
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of an NAD+ analogue with enhanced specificity for PARP1†
Xiao-Nan
Zhang‡
 a,
Albert T.
Lam‡
a,
Qinqin
Cheng‡
a,
Valentine V.
Courouble
b,
Timothy S.
Strutzenberg
b,
Jiawei
Li
a,
Yiling
Wang
a,
Hua
Pei
c,
Bangyan L.
Stiles
a,
Stan G.
Louie
c,
Patrick R.
Griffin
b and
Yong
Zhang
*adef
a,
Albert T.
Lam‡
a,
Qinqin
Cheng‡
a,
Valentine V.
Courouble
b,
Timothy S.
Strutzenberg
b,
Jiawei
Li
a,
Yiling
Wang
a,
Hua
Pei
c,
Bangyan L.
Stiles
a,
Stan G.
Louie
c,
Patrick R.
Griffin
b and
Yong
Zhang
*adef
aDepartment of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA 90089, USA. E-mail: yongz@usc.edu
bDepartment of Molecular Medicine, The Scripps Research Institute, Jupiter, FL 33458, USA
cTitus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA 90089, USA
dDepartment of Chemistry, Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
eNorris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA 90089, USA
fResearch Center for Liver Diseases, University of Southern California, Los Angeles, CA 90089, USA
First published on 27th January 2022
Abstract
Among various protein posttranslational modifiers, poly-ADP-ribose polymerase 1 (PARP1) is a key player for regulating numerous cellular processes and events through enzymatic attachments of target proteins with ADP-ribose units donated by nicotinamide adenine dinucleotide (NAD+). Human PARP1 is involved in the pathogenesis and progression of many diseases. PARP1 inhibitors have received approvals for cancer treatment. Despite these successes, our understanding about PARP1 remains limited, partially due to the presence of various ADP-ribosylation reactions catalyzed by other PARPs and their overlapped cellular functions. Here we report a synthetic NAD+ featuring an adenosyl 3′-azido substitution. Acting as an ADP-ribose donor with high activity and specificity for human PARP1, this compound enables labelling and profiling of possible protein substrates of endogenous PARP1. It provides a unique and valuable tool for studying PARP1 in biology and pathology and may shed light on the development of PARP isoform-specific modulators.
Introduction
Poly-ADP-ribose polymerase 1 (PARP1) catalyzes protein poly-ADP-ribosylation (PARylation) by utilizing nicotinamide adenine dinucleotide (NAD+) as a co-substrate. As a founding member of the PARP family, PARP1 accounts for the majority of PARylation activities in cells. PARP1-mediated PARylation plays vital roles in regulating genome stability, protein homeostasis, cell proliferation, differentiation, and apoptosis.1–4 Dysregulated PARP1 activity is strongly implicated in the pathology of many human diseases, including cancer, neurodegenerative diseases, immune disorders, and metabolic diseases.5–19Acting as the donor of ADP-ribose, NAD+ participates in cellular protein PARylation and mono-ADP-ribosylation (MARylation) catalyzed by PARP1 and many other ADP-ribosyltransferases. Chemically modifying NAD+ with distinct functional groups at varied positions have resulted in NAD+ mimics with great activities for protein ADP-ribosylation.20–30 These functional NAD+ analogues enable tracking, imaging, and profiling of ADP-ribosylated proteins. However, none of the reported NAD+ molecules display adequate specificity for a native PARP enzyme. Here we discovered an NAD+ analogue with high activity and specificity for human PARP1. Unlike NAD+ and other functional NAD+ analogues, this new compound characterized by an azido group at 3′-OH of the adenosine (ADO) moiety of NAD+ (ADO-3′-N3-NAD+) only shows considerable substrate activity for natural PARP1 and enables the identification of novel protein substrates for endogenous PARP1 (Fig. 1). The ADO-3′-N3-NAD+ may provide a valuable tool for studying biological and pathological functions of PARP1.
 | ||
| Fig. 1 ADO-3′-N3-NAD+ has high substrate activity and specificity for PARP1-catalyzed protein PARylation. | ||
Results
Considering the excellent activity for PARP1- and PARP2-catalyzed PARylation for a previously synthesized NAD+ with a 3′-azido substituted nicotinamide riboside (NR) (NR-3′-N3-NAD+),29 we reasoned that switching the azido group to the adenosine 3′-OH position may afford an NAD+ with notable activity for protein PARylation. The designed ADO-3′-N3-NAD+ was successfully generated using the same method as reported for the NR-3′-N3-NAD+ (Scheme S1†).The activity of ADO-3′-N3-NAD+ for protein ADP-ribosylation was first evaluated with full-length human PARP1 expressed from Escherichia coli through PARP1 automodification. Purified PARP1 together with activated DNA was incubated with NR-3′-N3-NAD+ or ADO-3′-N3-NAD+ in the absence or presence of olaparib, which can potently inhibit PARP1/PARP2 enzymatic activity and is also a moderate inhibitor for other PARP enzymes, such as PARP5a and PARP10.31–34 PARylated PARP1 was then labeled with biotin via click chemistry. Using a streptavidin-HRP for detection, immunoblot analysis indicated that as expected, the ADO-3′-N3-NAD+ exhibits marked activity for PARP1-catalyzed auto-PARylation though slightly lower than that of NR-3′-N3-NAD+ (Fig. 2A).
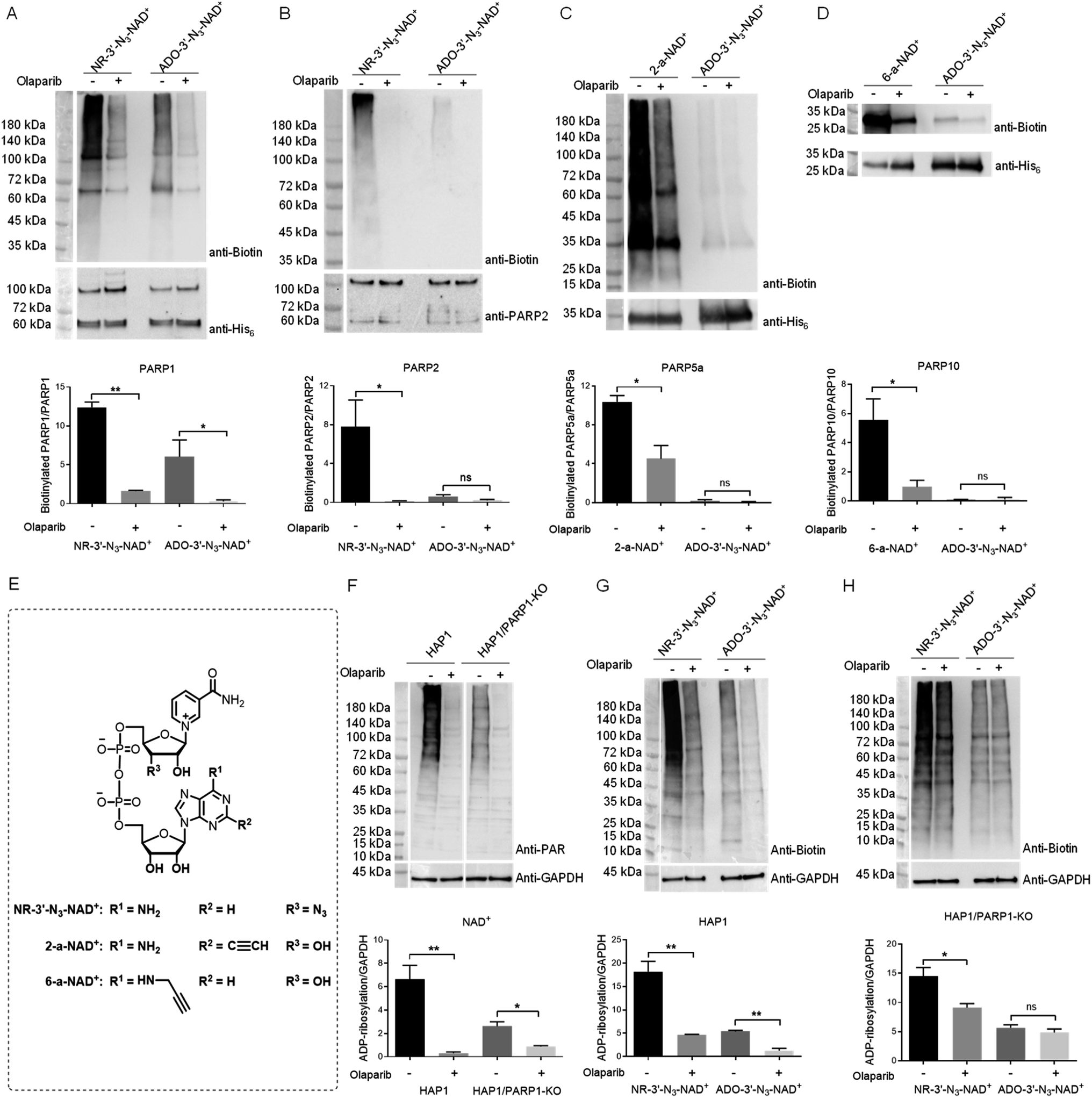 | ||
| Fig. 2 Substrate activities of NAD+ analogues for protein ADP-ribosylation. (A) and (B) Substrate activities of NR-3′-N3-NAD+ and ADO-3′-N3-NAD+ for human PARP1 (A) and PARP2 (B). (C) Substrate activities of 2-a-NAD+ and ADO-3′-N3-NAD+ for catalytic domain of human PARP5a. (D) Substrate activities of 6-a-NAD+ and ADO-3′-N3-NAD+ for catalytic domain of human PARP10. (E) Chemical structures of NR-3′-N3-NAD+, 2-a-NAD+, and 6-a-NAD+. (F) Protein ADP-ribosylation for HAP1 and HAP1/PARP1-KO cell lysates with NAD+. (G) and (H) Protein ADP-ribosylation for HAP1 (G) and HAP1/PARP1-KO (H) cell lysates with NR-3′-N3-NAD+ and ADO-3′-N3-NAD+. PARPs or cell lysates were incubated with NAD+ or NAD+ analogues in the absence or presence of olaparib at 30 °C for 2 hours for PARPs or overnight for cell lysates. The reactions with NAD+ analogues were then labeled with biotin through click chemistry, followed by immunoblot analysis as detected by the streptavidin-HRP conjugate (top panels). Immunoblots for the reactions with NAD+ were detected using an anti-PAR antibody. Middle panels: PARP or GAPDH loading controls as detected by an anti-His6 antibody, anti-PARP2 antibody, or anti-GAPDH antibody. Bottom panels: densitometric analysis of protein ADP-ribosylation normalized to loading controls. ns = not significant; *, p < 0.05; **, p < 0.01. | ||
As a close relative of PARP1, human PARP2 was next examined for automodification by ADO-3′-N3-NAD+. To our surprise, immunoblots showed that ADO-3′-N3-NAD+ has very weak substrate activity for PARP2-mediated auto-PARylation, unlike NR-3′-N3-NAD+ revealing significant activity for PARP2 under the same conditions (Fig. 2B). Furthermore, we investigated the activities of ADO-3′-N3-NAD+ for catalytic domains of human PARP5a and PARP10 (Fig. 2C and D). In contrast to 2-alkyne-NAD+ (2-a-NAD+) and 6-alkyne-NAD+ (6-a-NAD+) that are excellent substrates for PARP5a-catalyzed protein PARylation and PARP10-catalyzed MARylation,28,29,35 respectively (Fig. 2E), ADO-3′-N3-NAD+ displays no or very low activities for PARP5a and PARP10 according to immunoblot analyses. These results suggest that ADO-3′-N3-NAD+ possesses high activity and specificity for human PARP1.
To confirm substrate specificity of ADO-3′-N3-NAD+, lysates of human HAP1 cells without and with PARP1 knockout (KO) (Fig. S1†) were incubated with NAD+, NR-3′-N3-NAD+, or ADO-3′-N3-NAD+. Using an anti-poly-ADP-ribose (PAR) antibody for detection, immunoblot analysis revealed that both HAP1 and HAP1/PARP1-KO cell lysates have strong protein PARylation signals for NAD+ (Fig. 2F), which are sensitive to olaparib inhibitor. These results indicate PARylation activities from other PARP enzymes in those cell lysates. Following click chemistry-mediated biotin labeling for cell lysates incubated with NR-3′-N3-NAD+ or ADO-3′-N3-NAD+, immunoblots showed that NR-3′-N3-NAD+ gives rise to marked protein PARylation for both HAP1 and HAP1/PARP1-KO cell lysates, whereas ADO-3′-N3-NAD+ only exhibits significant PARylation activities in the presence of PARP1 expression (Fig. 2G and H). And the PARylation signals were inhibited by olaparib. These results support that ADO-3′-N3-NAD+ can function as a substrate with high specificity for PARP1.
Next, the kinetic parameters of NAD+ and ADO-3′-N3-NAD+ for human PARP1-catalyzed auto-PARylation (PARP activity) and hydrolysis (NADase activity) were determined by HPLC-based activity assays (Table 1). In comparison to NAD+ with a kcat of 26.0 min−1 and Km of 212.9 μM for PARP activity, ADO-3′-N3-NAD+ is characterized by a reduced kcat (3.8 min−1) and increased Km (524.8 μM) for PARP1 auto-PARylation. Similarly, ADO-3′-N3-NAD+ shows a lower kcat and higher Km than those of NAD+ for the NADase activity. Together with the immunoblot analyses, these data indicate considerable substrate activity of ADO-3′-N3-NAD+ for PARP1.
| Substrate | k cat (min−1) | K m (μM) | k cat/Km (min−1 M−1) | |
|---|---|---|---|---|
| PARP activity | NAD+ | 26.0 ± 2.3 | 212.9 ± 45.5 | 1.2 × 105 |
| ADO-3′-N3-NAD+ | 3.8 ± 1.1 | 524.8 ± 264.0 | 7.2 × 103 | |
| NADase activity | NAD+ | 2.4 ± 0.1 | 113.4 ± 17.2 | 2.1 × 104 |
| ADO-3′-N3-NAD+ | 1.4 ± 0.2 | 303.2 ± 83.4 | 4.6 × 103 |
To demonstrate its utility, ADO-3′-N3-NAD+ was applied to profile potential protein substrates of PARP1 by incubating with lysates of H2O2-treated HEK293T cells. Reactions containing NAD+ or ADO-3′-N3-NAD+ plus olaparib were included as controls. PARylated proteins were then labeled with biotin via click chemistry for enrichment and proteomic identification. Given that PARP1 is primarily a nuclear protein, hits present in nucleus were further analyzed. In total, 73 nuclear proteins were identified, of which 49 hits were known substrates of human PARP1, such as itself, DNA-(apurinic or apyrimidinic site)lyase (APEX1), and DNA damage-binding protein 1 (DDB1) (Fig. 3A and Table S1†).22,36,37
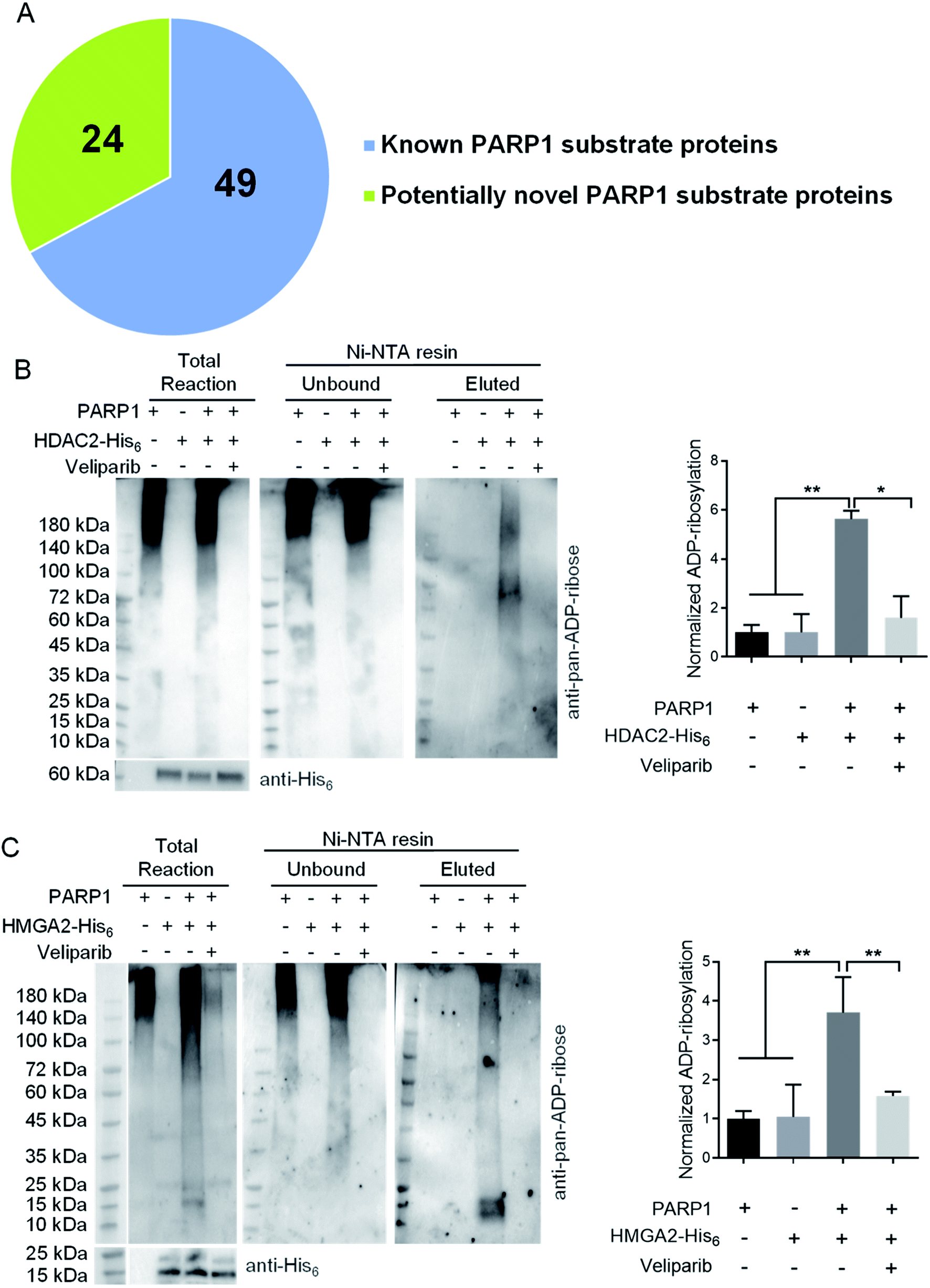 | ||
| Fig. 3 Potentially novel protein substrates of human PARP1 identified by ADO-3′-N3-NAD+. (A) A pie chart of protein substrates identified by ADO-3′-N3-NAD+. (B) and (C) ADP-ribosylation of HDAC2 (B) and HMGA2 (C) by human PARP1. HDAC2-His6 or HMGA2-His6 was incubated with NAD+ in the absence or presence of His6-tag free PARP1 without or with veliparib at 30 °C for overnight. Reactions with only PARP1 were incubated under the same conditions. The samples were then incubated with Ni-NTA beads to separate unbound PARP1 and elute HDAC2 or HMGA2 for immunoblot analysis as detected by an anti-pan-ADP-ribose binding reagent. The total reactions were detected by the anti-pan-ADP-ribose binding reagent (top panel) or anti-His6 antibody (bottom panels). Right panels: densitometric analysis of ADP-ribosylated proteins for eluted samples normalized to respective substrate proteins. *, p < 0.05; **, p < 0.01. | ||
Among identified 24 potentially novel PARP1 substrates, two were selected for validation, including histone deacetylase 2 (HDAC2) and high mobility group protein HMGI-C (HMGA2). Both HDAC2 and HMGA2 play important roles in regulating gene expression.38–42 Recombinant human HDAC2-His6 or HMGA2-His6 was incubated with NAD+ in the absence or presence of His6-tag free human PARP1 without or with veliparib inhibitor. Reactions with PARP1 alone were included as controls. Following enrichment of His6-tagged proteins by Ni-NTA resins, immunoblot analyses using an anti-pan-ADP-ribose binding reagent indicated significant levels of ADP-ribosylation for both HDAC2 and HMGA2 by PARP1, which are sensitive to veliparib treatment (Fig. 3B and C). These results support HDAC2 and HMGA2 as substrates of human PARP1.
Discussion
In comparison to adenine modifications on NAD+ that are well tolerated by PARPs, the azido substitution at NR 3′-OH resulted in the NR-3′-N3-NAD+ with high specificity for PARylation catalyzed by PARP1 and PARP2.29 Moving the azido group to 3′-OH of the adenosine moiety produced the ADO-3′-N3-NAD+ with unexpectedly enhanced selectivity for PARP1. Unlike adenine remote from the reaction center, both the ribosyl groups of NAD+ participate in the PARylation. PARP enzymes may therefore be more sensitive to functional replacements at 3′-OH positions. Notably, PARP2 shows little tolerance to the adenosyl 3′-azido substitution for PARylation despite high levels of similarity with PARP1 in sequence, structure, and function. This could be resulted from their different capabilities in capturing ADO-3′-N3-NAD+ and/or catalyzing the transfer of ADO-3′-N3-ADP-ribose. In addition, the selectivity of ADO-3′-N3-NAD+ for PARP1 over PARP2 may be attributed to structural differences in the vicinity of the substrate acceptor site.43–47 In comparison to the acceptor pocket of PARP1, the one for PARP2 features a uniquely extended loop (Leu547-Thr553), possibly affecting substrate activity of ADO-3′-N3-NAD+ for PARP2-catalyzed PARylation.Future studies will be needed to investigate determinant(s) for the substrate specificity of ADO-3′-N3-NAD+ for PARP1 as well as the effects of ribosyl 3′-OH modification on size and pattern of PAR polymers. Even though the functionalization on NAD+ riboses may cause potential loss of substrate activity, such derivatization may create opportunities for the discovery of PARP isoform-specific NAD+ substrates and inhibitors. It should be noted that ADO-3′-N3-NAD+ could potentially be recognized for catalysis by other PARPs that were not evaluated in this study. Further efforts will be required to study its activities with those enzymes.
In summary, introduction of an azido at adenosine 3′-OH affords an NAD+ with high activity and specificity for human PARP1. The resulting ADO-3′-N3-NAD+ represents a novel and important tool for studying PARP1 in human health and disease.
Experimental methods
Cell lines and recombinant proteins
Human HAP1 cell line and HAP1/human PARP1 knockout (KO) cell line 14 bp deletion were purchased from Horizon Discovery (catalog numbers: C631 and HZGHC003943c006; Waterbeach, United Kingdom). Baculovirus-expressed human PARP1 without a His6 tag was purchased from BioVision (catalog number: 4992; Milpitas, CA). Human PARP2 with a GST-tag and HDAC2 with a His6 tag were purchased from BPS Bioscience (catalog numbers: 80502 and 50002; San Diego, CA). Human HMGA2 with a His6 tag was purchased from Novus Biologicals (catalog number: NBP2-23122; Centennial, CO).Chemical synthesis and characterization of ADO-3′-N3-NAD+
The experimental details and results for synthesis and characterization of ADO-3′-N3-NAD+ are provided in the ESI.†Bacterial expression and purification of human PARP1
Five milliliters of LB broth with 50 μg mL−1 kanamycin was inoculated with BL21(DE3) Escherichia coli transformed with pET28a (+) vector expressing full-length human PARP1 and grown overnight in a temperature-controlled shaker at 37 °C at 250 rpm. Five mL of overnight culture was then inoculated into 1 L of LB broth with 50 μg mL−1 kanamycin and grown to OD600 = 0.8 in a temperature-controlled shaker at 37 °C at 250 rpm. Cultures were induced using a 500 mM stock of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 500 μM and grown overnight at 16 °C at 250 rpm. Cells were harvested by centrifugation for 30 minutes at 2700 × g and the supernatants were discarded. Cell pellets were resuspended in 30 mL of lysis buffer (20 mM Tris pH 7.5, 500 mM NaCl, 1 mM beta-mercaptoethanol (β-ME), 0.2% nonidet P-40, and 0.2% tween-20 with 1 mg mL−1 lysozyme, and 1 mM phenylmethylsulfonyl fluoride (PMSF)). Cells were lysed by running cells through a French press at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi three times. Cell debris was spun down by centrifugation at 27000 × g for 40 minutes at 4 °C.
000 psi three times. Cell debris was spun down by centrifugation at 27000 × g for 40 minutes at 4 °C.
One milliliter of Ni-NTA agarose resin was loaded onto a gravity flow column and washed with 15 column volumes (CVs) of water, followed by 15 CVs of equilibrium buffer (20 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole, and 1 mM β-ME) before adding soluble fractions of cell lysates. The column was then washed with 15 CVs of equilibrium buffer. Bound proteins were eluted with 15 CVs elution buffer (20 mM Tris pH 7.5, 500 mM NaCl, 400 mM imidazole, and 1 mM β-ME). Eluted proteins were concentrated with 30 kDa MWCO ultra-15 centrifugal filter units to 1 mL. The protein samples were then diluted in storage buffer (100 mM Tris pH 7.5, 150 mM NaCl, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM β-ME) at 1:10 before loading with a syringe onto a 5 mL heparin column equilibrated with heparin binding buffer (50 mM Tris pH 7.5, 250 mM NaCl, 0.1 mM EDTA, and 1 mM β-ME). After sample loading, the column was connected to an ÄKTA pure FPLC and weakly bound proteins were washed off with heparin binding buffer. Elution with heparin elution buffer (50 mM Tris pH 7.5, 1000 mM NaCl, 0.1 mM EDTA, and 1 mM β-ME) was done with a gradient of 20% to 80% over 2 CVs and fractions with protein were collected based on UV absorbance at 280 nm. Collected fractions were concentrated and loaded onto a Superdex 75 Increase 10/300 column pre-equilibrated with the storage buffer. An isocratic gradient with a flow rate of 0.5 mL min−1 was used to separate 0.5 mL fractions, which were run on SDS-PAGE gels to identify fractions containing the desired proteins. Fractions were combined and concentrated by centrifugal filter units. Protein concentrations were determined by UV absorbance at 280 nm using an extinction coefficient of 1.05.
Bacterial expression and purification of human PARP5a and PARP10
PARP5a and PARP10 were purified as previously described.29 cDNAs of human PARP5a (GE Healthcare Dharmacon: MHS6278-202806376) and PARP10 (MHS6278-213245042) were used as templates for polymerase chain reactions to amplify the catalytic domains (residues 1093-1327 and 809-1017 for PARP5a and PARP10, respectively) and add a His6 tag at the C-terminus. PCR products were purified by agarose gel electrophoresis and digested using XbaI and XhoI for 1 hour at 37 °C. pET-28a (+) plasmids were digested overnight using XbaI and XhoI at 37 °C and cut vector was purified from agarose gel electrophoresis. Digested PCR fragments and vector were ligated using T4 DNA Ligase (New England Biolabs) for 1 hour at room temperature prior to transformation into DH10B competent cells.Sequence-verified plasmids were transformed into BL21(DE3) E. coli for bacterial expression and purification. Five milliliters of LB broth with 50 μg mL−1 kanamycin were inoculated with transformed BL21(DE3) cells with the plasmids expressing human PARP5a or PARP10 grown overnight in a temperature-controlled shaker at 37 °C at 250 rpm. Each overnight culture was inoculated into 1 L of LB broth with 50 μg mL−1 kanamycin per recombinant protein and grown to OD600 = 0.8 in a temperature-controlled shaker at 37 °C at 250 rpm. Cultures were induced using a 500 mM stock of IPTG to a final concentration of 500 μM and grown overnight at 22 °C at 250 rpm. Cells were harvested by centrifuging for 30 minutes at 2700 × g and discarding the supernatants.
Cell pellets were resuspended in 30 mL of equilibrium buffer (20 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole, and 1 mM β-ME) with 1 mg mL−1 lysozyme, 10 μg mL−1 DNase I, 0.1 mM MgCl2, and 1 mM PMSF. For PARP10, the equilibrium buffer contained 1 mM tris(2-carboxyethyl)phosphine (TCEP) in place of β-ME. Cells were lysed by running cells through a French press at 25000 psi three times. Cell debris was spun down at 27000 × g for 40 minutes at 4 °C. One milliliter of Ni-NTA agarose resin was loaded onto a gravity flow column and washed with 15 CVs of water and 15 CVs of equilibrium buffer before running soluble cell lysates through. The column was then washed with 15 CVs of equilibrium buffer and 15 CVs of 20 mM Tris pH 7.5, 500 mM NaCl, 30 mM imidazole with 1 mM β-ME or 1 mM TCEP for PARP5a or PARP10, respectively. Bound proteins were eluted with 15 CVs of 20 mM Tris pH 7.5, 500 mM NaCl, 400 mM imidazole with 1 mM β-ME or 1 mM TCEP for PARP5a or PARP10, respectively. Proteins were concentrated with 10 kDa MWCO ultra-15 centrifugal filter units before running through an ÄKTA pure FPLC with a Superdex 75 Increase 10/300 column pre-equilibrated with 20 mM Tris pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM dithiothreitol (DTT). An isocratic gradient with a flow rate of 0.5 mL min−1 was used to separate 0.5 mL fractions, which were run on SDS-PAGE gels to identify fractions containing the desired proteins. Fractions were combined and concentrated by the centrifugal filter units. Protein concentrations were determined by UV absorbance at 280 nm using extinction coefficients of 0.753 and 0.762 for PARP5a and PARP10, respectively.
Preparation of cell lysates
Human HAP1 and HAP1/PARP1-KO cell lines were grown per manufacturer's protocols. Cells were grown to 70% confluency in T25 flasks prior to harvesting. Cell pellets were resuspended with 200 μL of NP-40 lysis buffer (25 mM Tris–HCl pH 7.5, 50 mM NaCl, 10% glycerol, 1% nonidet P-40, and halt protease inhibitor cocktail (Thermo Fisher Scientific: 78430)). Cells were shaken for 10 minutes at room temperature before spinning down cellular debris at 14000 × g at 4 °C for 15 minutes. Cell lysate concentrations of protein were measured using Bradford reagent. PARP1 expression in cell lines was examined with anti-human PARP1 monoclonal antibody (Cell Signaling Technologies: 46D11). And loading controls were detected using an anti-GAPDH antibody (Thermo Fisher Scientific: MA5-15738).
Copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC)
Automodified PARPs were conjugated where mentioned to alkyne-biotin or azide-biotin by CuAAC. Click reactions for purified proteins were carried out at room temperature for 1 hour with 500 μM tris-hydroxypropyltriazolylmethylamine (THPTA), 250 μM CuSO4, 2.5 mM sodium ascorbate, and the appropriate biotin-containing click reagent of biotin-PEG4-alkyne or biotin-PEG4 azide at 100 μM. Click reactions in cell lysates were performed by using 3× CuAAC buffer (1.5 mM THPTA, 750 μM CuSO4, 300 μM biotin-PEG4-alkyne, 7.5 mM sodium ascorbate) and incubating for 1 hour at room temperature.Substrate activities of ADO-3′-N3-NAD+ with PARPs and cell lysates
Bacteria-expressed human PARP1 (3 μM) and commercial human PARP2 (50 nM) were automodified for 2 hours at 30 °C with 600 μM of NAD+ analogues in 100 mM Tris–HCl pH 8.0, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, and 100 ng μL−1 activated DNA (Sigma-Aldrich: D4522). Purified PARP5a (10 μM) and PARP10 (20 μM) were automodified for 2 hours at 30 °C with 600 μM of NAD+ analogues in 50 mM Tris–HCl pH 7.5, 5 mM MgCl2 100 mM NaCl, and 200 μM DTT. Olaparib (100 μM) was included as a control. All reactions were stopped by the additions of 100 μM of olaparib after 2 hours.To evaluate activities in lysates of HAP1 and HAP1/PARP1-KO cells, 10 μg of cell lysates in 10 μL were incubated overnight with 600 μM of NAD+ or NAD+ analogues in PBS with 100 ng μL−1 of activated DNA and 10 μM of inhibitor of poly(ADP-ribose) glycohydrolase (PARG) PDD00017273 (Sigma-Aldrich) at 30 °C. Olaparib (100 μM) was included as a control. All reactions were stopped by the additions of 100 μM of olaparib after overnight.
ADP-ribosylated proteins were conjugated with biotin through CuAAC and the appropriate biotin-containing compound and run on precast SDS-PAGE gels. Proteins were transferred onto PVDF membranes and blocked with 3% bovine serum albumin (BSA) in PBS with 0.1% tween-20 (PBST) for 1 hour. After washing, detection was done with 1:200 streptavidin–horseradish peroxidase (HRP) conjugate in PBST. Blots were imaged using SuperSignal West Pico PLUS chemiluminescent substrate and ChemiDoc Touch Gel Imaging System. For detection of poly-ADP-ribosylation with NAD+, membranes were blocked in 5% milk before detection with an anti-poly-ADP-ribose (PAR) monoclonal antibody 10H at 1:3000 (Santa Cruz Biotechnology: sc-56198). Goat anti-mouse antibody conjugated to HRP (Thermo Fisher Scientific: G-21040) at 1:3000 was used as the secondary antibody.
Kinetic analysis of PARP1 with NAD+ and ADO-3′-N3-NAD+
Standards of NAD+ were run at 0 μM, 20 μM, 50 μM, 100 μM, 200 μM, 400 μM, and 600 μM. ADP-ribose (ADPr) was purchased from Sigma-Aldrich (A0752) and standards were run at 10 μM, 20 μM, and 50 μM. ADO-3′-N3-NAD+ standards were run at 0 μM, 50 μM, 100 μM, 200 μM, 400 μM, and 600 μM. ADO-3′-N3-ADPr was generated from cleavage of ADO-3′-N3-NAD+ using 7 N ammonia in overnight reactions and peaks were identified by mass spectrometric analysis.Automodification of PARP1 was carried out in 50 μL reactions per condition and time point. Bacteria expressed human PARP1 (0.9 μM) was added to reaction buffer (100 mM Tris–HCl pH 8.0, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, and 100 ng μL−1 activated DNA) and preincubated for 15 minutes. NAD+ or NAD+ analogues were then added at final concentrations of 20 μM, 50 μM, 100 μM, 200 μM, 400 μM, or 600 μM and incubated at 30 °C for 0, 3, or 6 minutes. Reactions were stopped at each time point by the additions of 20% trichloroacetic acid (TCA) to a final concentration of 10% TCA.
Reaction samples with NAD+ were run on a Waters HPLC using a C18 Kinetex column (5 μM, 100 Å, 150 × 10.0 mm, Phenomenex Inc, Torrance, CA) with mobile phase A: 0.1% formic acid (aq.) and mobile phase B: 0.1% formic acid in acetonitrile and detection of UV absorbance at 260 nm; flow rate = 2.0 mL min−1; 0–8 min: 0% B, 8–13 min: 0–2.5% B, 13–18 min: 2.5–40% B, 18–20 min: 40–80% B, 20–21 min: 80–0% B, 21–24 min: 0% B. Samples with ADO-3′-N3-NAD+ were run on the Waters HPLC using the C18 Kinetex column with mobile phase A: 0.1% formic acid (aq.) and mobile phase B: 0.1% formic acid in acetonitrile and detection of UV absorbance at 260 nm; flow rate = 2.0 mL min−1; 0–6 min: 0% B, 6–15 min: 0–20% B, 15–17 min: 20–50% B, 17–18 min: 50–0% B, 18–20 min: 0% B.
Substrate profiling in HEK293T lysates
HEK293T cells grown to 80% confluency in DMEM with 10% FBS in T75 flasks. Cells were then washed with PBS and DMEM with 10% FBS and 1 mM H2O2 was added to the cells for incubation at 37 °C for 10 minutes. Cells were then washed with PBS before collection by cell scraping. Cells were lysed by resuspending the cell pellets using 400 μL of NP-40 lysis buffer, followed by shaking for 10 minutes at room temperature before spinning down cellular debris at 14000 × g at 4 °C for 15 minutes. Cell lysates were collected, and the cellular debris was resuspended with another 100 μL of lysis buffer to repeat the cell lysis. Both fractions of cell lysate were combined. Cell lysate concentrations of protein were measured using Bradford reagent (Thermo Fisher Scientific: 23236).
ADO-3′-N3-NAD+ (200 μM) was added to 1 mg of HEK293T cell lysate in 400 μL and incubated for 2 hours at 30 °C. Control reactions included NAD+ in place of ADO-3′-N3-NAD+ and ADO-3′-N3-NAD+ with olaparib (100 μM). Samples were then treated with 3× CuAAC buffer for 1 hour at room temperature. Excess biotin-PEG4-alkyne was removed by buffer exchange into PBS using Amicron 10 kDa MWCO Ultra-4 centrifugal filter units (Millipore: UFC801024) by over 1000-fold dilution. Samples were added to 5 mL of PBS with 1% NP-40 and 100 mM NaCl before adding 100 μL of NeutrAvidin beads (Thermo Fisher Scientific: 29200) and incubated with head-over-end rotation overnight at room temperature. Beads were spun down at 2000 × g for 5 minutes and the supernatants were discarded. Beads were resuspended in 500 μL of PBS with 4 M urea (pH 7.4) and incubated with head-over-end rotation for 10 minutes. Beads were spun down, and the supernatant was discarded. This was repeated one additional time. Beads were then incubated in PBS with 1% NP-40 and incubated the same way for three times, 50 mM ammonium bicarbonate for two times, PBS for two times, 20% acetonitrile for two times, PBS for two times, and 50 mM ammonium bicarbonate for two times. For elution, beads were then resuspended in 100 μL of 0.1% rapigest, and the slurry was boiled at 98 °C for 10 minutes. The beads were spun down and the supernatants were collected. An additional 50 μL of 0.5% rapigest was added to the boiled beads and the beads were subjected to another round of boiling and the supernatants were collected and combined with the ones in 0.1% rapigest.
The eluted samples were first reduced by incubating in 10 mM DTT at 56 °C for 20 min. The resulting thiols were alkylated with 55 mM iodoacetamide in the dark for 15 minutes. Proteins were digested with trypsin (Promega: V5111) at a 1:50 (w:w) ratio (trypsin:protein) overnight at 37 °C. Peptides were acidified to 1% trifluoroacetic acid (TFA) and then desalted using C18 ZipTip (Millipore: ZTC18 5096). Dried peptides were resuspended in 10 μL of 0.1% TFA in water.
One microgram of sample was injected onto an UltiMate 3000 UHP liquid chromatography system (Thermo Fisher Scientific). Peptides were separated using a μPAC C18 trapping column (PharmaFluidics) in-line with a 50 cm μPAC column (PharmaFluidics). Peptides were eluted with a 90 min gradient (0–30% acetonitrile with 0.1% formic acid for 60 minutes, then 30–60% for 30 minutes) at a flow rate of 1 μL min−1 and electrosprayed into an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) with a Nanospray Flex ion source (Thermo Fisher Scientific). The source voltage was set to 2.5 kV and the S-Lens RF level was set to 30%. The instrument method consisted of one survey (full) scan from m/z 375 to 1500 at a resolution of 120000 in the Orbitrap mass analyzer, followed by data-dependent MS/MS scans of selected precursor ions in the linear quadrupole ion trap (LTQ) using the topN method. The AGC target value was set to 4 × 105, and the maximum injection time was set to 50 ms in the Orbitrap. The parameters were set to 2 × 104 and 120 ms in the LTQ with an isolation width of 1.2 Da and normalized collision energy of 28 for precursor isolation and MS/MS scanning. Precursor dynamic exclusion was enabled for a duration of 40 s.
Thermo.Raw files were imported into Proteome Discoverer 2.2 (Thermo Fisher Scientific) using default parameters. The search engine Sequest HT was used. Parameters for protein searching were defined as follows: database—Uniprot human protein database (updated October 2018); precursor mass tolerance—10 ppm; fragment mass tolerance—0.6 Da; digestion—trypsin with two missed cleavages allowed; fixed modification—carbamidomethylation (C); variable modification—oxidation (M) and N-terminal protein acetylation. The Percolator node was used for peptide validation based on the PEP score. For protein identification, a cut-off value of at least two unique high confidence peptides per protein with at 1% false discovery rate (FDR) was used. In total, each sample condition was repeated three times. Each hit from the samples with ADO-3′-N3-NAD+ was examined for appearance in the control samples. Each hit was considered reproducible if it was identified in at least two of the three replicates. Reproducible hits were examined against previously identified human PARP1 substrate proteins to identify novel protein substrates.21–23
Protein ADP-ribosylation by PARP1
Baculovirus-expressed human PARP1 (200 ng) without a His6 tag (BioVision: 4992) was incubated with 2 μg of HDAC2-His6 (BPS Bioscience: 50002) or 1 μg HMGA2-His6 (Novus Biologicals: NBP2-23122) in 20 μL reactions (600 μM NAD+, 100 mM Tris–HCl pH 8.0, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, and 100 ng μL−1 activated DNA) overnight at 30 °C. Ni-NTA agarose beads (20 μL) were added to the reactions and binding buffer (20 mM Tris–HCl pH 7.5, 500 mM NaCl, and 20 mM imidazole) was then added to a total volume of 500 μL for each condition in microcentrifuge tubes, followed by incubation at 4 °C with end-over-end rotation for 3 hours for HDAC2-containing reactions or 1 hour for HMGA2-containing reactions. Samples were spun down at 2000 × g for 2 minutes and the unbound supernatants were transferred to new tubes. Beads were resuspended in 100 μL of binding buffer and transferred to spin columns for washing the beads. Two additional washes were conducted with 150 μL and 100 μL of binding buffer. Elution was then performed with 100 μL of elution buffer (20 mM Tris–HCl pH 7.5, 500 mM NaCl, and 400 mM imidazole). Samples were diluted with 400 μL of PBS and concentrated before subjecting to immunoblots by using an anti-pan-ADP-ribose binding reagent (Sigma-Aldrich: MABE1016) and goat anti-rabbit secondary antibody for detection.Data availability
The data supporting the findings of this study are available from the corresponding author upon request.Author contributions
X. N. Z., A. T. L., Q. C. and Y. Z. designed research; X. N. Z., A. T. L., Q. C., V. V. C., T. S. S., J. L. and Y. W. performed research; H. P., B. L. S., S. G. L. and P. R. G. provided resources and critical insights; X. N. Z., A. T. L., Q. C. V. V. C., T. S. S, P. R. G. and Y. Z. analyzed data; and X. N. Z., A. T. L., Q. C. and Y. Z. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, Sharon L. Cockrell Cancer Research Fund, National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) grant R35GM137901 (to Y. Z.), and National Institute of Biomedical Imaging and Bioengineering (NIBIB) of NIH grant R01EB031830 (to Y. Z.).References
- B. A. Gibson and W. L. Kraus, New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs, Nat. Rev. Mol. Cell Biol., 2012, 13(7), 411–424 CrossRef CAS PubMed.
- P. O. Hassa, S. S. Haenni, M. Elser and M. O. Hottiger, Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going?, Microbiol. Mol. Biol. Rev., 2006, 70(3), 789–829 CrossRef CAS PubMed.
- M. O. Hottiger, Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics, Annu. Rev. Biochem., 2015, 84, 227–263 CrossRef CAS PubMed.
- K. W. Ryu, D. S. Kim and W. L. Kraus, New Facets in the Regulation of Gene Expression by ADP-Ribosylation and Poly(ADP-ribose) Polymerases, Chem. Rev., 2015, 115(6), 2453–2481 CrossRef CAS PubMed.
- A. Cardinale, E. Paldino, C. Giampa, G. Bernardi and F. R. Fusco, PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington's Disease, PLoS One, 2015, 10(8), e0134482 CrossRef PubMed.
- K. Gariani, D. Ryu, K. J. Menzies, H. S. Yi, S. Stein and H. Zhang, et al., Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease, J. Hepatol., 2016, 66(1), 132–141 CrossRef PubMed.
- F. Laudisi, M. Sambucci and C. Pioli, Poly(ADP-ribose) polymerase-1 (PARP-1) as immune regulator, Endocr., Metab. Immune Disord.: Drug Targets, 2011, 11(4), 326–333 CrossRef CAS PubMed.
- L. Lehtiö, N.-W. Chi and S. Krauss, Tankyrases as drug targets, FEBS J., 2013, 280(15), 3576–3593 CrossRef PubMed.
- S. Martire, A. Fuso, L. Mosca, E. Forte, V. Correani and M. Fontana, et al., Bioenergetic Impairment in Animal and Cellular Models of Alzheimer's Disease: PARP-1 Inhibition Rescues Metabolic Dysfunctions, JAD, J. Alzheimer's Dis., 2016, 54(1), 307–324 CAS.
- J. L. Riffell, C. J. Lord and A. Ashworth, Tankyrase-targeted therapeutics: expanding opportunities in the PARP family, Nat. Rev. Drug Discovery, 2012, 11(12), 923–936 CrossRef CAS PubMed.
- M. M. Rosado, E. Bennici, F. Novelli and C. Pioli, Beyond DNA repair, the immunological role of PARP-1 and its siblings, Immunology, 2013, 139(4), 428–437 CrossRef CAS PubMed.
- D. Ryu, H. Zhang, E. R. Ropelle, V. Sorrentino, D. A. Mazala and L. Mouchiroud, et al., NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation, Sci. Transl. Med., 2016, 8(361), 361ra139 Search PubMed.
- A. Sahaboglu, N. Tanimoto, J. Kaur, J. Sancho-Pelluz, G. Huber and E. Fahl, et al., PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function, PLoS One, 2010, 5(11), e15495 CrossRef PubMed.
- J. B. Strosznajder, G. A. Czapski, A. Adamczyk and R. P. Strosznajder, Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer's disease, Mol. Neurobiol., 2012, 46(1), 78–84 CrossRef CAS PubMed.
- I. Welsby, D. Hutin and O. Leo, Complex roles of members of the ADP-ribosyl transferase super family in immune defences: looking beyond PARP1, Biochem. Pharmacol., 2012, 84(1), 11–20 CrossRef CAS PubMed.
- S. Pazzaglia and C. Pioli, Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases, Cells, 2019, 9(1), 41 CrossRef PubMed.
- T. M. Kauppinen and R. A. Swanson, The role of poly(ADP-ribose) polymerase-1 in CNS disease, Neuroscience, 2007, 145(4), 1267–1272 CrossRef CAS PubMed.
- S. Martire, L. Mosca and M. d'Erme, PARP-1 involvement in neurodegeneration: A focus on Alzheimer's and Parkinson's diseases, Mech. Ageing Dev., 2015, 146–148, 53–64 CrossRef CAS PubMed.
- C. Liu and Y. Fang, New insights of poly(ADP-ribosylation) in neurodegenerative diseases: A focus on protein phase separation and pathologic aggregation, Biochem. Pharmacol., 2019, 167, 58–63 CrossRef CAS PubMed.
- A. Buntz, S. Wallrodt, E. Gwosch, M. Schmalz, S. Beneke and E. Ferrando-May, et al., Real-Time Cellular Imaging of Protein Poly(ADP-ribos)ylation, Angew. Chem. Int. Ed., 2016, 55(37), 11256–11260 CrossRef CAS PubMed.
- I. Carter-O'Connell, H. Jin, R. K. Morgan, L. L. David and M. S. Cohen, Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets, J. Am. Chem. Soc., 2014, 136(14), 5201–5204 CrossRef PubMed.
- B. A. Gibson, Y. Zhang, H. Jiang, K. M. Hussey, J. H. Shrimp and H. Lin, et al., Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation, Science, 2016, 353(6294), 45–50 CrossRef CAS PubMed.
- H. Jiang, J. H. Kim, K. M. Frizzell, W. L. Kraus and H. Lin, Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases, J. Am. Chem. Soc., 2010, 132(27), 9363–9372 CrossRef CAS PubMed.
- C. Krebs, W. Koestner, M. Nissen, V. Welge, I. Parusel and F. Malavasi, et al., Flow cytometric and immunoblot assays for cell surface ADP-ribosylation using a monoclonal antibody specific for ethenoadenosine, Anal. Biochem., 2003, 314(1), 108–115 CrossRef CAS PubMed.
- S. Laing, M. Unger, F. Koch-Nolte and F. Haag, ADP-ribosylation of arginine, Amino Acids, 2011, 41(2), 257–269 CrossRef CAS PubMed.
- A. T. Lam, X. N. Zhang, V. V. Courouble, T. S. Strutzenberg, H. Pei and B. L. Stiles, et al., A Bifunctional NAD(+) for Profiling Poly-ADP-Ribosylation-Dependent Interacting Proteins, ACS Chem. Biol., 2021, 16(2), 389–396 CrossRef CAS PubMed.
- S. Wallrodt, A. Buntz, Y. Wang, A. Zumbusch and A. Marx, Bioorthogonally Functionalized NAD(+) Analogues for In-Cell Visualization of Poly(ADP-Ribose) Formation, Angew. Chem., Int. Ed., 2016, 55(27), 7660–7664 CrossRef CAS PubMed.
- Y. Wang, D. Rosner, M. Grzywa and A. Marx, Chain-terminating and clickable NAD(+) analogues for labeling the target proteins of ADP-ribosyltransferases, Angew. Chem., Int. Ed., 2014, 53(31), 8159–8162 CrossRef CAS PubMed.
- X.-N. Zhang, Q. Cheng, J. Chen, A. T. Lam, Y. Lu and Z. Dai, et al., A ribose-functionalized NAD+ with unexpected high activity and selectivity for protein poly-ADP-ribosylation, Nat. Commun., 2019, 10(1), 4196 CrossRef PubMed.
- X. Shi, X. N. Zhang, J. Chen, Q. Cheng, H. Pei and S. G. Louie, et al., A poly-ADP-ribose polymer-based antibody-drug conjugate, Chem. Sci., 2020, 11(34), 9303–9308 RSC.
- A.-G. Thorsell, T. Ekblad, T. Karlberg, M. Löw, A. F. Pinto and L. Trésaugues, et al., Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors, J. Med. Chem., 2017, 60(4), 1262–1271 CrossRef CAS PubMed.
- C. C. Gunderson and K. N. Moore, Olaparib: an oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer, Future Oncol., 2015, 11(5), 747–757 CrossRef CAS PubMed.
- A. Min and S.-A. Im, PARP inhibitors as therapeutics: beyond modulation of PARylation, Cancers, 2020, 12(2), 394 CrossRef CAS PubMed.
- J. Murai, N. H. Shar-yin, B. B. Das, A. Renaud, Y. Zhang and J. H. Doroshow, et al., Trapping of PARP1 and PARP2 by clinical PARP inhibitors, Cancer Res., 2012, 72(21), 5588–5599 CrossRef CAS PubMed.
- J. Du, H. Jiang and H. Lin, Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD, Biochemistry, 2009, 48(13), 2878–2890 CrossRef CAS PubMed.
- R. Attar, C. Cacina, S. Sozen, E. Attar and B. Agachan, DNA repair genes in endometriosis, Genet. Mol. Res., 2010, 9(2), 629–636 CrossRef CAS PubMed.
- B. Iovine, M. L. Iannella and M. A. Bevilacqua, Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions, Int. J. Biochem. Cell Biol., 2011, 43(12), 1664–1667 CrossRef CAS PubMed.
- H. R. Ashar, L. Cherath, K. M. Przybysz and K. Chada, Genomic characterization of human HMGIC, a member of the accessory transcription factor family found at translocation breakpoints in lipomas, Genomics, 1996, 31(2), 207–214 CrossRef CAS PubMed.
- C. M. Trivedi, Y. Luo, Z. Yin, M. Zhang, W. Zhu and T. Wang, et al., Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity, Nat. Med., 2007, 13(3), 324–331 CrossRef CAS PubMed.
- B. Mansoori, A. Mohammadi, H. J. Ditzel, P. H. G. Duijf, V. Khaze and M. F. Gjerstorff, et al., HMGA2 as a Critical Regulator in Cancer Development, Genes, 2021, 12(2), 269 CrossRef CAS PubMed.
- U. Unachukwu, K. Chada and J. D’Armiento, High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia, Int. J. Mol. Sci., 2020, 21(9), 3151 CrossRef CAS PubMed.
- O. H. Kramer, HDAC2: a critical factor in health and disease, Trends Pharmacol. Sci., 2009, 30(12), 647–655 CrossRef PubMed.
- A. W. Oliver, J. C. Amé, S. M. Roe, V. Good, G. de Murcia and L. H. Pearl, Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2, Nucleic Acids Res., 2004, 32(2), 456–464 CrossRef CAS PubMed.
- Q. Chen, M. A. Kassab, F. Dantzer and X. Yu, PARP2 mediates branched poly ADP-ribosylation in response to DNA damage, Nat. Commun., 2018, 9(1), 1–13 CrossRef PubMed.
- E. Obaji, M. M. Maksimainen, A. Galera-Prat and L. Lehtio, Activation of PARP2/ARTD2 by DNA damage induces conformational changes relieving enzyme autoinhibition, Nat. Commun., 2021, 12(1), 3479 CrossRef CAS PubMed.
- J. W. Johannes, A. Balazs, D. Barratt, M. Bista, M. D. Chuba and S. Cosulich, et al., Discovery of 5-{4-[(7-Ethyl-6-oxo-5,6-dihydro-1,5-naphthyridin-3-yl)methyl]piperazin-1-yl}-N-methylpyridine-2-carboxamide (AZD5305): A PARP1-DNA Trapper with High Selectivity for PARP1 over PARP2 and Other PARPs, J. Med. Chem., 2021, 64(19), 14498–14512 CrossRef CAS PubMed.
- T. Karlberg, M. Hammarstrom, P. Schutz, L. Svensson and H. Schuler, Crystal structure of the catalytic domain of human PARP2 in complex with PARP inhibitor ABT-888, Biochemistry, 2010, 49(6), 1056–1058 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Results and experimental methods. See DOI: 10.1039/d1sc06256e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |