
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplate directed synthesis of antibody Fc conjugates with concomitant ligand release†
Viktoriia
Postupalenko
 a,
Léo
Marx
b,
David
Viertl
cd,
Nadège
Gsponer
a,
Natalia
Gasilova
e,
Thibaut
Denoel
c,
Niklaus
Schaefer
c,
John O.
Prior
c,
Gerrit
Hagens
a,
Frédéric
Lévy
f,
Patrick
Garrouste
b,
Jean-Manuel
Segura
a and
Origène
Nyanguile
*a
a,
Léo
Marx
b,
David
Viertl
cd,
Nadège
Gsponer
a,
Natalia
Gasilova
e,
Thibaut
Denoel
c,
Niklaus
Schaefer
c,
John O.
Prior
c,
Gerrit
Hagens
a,
Frédéric
Lévy
f,
Patrick
Garrouste
b,
Jean-Manuel
Segura
a and
Origène
Nyanguile
*a
aInstitute of Life Technologies, HES-SO Valais-Wallis, Rue de l'Industrie 23, CH-1950 Sion, Switzerland. E-mail: origene.nyanguile@hevs.ch
bDebiopharm Research & Manufacturing SA, Campus “après-demain”, Rue du Levant 146, 1920 Martigny, Switzerland
cDepartment of Nuclear Medicine and Molecular Imaging, Lausanne University Hospital, CH-1011 Lausanne, Switzerland
dIn Vivo Imaging Facility, Department of Research and Training, University of Lausanne, CH-1011 Lausanne, Switzerland
eEPFL Valais Wallis, MSEAP, ISIC-GE-VS, rue de l'Industrie 17, 1951 Sion, Switzerland
fDebiopharm International SA, Forum “après-demain”, Chemin Messidor 5-7, Case postale 5911, 1002 Lausanne, Switzerland
First published on 18th February 2022
Abstract
Antibodies are an attractive therapeutic modality for cancer treatment as they allow the increase of the treatment response rate and avoid the severe side effects of chemotherapy. Notwithstanding the strong benefit of antibodies, the efficacy of anti-cancer antibodies can dramatically vary among patients and ultimately result in no response to the treatment. Here, we have developed a novel means to regioselectively label the Fc domain of any therapeutic antibody with a radionuclide chelator in a single step chemistry, with the aim to study by SPECT/CT imaging if the radiolabeled antibody is capable of targeting cancer cells in vivo. A Fc-III peptide was used as bait to bring a carbonate electrophilic site linked to a metal chelator and to a carboxyphenyl leaving group in close proximity with an antibody Fc nucleophile amino acid (K317), thereby triggering the covalent linkage of the chelator to the antibody lysine, with the concomitant release of the carboxyphenyl Fc-III ligand. Using CHX-A′′-DTPA, we radiolabeled trastuzumab with indium-111 and showed in biodistribution and imaging experiments that the antibody accumulated successfully in the SK-OV-3 xenograft tumour implanted in mice. We found that our methodology leads to homogeneous conjugation of CHX-A′′-DTPA to the antibody, and confirmed that the Fc domain can be selectively labeled at K317, with a minor level of unspecific labeling on the Fab domain. The present method can be developed as a clinical diagnostic tool to predict the success of the therapy. Furthermore, our Fc-III one step chemistry concept paves the way to a broad array of other applications in antibody bioengineering.
Introduction
Cancer is the second cause of death worldwide.1 The emergence of precision medicine and the development of new treatment modalities such as targeted therapies2 and immune checkpoint inhibitors (ICIs) have resulted in significantly improved clinical benefits, albeit for a minority of patients.3–5 In the case of targeted therapies, stratifying biomarkers is essential to determine the choice of the appropriate drug. These biomarkers are often extracted from biopsies taken at various times before the onset of the treatment and the biopsies are normally sampled from few tumor lesions in the case of metastatic patients.6,7 For ICIs, the presence of programmed death ligand 1 (PD-L1)-positive tumors is determinant for the prescription of currently FDA-approved antibodies.8,9 Additionally, recurrences emerge for most patients treated with targeted therapies.10It would therefore be most useful for the oncologist to benefit from a diagnostic tool that would be antigen-specific and would enable the non-invasive, whole-body imaging of patients at different time points just before and during the treatment to select the best treatment modality and identify as soon as possible the emergence of resistant tumor lesions.11 This could be achieved by injecting low doses of the radiolabeled therapeutic antibody to the patient to assess binding of the antibody to the antigen by positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging.12
To fulfill all these criteria, the following challenges should be addressed: (i) the technology should be applicable to any commercial monoclonal antibody (mAb) directly off-the-shelf without prior purification and/or chemical modification, and (ii) the labeling procedure should not affect the binding and biophysical properties of the mAb. The extent of these challenges can be best appreciated by considering the bioconjugation chemistries that are applied to monoclonal antibodies. Most antibody conjugates developed to date have been prepared using random conjugation at lysines or cysteines. However, given that antibodies carry around 40 lysines exposed at their surface and several cysteines, the ligation of the payload usually results in a highly heterogeneous mixture.13 This is a major challenge with regard to process consistency and product characterization in the context of drug production for medical applications. Random conjugation may also affect the efficacy, safety, pharmacokinetics, and immunogenicity of the conjugated antibody.14 This well-known issue is directing the field toward developing means to attach the payload at a specific site of the antibody. Several site-specific conjugations have recently been developed.14–28 Although all these site-specific approaches have been successfully used for various applications, they all require genetic engineering or chemical modification of the antibody prior to the conjugation step, which makes them unsuitable for the development of a generic antibody labeling method. Therefore, the regioselectivity of the chemical modification should be mediated through the labeling moiety, and not by engineering or modifying chemically the antibody. Template directed ligation is emerging as powerful means to site specifically label unmodified antibodies.29,30 Through templating the chemical reaction with an aptamer,31 a protein,32 a small molecule33 or a peptide,34,35 authors were able to regioselectively label unmodified proteins. However, several drawbacks still remain to be solved because of the need to use UV irradiation, possible immunogenicity due to the covalent linkage of the protein or peptide ligand, or additional chemical steps to remove the ligand.
Here, we report for the first time a regioselective approach to attach the radionuclide chelator in a single step chemistry to the Fc domain of any commercial therapeutic antibody.
Results and discussion
Design of the Fc-III reactive conjugate
Regioselectivity was achieved with Fc-III, a 13-mer cyclic peptide binding with high affinity to the IgG-Fc domain of antibodies.36,37 The Fc domain hinge region is a consensus binding site found on the constant fragment (Fc) of immunoglobulin G (IgG), which interacts with four different proteins: protein A, protein G, rheumatoid factor, and neonatal Fc-receptor.36 We designed a strategy where an endogenous amino acid nucleophile of the IgG-Fc domain is targeted to react selectively with a chemically reactive site engineered within Fc-III. In this approach, Fc-III is used as a bait to bring a chemically reactive site linked to the chelator in close proximity with an antibody Fc nucleophile amino acid (lysine – K), thereby triggering the covalent linkage of the chelator to the antibody lysine, with the concomitant release of the Fc-III ligand. The driving forces of this reaction are (i) the proximity of the two reactive moieties, which increases dramatically their local effective concentration (typically around 30 mM, see the Materials and Methods) and (ii) the nature of the chemically reactive site carrying an electrophilic site X and a leaving group Z (Fig. 1A). | ||
| Fig. 1 (A) Schematic representation of the one-step antibody labeling approach. The payload is attached to the Fc-III peptide via a chemically reactive site “X–Z”. X in red refers to the electrophilic site, and Z in violet refers to the leaving group. Upon binding of the Fc-III reactive conjugate to IgG-Fc, the antibody lysine is poised to attack the electrophilic center of the chemically reactive site X, and the peptide vector is detached from the payload thanks to the engineered leaving group Z. This strategy allows subsequent wash out of the ligand without any additional steps. (B) Representation of the binding of Fc-III to the Fc domain of IgG based on the X-ray structure. The surface of IgG-Fc is shaded grey, and Fc-III is shown in a green ribbon structure. The antibody K317 is shown with a pink sphere. The PEG spacer is linked to a carbonate reactive site via a reactivity modulator that jointly ensures high reactivity in the presence of lysine residues and adequate stability of the reactive site towards hydrolysis. The payload (yellow star), typically a fluorescent label (fluorescein), a radiolabel, or a chelator (DOTA, DTPA, and DFO), is chemically bound to the other side of the carbonate reactive group via a short linker. (C) Chemical structure of the payload-PEGn-Fc-III reactive conjugate using the same color codes as in A and B. | ||
To identify a lysine suitable to react with the Fc-III reactive conjugate, we performed a molecular dynamics simulation to explore the dynamics of the solvent-exposed IgG-Fc lysines based on the available X-ray crystallographic structure of the Fc-III/IgG-Fc complex (PDB ID: 1DN2). As expected, the solvent-exposed lysines showed a great extent of flexibility around the X-ray structure. Among the 18 residues studied, the less flexible lysines were at positions 248, 317, 320, 322, 338, and 370 (Fig. S1†). K317 is at a distance fluctuating between 17 Å and 26 Å to the active site (Fig. 1B). The salt bridge of K317 with D280 reduces the transient fluctuation of this amino acid. As a result, the addition of a spacer spanning this distance to Fc-III should bring the chemically reactive site in proximity to K317. The N-terminal and C-terminal moieties of Fc-III are exposed to the solvent, thereby suggesting that the spacer can be chemically attached to both these positions without interfering with the binding of Fc-III to IgG-Fc.
The Fc-III reactive cleavable conjugate consists of the Fc-III ligand, a spacer at its N-terminus, to which a reactivity modulator, a carbonate chemically reactive site, a linker, and finally the payload are covalently attached (Fig. 1B and C). To bring the chemically reactive site in proximity to K317 a flexible PEG20 spacer was introduced at the N-terminus of the Fc-III peptide. The average length of the PEG spacer was estimated to be 23.8 Å based on the WLC model. The carbonate moiety was selected as the chemically reactive site to confer sufficient stability to the Fc-III reactive conjugate in water (contrary to succinimidyl esters) and to yield a carbamate linkage between the antibody and the payload, which has shown good stability in clinically relevant ADC drug linkers such as vedotin (estimated half-life of 4 to 6 days in humans).38 Next to the chemically reactive site, a p-carboxy phenyl reactivity modulator was engineered to jointly ensure the high reactivity of the carbonate, as well as sufficient stability towards aqueous hydrolysis. For example, when an m-nitro moiety was added to the p-carboxy phenyl, the carbonate reactivity was highly increased, but the susceptibility to water hydrolysis became too high to be used successfully (data not shown). Most importantly, the p-carboxy phenyl moiety also plays the role of a leaving group as it enables the release of the Fc-III ligand during the ligation of the chelator to the antibody, and not inversely, a side reaction that would happen otherwise given the symmetry of the carbonate (Fig. 1C). Altogether, the Fc-III reactive conjugate presented here is designed to regioselectively label the antibody at K317 with a payload, together with the concomitant release of Fc-III.
Synthesis and characterization of the FITC-PEG20-Fc-III reactive conjugate
Fc-III peptide was synthesized by solid-phase peptide synthesis using standard Fmoc/tBu chemistry. Following cleavage of the amino acid protecting groups and release from the resin, Fc-III was cyclized under mild oxidizing conditions and purified using RP-HPLC. Next, a PEG20 spacer was added to Fc-III by HATU coupling of Fmoc-PEG20-OH to the free N-terminal amino group of Fc-III, followed by Fmoc deprotection yielding NH2-PEG20-Fc-III (Scheme 1A). To assess if the resulting conjugate can efficiently bind to the Fc domain of an antibody, we developed a fluorescence polarization assay using recombinant trastuzumab and Fc-III labeled with fluorescein (Fc-III-FAM). Briefly, when Fc-III-FAM binds to trastuzumab, the rapid rotation of the fluorescein fluorophore will be dramatically reduced resulting in the increase of the fluorescence polarization signal. The affinity of Fc-III-FAM to trastuzumab was measured in a binding isotherm experiment yielding a Kd of 14 nM (Fig. 2A), consistent with the previously reported value.36 Next, we used a fluorescence polarization competition experiment to assess whether NH2-PEG20-Fc-III was able to displace Fc-III-FAM from trastuzumab. As shown in Fig. 2B, NH2-PEG20-Fc-III competed efficiently with Fc-III FAM with similar potency to wild-type Fc-III (50% inhibitory concentrations [IC50], 91 ± 12 nM and 58 ± 15 nM, respectively, Fig. 2B). This confirmed our hypothesis that the addition of a spacer at the N-terminus of Fc-III should not affect the binding of NH2-PEG20-Fc-III to IgG-Fc. | ||
| Scheme 1 Synthesis of the FITC-PEG20-Fc-III reactive conjugate: (a) HATU, DIEA, DMF, and then Fc-III peptide; (b) 20% piperidine, DMF; (c) compound 4, Et3N in ACN at 40 °C; (d) compound 6, DMAP in DCM at 25 °C; (e) TFA/DCM (1/3); (f) FITC, DIEA in ACN/DMF 1/1 at 25 °C; (g) HATU, DIEA, DMF, and NH2-PEG20-Fc-III peptide. | ||
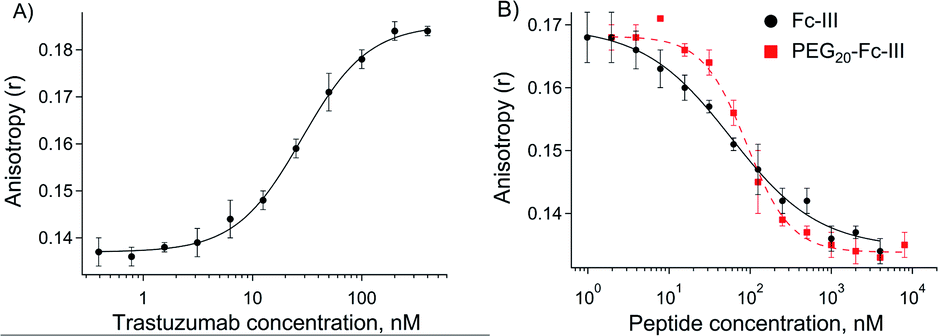 | ||
| Fig. 2 (A) Binding isotherm of Fc-III-FAM to trastuzumab measured by fluorescence anisotropy. The trastuzumab concentration is gradually increased while the concentration of the Fc-III-FAM labeled peptide was kept constant (5 nM). The fluorescence polarization signal of the trastuzumab/peptide bound complexes is reported on the y axis. The black solid line corresponds to the fit of the data using the Hill equation, yielding the half-maximal effective concentration (EC50). (B) Competition assays of Fc-III and PEG20-Fc-III for trastuzumab against Fc-III-FAM. Black solid (Fc-III) and red dashed lines (PEG20-Fc-III) are fittings of the data using the Hill equation yielding the half-maximal inhibitory concentration (IC50). | ||
Next, NH2-PEG20-Fc-III was converted into a Fc-III reactive conjugate (Scheme 1) bearing fluorescein (FITC) as a model payload. The payload was equipped with a linker, a carbonate reactive center, and a reactivity modulator with a carboxylic function at the end of the molecule to yield compound 9 to which was coupled the peptide with a spacer via an amide bond (Scheme 1). The resulting FITC-PEG20-Fc-III reactive conjugate was purified by RP-HPLC, and the correct mass was determined using UPLC-MS. The advantage of this strategy is that the synthetic steps required to prepare the activated payload carbonate are performed independently (compound 9 in Scheme 1B) and then the building block can be coupled to Fc-III in a single step using standard peptide chemistry.
Fluorescein labeling of trastuzumab
The reaction conditions to conjugate fluorescein to trastuzumab using FITC-PEG20-Fc-III were optimized by varying the reaction time, peptide equivalents (eq.), and pH. First, a time-course study was performed at pH 8.5 with FITC-PEG20-Fc-III and FITC-carbonate (compound 9 in Scheme 1B), the latter serving as a negative control. Trastuzumab, directly sampled from the commercial vial, was treated with 1 equivalent of FITC-PEG20-Fc-III or FITC-carbonate and incubated for 20 min, or 1, 2, 3, or 4 h. Each resulting reaction mixture was then directly loaded onto a reducing SDS-PAGE gel without any purification step. As seen in Fig. S2,† the Coomassie-stained SDS-PAGE showed easily distinguishable bands corresponding to the light (Lc) and heavy chains (Hc) of the antibody. Fluorescence imaging was then used to investigate the extent of antibody labeling. As expected, the Fc-III reactive conjugate yielded stronger and more rapid labeling of the heavy chain already after 1 h of incubation (Fig. 3A, lanes 1–5 and 3B) compared to the negative control FITC-carbonate, which showed no fluorescence except for a very faint signal after 4 h of incubation, indicative of a very low level of non-specific conjugation (Fig. 3B and S2,† lanes 6–10). In support of these data, the amount of unconjugated fluorescent peptide was significantly more important at the bottom of the gel (Fig. S2†). Unexpectedly, a small extent of unspecific labeling of the Lc domain was observed using FITC-PEG20-Fc-III (Fig. 3A). Altogether these data confirm that Fc-III increases the reaction kinetics by bringing the reactive site in proximity to the antibody lysine and selectively controls labeling of the heavy chain. For further experiments, 2 h of incubation were selected as the most optimal time for antibody conjugation (Fig. 3A, lane 3; 3B, 120 min).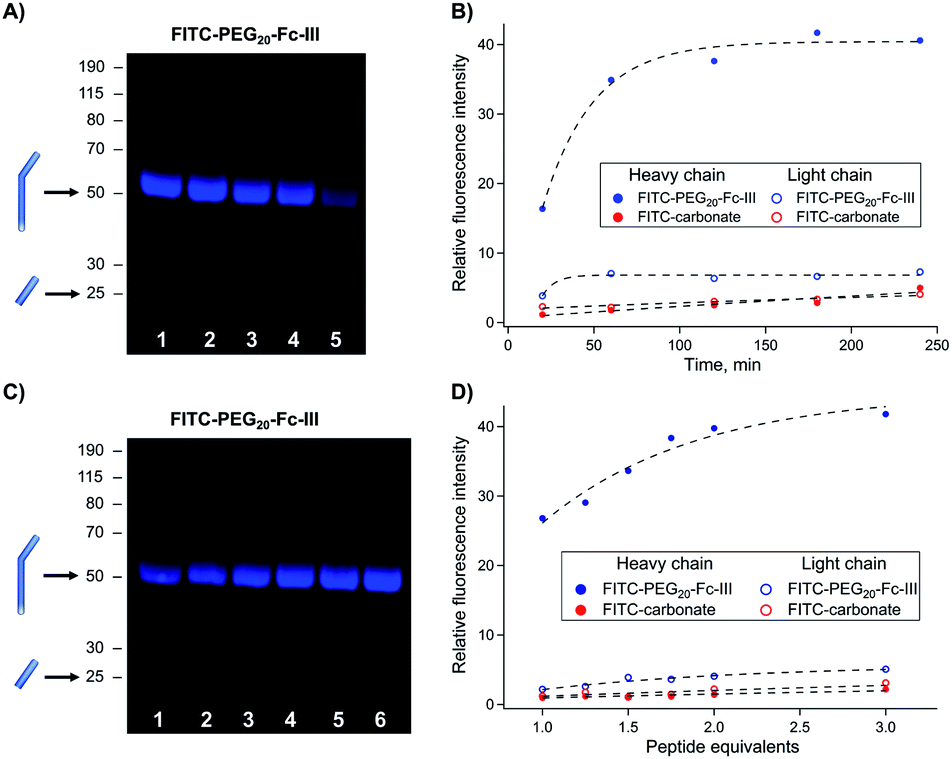 | ||
| Fig. 3 Optimization of the reaction conditions for trastuzumab conjugation with FITC-PEG20-Fc-III. (A and C) SDS gel electrophoresis of fluorescently labeled antibody obtained after increasing the incubation time (top panel) and peptide equivalents (bottom panel). Derivatized mAbs were run under the reducing conditions, and fluorescence was measured using the FluoroM bio-imaging system. The bands next to the 50 and 25 kDa marker correspond to Hc and Lc, respectively. (B and D) Quantification of the band fluorescence intensities of the SDS gels of panels A and C, respectively. The incubation time and peptide equivalents of each band of panels A (from right to left) and C correspond to the data points of panels B and D , respectively (Fig. S2†). The FITC-carbonate data (B and D) were extracted from the gels in Fig. S2.† | ||
Second, we varied the peptide:trastuzumab ratio (from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:1), performed the reaction under the same conditions as mentioned above (2 h incubation), and analyzed the reactions by reducing SDS PAGE. The gel showed that 2 equivalents of the Fc-III reactive conjugate were sufficient to bring the reaction to completion (Fig. 3C, lane 5; 3D, 2 eq.) consistent with the presence of two Fc-III IgG binding sites.
:1 to 3:1), performed the reaction under the same conditions as mentioned above (2 h incubation), and analyzed the reactions by reducing SDS PAGE. The gel showed that 2 equivalents of the Fc-III reactive conjugate were sufficient to bring the reaction to completion (Fig. 3C, lane 5; 3D, 2 eq.) consistent with the presence of two Fc-III IgG binding sites.
Last, the influence of pH on the labeling of trastuzumab was studied. A 2-fold excess of FITC-PEG20-Fc-III was incubated with trastuzumab for 2 h. We found that the level of heavy chain conjugation increased with pH (Fig. S3†). No significant reaction occurred between pH 6.0 and 8.0. A higher pH (8.5–9.0) improved substantially the conjugation efficiency (Fig. S3,† lanes 7–10) without any dependence on the buffer composition (Fig. S3†). Interestingly no reaction occurred when lysine or non-acetylated Fc-III peptide was incubated under the same reaction conditions with FITC-carbonate, while FITC carbonate remained stable throughout the whole incubation time (17 h, data not shown). Given the pKa of the α and γ-amino groups of lysine (9.2 and 10.8), the lack of reaction at the γ-amino function is expected, but some reaction would be expected at the α-amino function both of lysine and Fc-III. This suggests that the carbonate reactivity is too low to undergo a reaction under these conditions without a significant decrease in the pKa value of the amino group as seen in proteins39 and/or by the increase of the local concentration as sought by our approach. In summary, we found that the optimal antibody labeling conditions are 33 μM antibody, 2 equivalents of the Fc-III reactive conjugate, 2 h incubation time, and a pH of 9.0. These conditions were used for all further experiments.
We then confirmed the specificity of the labeling reaction: we designed two negative controls, PEG20-Fc-III-V10W-W11A and PEG20-Fc-III-C2A-C12A, where the binding affinity to IgG Fc has been impaired. The V10W substitution was designed to introduce a steric clash at the antibody binding site, and W11A was reported previously to decrease the Fc-III binding affinity.40 The C2A and C12A modifications remove the disulfide bond, which should affect the binding of Fc-III to the antibody since the macrocyclic constraint is lost. The negative controls were prepared and tested using the fluorescence competition assay described above. As expected, the binding affinity of both negative controls was abrogated (data not shown). Next, fluorescent Fc-III reactive conjugates FITC-PEG20-Fc-III-V10W-W11A and FITC-PEG20-Fc-III-C2A-C12A were prepared, and a labeling reaction was attempted with trastuzumab. No fluorescent band could be observed by SDS PAGE, indicating that trastuzumab could not be efficiently labeled with the Fc-III negative controls (data not shown). This confirms that the proximity induced by Fc-III at the IgG Fc binding site is required for efficient antibody labeling.
DOTA labeling of trastuzumab
DOTA (dodecane tetraacetic acid) is widely used to chelate 111In and 177Lu and is of particular interest for SPECT imaging41 and radioimmunotherapy because of the high thermodynamic stability and slow decomposition kinetics of the DOTA-radionuclide complex. To label trastuzumab with DOTA, the DOTA-PEG20-Fc-III reactive conjugate was prepared by coupling DOTA-tris(tBu)ester NHS ester 11 to reactivity modulator 8 yielding carbonate activated DOTA 12 (Scheme S1†), which was then coupled to NH2-PEG20-Fc-III 2 as described in Scheme 1 for the preparation of FITC-PEG20-Fc-III 10. Following the removal of the tBu protecting groups and RP-HPLC purification of the DOTA-PEG20-Fc-III reactive conjugate, the antibody DOTA conjugate was prepared by incubation of trastuzumab with a 2-fold molar excess of DOTA-PEG20-Fc-III. The Fc-III peptide was detached from the antibody by acidification with a glycine solution at pH 2.5, and the labeled antibody was purified by gel filtration chromatography, followed by neutralization with phosphate buffer, pH 8.5.42 To determine the extent of labeling of our one-step method, the DOTA labeled trastuzumab was deglycosylated with EndoS enzyme to facilitate the intact mass measurement of the resulting conjugates by liquid chromatography coupled with Orbitrap Fourier transform mass spectrometry (LC-FTMS) (Fig. 4A). The antibody conjugate was characterized by LC-FTMS under denaturing conditions, providing accurate estimates of the molecule loading profile and allowing calculation of the average degree of conjugation (DoC) of the payload. Deconvolution of the FTMS data confirmed the successful conjugation of DOTA to trastuzumab, with one, two, three, and four DOTA molecules attached to the antibody as observed with the +517 Da adducts in D1, D2, D3, and D4, respectively, the most abundant species being D0, D1, and D2 (Fig. 4A). The average DoC was found to be 1.09; the DoC was calculated as a weighted average of each DOTA-load (D1–D4), based on the relative intensities of each peak in the deconvolved mass spectra over the total amount of antibody, as described in the Materials and Methods. The ionization efficiency was assumed to be identical for D0–D4 species. Next, the deglycosylated sample was reduced with TCEP to yield light (Lc) and heavy chains (Hc) and then analyzed by LC-FTMS. Analysis of the deconvolved spectra revealed ∼1% labeling of Lc and 45% labeling of Hc in the reduced form with various degrees of conjugation (D1–D2, Fig. 4B and C). To assess the Fc regioselectivity of the conjugation, the intact sample was digested with GingisKHAN™ or FabALACTICA™ enzymes. These enzymes are selective towards IgG1 and cleave the antibody in a highly specific and reproducible manner into Fc and Fab subunits. The enzymatically digested samples were then analyzed by LC-FTMS providing a DoC of Fc and a DoC of two Fab subunits (F(ab')2) (Fig. 4D, E, Table S3 and Fig. S5†), resulting in a Fc/F(ab')2 selectivity of 3.2 or 3.8 with GingisKHAN™ or FabALACTICA™, respectively (Table S3†).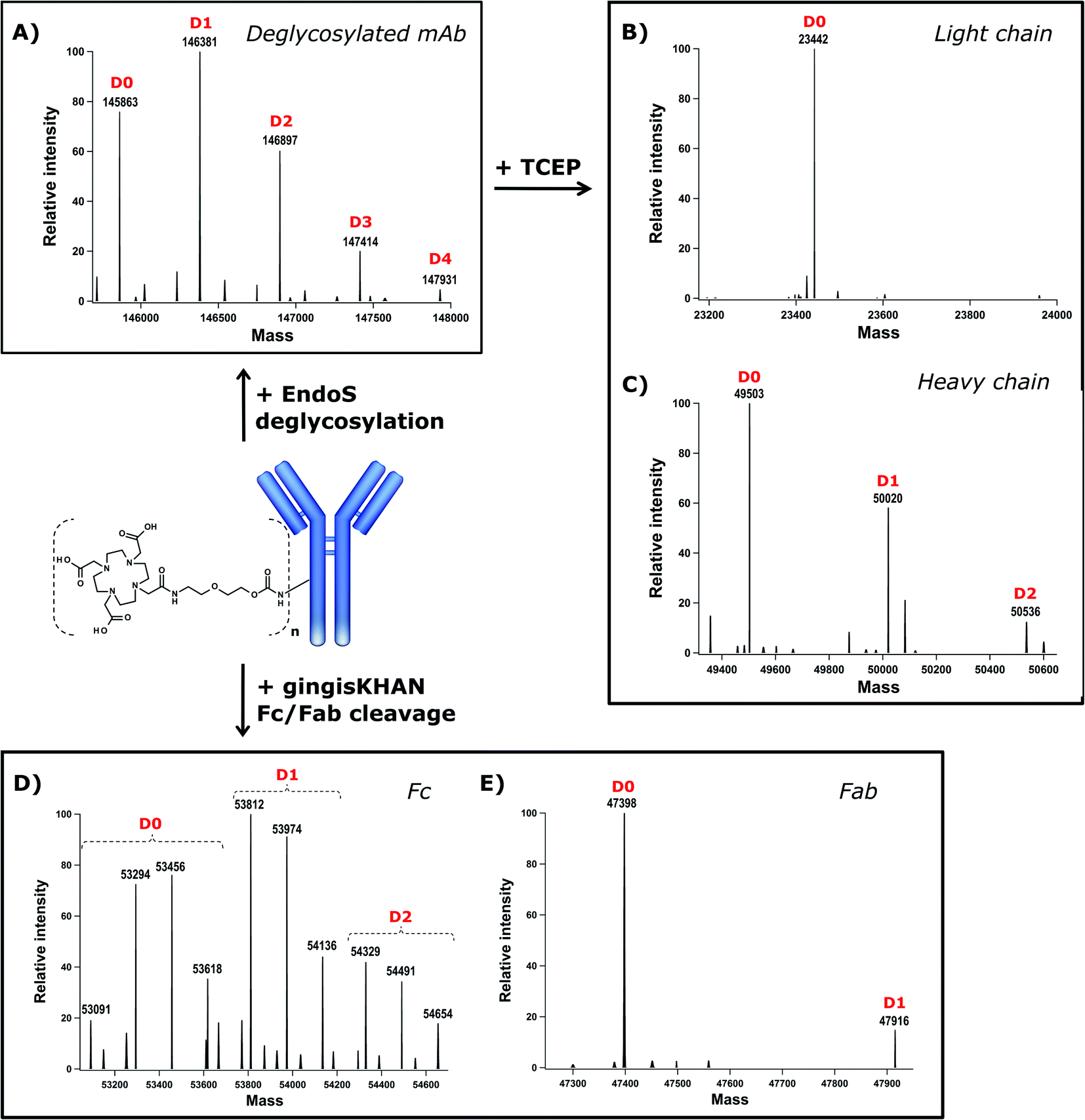 | ||
| Fig. 4 Intact mass analysis of the trastuzumab-DOTA conjugate and its subunits by LC-FTMS. The deconvolved mass spectra of the intact deglycosylated sample (A) and deglycosylated TCEP reduced sample, yielding light (B) and heavy chains (C); GingisKHAN™ derived Fc (D) and Fab (E) subunits. The D0–D4 peaks correspond to the antibody or antibody fragment with different degrees of conjugation, where Dn is the number of attached DOTA molecules. | ||
The complete removal of the free chelator in the purified conjugate is a critical parameter in radio imaging to ensure high radiochemical purity. For this reason, high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) was used to determine if residual amounts of unconjugated DOTA-PEG20-Fc-III, cleaved/free DOTA or cleaved residual peptide impurities remained in the final product after gel filtration chromatography. HR-ESI-MS analysis indicated that the DOTA-PEG20-Fc-III conjugate was fully consumed under the conditions of the conjugation process. No above-mentioned impurities were found in the final trastuzumab-DOTA conjugate (data not shown). In conclusion, the one step labeling method provides the means to regioselectively label the Fc domain of trastuzumab with DOTA with a DoC value of 1.09 and a Fc/F(ab')2 selectivity of 3.2.
Optimization of the PEG spacer length
Given that the distance of targeted K317 to the Fc-III binding site was estimated by molecular dynamics simulation and that the average length of the PEG spacer was calculated with the WLC model, it is uncertain whether PEG20 is the most appropriate spacer to reach K317. For these reasons, we synthesized and evaluated Fc-III reactive conjugates using different spacer lengths, ranging from 2 to 36 PEG units. DOTA-PEGn-Fc-III reactive conjugates were prepared as described above for DOTA-PEG20-Fc-III. The effect of the spacer length was investigated by labeling trastuzumab with DOTA using the DOTA-PEGn-Fc-III reactive conjugates and analyzing the resulting labeled antibodies by LC-FTMS. As can be seen in Table S4,† the efficiency of antibody labeling increased simultaneously with the length of the spacer (from 2 to 10 PEG units, DoC of 0.33 to 1.40), and then reached a plateau with longer PEGs (12, 20, and 36 units). These results indicate that at least 10 PEG units must be used for maximum labeling. Similarly, the Fc/F(ab')2 selectivity ratio increased up to 4.1 (corresponding to 10 PEG units) but then decreased when longer spacer lengths were tested (Fig. 5). As a result, it was concluded that PEG10 is the optimal spacer for the highest DoC and Fc/F(ab')2 selectivity ratio and was therefore selected for further studies. | ||
| Fig. 5 Effect of the spacer length on Fc/F(ab')2 selectivity. The spacer length calculated with the WLC model is plotted on the x-axis relative to the Fc/F(ab')2 selectivity ratio. The number of PEG units is color coded in the legend box according to its length. | ||
To show the universality of our approach, atezolizumab (IgG1), the ICI targeting programmed death ligand 1 (PD-L1 or CD274 antigen)43 was labeled using DOTA-PEG10-FcIII under the same conditions as described above for trastuzumab. LC-FTMS of the conjugate revealed that atezolizumab was labeled successfully with similar efficiency (DoC of 1.07 and Fc/F(ab')2 selectivity of 5.5, Table 1).
| Peptide conjugates | Antibody | DoC mAb | Labeled mAb | Selectivity Fc/F(ab')2 | DoC Fc | DoC F(ab')2 |
|---|---|---|---|---|---|---|
| a 6eq. of DFO-PEG10-Fc-III were used. b Fc loading was extrapolated. | ||||||
| DOTA-PEG10-Fc-III | Trastuzumab | 1.20 | 72% | 4.1 | 0.90 | 0.22 |
| DOTA-PEG10-Fc-III | Atezolizumab | 1.07 | 70% | 5.5 | 0.90 | 0.16 |
| DFO-PEG10-Fc-III | Trastuzumab | 0.47 | 39% | 0.4 (2.4b) | 0.05 (0.33b) | 0.14 |
| DFO-PEG10-Fc-IIIa | Trastuzumab | 1.52 | 79% | 1.3 (9.2b) | 0.20 (1.37b) | 0.15 |
| DTPA-PEG10-Fc-III | Trastuzumab | 0.87 | 63% | 3.7 | 0.37 | 0.10 |
Middle-down mass spectrometry analysis of the trastuzumab-DOTA conjugate
The complementary middle-down mass spectrometry (MS/MS) approach is gaining increasing attention nowadays for antibodies and ADC analysis44–46 and was used to identify the antibody conjugation site(s). The trastuzumab-DOTA conjugate was digested with FabRICATOR™ (IdeS) protease, and the disulfide bonds were subsequently reduced with TCEP, yielding 25 kDa antibody fragments, i.e., Lc, Fd' and Fc/2. The resulting digests were then analyzed by LC-FTMS/MS with high-energy collision-induced dissociation (HCD) as a fragmentation technique (more details in the Materials and Methods). Using this approach, we found that K317 appeared to be quantitatively labeled for FabRICATOR™-derived Fc/2 subunits (Fig. S14A†). These data confirmed our hypothesis and indicated that this approach could be used to regioselectively label the IgG Fc domain. However, we also observed the unexpected labeling of K326 (Fig. S14B†), although this appeared to be a minor secondary product, as it was observed mostly for the antibody fractions that were labeled with three DOTAs (two DOTAs conjugated to two K317 and one to K326). K326 is also located in the IgG Fc domain; molecular dynamics simulation suggested that distance fluctuations are not large enough to make the experimentally observed labeling of K326 compatible with the Fc-III peptide being simultaneously bound in its canonical binding site (data not shown). Given that K326 displays greater structural flexibility, solvent accessibility, and no involvement in H-bonding (Fig. S1†), the DOTA-PEG10-Fc-III reactive conjugate may preferably react with this amino acid when it is in an unbound transient state. To further confirm the antibody labeling site, we also performed a targeted peptide mapping experiment of the trypsin/Lys-C digested trastuzumab-DOTA conjugate, which confirmed K317 as the main labeling site (Fig. S15†).Application to other chelators
Slow biological processes, such as the biodistribution of antibodies, are monitored by PET or SPECT using different radiometals. PET imaging provides sensitive, quantitative, and non-invasive images, while SPECT is a more ubiquitous imaging modality.47 Bioconjugates labeled with 68Ga (t1/2 ∼ 1.1 h), 64Cu (t1/2 ∼ 12.7 h), and 89Zr (t1/2 ∼ 78.4 h) for PET imaging or 99mTc (t1/2 ∼ 6.0 h), 111In (t1/2 ∼ 67.4 h), and 177Lu (t1/2 ∼ 6.6 days) for SPECT represent the most promising tracers in chelator-based radiopharmaceuticals.41,4889Zr has raised interest as a potential isotope to label monoclonal antibodies for in vivo PET imaging.49 Its long half-life matches well with the pharmacokinetics of monoclonal antibodies that typically show optimal tumor-to-blood ratios several days after injection. To date, desferrioxamine (DFO) is a chelator of choice for 89Zr radiolabeling in the clinic. The routinely used random DFO conjugation approaches involving isothiocyanate/activated ester or maleimide groups are far from being ideal due to poor solubility of the DFO derivatives or synthetically demanding conjugation procedures.49–51 To prepare the DFO-PEG10-Fc-III reactive conjugate, the reactivity modulator and carbonate cleavable sites were first attached to NH2-PEG10-Fc-III (Scheme S2†), and the resulting activated derivative 15 was then coupled to DFO isothiocyanate, yielding DFO-PEG10-Fc-III (see the ESI†). The route of synthesis was changed compared to Scheme 1 to avoid unwanted side reactions caused by the presence of hydroxylamine reactive groups. Next, DFO-PEG10-Fc-III was used to label trastuzumab using the same one-step procedure as described above for trastuzumab-DOTA, yielding a DoC of 0.47 (Table 1). This result was unexpected when compared to the data obtained with DOTA-PEG10-Fc-III (DoC of 1.14). We hypothesized that this lower value may be due to the instability of the DFO-PEG10-Fc-III conjugate during the conjugation process due to hydrolysis of the carbonate linkage by the hydroxylamine of DFO. Consistent with this, UPLC-MS analysis of a DFO activated Fc-III conjugate revealed that 73% of the peptide is already hydrolyzed at t = 0 in the conjugation buffer at pH 9 (data not shown). To improve the labeling efficiency, an excess of DFO-PEG10-Fc-III (6 eq.) was added to trastuzumab, resulting in an increased DoC of 1.52. The conjugate was then cleaved into Fc/2 and F(ab')2 by the FabRICATOR™ enzyme to calculate the Fc/F(ab')2 selectivity value. Unexpectedly, the sum of the obtained DoC of Fc and F(ab')2 was much lower than the DoC of intact mAb, suggesting a cleavage of the DFO molecules from the antibody conjugate by the FabRICATOR™ enzyme. Taking into account that the F(ab')2 DoC of DOTA-trastuzumab conjugates had always been between 0.16 and 0.22, which corresponded well to the observed value of 0.15 for the F(ab')2 DoC of DFO-trastuzumab, we hypothesized that the DFO cleavage occurred on the Fc domain. As a result, the DoC of Fc was underestimated, leading to a selectivity of 1.3. Therefore, the Fc DoC values were extrapolated by subtraction of the F(ab')2 DoC from DoC mAb, yielding an extrapolated selectivity of 9.2 (Table 1).Diethylenetriaminepentaacetic acid (DTPA) is an acyclic chelator that is often used for labeling with various radiometals such as 64Cu, 68Ga, 111In, and 177Lu under mild conditions.41 In this study we used CHX-A′′-DTPA, a DTPA derivative with a trans-1,2-diaminocyclohexane backbone because of its improved metal chelation stability (DTPA will refer to CHX-A′′-DTPA throughout the text that follows). DTPA was conjugated to PEG10-Fc-III as described above for DFO, and the resulting DTPA-PEG10-Fc-III reactive conjugate was used to label trastuzumab. A DoC of 0.87 and Fc/F(ab')2 selectivity of 3.7 were found by HRMS (Table 1). These data are consistent with the DOTA DoC values and support our extrapolation of the DFO DoC values. For diagnosis efficacy, the remaining 37% of the unlabeled antibody is not a problem. In nuclear medicine, cold antibody is generally injected in combination with the radiolabeled antibody to saturate the neonatal Fc receptor and decrease the so called antigen sink effect where antibodies are withdrawn from the circulation by the hepatocytes.52
111In radiolabeling of trastuzumab
Next, the antibody-DTPA conjugate was radiolabeled with [111In]indium (170 Mbq of [111In]indium chloride in 0.02 M HCl solution) by incubation in 0.4 M acetate buffer pH 5.5 at 37 °C for 1 h, and [111In]In-DTPA-trastuzumab was obtained without further purification with >99% radiochemical purity as measured by instant thin-layer chromatography (iTLC, Fig. S20†). No aggregation was observed by radio-SEC (Fig. S21†) or SEC (data not shown). The stability of radioimmunoconjugates is an important parameter to assess, first because of the long circulation time of the antibody in vivo and second because free 111In circulating in the blood (due to decomplexation of 111In) will negatively affect the specificity of the signal. In vitro stability experiments were performed in human serum at different time points (24, 48, 72, and 144 h) by incubating [111In]In-DTPA-trastuzumab at 37 °C under agitation, and the stability of the activated antibody was measured by iTLC and radio-SEC. We found that the radiochemical purity of the activated antibody was 91% after 48 h (Fig. S22†) and 87% after 6 days. These results are consistent with the reported value for [111In]In-DTPA-trastuzumab and [177Lu]Lu-DOTA-trastuzumab53 and support in vivo applications.The immunoreactivity of [111In]In-DTPA-trastuzumab is not affected by the radiolabeling procedure
To verify that [111In]In-DTPA-trastuzumab retained its affinity to the HER2 antigen after DTPA conjugation and radiolabeling, the immunoreactive fraction of the radioconjugate was quantified by cell-binding assays in human ovarian carcinoma SK-OV-3 (HER2-positive) and human breast adenocarcinoma MDA-MB-231 (HER2-negative) cells using an infinite antigen excess.54,55 The binding specificity of [111In]In-DTPA-trastuzumab to SK-OV-3 and MDA-MB-231 cells was measured after incubating a constant amount of [111In]In-DTPA-trastuzumab (15 ng mL−1) with increasing numbers (0.25–8 × 106) of cells. We found that the immunoreactive fractions were 81% for the HER2-positive SK-OV-3 and 15% for the HER2-negative MDA-MB-231 (Fig. 6), thereby confirming that the immunoreactivity of [111In]In-DTPA-trastuzumab is preserved with the DTPA conjugation and under radiolabeling conditions used in this study (buffers, pH and temperature, consistent with previous studies).56,57 These data suggest that the integrity of trastuzumab was not affected by the reaction conditions used to conjugate DTPA. Together with the radiochemical purity and stability results, these data warrant further studies of [111In]In-DTPA-trastuzumab in vivo.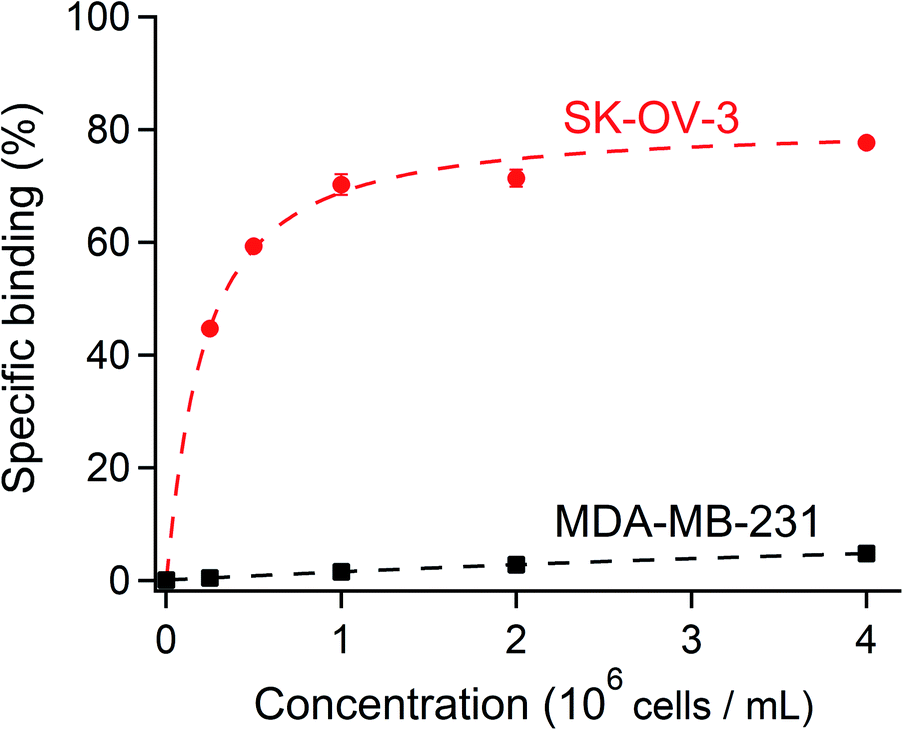 | ||
| Fig. 6 A constant amount of the radioligand was incubated with an increasing concentration of SK-OV-3 (HER2-positive) and MDA-MB-231 (HER2-negative) cells. The specific cell binding is expressed as a function of the number of cells. A binding curve (dashed line) is fitted using nonlinear regression, and the immunoreactive fraction corresponding to the extrapolation to infinite antigen excess is calculated using Graphpad Prism 7 software.34 81% of the immunoreactive fraction of [111In]In-DTPA-trastuzumab for SK-OV-3 showed a conserved affinity for HER2 after DTPA regiospecific conjugation and radiolabeling with [111In]indium. The immunoreactivity decreased to 15% with MDA-MB-231 (HER2-negative). | ||
[111In]In-DTPA-trastuzumab biodistribution study
[111In]In-DTPA-trastuzumab (20 kBq, corresponding to 0.05 μg [111In]In-DTPA-trastuzumab completed with 49.95 μg cold trastuzumab to saturate nonspecific Fc receptors involved in the turnover of antibodies) was administered by intravenous injection to mice bearing SK-OV-3 tumor xenograft implanted subcutaneously. Mice were sacrificed at different time points (2, 72, and 144 h), and selected organs were collected for radioactivity measurement to further study the level of antibody biodistribution (Table 2). As expected, we observed a high distribution of [111In]In-DTPA-trastuzumab in blood (36.6 ± 5.4% IA per g) at 2 h and well-vascularized organs (lungs 19.3 ± 9.5% IA per g, kidney 11.6 ± 2.2% IA per g and heart 9.67 ± 0.9% IA per g for example). These data are consistent with the pharmacokinetics of antibodies. Accumulation of [111In]In-DTPA-trastuzumab in the SK-OV-3 xenograft increased over time, reaching maximal accumulation (39.9 ± 15.4% IA per g) after 72 h. At 144 h, the antibody accumulation in normal organs was at least 3.9 times lower than in the engrafted tumour (Table 2). At 144 h the tumour to blood ratio was better than that at 72 h (2.1 and 3.9, respectively) giving potentially the best signal to noise ratio. In contrast, [111In]In-DTPA-trastuzumab accumulated to much lower extent (20.7 ± 7.7% IA per g) in mice engrafted with the HER2-negative MDA-MB-231 xenograft tumour at 72 h (1.9-fold less than the SK-OV-3 tumours), and the biodistribution of [111In]In-DTPA-trastuzumab was higher in the blood (27 ± 8% IA per g) and well-vascularized organs such as liver (18.0 ± 8.5% IA per g), indicating a higher circulation of [111In]In-DTPA-trastuzumab and lower specific uptake by the HER2-negative tumour.| Organ | SK-OV-3 [%IA per g] (HER2 positive) | MDA-MB-231 [%IA per g] (HER2 negative) | |||
|---|---|---|---|---|---|
| 2 h | 72 h | 144 h | Blocking (72 h) | 72 h | |
| Tumor | 7.16 ± 2.1 | 39.9 ± 15.4 | 30.3 ± 6.7 | 11.9 ± 2.4 | 20.7 ± 7.7 |
| Liver | 8.4 ± 2.0 | 5.0 ± 0.7 | 2.5 ± 0.1 | 4.9 ± 2.8 | 18.0 ± 8.5 |
| Kidney | 11.6 ± 2.2 | 7.1 ± 0.7 | 2.8 ± 0.7 | 4.4 ± 1.1 | 11.0 ± 4.6 |
| Adrenal glands | 14.0 ± 3.9 | 7.3 ± 1.8 | 5.0 ± 0.7 | 6.5 ± 1.5 | 12.5 ± 3.6 |
| Lungs | 19.3 ± 9.5 | 12.0 ± 1.0 | 4.0 ± 1.2 | 6.6 ± 1.9 | 14.6 ± 3.1 |
| Spleen | 8.1 ± 2.3 | 6.2 ± 0.8 | 2.7 ± 1.0 | 5.0 ± 2.3 | 15.2 ± 6.5 |
| Heart | 9.7 ± 0.9 | 5.8 ± 0.7 | 2.6 ± 0.7 | 3.7 ± 1.2 | 8.59 ± 4.3 |
| Muscle | 1.8 ± 0.2 | 2.0 ± 0.3 | 1.0 ± 0.2 | 1.3 ± 0.4 | 2.8 ± 0.6 |
| Bone | 3.7 ± 0.5 | 3.1 ± 0.3 | 1.5 ± 0.3 | 1.8 ± 0.5 | 5.0 ± 2.7 |
| Skin | 3.5 ± 0.6 | 5.2 ± 0.8 | 2.4 ± 0.7 | 3.4 ± 1.1 | 12.9 ± 2.3 |
| Stomach | 1.4 ± 0.5 | 1.3 ± 0.3 | 0.7 ± 0.2 | 0.8 ± 0.1 | 1.8 ± 0.0 |
| Small intestine | 4.6 ± 1.0 | 1.9 ± 0.2 | 0.9 ± 0.2 | 1.3 ± 0.4 | 5.4 ± 0.3 |
| Large intestine | 4.3 ± 1.1 | 1.4 ± 0.1 | 0.7 ± 0.1 | 1.6 ± 0.6 | 5.6 ± 1.8 |
| Pancreas | 3.8 ± 1.8 | 2.7 ± 0.6 | 1.1 ± 0.0 | 1.5 ± 0.5 | 6.0 ± 0.4 |
| Uterus | 5.2 ± 2.2 | 7.0 ± 3.3 | 2.1 ± 0.9 | 2.8 ± 1.1 | 15.3 ± 1.8 |
| Blood | 36.56 ± 5.4 | 18.7 ± 2.3 | 7.7 ± 2.6 | 9.4 ± 1.9 | 27.2 ± 8 |
Finally, the specificity of [111In]In-DTPA-trastuzumab was confirmed by an inhibition competition experiment (blocking) where mice bearing SK-OV-3 tumors were concomitantly injected with a vast molar excess of cold trastuzumab (2 mg) in addition to [111In]In-DTPA-trastuzumab. Under these conditions, the uptake of [111In]In-DTPA-trastuzumab in the SK-OV-3 tumour at 72 h was reduced to 11.9 ± 2.4% IA per g (3.4-times less than that without blocking) confirming the in vivo specificity of [111In]In-DTPA-trastuzumab for HER2 (Table 2). Consistent with the immunoreactivity experiment, the biodistribution and inhibition competition experiment suggest that the integrity of trastuzumab was not affected by the DTPA conjugation conditions. These data are consistent with the previously published study of Lub-de Hooge et al.56 and therefore, confirm that the site-specific chelator conjugation approach used in this study shows comparable results with conventional conjugated DTPA antibodies.
[111In]In-DTPA-trastuzumab SPECT/CT imaging study
SPECT and CT images of mice bearing a SK-OV-3 xenograft tumour were acquired using an Albira Si PEC/SPECT/CT. [111In]In-DTPA-trastuzumab (18 Mbq corresponding to 26 μg of [111In]In-DTPA-trastuzumab completed with 24 μg of cold DTPA-trastuzumab) was injected intravenously, and mice were visualized by SPECT and CT scans at 3, 72, and 144 h post-injection. As can be seen in Fig. 7, the specific tumour uptake of the antibodies increased over time, whereas the background activity decreased, resulting in an increased contrast of the tumour over time with an optimum at 144 h consistent with the biodistribution experiments. This preclinical SPECT/CT study is a proof of concept that the same approach could be used in a clinical context to obtain an image of tumours by nuclear medicine technologies. The present data confirms that a non-invasive method can be used to measure the specific uptake of antibodies by tumors and suggest that the method is applicable in the clinics. | ||
| Fig. 7 Coronal MIP fused SPECT and CT images at 3, 72, and 144 h post-injection of 18 MBq [111In]In-DTPA-trastuzumab in the same mouse bearing an SK-OV-3 tumour (HER2-positive). Uptake of [111In]In-DTPA-trastuzumab was observed in the tumour (T) and liver (L). The longer blood circulation of [111In]In-DTPA-trastuzumab leads to an increased specific tumour uptake over time. | ||
Conclusions
Here, we have developed a generic technology to label any antibody with a radionuclide chelator in a single step chemistry. Through engineering the Fc-III peptide ligand with a carbonate reactive site, a PEG10 spacer, and a chelator, we have shown that the resulting preactivated Fc-III conjugate is capable of efficiently and selectively labelling trastuzumab with the concomitant release of Fc-III. Successful labeling was achieved with FITC, DOTA, DFO, and DTPA payloads. We demonstrated that our radionuclide chelator approach leads to homogeneous labeling of trastuzumab with a DoC in the range of 1 to 2, and the regioselective labeling of the IgG-Fc domain mainly at K317. This will greatly facilitate the analytics required by the regulatory authorities to approve GMP production of the API for clinical applications.In comparison to the many other site-specific conjugation chemistries that have been developed to date, we believe that our one step Fc-targeted labeling approach is a major advancement in the field because (i) it requires no prior purification or modification of the antibody, (ii) the chemical reaction occurs spontaneously upon mixing the antibody with the Fc-III-reactive conjugate and is completed within 2 hours, and (iii) there is no need of any additional chemical step. Given that Fc-III binds to any antibody isotypes in a region that is not involved in antigen binding, we believe that this will greatly facilitate bioconjugation, since any chelators, and as a matter of fact any payload, can be chemically pre-activated with Fc-III. This paves the way to a broad array of applications in antibody engineering such as diagnostics, ADCs, Fc-fusion proteins, and so on. Other important perspectives of this technology may be the in vivo non-invasive determination of tissue distribution, tumor accumulation and retention, and target engagement.
Ethical statement
All animal experiments were performed in accordance with the Swiss legislation for the care and use of laboratory animals under the license VD-2993 (09/2018) delivered after approbation by the Veterinarian Office of the canton of Vaud and the ethics committee.Data availability
The molecular dynamics simulation data performed in this work is available upon request.Author contributions
V. P, N. G, N. G, T. D, D. V, N. S, J. O. P, and G. H devised and performed the experiments. V. P, D. V, F. L and O. N wrote the paper. O. N, J. M. S, F. L, N. S, and L. M devised the concept. P. G, D. V, L. M and O. N managed the project. O. N. and J. M. S prepared the Innosuisse grant application. All authors revised and approved the last version of the paper.Conflicts of interest
There are no conflicts to declare. V. P, L. M, F. L, P. G, J.-M. S, and O. N are coinventors of application WO2021110860 A1 filed on 3 December 2019, entitled “Reactive conjugates”.Acknowledgements
This work was co-financed by Innosuisse (project number 26695.2 PFLS-LS) and Debiopharm International SA, Switzerland, whose support we gratefully acknowledge. We also would like to thank Genochem (Grasse, France) for the synthesis of the key building blocks. We are most grateful to Dr Michel A Cuendet from the Swiss Institute of Bioinformatics for the molecular dynamics simulation.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- B. A. Chabner and T. G. Roberts Jr, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- A. M. Tsimberidou, E. Fountzilas, M. Nikanjam and R. Kurzrock, Cancer Treat. Rev., 2020, 86, 102019 CrossRef CAS PubMed.
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- M. Dietel, K. Johrens, M. V. Laffert, M. Hummel, H. Blaker, B. M. Pfitzner, A. Lehmann, C. Denkert, S. Darb-Esfahani, D. Lenze, F. L. Heppner, A. Koch, C. Sers, F. Klauschen and I. Anagnostopoulos, Cancer Gene Ther., 2015, 22, 417–430 CrossRef CAS PubMed.
- M. Schwaederle, M. Zhao, J. J. Lee, V. Lazar, B. Leyland-Jones, R. L. Schilsky, J. Mendelsohn and R. Kurzrock, JAMA Oncol., 2016, 2, 1452–1459 CrossRef PubMed.
- E. R. Malone, M. Oliva, P. J. B. Sabatini, T. L. Stockley and L. L. Siu, Genome Med., 2020, 12, 8 CrossRef PubMed.
- D. B. Doroshow, S. Bhalla, M. B. Beasley, L. M. Sholl, K. M. Kerr, S. Gnjatic, I. I. Wistuba, D. L. Rimm, M. S. Tsao and F. R. Hirsch, Nat. Rev. Clin. Oncol., 2021, 18, 345–362 CrossRef CAS PubMed.
- D. a. F. F. A. Drugs, https://www.accessdata.fda.gov/scripts/cder/daf/ Search PubMed.
- R. Nahta, D. Yu, M. C. Hung, G. N. Hortobagyi and F. J. Esteva, Nat. Clin. Pract. Oncol., 2006, 3, 269–280 CrossRef CAS PubMed.
- C. L. Ventola, Pharm. Ther., 2017, 42, 514–521 Search PubMed.
- J. Dewulf, K. Adhikari, C. Vangestel, T. V. D. Wyngaert and F. Elvas, Cancers, 2020, 12, 1868 CrossRef CAS PubMed.
- O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 CrossRef CAS PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grunberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed. Engl., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS PubMed.
- T. Botzanowski, S. Erb, O. Hernandez-Alba, A. Ehkirch, O. Colas, E. Wagner-Rousset, D. Rabuka, A. Beck, P. M. Drake and S. Cianferani, mAbs, 2017, 9, 801–811 CrossRef CAS PubMed.
- P. M. Drake, A. E. Albers, J. Baker, S. Banas, R. M. Barfield, A. S. Bhat, G. W. de Hart, A. W. Garofalo, P. Holder, L. C. Jones, R. Kudirka, J. McFarland, W. Zmolek and D. Rabuka, Bioconjugate Chem., 2014, 25, 1331–1341 CrossRef CAS PubMed.
- T. Hofer, L. R. Skeffington, C. M. Chapman and C. Rader, Biochemistry, 2009, 48, 12047–12057 CrossRef CAS PubMed.
- T. Hofer, J. D. Thomas, T. R. Burke Jr and C. Rader, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12451–12456 CrossRef CAS PubMed.
- X. Li, J. Yang and C. Rader, Methods, 2014, 65, 133–138 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- V. F. C. Ferreira, B. L. Oliveira, A. D'Onofrio, C. M. Farinha, L. Gano, A. Paulo, G. J. L. Bernardes and F. Mendes, Bioconjugate Chem., 2021, 32, 121–132 CrossRef CAS PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Companon, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jimenez-Oses, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef CAS PubMed.
- M. Farleigh, T. T. Pham, Z. Yu, J. Kim, K. Sunassee, G. Firth, N. Forte, V. Chudasama, J. R. Baker, N. J. Long, C. Rivas and M. T. Ma, Bioconjugate Chem., 2021, 32, 1214–1222 CrossRef CAS PubMed.
- J. P. M. Nunes, V. Vassiileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- E. von Witting, S. Hober and S. Kanje, Bioconjugate Chem., 2021, 32, 1515–1524 CrossRef CAS PubMed.
- K. L. Wu, C. Yu, C. Lee, C. Zuo, Z. T. Ball and H. Xiao, Bioconjugate Chem., 2021, 32, 1947–1959 CrossRef CAS PubMed.
- C. Cui, H. Zhang, R. Wang, S. Cansiz, X. Pan, S. Wan, W. Hou, L. Li, M. Chen, Y. Liu, X. Chen, Q. Liu and W. Tan, Angew. Chem., Int. Ed. Engl., 2017, 56, 11954–11957 CrossRef CAS PubMed.
- Y. Jung, J. M. Lee, J. W. Kim, J. Yoon, H. Cho and B. H. Chung, Anal. Chem., 2009, 81, 936–942 CrossRef CAS PubMed.
- N. J. Alves, N. Mustafaoglu and B. Bilgicer, Biosens. Bioelectron., 2013, 49, 387–393 CrossRef CAS PubMed.
- T. B. Nielsen, R. P. Thomsen, M. R. Mortensen, J. Kjems, P. F. Nielsen, T. E. Nielsen, A. L. B. Kodal, E. Clo and K. V. Gothelf, Angew. Chem., Int. Ed. Engl., 2019, 58, 9068–9072 CrossRef CAS PubMed.
- K. Yamada, N. Shikida, K. Shimbo, Y. Ito, Z. Khedri, Y. Matsuda and B. A. Mendelsohn, Angew. Chem., Int. Ed. Engl., 2019, 58, 5592–5597 CrossRef CAS PubMed.
- W. L. DeLano, M. H. Ultsch, A. M. de Vos and J. A. Wells, Science, 2000, 287, 1279–1283 CrossRef CAS PubMed.
- B. Nilsson, T. Moks, B. Jansson, L. Abrahmsen, A. Elmblad, E. Holmgren, C. Henrichson, T. A. Jones and M. Uhlen, Protein Eng., 1987, 1, 107–113 CrossRef CAS PubMed.
- H. Li, T. H. Han, N. N. Hunder, G. Jang and B. Zhao, J. Clin. Pharmacol., 2017, 57, 1148–1158 CrossRef CAS PubMed.
- D. G. Isom, C. A. Castaneda, B. R. Cannon and B. Garcia-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- M. S. Dennis, H. B. Lowman, W. L. DeLano, Methods and compositions for prolongingelimination half-times of bioactive compounds, US Pat., US7608681B2, 10/149s35, Genentech, Inc., So. San Franclsco, CA, 2009 Search PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- J. B. Fishman and E. A. Berg, Cold Spring Harb. Protoc., 2019, 331–344 Search PubMed.
- A. Markham, Drugs, 2016, 76, 1227–1232 CrossRef CAS PubMed.
- F. Lermyte, Y. O. Tsybin, P. B. O'Connor and J. A. Loo, J. Am. Soc. Mass Spectrom., 2019, 30, 1149–1157 CrossRef CAS PubMed.
- K. Srzentić, K. O. Nagornov, L. Fornelli, A. A. Lobas, D. Ayoub, A. N. Kozhinov, N. Gasilova, L. Menin, A. Beck, M. V. Gorshkov, K. Aizikov and Y. O. Tsybin, Anal. Chem., 2018, 90, 12527–12535 CrossRef PubMed.
- K. Srzentić, L. Fornelli, Y. O. Tsybin, J. A. Loo, H. Seckler, J. N. Agar, L. C. Anderson, D. L. Bai, A. Beck, J. S. Brodbelt, Y. E. M. van der Burgt, J. Chamot-Rooke, S. Chatterjee, Y. Chen, D. J. Clarke, P. O. Danis, J. K. Diedrich, R. A. D'Ippolito, M. Dupré, N. Gasilova, Y. Ge, Y. A. Goo, D. R. Goodlett, S. Greer, K. F. Haselmann, L. He, C. L. Hendrickson, J. D. Hinkle, M. V. Holt, S. Hughes, D. F. Hunt, N. L. Kelleher, A. N. Kozhinov, Z. Lin, C. Malosse, A. G. Marshall, L. Menin, R. J. Millikin, K. O. Nagornov, S. Nicolardi, L. Paša-Tolić, S. Pengelley, N. R. Quebbemann, A. Resemann, W. Sandoval, R. Sarin, N. D. Schmitt, J. Shabanowitz, J. B. Shaw, M. R. Shortreed, L. M. Smith, F. Sobott, D. Suckau, T. Toby, C. R. Weisbrod, N. C. Wildburger, J. R. Yates, S. H. Yoon, N. L. Young and M. Zhou, J. Am. Soc. Mass Spectrom., 2020, 31, 1783–1802 CrossRef PubMed.
- A. Rahmim and H. Zaidi, Nucl. Med. Commun., 2008, 29, 193–207 CrossRef PubMed.
- D. S. MacPherson, K. Fung, B. E. Cook, L. C. Francesconi and B. M. Zeglis, Dalton Trans., 2019, 48, 14547–14565 RSC.
- S. Heskamp, R. Raavé, O. Boerman, M. Rijpkema, V. Goncalves and F. Denat, Bioconjugate Chem., 2017, 28, 2211–2223 CrossRef CAS PubMed.
- I. Verel, G. W. Visser, R. Boellaard, M. Stigter-van Walsum, G. B. Snow and G. A. van Dongen, J. Nucl. Med., 2003, 44, 1271–1281 CAS.
- L. R. Perk, M. J. Vosjan, G. W. Visser, M. Budde, P. Jurek, G. E. Kiefer and G. A. van Dongen, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 250–259 CrossRef CAS PubMed.
- J. S. Jaggi, J. A. Carrasquillo, S. V. Seshan, P. Zanzonico, E. Henke, A. Nagel, J. Schwartz, B. Beattie, B. J. Kappel, D. Chattopadhyay, J. Xiao, G. Sgouros, S. M. Larson and D. A. Scheinberg, J. Clin. Invest., 2007, 117, 2422–2430 CrossRef CAS PubMed.
- S. Rasaneh, H. Rajabi, M. H. Babaei, F. J. Daha and M. Salouti, Nucl. Med. Biol., 2009, 36, 363–369 CrossRef CAS PubMed.
- T. Lindmo, E. Boven, F. Cuttitta, J. Fedorko and P. A. Bunn Jr, J. Immunol. Methods, 1984, 72, 77–89 CrossRef CAS PubMed.
- T. Denoël, L. Pedrelli, G. Pantaleo and J. O. Prior, Pharmaceuticals, 2019, 12, 177 CrossRef PubMed.
- M. N. Lub-de Hooge, J. G. Kosterink, P. J. Perik, H. Nijnuis, L. Tran, J. Bart, A. J. Suurmeijer, S. de Jong, P. L. Jager and E. G. de Vries, Br. J. Pharmacol., 2004, 143, 99–106 CrossRef CAS PubMed.
- M. M. Deken, D. L. Bos, W. Tummers, T. L. March, C. J. H. van de Velde, M. Rijpkema and A. L. Vahrmeijer, EJNMMI Res., 2019, 9, 98 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06182h |
| This journal is © The Royal Society of Chemistry 2022 |