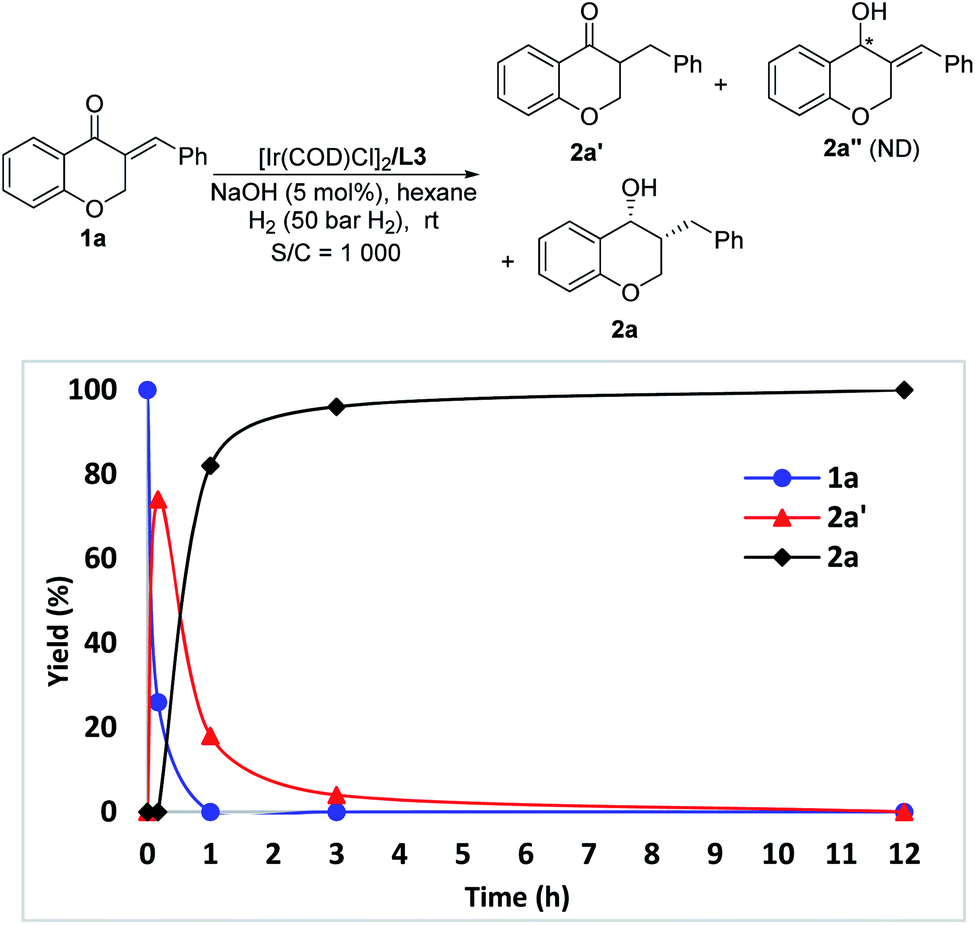 | ||
| Fig. 1 Plots of product distribution with reaction time. | ||
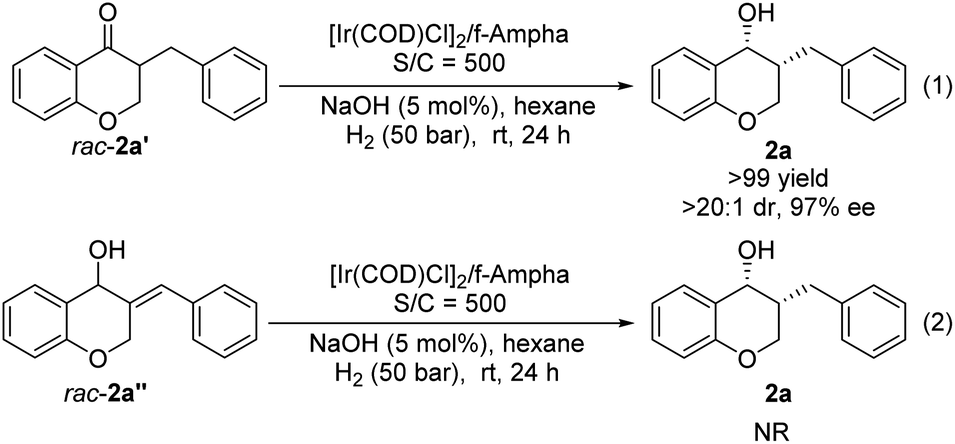 | ||
| Scheme 3 The hydrogenation of the possible intermediates 2a′ and 2a′′. | ||
DFT calculations were also conducted to understand the reaction pathway and the enantioselectivity in the DKR process. The hydrogenation of 1a to 2a was considered in our DFT calculations. Referring to the above mechanistic discussion and our related work,13 an outer sphere pathway for 1,4-addition to produce 2a′, followed by hydrogenation of CO is proposed (Scheme 4). The role of alkali cation in hydrogenation reactions has been demonstrated in recent publications14 and our previous study.9 The trihydride IrH3(f-Ampha) complex I involving an alkali cation Na+ was proposed as the starting point in our DFT study. Complex I attracts 1a and gives complex II, and the reaction is exothermic by 10.7 kcal mol−1. The Ir(III)-hydride can make a nucleophilic attack on the carbonyl carbon or the β-carbon on the Re/Si face of 1a. DFT calculations indicate that hydride transfer to the β-carbon of the 1a Re face gives the most favorable transition state (TSII–III) with a barrier of 9.4 kcal mol−1 (with respect to complex II) (see the ESI, Fig. S1 and S2†). It is understandable that the carbonyl as a π withdrawing group increases the contribution of the β-carbon to the LUMO, thus promoting the hydride transfer to the β-carbon. A similar mechanism of nucleophilic attack of the Cu–B σ bond on α,β-unsaturated carbonyl compounds was well studied.15 After hydride transfer, molecular H2 coordinates to the Ir(III) and forms a dihydrogen Ir(III)(η2-H2) complex III+H2. The H2 activation between the Ir(III) and the enolate anion (formed by hydride transfer to 1a) is barrierless and gives an enol compound 2a′–1. The compound 2a′–1 undergoes a keto-to-enol isomerization to the more stable keto 2a′. Compound 2a′ enters the cycle B. The hydrogenation of 2a′ to the β-benzyl cyclic alcohol 2a is also achieved through hydride transfer and H2 activation processes (see the ESI, Fig. S3†). The hydride transfer to the carbonyl carbon is the enantio-determining step. The energy barrier leading to the major product 2a(R,R) is 10.9 kcal mol−1 from complex V, which is lower than the barriers to 2a(S,R), 2a(S,S) and 2a(R,S) by 2.2, 4.3 and 7 kcal mol−1, respectively (see the ESI, Fig. S4†). The DFT-calculated ee and dr values are 99.9% and 40:1, which are well consistent with the experimental results (ee 99%, dr > 20:1). The unique chair pocket of the Ir(III)/f-Ampha catalyst can account for the excellent enantioselectivities and diastereoselectivities in the DKR process.
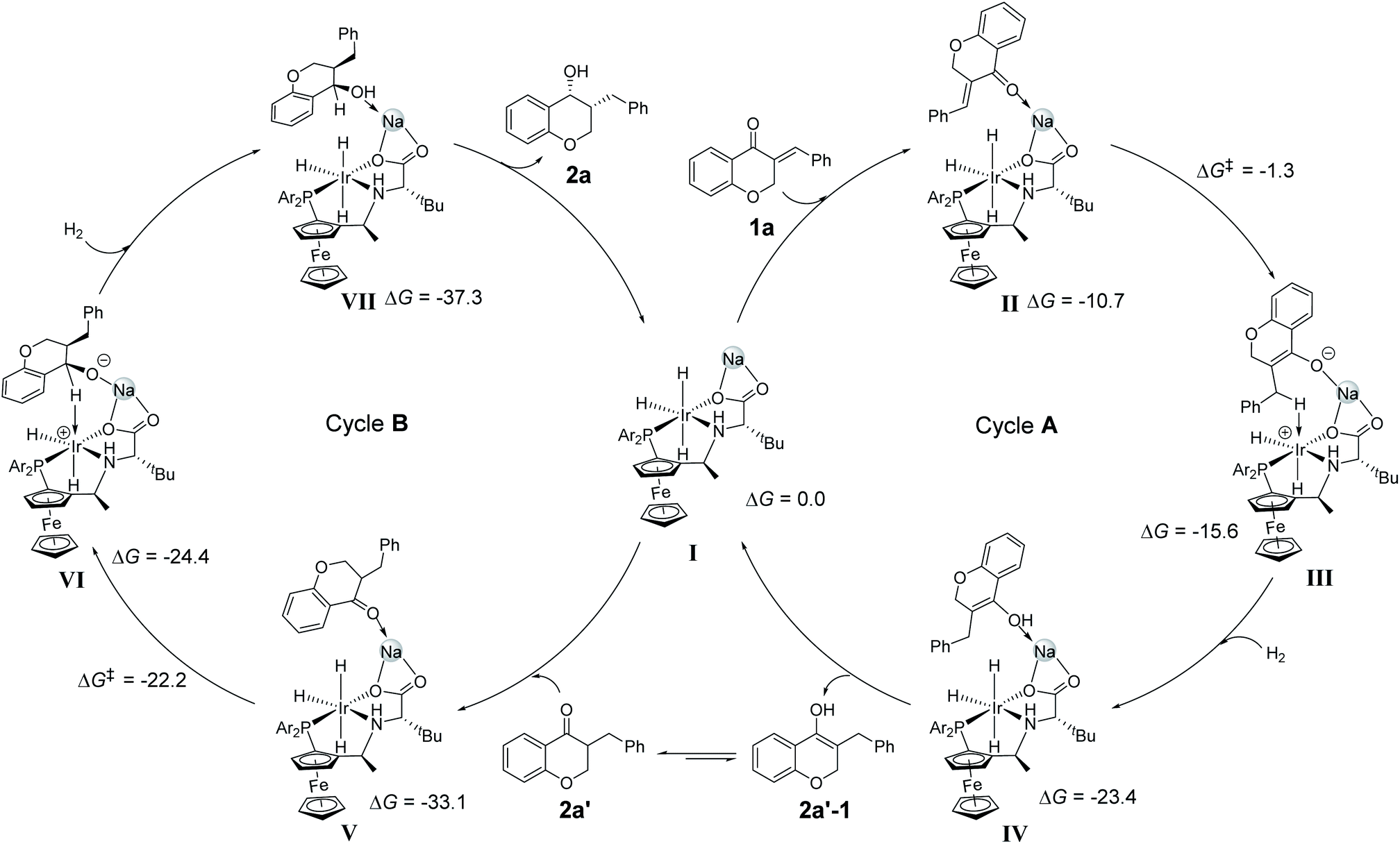 | ||
| Scheme 4 Proposed catalytic cycles. Cycle A: 1,4-addition of 1a; cycle B: DKR to form 2a. Relative free energies are given in kcal mol−1. | ||
Conclusions
In summary, we have developed a highly efficient asymmetric sequential hydrogenation of α,β-unsaturated ketones via an Ir/f-ampha complex catalyzed DKR process, which provides a general and practical access to chiral alcohols with two contiguous stereocenters. Remarkably, this method not only exhibited excellent stereo-control (up to >99% ee and >20:1 dr, TON up to 18200), but also showed excellent substrate compatibility with various enones, including exocyclic α,β-unsaturated ketones, endocyclic α,β-unsaturated ketones and acyclic enones, which make this method particularly valuable in organic synthesis. In addition, the experimental and theoretical calculation investigations of the mechanism revealed that the CC and CO double bonds of the enones were hydrogenated sequentially through an outer sphere pathway. Furthermore, the synthetic utility of this protocol was demonstrated by a concise synthetic route to a key chiral intermediate of the antiasthmatic drug CP-199,330. Further studies on the extension of this novel catalytic system are currently underway in our laboratory.
Data availability
All experimental procedures, characterization data, computational methods and detailed energy profiles in this article are available in the ESI.†Author contributions
H. L. conceived the project. W. L., N. S., R. L. and J. L. performed the experiments. T. Y. performed DFT calculations. W. L. and H. L. wrote the manuscript, H. L and X. Z. provided useful suggestions for this work. All the authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 22071188, 21871212), the open foundation of CAS Key Laboratory of Molecular Recognition and Function, the “Double First-Class” project of Shihezi University.Notes and references
- Selected reviews for recent advances on transition-metal catalyzed asymmetric hydrogenation, see: (a) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278–1290 CrossRef CAS PubMed; (b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (c) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed; (d) G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101–1112 CrossRef CAS PubMed; (e) Z. Zhang, N. A. Butt, M. Zhou, D. Liu and W. Zhang, Chin. J. Chem., 2018, 36, 443–454 CrossRef CAS; (f) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed; (g) Q.-A. Chen, Z.-S. Ye, Y. Duan and Y.-G. Zhou, Chem. Soc. Rev., 2013, 42, 497–511 RSC; (h) Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769–14827 CrossRef CAS PubMed; (i) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427–1438 CrossRef CAS; (j) Y. Liu, Z. Wang and K. Ding, Acta Chim. Sin., 2012, 70, 1464–1470 CrossRef CAS; (k) S. Kraft, K. Ryan and R. B. Kargbo, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS PubMed; (l) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS PubMed; (m) F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed; (n) C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346–1356 CrossRef CAS PubMed; (o) P. Tang, H. Wang, W. Zhang and F.-E. Chen, Green Synth. Catal., 2020, 1, 26–41 CrossRef.
- (a) J. Li, Y. Zhu, Y. Lu, Y. Wang, Y. Liu, D. Liu and W. Zhang, Organometallics, 2019, 38, 3970–3978 CrossRef CAS; (b) J. Li, Y. Lu, Y. Zhu, Y. Nie, J. Shen, Y. Liu, D. Liu and W. Zhang, Org. Lett., 2019, 21, 4331–4335 CrossRef CAS PubMed; (c) J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538–4539 CrossRef CAS PubMed; (d) F. Chen, Y. Zhang, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2017, 56, 2022–2025 CrossRef CAS PubMed; (e) R. Moser, Ž. V. Bošković, C. S. Crowe and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 7852–7853 CrossRef CAS PubMed; (f) J. Kim, J. Bruning, K. E. Park, D. J. Lee and B. Singaram, Org. Lett., 2009, 11, 4358–4361 CrossRef CAS PubMed; (g) Y. Wang, G. Yang, F. Xie and W. Zhang, Org. Lett., 2018, 20, 6135–6139 CrossRef CAS PubMed.
- (a) S.-M. Lu and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 8920–8923 CrossRef CAS PubMed; (b) W.-J. Lu, Y.-W. Chen and X.-L. Hou, Angew. Chem., Int. Ed., 2008, 47, 10133–10136 CrossRef CAS PubMed; (c) F. Tian, D. Yao, Y. Liu, F. Xie and W. Zhang, Adv. Synth. Catal., 2010, 352, 1841–1845 CrossRef CAS; (d) X. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 936–940 CrossRef CAS PubMed; (e) X. Liu, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2014, 53, 1978–1982 CrossRef CAS PubMed; (f) Q. Li, P. Wan, Y. He, Y. Zhou, L. Li, B. Chen, K. Duan, R. Cao, Z. Zhou and L. Qiu, Asian J. Org. Chem., 2014, 3, 774–783 CrossRef CAS; (g) J. Xia, Y. Nie, G. Yang, Y. Liu, I. D. Gridnev and W. Zhang, Chin. J. Chem., 2018, 36, 612–618 CrossRef CAS; (h) B. Gao, X. Feng, W. Meng and H. Du, Angew. Chem., Int. Ed., 2020, 59, 4498–4504 CrossRef CAS PubMed; (i) J. Yang, X. Li, C. You, S. Li, Y.-Q. Guan, H. Lv and X. Zhang, Org. Biomol. Chem., 2020, 18, 856–859 RSC; (j) J. Xia, Y. Nie, G. Yang, Y. Liu, I. D. Gridnev and W. Zhang, Chin. J. Chem., 2018, 36, 612–618 CrossRef CAS; (k) B. B. C. Peters, J. Jongcharoenkamol, S. Krajangsri and P. G. Andersson, Org. Lett., 2021, 23, 242–246 CrossRef CAS PubMed.
- (a) L. Tang, Z. Lin, Q. Wang, X. Wang, L. Cun, W. Yuan, J. Zhu and J. Deng, Tetrahedron Lett., 2012, 53, 3828–3830 CrossRef CAS; (b) D. Zhao, B. Beiring and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 8454–8458 CrossRef CAS PubMed; (c) Q. Hu, J. Chen, Z. Zhang, Y. Liu and W. Zhang, Org. Lett., 2016, 18, 1290–1293 CrossRef CAS PubMed; (d) Y.-T. Liu, J.-Q. Chen, L.-P. Li, X.-Y. Shao, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2017, 19, 3231–3234 CrossRef CAS PubMed; (e) Y. Ma, J. Li, J. Ye, D. Liu and W. Zhang, Chem. Commun., 2018, 54, 13571–13574 RSC; (f) N. Arai, H. Satoh, R. Komatsu and T. Ohkuma, Chem.–Eur. J., 2017, 23, 8806–8809 CrossRef CAS PubMed.
- (a) R. J. Chambers, A. Marfat, G. W. Antognoli, J. B. Cheng, D. B. Damon, A. V. Kuperman, T. C. Liston, C. Mebus, J. S. Pillar, J. T. Shirley and J. W. Watson, Bioorg. Med. Chem. Lett., 1999, 9, 2773–2778 CrossRef CAS PubMed; (b) P. P. Deshpande, F. Tagliaferri, S. F. Victory, S. Yan and D. C. Baker, J. Org. Chem., 1995, 60, 2964–2965 CrossRef CAS; (c) T. Ma, L. Liu, H. Xue, L. Li, C. Han, L. Wang, Z. Chen and G. Liu, J. Med. Chem., 2008, 51, 1432–1446 CrossRef CAS PubMed; (d) C. Conti and N. Desideri, Bioorg. Med. Chem., 2009, 17, 3720–3727 CrossRef CAS PubMed; (e) K. Koch, L. S. Melvin, L. A. Reiter, M. S. Biggers, H. J. Showell, R. J. Griffiths, E. R. Pettipher, J. B. Cheng and A. J. Milici, J. Med. Chem., 1994, 37, 3197–3199 CrossRef CAS PubMed; (f) E. G. Andrews, G. W. Antognoli, R. Breslow, M. P. Carta, T. J. Carty, R. J. Chambers, J. B. Cheng, V. L. Cohan, J. L. Collins, D. B. Damon, J. Delehunt, J. F. Eggler, J. D. Eskra, K. W. Freiert, W. A. Hada, A. Marfat, H. Masamune, L. S. Melvin, C. J. Mularski, B. A. Naclerio, C. J. Pazoles, J. S. Pillar, L. A. Rappach, P. Reiche, F. W. Rusek, H. Sherman, J. T. Shirley, F. J. Sweeney, J. E. Tickner, J. W. Watson and C. F. Wright, Bioorg. Med. Chem. Lett., 1995, 5, 1365–1370 CrossRef CAS; (g) R. J. Chambers, G. W. Antognoli, J. B. Cheng, A. Marfat, J. S. Pillar, J. T. Shirley and J. W. Watson, Bioorg. Med. Chem. Lett., 1998, 8, 1791–1796 CrossRef CAS PubMed; (h) L.-G. Lin, H. Xie, H.-L. Li, L.-J. Tong, C.-P. Tang, C.-Q. Ke, Q.-F. Liu, L.-P. Lin, M.-Y. Geng, H. Jiang, W.-M. Zhao, J. Ding and Y. Ye, J. Med. Chem., 2008, 51, 4419–4429 CrossRef CAS PubMed; (i) T. Janeczko, J. Dmochowska-Gładysz, A. Szumny and E. Kostrzewa-Susłow, J. Mol. Catal. B: Enzym., 2013, 97, 278–282 CrossRef CAS.
- W. Chiang, Y. Lin, C. Chung and H. Chen, US Pat., 2013/0158107A1, 2013 Search PubMed.
- In the preparation of this manuscript, asymmetric sequential reduction of enones using transfer hydrogenation was reported, see: (a) R. Molina Betancourt, P. Phansavath and V. Ratovelomanana-Vidal, Org. Lett., 2021, 23, 1621–1625 CrossRef CAS PubMed; (b) G. S. Caleffi, J. d. O. C. Brum, A. T. Costa, J. L. O. Domingos and P. R. R. Costa, J. Org. Chem., 2021, 86, 4849–4858 CrossRef CAS PubMed.
- (a) W. Wu, S. Liu, M. Duan, X. Tan, C. Chen, Y. Xie, Y. Lan, X.-Q. Dong and X. Zhang, Org. Lett., 2016, 18, 2938–2941 CrossRef CAS PubMed; (b) Y. Zheng, X. Zhang, R. Zhang and B. Ma, Green Synth. Catal., 2021, 2, 393–396 CrossRef.
- J. Yu, J. Long, Y. Yang, W. Wu, P. Xue, L. W. Chung, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 690–693 CrossRef CAS PubMed.
- J. Yu, M. Duan, W. Wu, X. Qi, P. Xue, Y. Lan, X.-Q. Dong and X. Zhang, Chem.–Eur. J., 2017, 23, 970–975 CrossRef CAS PubMed.
- (a) F. Thuaud, Y. Bernard, G. Türkeri, R. Dirr, G. Aubert, T. Cresteil, A. Baguet, C. Tomasetto, Y. Svitkin, N. Sonenberg, C. G. Nebigil and L. Désaubry, J. Med. Chem., 2009, 52, 5176–5187 CrossRef CAS PubMed; (b) M. Itoigawa, C. Ito, H. T. W. Tan, M. Okuda, H. Tokuda, H. Nishino and H. Furukawa, Cancer Lett., 2001, 174, 135–139 CrossRef CAS PubMed.
- R. J. Chambers, A. Marfat, G. W. Antognoli, J. B. Cheng, D. B. Damon, A. V. Kuperman, T. C. Liston, C. Mebus, J. S. Pillar, J. T. Shirley and J. W. Watson, Bioorg. Med. Chem. Lett., 1999, 9, 2773–2778 CrossRef CAS PubMed.
- (a) G. Gu, T. Yang, J. Lu, J. Wen, L. Dang and X. Zhang, Org. Chem. Front., 2018, 5, 1209–1212 RSC; (b) Z. Liang, T. Yang, G. Gu, L. Dang and X. Zhang, Chin. J. Chem., 2018, 36, 851–856 CrossRef CAS.
- (a) P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS; (b) P. A. Dub, Eur. J. Inorg. Chem., 2021, 2021, 4884–4889 CrossRef CAS; (c) P. A. Dub, N. J. Henson, R. L. Martin and J. C. Gordon, J. Am. Chem. Soc., 2014, 136, 3505–3521 CrossRef CAS PubMed.
- L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443–4454 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05963g |
| ‡ W. L. and T. Y. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |