
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrtho-selective C–H arylation of phenols with N-carboxyindoles under Brønsted acid- or Cu(I)-catalysis†
Nguyen H.
Nguyen‡
 a,
Soo Min
Oh‡
a,
Cheol-Min
Park
b and
Seunghoon
Shin
*a
a,
Soo Min
Oh‡
a,
Cheol-Min
Park
b and
Seunghoon
Shin
*a
aDepartment of Chemistry, Center for New Directions in Organic Synthesis (CNOS), Research Institute for Natural Sciences, Hanyang University, Seoul 04763, Korea. E-mail: sshin@hanyang.ac.kr
bDepartment of Chemistry, UNIST (Ulsan National Institute of Science and Technology), Ulsan 44919, Korea
First published on 27th December 2021
Abstract
Control over chemo- and regioselectivity is a critical issue in the heterobiaryl synthesis via C–H oxidative coupling. To address this challenge, a strategy to invert the normal polarity of indoles in the heterobiaryl coupling was developed. With N-carboxyindoles as umpoled indoles, an exclusively ortho-selective coupling with phenols has been realized, employing a Brønsted acid- or Cu(I)-catalyst (as low as 0.01 mol%). A range of phenols and N-carboxyindoles coupled with exceptional efficiency and selectivity at ambient temperature and the substrates bearing redox-active aryl halides (–Br and –I) smoothly coupled in an orthogonal manner. Notably, preliminary examples of atropselective heterobiaryl coupling have been demonstrated, based on a chiral disulfonimide or a Cu(I)/chiral bisphosphine catalytic system. The reaction was proposed to occur through SN2′ substitution or a Cu(I)–Cu(III) cycle, with Brønsted acid or Cu(I) catalysts, respectively.
Introduction
Bi(hetero)aryl motifs are widely found in bioactive natural products and are commonly utilized as chiral auxiliaries and ligands.1 These compounds have been synthesized by transition metal-catalyzed coupling between aryl halides (RX) and aryl metal species (RM) which requires pre-functionalization of the parent (hetero)arenes. Transition metal-catalyzed dehydrogenative C–H/C–H coupling may provide an appealing option.2 In addition, several non-metal protocols,3 such as hypervalent iodine chemistry,4 electrochemical oxidations,5 and photoredox catalysis,6 have also emerged. In many cases, however, control of chemo (homo- vs. heterocoupling)- or regio-selectivity remains challenging. For instance, oxidative functionalization of phenols may lead to homo-coupling and/or quinone formation.7 Control of site-selectivity of phenols (o-, m-, p-, and O-)8 and indoles9 also presents a prominent challenge in the absence of directing groups for which extra steps are required for the installation and removal.10We anticipated that inverting the polarity of nucleophilic indoles may enable precise control of chemoselectivity for the oxidative C–H heterobiaryl coupling.11 In the past, C3-umpoled indoles were accessed through electrophilic activation,12a,b oxidation,12c,d or hydroarylation of N-acylindole derivatives.12e–g More recently, the groups of Kita, Yorimitsu, Procter, and Maulide have developed metal-free C–H heteroarylation based on the activation of sulfoxides.13,14 In this context, Yorimitsu and coworkers reported an ortho-selective coupling of phenols with the activated (hetero)aryl sulfoxides that was proposed to proceed through an interrupted Pummerer attack to form the sulfoxonium I and charge-accelerated [3,3]-sigmatropic rearrangement to afford the biaryl compound.15a Procter and coworkers developed heteroarylation of phenols with activated benzothiophene-S-oxides,15b–d which occurred selectively at the ortho-15c or para-position15d of phenols (Scheme 1A).
 | ||
| Scheme 1 Inversion of polarity in the heterobiaryl coupling. | ||
In continuation of our interest in aliphatic Umpolung chemistry based on N–O bond redox,16 we turned our attention to the aromatic Umpolung by way of N-hydroxyindoles which was pioneered by Somei and coworkers.17 Somei and others disclosed C3-arylation of indoles via [3,3]-sigmatropic rearrangement of N-carboxyindoles18 that were accessed by nucleophilic aromatic substitution (SNAr)18a,b or by the O-arylation of N-hydroxyindoles with biaryliodonium salt.18c SN2′ substitution N-hydroxyindoles with indoles and pyrroles was also demonstrated (Scheme 1B).18d,e Nonetheless, these biaryl coupling had extremely limited scope and occurred with only modest yields and selectivity. More recently, Buchwald and coworkers demonstrated Cu-catalyzed asymmetric alkylation of N-carboxyindoles.19 Despite these advances, the development of a general and selective oxidative C–H arylation protocol based on the N-hydroxyindole derivatives as umpoled coupling partners remains elusive. We disclose herein two catalytic systems for the union of umpoled indoles 1 with phenols 2, viz. HNTf2 or [Cu(OTf)]2·C6H6, that operate at ambient temperature with exceptional efficiency, generality, and chemo- and regioselectivity (Scheme 1D). Details of this discovery, preliminary enantioselective control of atropisomerism, and the mechanistic study are described.
Results and discussion
The requisite N-hydroxyindole derivatives were synthesized according to the literature.20 Particularly, for the C2-substituted N-hydroxyindoles used in this study, a sequence comprising Sonogashira coupling of o-halonitrobenzenes, partial reduction into hydroxylamines, and Larock-type cyclization, were found general.20d With an N-carboxyindole 1a and 3,5-dimethoxyphenol 2a as prototypical substrates, we set out to explore the possibility of a non-metal catalyzed C–H/C–H coupling, employing various Brønsted acids (entries 1–6, Table 1).21,22 Weak acids underwent a sluggish conversion to 3aa (entries 1 and 2). The product 3aa had two inequivalent methoxy groups in the 1H and 13C NMR spectra and was assigned as an ortho-isomer to the OH, which was later confirmed by the single-crystal X-ray diffraction analysis of a related 3ba.22 Superacids, such as TfOH, HBF4, or HNTf2 significantly accelerated the formation of 3aa, reducing the reaction time to several hours (entries 3–5). Unlike TfOH and HBF4 which were not completely soluble, HNTf2 formed a homogeneous solution in chloroform and gave more reproducible results. The reaction was further accelerated at a higher concentration (entry 6). Reduction of the catalyst loading to 1 mol% still maintained significant activity, furnishing 3aa in 85% yield in a day (entry 7).|
|
|||
|---|---|---|---|
| Entry | Catalyst | Time | 3aa (%) |
| a 1a (0.1 mmol), 2a (0.2 mmol) and catalyst (10 mol%) in CHCl3 (0.1 M). b Determined by 1H NMR with CH2Br2 as an internal standard. c Starting 1a remained. d 0.2 M in CHCl3. e The catalyst was added as a stock solution in CHCl3 (entry 7) and in EtOAc (entries 16–18). f Messy mixture due to decomposition of 1a. g 2-Ph-indole 4a (10%) was observed as byproduct. h CHCl3 (0.05 M). i 2a (1.2 equiv.). | |||
| 1 | (PhO)2P(O)OH | 4 days | 26c |
| 2 | CF3CO2H | 3.5 days | 43c |
| 3 | TfOH | 12 h | 84 |
| 4 | HBF4·OEt2 | 8 h | 86 |
| 5 | HNTf2 | 16 h | 83 |
| 6 | HNTf2d | 8 h | 85 |
| 7 | HNTf2 (1 mol%)d,e | 22 h | 85 |
| 8 | AuCl(PPh3), AgOTf | 2 days | 10 |
| 9 | Fe(OTf)3 | 1 day | 96 |
| 10 | Zn(OTf)2 | 2.5 days | 96 |
| 11 | Cu(OTf)2 | 0.5 h | 53f |
| 12 | Cu(OTf)2 (1 mol%) | 3 h | 69g |
| 13 | CuBr | 5 h | 96 |
| 14 | Cu(MeCN)4·BF4 | 10 min | 74 |
| 15 | [Cu(OTf)]2·C6H6 | 5 min | 76f |
| 16h | [Cu(OTf)]2·C6H6 (0.1 mol%)e | 0.5 h | 93 |
| 17h | [Cu(OTf)]2·C6H6 (0.01 mol%)e | 1 h | >99 |
| 18h,i | [Cu(OTf)]2·C6H6 (0.1 mol%)e | 8 h | 89 |

Although HNTf2 provided an efficient metal-free catalytic system, we went on to examine Lewis acids for more efficient cross-coupling (entries 8–18, Table 1; see Table S1† for full details of optimization).22 Lewis acids, such as Fe(OTf)3 and Zn(OTf)2 gave excellent yields of 3aa, although it took several days (entries 7–9). A dramatic acceleration could be observed with Cu(OTf)2, but the reaction was accompanied by the extensive decomposition of starting materials and 3aa was obtained in only 53% yield (entry 11). Reducing the amount of Cu(OTf)2 gave marginally improved yield (entry 12). On the other hand, Cu(I) catalysts were significantly more efficient (entries 13–15). For instance, the reaction with [Cu(OTf)]2·C6H6 was complete only in 5 min at room temperature, delivering 3aa in 76% yield (entry 15). To our initial surprise, lowering the catalyst loading and diluting the reaction mixture led to a higher yield (entries 16 and 17; Table S4†). Presumably, this is due to poor solubility of [Cu(OTf)]2·C6H6 in CHCl3 and only a tiny amount of soluble catalyst mediated the reaction, while excess Cu-catalyst led to unselective decomposition of 1a. This observation led us to lower the catalyst loading to 0.01 mol% of [Cu(OTf)]2·C6H6, to obtain 3aa in quantitative yield (entry 17). When the amount of nucleophile 2a was reduced to 1.2 equivalent, 89% of 3aa was still obtained, attesting to the remarkable chemoselectivity of this protocol (entry 18).
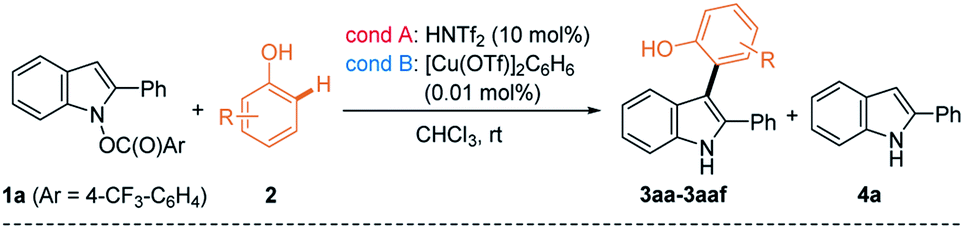
With two different cross-coupling conditions established (conditions A: 10 mol% of HNTf2 and conditions B: 0.01 mol% of [Cu(OTf)]2·C6H6), we explored the general applicability of the phenol nucleophiles in the heterobiaryl coupling with umpoled indole 1a (Table 2). Initially, monocyclic phenols (2a–2p) were employed in the reaction. Not surprisingly, electron-richer phenols underwent more facile coupling (e.g.2avs.2b; 2ivs.2j; 2kvs.2l). Phenols having ortho-substituents (2e–2g) underwent markedly slower reaction, suggesting developing steric congestion in their transition states. Interestingly, an electron-donating group at the m-position of phenols, accelerated the coupling reaction, compared to the p-position (e.g.2ivs.2k; 2jvs.2l). This is presumably because the m-substituent is placed para- to the reacting center, increasing the electron-density in the proposed [3,3]-sigmatropic rearrangement (e.g. in IV to 3, Scheme 1C). Likewise, the arylation of 2c preferentially occurred at Ha having -OMe (rather than Me) at the p-position. Notably, in case regioisomeric mixture was obtained (2c, 2h–j, 2n, and 2t),23 similar ratios were noted in both catalytic systems, which led us to posit that they partly share the same catalytic manifold. In general, Cu(I)-catalyst had the more general scope and can be reliably employed for substrates that are challenging with HNTf2 catalyst (e.g.2f and 2p). Fused phenols (2q–2ad) were then examined and they also underwent smooth coupling with 1a with exclusive selectivity for the ortho-position. For example, 1-naphthol 2q, H4-1-naphthol 2r, and 2-naphthol 2s underwent uneventful coupling to form 3aq–3as, as their single respective regioisomers. Unexpectedly, H4-2-naphthol 2t slightly favored substitution at the less activated C1 over C3 (cf.2d), which is presumably due to a subtle conformational influence. Easily oxidized catechol derivatives (2p and 2v), and phenols embedded within indoles (2w and 2x), carbazole (2y), and phenanthrol (2z) smoothly reacted. Functional groups such as esters (2p), bromo- (2aa, 2ab), chloro- (2ac), and iodoarenes (2ad) furnished respective products uneventfully. Compatibility of these aryl halides suggested that the current protocol could be used in an orthogonal fashion to other transition metal-catalyzed coupling. The current ortho-selective indolylation provided a unique opportunity for the modification of phenolic natural products. For example, TBS-protected resveratrol 2ae24 coupled with 1a only at Ha using HNTf2 (condition A), although the yield of 3aae was modest (28%). Alternatively, with Cu(I) catalyst (condition B), exclusive indolylation at Hb was obtained to give its regioisomer 3aae′ in 71% yield.23 Although a proper rationale for this divergence could not be found at the moment, the ability to switch regioselectivity in a catalyst-dependent manner must be highly useful. With some phenols (2m and 2w–2y), a reduced indole 4a from 1a was observed in varying amounts. However, even in these cases, side reactions such as oxidative dimerization into 2,2′-biphenol or 3,3′-bisindole derivatives could not be observed, which suggested highly chemoselective coupling of 1a. To demonstrate the practicality of the current method, synthesis of 3aa and 3ak was conducted in a gram scale (3 mmol of 1a), giving comparable yields (3aa: 75% (condition A) and 97% (condition B); 3ak: 48% (condition A) and 68% (condition B)). Lastly, 2,6-disubstituted 2af with both ortho-positions blocked underwent p-arylation instead, but very slowly. It should be pointed out that the current coupling requires unprotected phenol and did not work with anisole derivative such as 2ag, even under forcing conditions (60 °C, both conditions A & B). Likewise, other electron-rich (hetero)arenes, such as indoles, pyrrole, and benzofurans, failed to provide the corresponding coupled product, under both conditions A and B (for a list of unsuccessful nucleophiles, see Chart S1†).
a Reaction conditions: 1a (0.1 mmol) and 2 (2 equiv.) in CHCl3; conditions A (10 mol% of HNTf2, in CHCl3 (0.2 M)) and conditions B (0.01 mol% of [Cu(OTf)]2·C6H6 in CHCl3 (0.05 M)); isolated yield after chromatography.
b Regioisomeric ratio (Ha![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Hb).
c 20 mol% of HNTf2.
d 0.1 mol% of [Cu(OTf)]2·C6H6.
e At 60 °C. :Hb).
c 20 mol% of HNTf2.
d 0.1 mol% of [Cu(OTf)]2·C6H6.
e At 60 °C.
|
|---|
|

|
We then inspected substituted indoles for generality, using 2a as a phenolic partner (Table 3). The presence of various aryl groups at C2 having different electron-density were well tolerated as in the formation of 3ba–3fa. Various groups at C2, such as 2-naphthyl (3ga), 1-naphthyl (3ha), and alkyl groups (3ia–3la) were also allowed. Next, substitution at the C4–C7 of indoles was investigated. Those with 6-CF3 (1m), 6-Me (1n), 5-F/6-F/7-F (1o, 1p, and 1q), 4-Br (1r), and 6-CO2Me (1s) all reacted smoothly. Despite steric hindrance, indoles having a 4-Me-substituent (1t–1v) delivered the corresponding products 3ta–3va uneventfully.
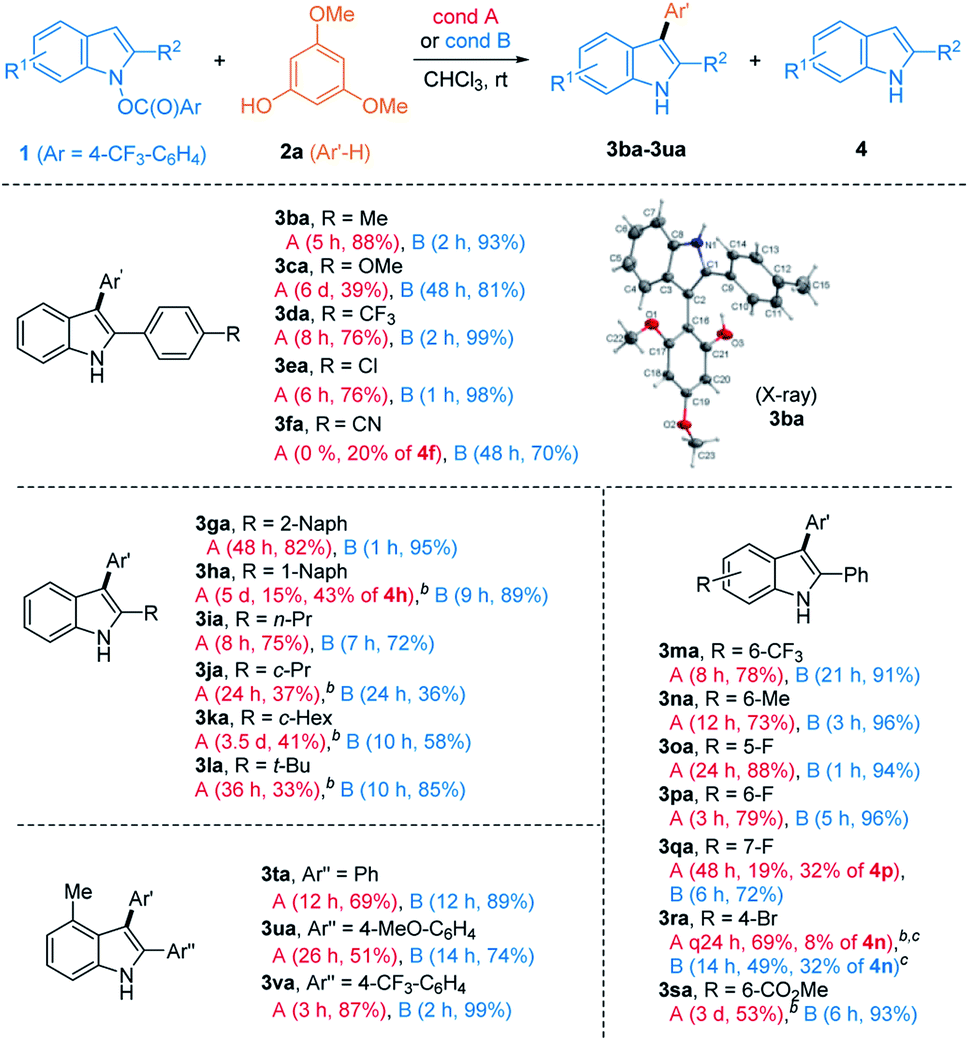
Notwithstanding the generality demonstrated in Table 3, the feasibility of the coupling with phenols requires the presence of C2-substitution. For example, C2/C3-unsubstituted indole derivative 1w failed to give the desired product under both conditions. Instead, with HNTf2 catalyst, a mixture of N-substitution products 5wa and 5wa′ were obtained in modest yield (eqn (1), Scheme 2). Presumably, in the absence of flanking C2-substituents, a C-attack of phenols that is sterically more demanding than an O-attack may be preferred. Substrate 1x having C3–Me moiety underwent C3-selective coupling followed by cyclization, providing a benzofuroindoline 6xa in low yield (eqn (2)). In contrast, 1y with a C3-Ph group failed to couple with 2a but instead underwent unprecedented 1,5-carboxy rearrangement to afford 7y in modest yield (eqn (3)).
 | ||
| Scheme 2 Reactions of differently substituted N-carboxyindoles. | ||
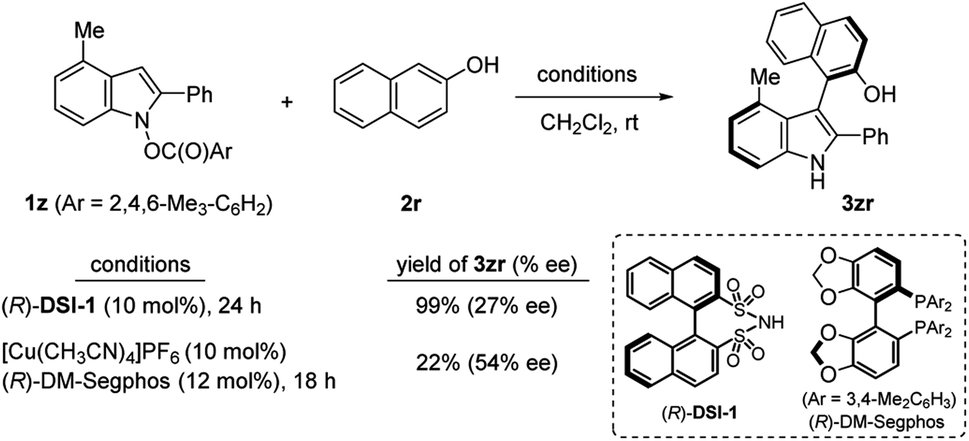
Then we turned our attention to atrop-selective indolylation of phenols. Synthesis of axially chiral indolyl biaryl compounds has been previously realized by chiral Brønsted acid,25 and transition metal catalysts.26 However, given the unprecedented reversed polarity of indoles, it is highly intriguing whether the current heterobiarylation protocol can be rendered enantioselective. Toward this end, 1z having 4-Me and N-mesitylene carboxylate as a leaving group was prepared (Scheme 3). Two catalytic systems appeared as viable solutions: a disulfonimide (R)-DSI-1 (10 mol%) delivered the coupled product 3zr in almost quantitative yield, albeit with a low 27% ee. To our delight, Cu(I)/DM-Segphos combination delivered 3zr in 54% ee. The modest yield in the latter case was due to the extensive decomposition of 1z into the reduced indole 4z (68%) and 1,1′-bi(2-naphthol) (29%). These preliminary results offer an outstanding prospect of successful enantio-control in the future.
 | ||
| Scheme 3 Preliminary atropselective indolylation of 2-naphthol. | ||
Having established the generality in the indolylation of phenols as well as the feasibility of the asymmetric synthesis, we then conducted a kinetic analysis of the Cu(I)-catalyzed system that gave consistently high yield (>95%) with little side-products. The reaction was determined to be the first order in both 1a and 2a, and a half order in dimeric [Cu(OTf)]2·C6H6. The latter result suggested that the dominant form of the Cu-species is an inactive off-cycle dimer that is in equilibrium with an active monomeric Cu(I) complex.27
The most enlightening piece of evidence was obtained from the deuterium labeling study (Scheme 4). For the measurement of the kinetic isotope effect, we prepared d-1a and d3-2a,22 and the kH/kD was determined from the rate constants kobs (s−1) measured in separate reactions.28 The reaction of d-1a with 2a showed a significant primary kinetic isotope effect (kH/kD = 1.72) and in the reaction of 1a and d3-2a, kH/kD (2.08) were observed (eqn (4) and (5)). When both d-1a and d3-2a are deuterated (eqn (6)), a combined isotope effect was manifested. These kinetic isotope effects suggested that re-aromatization step (IV to 3) in which both the C3–H bond of the indole d-1a and the ortho-C–H bond of the phenol d3-2a are cleaved may, at least in part, limit the turnover. Interestingly, significant incorporation of deuterium at the N1 of 3aa was observed when deuterated phenol d3-2a was employed. For instance, employing d-1a as the only deuterium source resulted in a minimal introduction of d-atom (eqn (4)), but the use of d3-2a resulted in a larger degree of d-incorporation at N1 (eqn (5)). This indicated the N1–H hydrogen mostly came from the ortho-hydrogen of the phenol 2a.
 | ||
| Scheme 4 Kinetic isotope effect. | ||
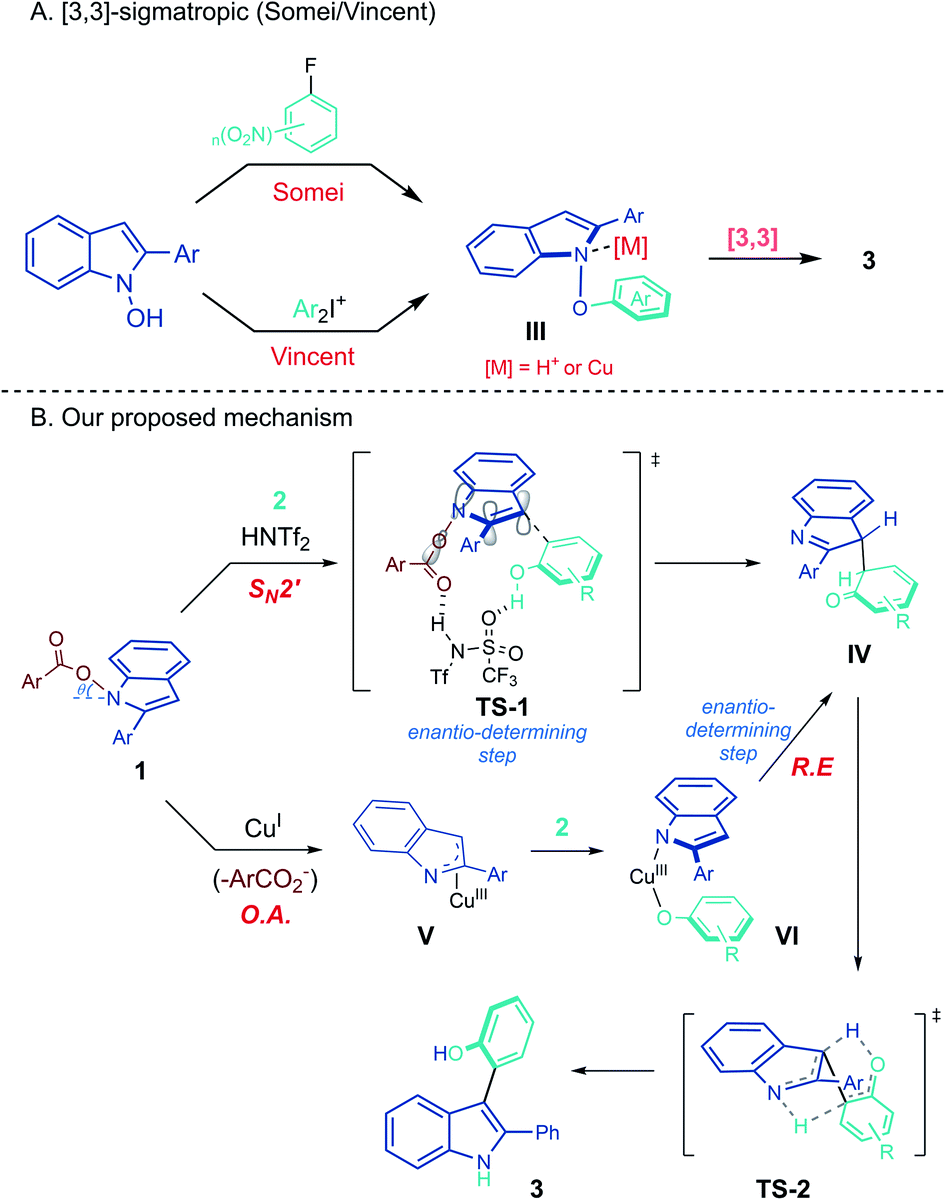
Based on the above data, the mechanism of the heterobiaryl coupling of umpoled indoles was put forward as in Scheme 5. From the precedents of [3,3]-sigmatropic rearrangement of N-phenoxyindoles III to the product 3 (Scheme 5A), we initially considered similar routes for both Brønsted acid- and Cu(I)-catalysis. However, the required SN2 attack forming a weak N–O bond (50–60 kcal mol−1) was difficult to imagine.29 To this end, we attempted to prepare N-phenoxy-2-Ph-indole 9 from the reaction of N-hydroxy-2-Ph-indole 8 with a benzyne precursor (eqn (7)). We could not observe 9 during the reaction, and the corresponding [3,3]-sigmatropic rearrangement product 10 was directly obtained in 48% yield in 2 h. This result suggested that the [3,3]-rearrangement of the intermediate 9 is not catalyzed by H+ or Cu(I). Furthermore, in contrast to our current protocols which required electron-rich phenols for efficient conversion, unsubstituted phenol underwent smooth [3,3]-rearrangement at rt. These led us to consider an alternative mechanism.
 | (1) |
 | ||
| Scheme 5 A proposed mechanism. | ||
During the study on the asymmetric heterobiarylation (Scheme 3), we inspected the effect of N-carboxylate leaving groups on the %ee of the product 3zr, employing a set of N-carboxylates of varying steric and electronic demand (4-CF3C6H4CO2–, 4-MeO–C6H4CO2–, and 2-Naph–CO2–; Tables S5 and S6 in ESI†). Interestingly, with Brønsted acid (R)-DSI-1, the %ee of 3zr varied widely from 13% ee to 19% ee, depending on the carboxylates. In contrast, the %ee of 3zr with Cu(I)/DM-Segphos system remained almost similar (67–69% ee) irrespective of the leaving carboxylates. This suggest that the carboxylates play a role in the enantio-determining step of Brønsted acid-catalysis but not in the Cu(I)-catalysis. A concerted SN2′ mechanism18d,e where the sulfonimides bind both to the N-carboxylate and the phenols (TS-1) may account for such an outcome (Scheme 5B).30 In contrast, with Cu(I)/L* system, the enantio-determining step may come after the liberation of the carboxylate and the %ee of 3 may be insensitivity to the carboxylates of 1. Oxidative addition of Cu(I) into N–O bond in 1 to from aza-allyl Cu(III) V,19 followed by ligand exchange with phenols, and reductive elimination from VI to IV may account for the observation. In the reaction of 1 with a stoichiometric amount of Cu-complex (1:Cu = 1:1) in the absence of phenols, immediate (<1 h) decomposition of 1 was noted, further suggesting an oxidative addition of Cu(I) into 1.
As Somei suggested,18e the carboxylate O-atom deviates from the indole plane (θ) as a result of repulsion between N- and O-lone pairs, which becomes more pronounced with a bulky C2-substituent on the indole 1 as in our case. The resulting sp3 like N1-atom may assist the proposed SN2′ substitution by aligning the σ*(N–O) parallel to the π*(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) for facile nucleophilic attack at C3 or may facilitate the oxidative addition by allowing coordination of Cu(d) with both σ*(N–O) and π*(CC) orbital.31
C) for facile nucleophilic attack at C3 or may facilitate the oxidative addition by allowing coordination of Cu(d) with both σ*(N–O) and π*(CC) orbital.31
In the atrop-selective processes (Scheme 3), the incipient centered chirality in IV may be transferred to the axial chirality in 3via a TS-2. The observation of small but discernible primary kinetic isotope effect with both d-1a and d3-2a could be rationalized by the simultaneous intramolecular proton transfers in the transition state such as TS-2.
Conclusions
In summary, we have developed an efficient ortho-indolylation of phenols, based on Brønsted acid- or Cu(I)-catalyzed reaction of N-carboxyindole derivatives. We demonstrated that such polarity-inverted indoles participate in the C–H arylation with phenols, which is unprecedented to the best of our knowledge. We proposed that the reaction proceeded through an SN2′ substitution for Brønsted acid catalysis or through oxidative addition for Cu(I)-catalysis. The sp3 hybridized nitrogen of the N-carboxyindoles was proposed to facilitate both processes. We also demonstrated a preliminary atropselective biaryl synthesis. Efforts to improve the enantioselectivity and to understand the mechanistic underpinning more precisely are currently underway and will be reported in due course.Author contributions
C.-M. P. discovered the transformation and performed the initial study with Cu(OTf)2 catalyst. N. H. N. and S. M. O. performed all the experimental work. S. S. conceived Brønsted acid- and Cu(I) catalysis, directed the project, wrote the paper, and designed mechanistic study. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This paper is dedicated to Professor Phil Ho Lee on the occasion of his 60th birthday. The authors gratefully thank the National Research Foundation of Korea (NRF-2021R1A5A6002803 and NRF-2021R1A2C3010552) for generous financial support.Notes and references
- (a) T. Wezeman, S. Bräse and K.-S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC; (b) W. Hüttel and M. Müller, Nat. Prod. Rep., 2021, 38, 1011–1043 RSC; (c) A. A. El-Gamal, W.-L. Wang and C.-Y. Duh, J. Nat. Prod., 2005, 68, 815–817 CrossRef CAS PubMed; (d) L. H. Franco, E. B. D. K. Joffe, L. Puricelli, M. Tatian, A. M. Seldes and J. A. Palermo, J. Nat. Prod., 1998, 61, 1130–1132 CrossRef CAS PubMed; (e) C. Bailly, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 363–378 CrossRef CAS PubMed; (f) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- (a) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (b) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (c) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS PubMed.
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed; (b) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed.
- (a) T. Dohi, M. Ito, K. Morimoto, M. Iwata and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 1301–1304 CrossRef CAS PubMed; (b) Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668–1669 CrossRef CAS PubMed; (c) K. Morimoto, K. Sakamoto, T. Ohshika, T. Dohi and Y. Kita, Angew. Chem., Int. Ed., 2016, 55, 3652–3656 CrossRef CAS PubMed.
- (a) T. Morofuji, A. Shimizu and J.-i. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 7259–7262 CrossRef CAS PubMed; (b) B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 5210–5213 CrossRef CAS PubMed; (c) S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 13325–13329 CrossRef CAS PubMed.
- (a) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed; (b) Z. Xu, L. Gao, L. Wang, M. Gong, W. Wang and R. Yuan, ACS Catal., 2015, 5, 45–50 CrossRef CAS; (c) L. Marzo, I. Ghosh, F. Esteban and B. König, ACS Catal., 2016, 6, 6780–6784 CrossRef CAS. For a combination of interrupted Pummerer and photoredox: (d) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS.
- (a) S. Narute, R. Parnes, F. D. Toste and D. Pappo, J. Am. Chem. Soc., 2016, 138, 16553–16560 CrossRef CAS PubMed; (b) K. V. N. Esguerra, Y. Fall, L. Petitjean and J.-P. Lumb, J. Am. Chem. Soc., 2014, 136, 7662–7668 CrossRef CAS PubMed; (c) H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633–13635 CrossRef CAS PubMed; (d) X. Li, J. B. Hewgley, C. A. Mulrooney, J. Yang and M. C. Kozlowski, J. Org. Chem., 2003, 68, 5500–5511 CrossRef CAS PubMed.
- (a) A. Libman, H. Shalit, Y. Vainer, S. Narute, S. Kozuch and D. Pappo, J. Am. Chem. Soc., 2015, 137, 11453–11460 CrossRef CAS PubMed; (b) Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782–6785 CrossRef CAS PubMed; (c) A. Dyadyuk, K. Sudheendran, Y. Vainer, V. Vershinin, A. I. Shames and D. Pappo, Org. Lett., 2016, 18, 4324–4327 CrossRef CAS PubMed.
- (a) Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 CrossRef CAS PubMed; (b) L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- (a) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS PubMed; (b) C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef CAS PubMed. For the “temporary directing group” strategy, see: (c) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS; (d) R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669–8682 CrossRef CAS PubMed.
- For excellent reviews on C3-arylation via umpoled indoles in total synthesis: (a) N. Denizot, T. Tomakinian, R. Beaud, C. Kouklovsky and G. Vincent, Tetrahedron Lett., 2015, 56, 4413–4429 CrossRef CAS; (b) R. Beaud, T. Tomakinian, N. Denizot, A. Pouihès, C. Kouklovsky and G. Vincent, Synlett, 2015, 432–440 CAS.
- Umpoled reactivity of indoles at C3-position have been accessed through electrophilic activation, oxidation, or hydroarylation of N-acylindole derivatives: (a) K. Matsumoto, H. Tokuyama and T. Fukuyama, Synlett, 2007, 3137–3140 CAS; (b) H. Ueda, H. Satoh, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2009, 48, 7600–7603 CrossRef CAS PubMed; (c) H. Ishikawa, M. Kitajima and H. Takayama, Heterocycles, 2004, 63, 2597–2604 CrossRef CAS; (d) H. Ishikawa, H. Takayama and N. Aimi, Tetrahedron Lett., 2002, 43, 5637–5639 CrossRef CAS; (e) N. Tajima, T. Hayashi and S.-i. Nakatsuka, Tetrahedron Lett., 2000, 41, 1059–1062 CrossRef CAS; (f) R. Beaud, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2012, 51, 12546–12550 CrossRef CAS PubMed; (g) T. Tomakinian, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2014, 53, 11881–11885 CrossRef CAS PubMed.
- For reviews: (a) S. Akai and Y. Kita, Top. Curr. Chem., 2006, 274, 35–76 CrossRef; (b) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed; (c) H. Yorimitsu, Chem. Rec., 2017, 17, 1156–1167 CrossRef CAS PubMed; (d) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- For early examples: (a) S. Akai, N. Kawashita, H. Satoh, Y. Wada, K. Kakiguchi, I. Kuriwaki and Y. Kita, Org. Lett., 2004, 6, 3793–3796 CrossRef CAS PubMed; (b) S. Akai, N. Kawashita, Y. Wada, H. Satoh, A. H. Alinejad, K. Kakiguchi, I. Kuriwaki and Y. Kita, Tetrahedron Lett., 2006, 47, 1881–1884 CrossRef CAS; (c) S. Yoshida, H. Yorimitsu and K. Oshima, Org. Lett., 2009, 11, 2185–2188 CrossRef CAS PubMed; (d) T. Kobatake, D. Fujino, S. Yoshida, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2010, 132, 11838–11840 CrossRef CAS PubMed; (e) X. Huang and N. Maulide, J. Am. Chem. Soc., 2011, 133, 8510–8513 CrossRef CAS PubMed; (f) X. Huang, M. Patil, C. Farés, W. Thiel and N. Maulide, J. Am. Chem. Soc., 2013, 135, 7312–7323 CrossRef CAS PubMed.
- (a) T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582–14585 CrossRef CAS PubMed; (b) Z. He, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2019, 58, 7813–7817 CrossRef CAS PubMed; (c) H. J. Shrives, J. A. Fernández-Salas, C. Hedtke, A. O. Pulis and D. J. Procter, Nat. Commun., 2018, 8, 14801–14807 CrossRef PubMed; (d) Z. He, T. Biremond, G. J. Perry and D. J. Procter, Tetrahedron, 2020, 76, 131315–131319 CrossRef CAS.
- (a) R. Kumar, Q. H. Nguyen, T.-W. Um and S. Shin, Chem. Rec., 2021 DOI:10.1002/tcr.202100172; (b) D. V. Patil and S. Shin, Asian J. Org. Chem., 2019, 8, 63–73 CrossRef CAS; (c) A. Bauer and N. Maulide, Chem. Sci., 2021, 12, 853–864 RSC; (d) A. de la Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef CAS PubMed; (e) D. V. Patil, S. W. Kim, Q. H. Nguyen, H. Kim, S. Wang, T. Hoang and S. Shin, Angew. Chem., Int. Ed., 2017, 56, 3670–3674 CrossRef CAS PubMed; (f) Q. H. Nguyen, N. H. Nguyen, H. Kim and S. Shin, Chem. Sci., 2019, 10, 8799–8805 RSC; (g) J. Im, S. I. Shin, C.-G. Cho and S. Shin, J. Org. Chem., 2020, 85, 6935–6950 CrossRef CAS PubMed; (h) S. W. Kim, T.-W. Um and S. Shin, J. Org. Chem., 2018, 83, 4703–4711 CrossRef CAS PubMed; (i) T.-W. Um, G. Lee and S. Shin, Org. Lett., 2020, 22, 1985–1990 CrossRef CAS PubMed.
- M. Somei, Heterocycles, 1999, 50, 1157–1211 CrossRef CAS.
- (a) M. Somei, T. Kawasaki, Y. Fukui, F. Yamada, T. Kobayashi, H. Aoyama and D. Shinmyo, Heterocycles, 1992, 34, 1877–1884 CrossRef CAS; (b) Y. Fukui, T. Kobayashi, T. Kawasaki, F. Yamada and M. Somei, Heterocycles, 2019, 99, 465–483 CrossRef CAS; (c) T. Tomakinian, C. Kouklovsky and G. Vincent, Synlett, 2015, 26, 1269–1275 CrossRef CAS; (d) F. Yamada and M. Somei, Heterocycles, 2000, 53, 1255–1258 CrossRef CAS; (e) K. Yoshino, F. Yamada and M. Somei, Heterocycles, 2008, 76, 989–994 CrossRef CAS.
- Y. Ye, S.-T. Kim, J. Jeong, M.-H. Baik and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 3901–3909 CrossRef CAS PubMed . It is noteworthy that the scope of this Cu-catalyzed alkylation is very different from the current work. For example, Buchwald's alkylation employed unsubstituted (both at C2 and C3) N-oxyindoles. In contrast, such substrates afforded N-arylation products in the current heterobiarylation (see eqn (1), Scheme 2)..
- (a) M. Somei and T. Shoda, Heterocycles, 1981, 16, 1523–1525 CrossRef CAS; (b) M. Somei, T. Kawasaki, K. Shimizu, Y. Fukui and T. Ohta, Chem. Pharm. Bull., 1991, 39, 1905–1907 CrossRef CAS; (c) Y. Zhao, H. Zhu, S. Sung, D. J. Wink, J. M. Zadrozny and T. G. Driver, Angew. Chem., Int. Ed., 2021, 60, 19207–19213 CrossRef CAS PubMed; (d) S. M. Oh and S. Shin, Bull. Korean Chem. Soc., 2021, 42, 925–928 CrossRef CAS ; and ref. 18(e)..
- For organocatalytic biarylation and the mechanistic discussion: (a) G.-Q. Li, H. Gao, C. Keene, M. Devonas, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2013, 135, 7414–7417 CrossRef CAS PubMed; (b) H. Gao, D. H. Ess, M. Yousufuddin and L. Kürti, J. Am. Chem. Soc., 2013, 135, 7086–7089 CrossRef CAS PubMed; (c) H. Gao, Q.-L. Xu, C. Keene, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2016, 55, 566–571 CrossRef CAS PubMed; (d) J.-Z. Wang, J. Zhou, C. Xu, H. Sun, L. Kürti and Q. L. Xu, J. Am. Chem. Soc., 2016, 138, 5202–5205 CrossRef CAS PubMed.
- See ESI for details. X-ray crystallographic data of 3ba (CCDC 2073434) could be obtained from Cambridge Crystallographic Data Center. Further details of optimization, including the effect of solvent (Table S2) and the carboxylate leaving group (Table S3) could be found in the ESI.†.
- Regioisomeric identities were determined from the splitting pattern in the 1H NMR, symmetry elements in the 13C NMR spectra along with the COSY and 1D-NOE experiments, details of which can be found in the ESI.†.
- Unprotected resveratrol was insoluble in chloroform..
- (a) L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58–64 CrossRef CAS PubMed; (b) C. Ma, F. Jiang, F.-T. Sheng, Y. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014–3020 CrossRef CAS PubMed; (c) F. Jiang, K.-W. Chen, P. Wu, Y.-C. Zhang, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 15104–15110 CrossRef CAS PubMed; (d) L. Peng, K. Li, C. Xie, S. Li, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 17199–17204 CrossRef CAS PubMed.
- [Pd] (a) C. Fe, M. Hou, Z. Zhu and Z. Gu, ACS Catal., 2017, 7, 5316–5320 CrossRef; (b) Y. P. He, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 2105–2109 CrossRef CAS PubMed; [Rh] (c) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed; [Fe] (d) R. R. Surgenor, X. Liu, W. Myers and M. D. Smith, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-zprv7.
- (a) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed; (b) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- For the kobs (s−1) measurement, addition of 4-CF3C6H4CO2H (1 equiv.) reduced the induction period, likely by dissociating the dimeric Cu(I); during the reaction, there was no reduction of D-content in the remaining d-1a throughout the reaction. However, there was <25% reduction in D-content in the ortho-proton of the remaining d3-2a at 84% conversion..
- (a) T. Okawara, N. Ikeda, T. Yamasaki and M. Furukawa, Chem. Pharm. Bull., 1988, 36, 3628–3631 CrossRef CAS; (b) M. Itoh, D. Hagiwara and J. Notani, Synthesis, 1975, 1975, 456–458 CrossRef.
- M. Shimizu, J. Kikuchi, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 5747–5757 RSC.
- E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1984, 25, 3063–3066 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of all products. CCDC 2073434. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06157g |
| ‡ There authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |