
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuFEx-enabled, chemoselective synthesis of triflates, triflamides and triflimidates†
Bing-Yu
Li
 a,
Lauren
Voets
a,
Ruben
Van Lommel
ab,
Fien
Hoppenbrouwers
a,
Mercedes
Alonso
b,
Steven H. L.
Verhelst
cd,
Wim M.
De Borggraeve
*a and
Joachim
Demaerel
*ac
a,
Lauren
Voets
a,
Ruben
Van Lommel
ab,
Fien
Hoppenbrouwers
a,
Mercedes
Alonso
b,
Steven H. L.
Verhelst
cd,
Wim M.
De Borggraeve
*a and
Joachim
Demaerel
*ac
aMolecular Design and Synthesis, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, Box 2404, 3001 Leuven, Belgium. E-mail: joachim.demaerel@kuleuven.be; wim.deborggraeve@kuleuven.be
bEenheid Algemene Chemie (ALGC), Department of Chemistry, Vrije Universiteit Brussel (VUB), Pleinlaan 2, 1050 Brussels, Belgium
cLaboratory of Chemical Biology, Department of Cellular and Molecular Medicine, KU Leuven, O&N I bis, Herestraat 49, box 901, 3000 Leuven, Belgium
dLeibniz Institute for Analytical Sciences ISAS, e.V., Otto-Hahn-Str. 6b, 44227 Dortmund, Germany
First published on 5th January 2022
Abstract
Sulfur(VI) Fluoride Exchange (SuFEx) chemistry has emerged as a next-generation click reaction, designed to assemble functional molecules quickly and modularly. Here, we report the ex situ generation of trifluoromethanesulfonyl fluoride (CF3SO2F) gas in a two chamber system, and its use as a new SuFEx handle to efficiently synthesize triflates and triflamides. This broadly tolerated protocol lends itself to peptide modification or to telescoping into coupling reactions. Moreover, redesigning the SVI–F connector with a S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O → SNR replacement furnished the analogous triflimidoyl fluorides as SuFEx electrophiles, which were engaged in the synthesis of rarely reported triflimidate esters. Notably, experiments showed H2O to be the key towards achieving chemoselective trifluoromethanesulfonation of phenols vs. amine groups, a phenomenon best explained—using ab initio metadynamics simulations—by a hydrogen bonded termolecular transition state for the CF3SO2F triflylation of amines.
O → SNR replacement furnished the analogous triflimidoyl fluorides as SuFEx electrophiles, which were engaged in the synthesis of rarely reported triflimidate esters. Notably, experiments showed H2O to be the key towards achieving chemoselective trifluoromethanesulfonation of phenols vs. amine groups, a phenomenon best explained—using ab initio metadynamics simulations—by a hydrogen bonded termolecular transition state for the CF3SO2F triflylation of amines.
Introduction
Recent interest in high-valent sulfur species has brought about an increasing number of SVI–F bond-containing connective hubs. In the framework of Sulfur(VI)–Fluoride Exchange (SuFEx) chemistry—an umbrella term for substitution events replacing fluoride at the electrophilic sulfur center—these ‘molecular plugins’ allow selective and efficient installation of linkages around the SVI core. Especially in the last seven years, various research groups have demonstrated the potential of SuFEx hubs such as sulfonyl fluorides (R–SO2F),1 sulfuryl fluoride (SO2F2),2 thionyl tetrafluoride (SOF4),3 ethenesulfonyl fluoride (ESF, CH2CH–SO2F),2a,4 and others.5 The chemoselective and straightforward nature of SuFEx chemistry has enabled a range of applications in synthesis and materials.6
A particularly intriguing aspect of SuFEx chemistry is its ability to activate oxygen nucleophiles. Various OH-containing materials of different acidities and nucleophilicities have been shown to react cleanly at the sulfur center, and subsequently transform them into useful electrophiles for further derivatization. For example, through SO2F-containing reagents, aliphatic alcohols have been converted into alkyl fluorides7 or alkylating agents,8 carboxylic acids into acyl fluorides,9 and silyl ethers into sulfonate esters10 (Scheme 1B). A unique role in this collection is reserved for aromatic alcohols, which in reaction with SO2F2 selectively form the valuable aryl fluorosulfates in the presence of various other nucleophiles.2a,11 By far, the most commonly employed category of O-based pseudohalides consists of aryl triflates.12 Even though a number of ways to prepare aryl triflates exist,13,14,15,16,17 a broadly applicable protocol that uses an inexpensive and atom-economic [CF3SO2] precursor in a chromatography-free and water-tolerable fashion is still missing from the toolbox.
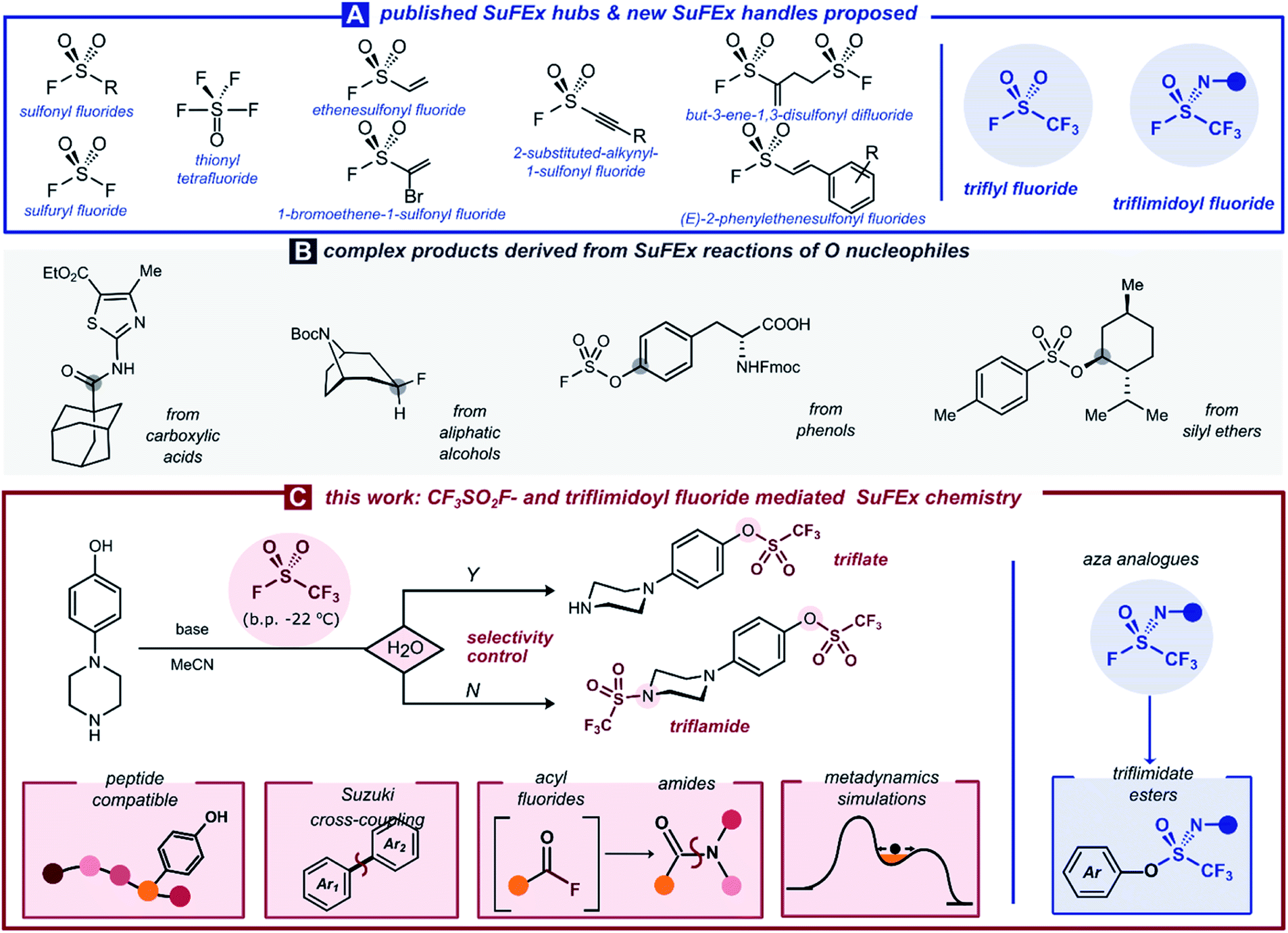 | ||
| Scheme 1 (A) Selected published SuFEx hubs and new SuFEx handles proposed; (B) complex products derived from SuFEx reactions of O nucleophiles; (C) this work: CF3SO2F-mediated and N-substituted triflimidoyl fluoride-mediated SuFEx chemistry. | ||
Herein, we set out to investigate whether SuFEx chemistry can provide this general way of [CF3SO2] transfer onto complex organic molecules. Building on our previous work on sulfuryl fluoride,2b we propose trifluoromethanesulfonyl fluoride gas, CF3SO2F (b.p. −22 °C), as a new electrophilic SuFEx hub, easily generated via two-chamber reactor technology and which reacts efficiently with phenols. Other nucleophiles such as carboxylic acids and amines reacted smoothly with the gas under dry conditions, identifying water as a key additive to obtain complete chemoselectivity for aromatic alcohols (Scheme 1C). Moreover, to shed some light on the mechanistic origins of this chemoselectivity, we relied on ab initio metadynamics simulations to gain fundamental insight into the key SuFEx transition state. Finally, we report a general synthesis of aryl trifluoromethanesulfonimidate (triflimidate) esters: the rarely reported aza analogs of the ArOTf scaffold. Triflimidoyl fluorides show potential as weakly electrophilic SuFEx hubs, which could have unexplored applications as covalent warheads.
Results and discussion
Triflyl fluoride gas was first reported in 1956 by Gramstad for the synthesis of trifluoromethanesulfonic acid derivatives.18 This smallest perfluoroalkanesulfonyl fluoride is gaseous above −22 °C, and its atmospheric chemistry is relatively innocuous.19 The most relevant industrial preparation consists of the electrolytic fluorination of methanesulfonic acid or methanesulfonyl fluoride, and the resulting gas serves as the precursor to all other [CF3SO2]-containing bulk chemicals such as TfOH or Tf2O.20 Other authors have prepared triflyl fluoride on a semibulk scale, by reacting CF3SO2Cl19,21 or Tf2O22 with a fluoride source.23 Recently, Pees and co-workers have developed CF3SO218F as a carrier gas for nucleophilic [18F]-fluoride, evolving it from PhNTf2 as a precursor.24We envisaged the generation of CF3SO2F in a two-chamber reactor as the most convenient way to employ this gas safely on lab scale.25 Even though a higher-MW precursor adds to the process mass intensity of this procedure, the results obtained with ex situ CF3SO2F gas remain true on larger scales in which case the precursor would be abandoned for direct use of gas bottles. Inspired by the aforementioned results, we set out to develop a CF3SO2F gas generation method using PhNTf2 as a bench-stable and easily handled solid precursor (for optimization, see ESI Section 3†). To our delight, the final reaction conditions allowed conversion of the model substrate 4-fluoro-4′-hydroxybiphenyl into product 1 in 85% yield after 4 hours at room temperature (Scheme 2A). With optimized conditions of method A in hand, a variety of readily accessible phenol derivates was examined to further explore the scope of this methodology (Scheme 2). First, monosubstituted electron-rich and deficient phenols were successfully transformed into their corresponding triflates (2–8). Sterically hindered triflates 8, 12 and 27 were also formed efficiently. Although 19F NMR monitoring of catechols showed a high degree of ditriflation at the reaction onset, they nevertheless converged to the monotriflates (14 and 16) after longer reaction times, most likely due to subsequent hydrolysis (see ESI Section 5.1†). With a few experimental adaptations and shorter reaction times, however, it was possible to get the less stable ditriflates 15 and 17 in a fair isolated yield. The triflation of two L-tyrosine derivatives not only offered corresponding products in excellent yields (24 and 25), but also without loss of enantiopurity (25). When it comes to naturally occurring phenols, all afforded the corresponding monotriflates in good to excellent yields (4, 9, 10, 19, 20, 26, 27 and 29). In addition, three heteroaryl triflates were obtained in good to excellent yields (21, 22 and 23). It is worth pointing out that in many cases, the two-chamber reactor method afforded the triflates in sufficiently pure form after extractive work-up, without the need for column chromatography.
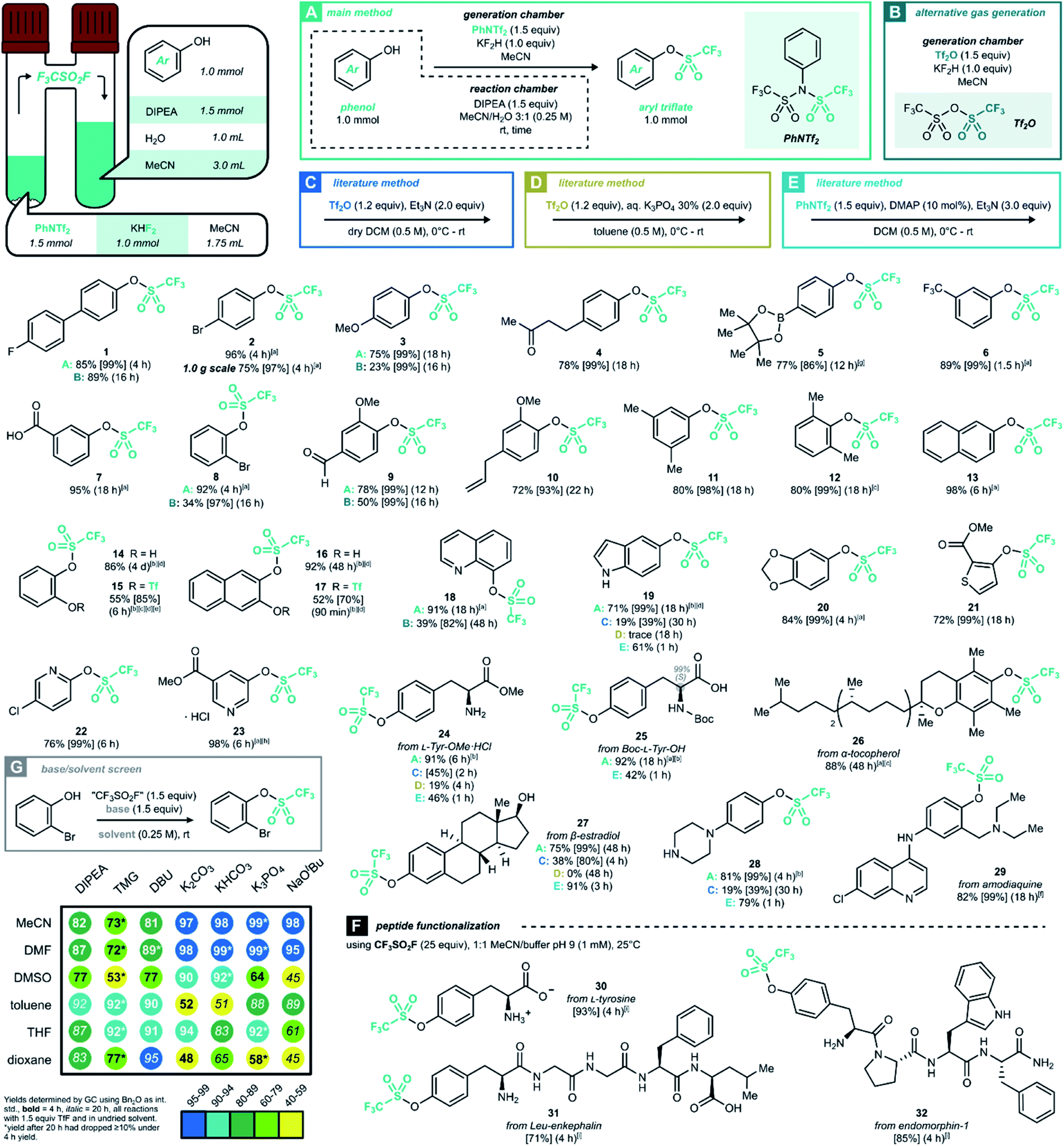 | ||
| Scheme 2 Synthesis of aryl triflates through ex situ generation of CF3SO2F gas in a two-chamber reactor. Unless stated otherwise, method A was used. Generation chamber: N-phenyltrifluoromethanesulfonimide (PhNTf2, 1.5 equiv.), KHF2 (1.0 equiv.) and MeCN (0.86 M, 1.75 mL) at room temperature. Reaction chamber: (hetero)aryl alcohol (1.0 mmol, 1.0 equiv.), N,N-diisopropylethylamine (DIPEA, 1.5 equiv.) in 3.0 mL of MeCN and 1.0 mL of H2O. Reaction details see ESI Section 4.† Isolated yield after column chromatography unless stated otherwise. Between brackets is given the 19F NMR yield using PhCF3 as internal standard, between parentheses the reaction time. [a] Isolated yield after aqueous work-up. [b] 2.5 equiv. of DIPEA were used in the reaction chamber. [c] 3 mL MeCN was used in the reaction chamber as solvent, and the crude reaction mixture was purified on silica directly without aqueous work-up. [d] 2.5 equiv. of PhNTf2 and 1.67 equivalents of KHF2 were used in the generation chamber. [e] The reaction was set under Argon atmosphere. [f] Et3N (3.5 equiv.) and DMSO (0.25 M, 4.0 mL) were used in the reaction chamber. [g] The corresponding boronic acid was used as the starting material, and protected afterwards with pinacol. [h] Yield corresponds to product isolated as an HCl salt. [i] The assay yield is reported (average over two runs), defined by dividing the [M + 132] peak area by the total AUC of the HPLC-MS TIC chromatogram. | ||
In parallel to this method, a different set of conditions was developed using Tf2O as the gas precursor,22 a less expensive and commonly available chemical (method B). Even though good results were obtained for simple phenols (1), the unpleasant nature of this fuming and sensitive liquid, and the reduced yields for more complex phenols (3, 8, 9 and 18) make this method less ideal. Next, in order to further assess the validity of CF3SO2F as a triflating agent, our method was benchmarked against other known triflation methods (for details, see ESI Section 4.1.3†). Four representative phenols were treated according to three literature triflation protocols: adding Tf2O to a solution of phenol and organic base (method C);26 adding Tf2O under Frantz' aqueous conditions (method D)27 and using the PhNTf2 reagent directly (method E).28 Even though the gas-free methods required a shorter reaction time, the corresponding triflates were almost universally obtained in lower yield than with CF3SO2F. Not only did the literature methods require more careful temperature control or moisture exclusion, also the chemoselectivity was usually inferior when the phenol starting materials contained indoles (19), aliphatic amines (24 and 28), carboxylic acids (25) or aliphatic alcohols (27). Moreover, amine 28 did not show any trace of sulfonamide formation, even with 2.5 equivalents of gas (see ESI Section 5.2.1†). To sum up, our CF3SO2F gas-based two-chamber system allowed triflation to proceed in a stable, productive and chemoselective fashion.
During the development of this work, it was observed that the aryl triflate synthesis was relatively insensitive towards the choice of solvent or base. To further showcase the versatility of this SuFEx reaction, a series of solvent–base combinations was explored (Scheme 2G). While maintaining the original gas generation using PhNTf2, a set of 7 bases (organic and inorganic) was screened against a set of 6 commonly used reaction solvents. In almost all cases, the reactions had reached >50% conversion after 20 h, and the majority even >80% under unoptimized conditions. While some of the stronger bases were more prone to cause product degradation, nevertheless this broad compatibility enables a subsequent reaction step without intermediate ArOTf isolation.
Given the variety of allowed solvent/base combinations, we wondered whether the triflation method can reach further synthetic utility in a one-pot Suzuki–Miyaura cross-coupling reaction. Based on a literature protocol,29 we found that the (hetero)aryl triflates underwent efficient cross-coupling by transferring the reaction mixture to a vial with the (hetero)aryl boronic acid, palladium(II) acetate and tricyclohexylphosphine (Scheme 3A). With this protocol, biaryls 33–37 were synthesized under mild conditions with good to near-quantitative isolated yield over two steps. The more challenging bipyridine 35 was prepared in a 1,4-dioxane/H2O mixture in 63% yield, which was higher than the 42% yield reported in literature.30 In addition, this Suzuki cross-coupling afforded 2-methyl-5-(3-fluoro phenyl) pyridine 36, the pharmacophore of vorapaxar31 in 80% yield without purifying the intermediate triflate.
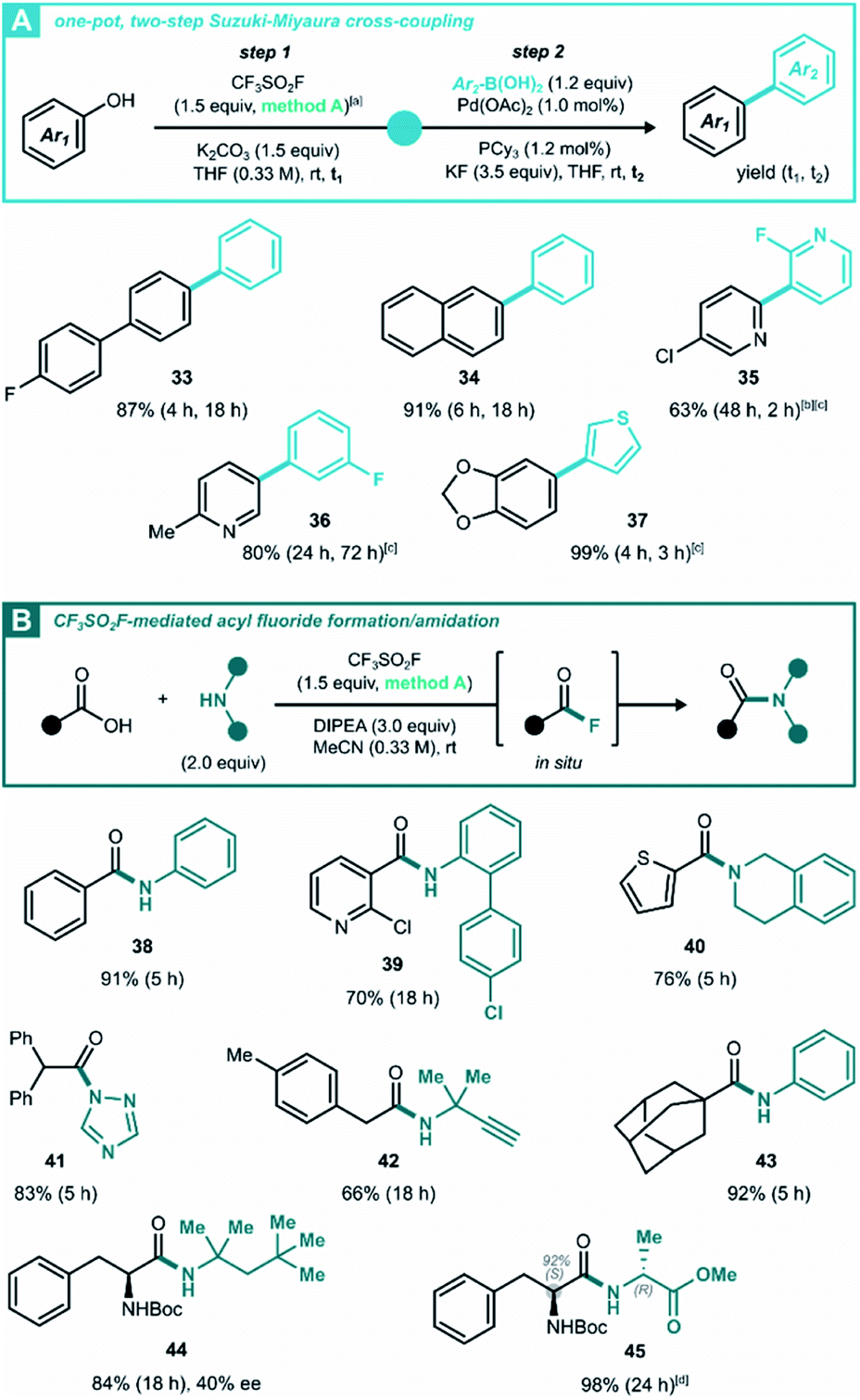 | ||
Scheme 3 One-pot reactions enabled by CF3SO2F gas generation. (A) One-pot, two-step method of aryl triflate generation followed by Suzuki–Miyaura cross-coupling. (B) Amide synthesis with in situ generated acyl fluorides. The yield corresponds in all cases to the isolated yield after column chromatography without isolation of the intermediates; the enantiomeric excess (ee) was determined by HPLC analysis. [a] DMF was used in the generation chamber instead of MeCN for volatility reasons. [b] NaHCO3 was used as the only base (1.5 + 2.2 equiv. in step 1 and 2, resp.), with 1,4-dioxane/H2O 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the solvent, step 2 was heated to 80 °C. [c] Pd(OAc)2 (2.0 mol%) and PCy3 (2.4 mol%) were used. [d] The product was isolated as a 92:8 mixture of diastereoisomers, which was detected by 1H NMR. :1 as the solvent, step 2 was heated to 80 °C. [c] Pd(OAc)2 (2.0 mol%) and PCy3 (2.4 mol%) were used. [d] The product was isolated as a 92:8 mixture of diastereoisomers, which was detected by 1H NMR. | ||
Another class of oxygen nucleophiles that was subjected to CF3SO2F-enabled post-transformations, consists of carboxylic acids. In line with Moses'9c and Qin's9b work on SuFEx-mediated carboxylic acid activation, we aimed to develop a new method based on generating acyl fluoride intermediates via CF3SO2F gas (Scheme 3B). Without isolating the acyl fluorides, they were reacted immediately to build amides with various degrees of steric congestion. Where the biphasic conditions developed in Scheme 2A left carboxylic acids untouched (products 7 and 25), simply shifting to a pure organic solvent led to smooth deoxofluorination. To explore the substrate scope and functional group tolerance of the amidation process, a variety of aromatic and aliphatic carboxylic acids were examined for coupling with different kinds of amines, including anilines, primary and secondary alkylamines and azoles. All coupling reactions proceeded in fair to excellent yields (Scheme 3, 38–44). This work could be extended to peptide formation, and dipeptide 45 was obtained in 98% isolated yield, while retaining 84% diastereomeric ratio. Especially noteworthy is the procedure's tolerance of bulky coupling partners, a known feature of acyl fluorides.32
After investigating the chemistry of CF3SO2F with oxygen nucleophiles, we were curious to see whether SN analogs uphold the same substitution reactions. By replacing a single oxo-group with a substituted nitrogen in the SVI–F hub, trifluoromethanesulfonimidoyl (triflimidoyl) fluorides are obtained. These chiral molecules are characterized by a milder electrophilicity compared to CF3SO2F, due to the increased electron density around the sulfur atom. Since the first description of triflimidoyl fluorides in 2002,33 the recent report by Oehlrich and co-workers is the only example of triflimidoyl fluorides reacting with phenols to form trifluoromethanesulfonimidate (triflimidate) esters.34 Given that only two examples were made under strongly basic conditions, we surmised that an improved synthesis under mild SuFEx conditions should be possible.3b,35 We synthesized three different triflimidoyl fluoride compounds containing N-aryl or N-alkyl substituents (for preparation, see ESI Section 4.5†). These electrophiles were engaged in SuFEx reactions with various phenols to generate a small library of triflimidate esters. The N-aryl substituted triflimidoyl fluorides reacted efficiently under mild conditions to afford the corresponding products in moderate to excellent yields (Scheme 4, 46–51). The N-alkyl counterparts, which are less electrophilic,3b,36 required DBU as a stronger base and an elevated reaction temperature of 50 °C. Naturally occurring phenols such as vanillin (47), eugenol (50), L-tyrosine methyl ester (53), raspberry ketone (54), as well as sterically hindered 2-bromophenol (51) and thiophen-2-ol (48) were all well tolerated (Scheme 4).
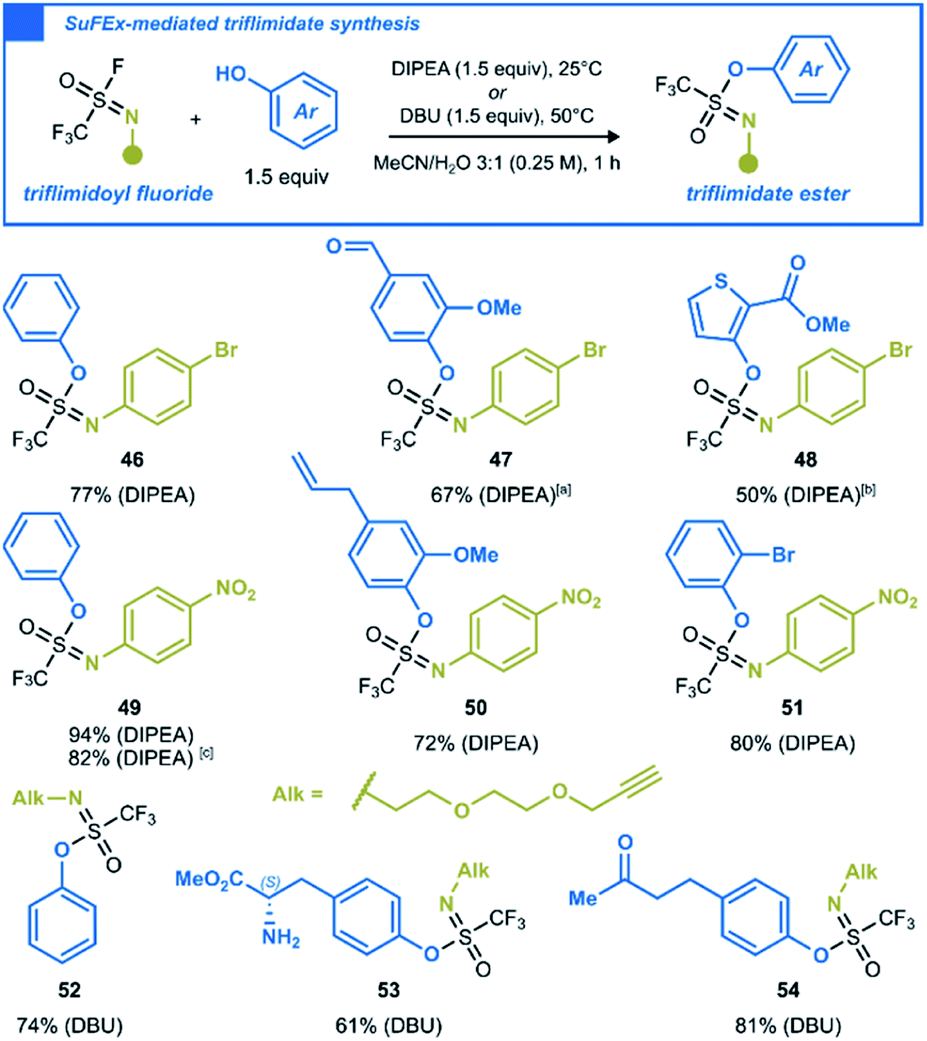 | ||
| Scheme 4 SuFEx-enabled synthesis of aryl triflimidate esters from triflimidoyl fluorides. Reactions were carried out at a 0.2 mmol scale. [a] 1.1 equiv. of ArOH was used. [b] 1.2 equiv. of ArOH used, reaction time was 3 h. [c] Reaction was carried out at a 2.0 mmol scale. | ||
After the transformation of various oxygen nucleophiles into reactive handles with CF3SO2F, we also wanted to investigate nitrogen nucleophiles. To this end, a range of aliphatic amines, anilines and azoles was engaged in a triflylating reaction to form the trifluoromethanesulfonamides (triflamides) (Scheme 5). Based on a literature SuFEx reaction between SO2F2 and secondary amines,2a we selected DMAP as a stoichiometric base, although we found later that Et3N furnishes the same products in equal reaction times and yields with the dry MeCN served as the solvent.
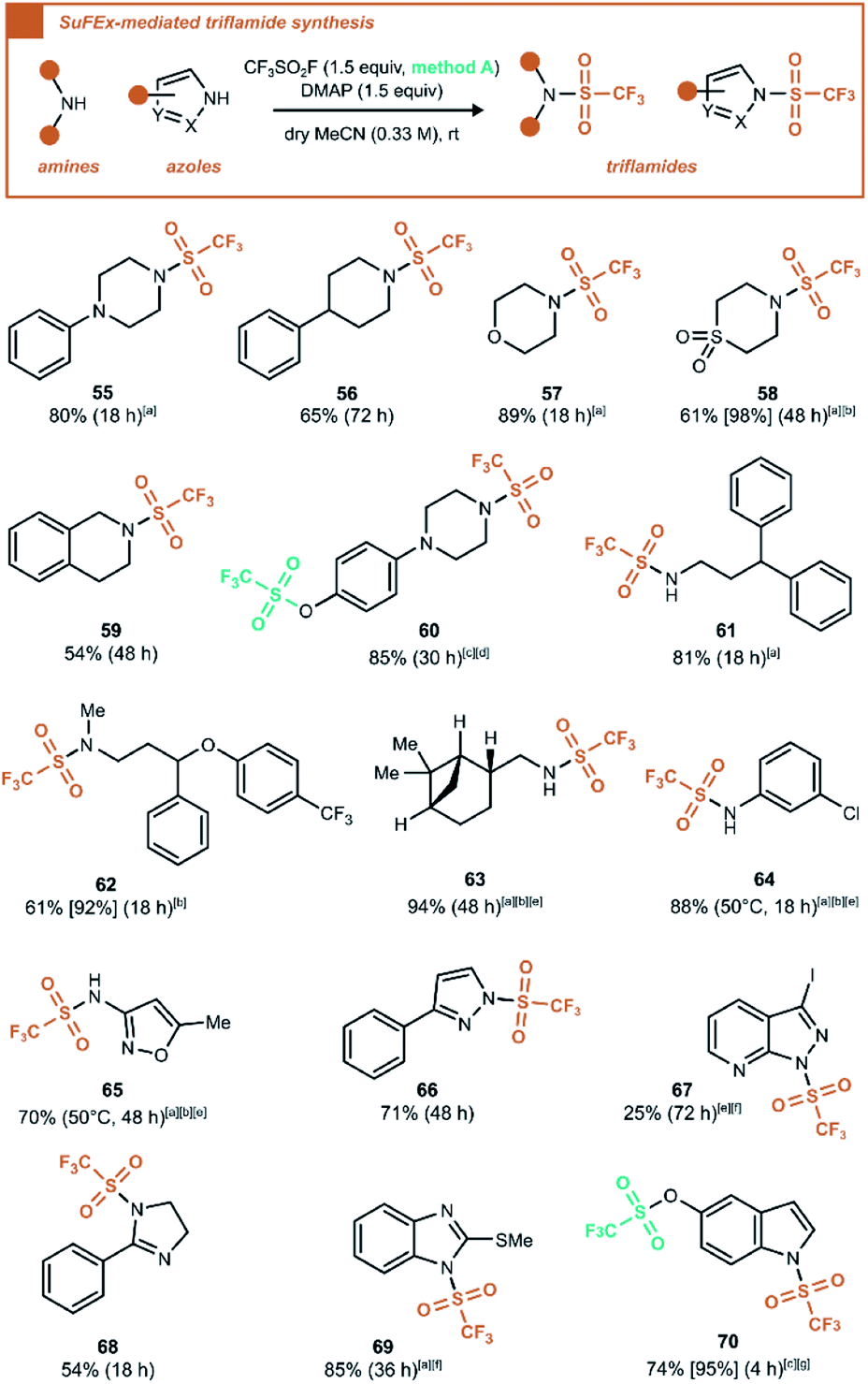 | ||
| Scheme 5 Synthesis of triflamides by reaction of CF3SO2F with amines and azoles. Reaction was carried out on 1.0 mmol scale and reported yields are after column chromatography unless noted otherwise. Between brackets is given the 19F NMR yield using PhCF3 as internal standard, between parentheses the reaction time. [a] Isolated yield of pure material after aqueous work-up. [b] 3.0 equiv. of base was used. [c] 2.5 equiv. of CF3SO2F gas was used. [d] 3.5 equiv. of base was used. [e] 2.0 equiv. of CF3SO2F was generated. [f] K2CO3 was used as the base. [g] 2.5 equiv. of base was used. | ||
Under these conditions, secondary amines (55–60, 62) reacted efficiently to form the tertiary sulfonamides. Also, primary amines (61, 63–65) were suitable reaction partners to form N-monosubstituted triflamides, an interesting contrast with monosubstituted sulfamoyl fluorides, which cannot be formed under basic conditions.2a Finally, except for a few unsuccessful substrates (see ESI Section 7.7†), various N-triflyl heterocycles were prepared in the same manner in fair to good yields (66–70). It is worth noting that the N,O-bis(trifluoromethanesulfonyl) compound 60 was formed in high yield using 2.5 equivalents of the generated gas. This stands in contrast to the reaction leading to 28, where no trace of N-triflyl product was observed. The same discrepancy was observed for 70vs.19. It was also verified that N-triflyl compounds 60 and 70 were not hydrolysed by water (see ESI Section 5.3 and 5.4†). Since the only difference between these reaction conditions is the presence or absence of water, it seems that water influences the mechanism in such a way that it plays a decisive role in the reaction outcome. Ultimately, a direct reactivity comparison of phenol and amine groups in compound 60 was evaluated using only 1.0 equiv. of CF3SO2F gas. Regardless of choice of base, the product mixtures resulting from trifluoromethanesulfonation in anhydrous MeCN invariably lacked N–SO2CF3 monotriflylated product, indicating highest reactivity for the phenol group (see ESI Section 5.2.2†).
Having established a robust procedure for installing a triflyl group through our CF3SO2F SuFEx hub, we turned towards the mechanism of this reaction. More specifically, we investigated the base-mediated triflylation of secondary amines, aiming to elucidate the reaction pathway and the specific role of the base. As a result, we hope to shed light on the observed chemoselectivity, by comparing our simulations for secondary amines with the better-studied mechanism of phenol SuFEx reactions.35,37 To achieve this goal, we use ab initio metadynamics (AIMtD) to retrieve the mechanism as well as quantify the associated activation barriers.38 In contrast to static DFT computations, AIMtD usually includes all molecules in the simulation box, meaning explicit interactions between reactants and additives or solvents are accurately modelled, with the trade-off of a significant increase in computational workload (for theoretical background, see ESI Section 8.1†). We, among others, have previously shown the ability of AIMtD to elucidate reaction mechanisms, quantify reaction barriers and unveil solvation effects.39 Here, piperidine served as a case study for the computationally modelled CF3SO2F triflylation reaction (Fig. 1A). In parallel, a series of experimental studies was performed, to complement the in silico findings (Fig. 1B).40 Initially, three different systems were considered. A single CF3SO2F and one piperidine molecule were placed in the simulation box together with explicit acetonitrile (I), or with DMAP (II) or Et3N (III) included as a base (Fig. 1A). All simulations in this study followed the Born–Oppenheimer molecular dynamics scheme at the DFT level of theory, with the GGA PBE functional and DZVP-MOLOPT-GTH plane wave basis set.41 Additionally, the description of long-range dispersion interactions was improved by Grimme's D3 dispersion correction.42 The CP2K code (version 6.1) was used together with the Quickstep implementation (for full computational details see ESI Section 8.1†).43
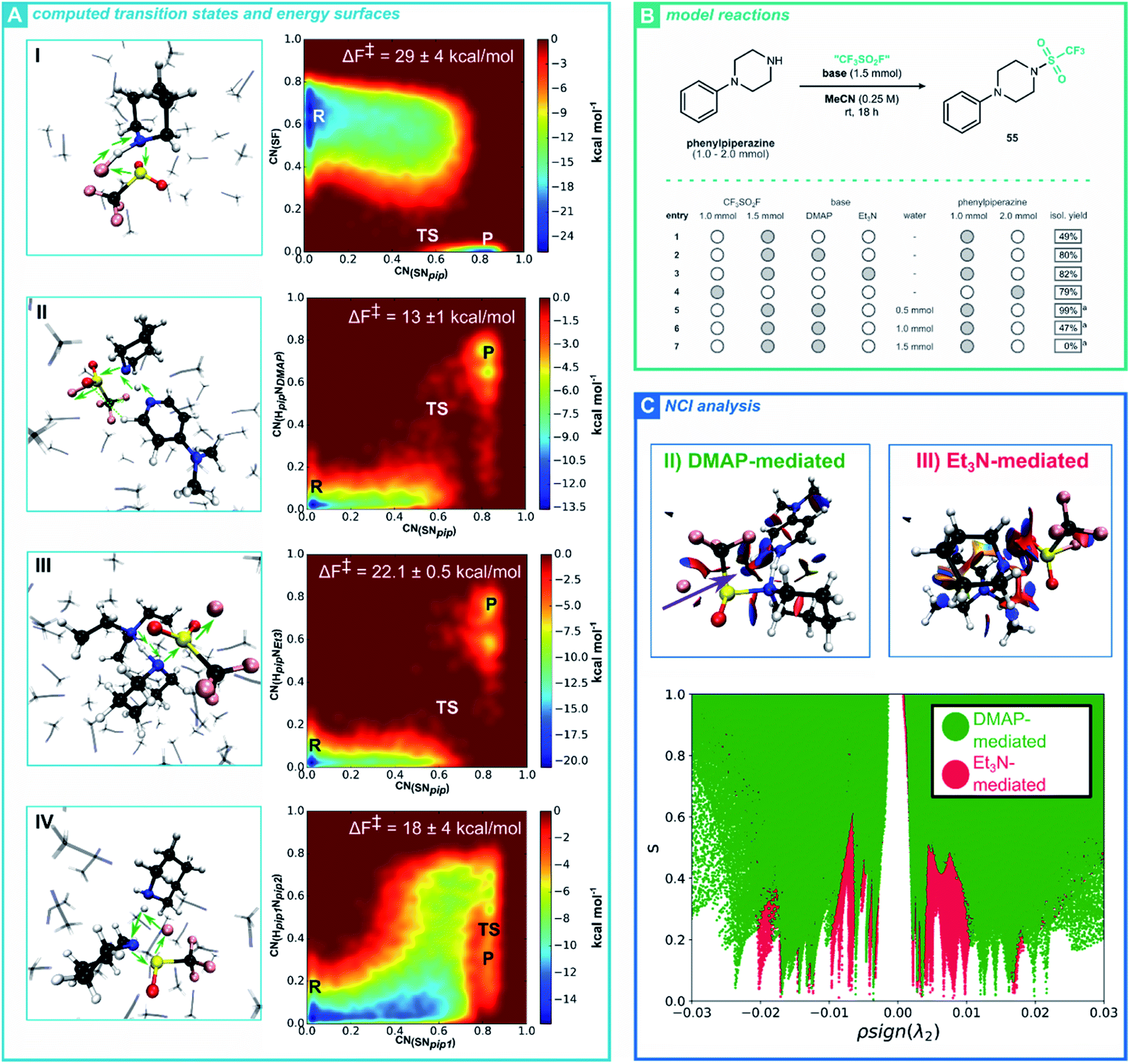 | ||
| Fig. 1 (A) Transition states obtained through metadynamics simulations for: (I) the non-activated CF3SO2F-triflylation of piperidine in acetonitrile, (II) the DMAP-activated CF3SO2F-triflylation of piperidine in acetonitrile, (III) the Et3N-activated CF3SO2F-triflylation of piperidine in acetonitrile and (IV) the non-activated CF3SO2F-triflylation of piperidine in acetonitrile including two molecules of piperidine. In all cases, electron displacement is schematically illustrated using green arrows. During the simulations, Gaussian shaped potentials were placed along two coordination numbers, resulting in a free energy surface and Helmholtz free energy of activation (ΔF‡). Simulations were performed in triplicate. (B) Triflylation of phenylpiperazine as model reaction varying the base, solvent and relative amounts of substrate and CF3SO2F. Isolated yields are provided unless stated otherwise. [a] 19F NMR yield relative to int. std. after 72 h reaction time. (C) NCI analyses were performed on the transition states of the DMAP-mediated CF3SO2F triflylation (II, green) and Et3N-mediated CF3SO2F triflylation (III, red). Analyses were performed in absence of the solvent to focus on the noncovalent interactions present in and between the reactive species. Top; 3D NCI isosurfaces (s = 0.5) visualized for both reactive systems. An RGB-scale is used to differentiate between repulsive (red) and attractive (green) interactions, set from −0.005 a.u. to 0.005 a.u. For the DMAP-mediated triflylation, a non-classical CH⋯O hydrogen bond is observed as an attractive blue surface, which connects DMAP with CF3SO2F (purple arrow). Bottom; an overlay plot of s against ρsign(λ2) is presented for both NCI analyses. | ||
From analysing the trajectory obtained for the non-activated CF3SO2F-triflylation of piperidine (I), a concerted bimolecular reaction mechanism was observed, akin to an SN2-type pathway (see ESI Movie†). Indeed, bond length analysis shows a simultaneous S–F bond breaking and S–Npip bond formation (see ESI Section 8.1†) and the free energy surface displays a reactant and product phase, without an additional intermediate basin (Fig. 1A, I). Notably, without a base, the piperidine nucleophile attacks the sulfur-center from the frontside, which for most SN2 reactions would be less favourable compared to the corresponding backside pathway.44 Herein, frontside attack allows F− to directly scavenge the amine hydrogen of piperidine.
While this mechanism coincides with the findings of Luy and Tonner, the AIMtD simulations result in a Helmholtz free energy of activation (ΔF‡) of 29 ± 4 kcal mol−1, which exceeds a barrier that can readily be crossed at ambient conditions.37 As the non-activated triflylation of 55 yielded 49% of product at room temperature after 18 hours (Fig. 1B, entry 1), the obtained high activation barrier raises questions on the validity of this mechanism. When adding a base such as DMAP (A, II) or Et3N (A, III) to the simulation box, a significantly reduced ΔF‡ is observed (13 ± 1 kcal mol−1 and 22.1 ± 0.05 kcal mol−1, respectively, Fig. 1A). These activation barriers are reasonable, given the high experimental yields obtained for the base-mediated triflylation of 55 (entries 2–3). Mechanistically, the reaction occurs concertedly when DMAP or Et3N are used, similar to the non-activated CF3SO2F-triflylation of piperidine (see ESI Section 8.1 and Movie†). Moreover, the trajectory indicates that the base forms a Lewis adduct with piperidine through a hydrogen bond, enhancing the nucleophilicity of Npip. Collectively, these observations indicate that the transition state has a termolecular nature, meaning the reaction follows an SN3-type pathway. While initially these findings might seem surprising, such SN3 pathways have previously been proposed as mechanisms for substitution reactions on sulfonyl substrates.45 Moreover, when the reaction is activated by DMAP or Et3N, backside attack of the nucleophile is preferred.
Another intriguing observation was the difference between ΔF‡ of the DMAP and Et3N activated triflylation. One would expect that a stronger base would activate the nucleophile more efficiently and thus further decrease the activation barrier. Nevertheless, our AIMtD simulations resulted in a value for ΔF‡ of 13 ± 1 kcal mol−1 and 21.9 ± 0.5 kcal mol−1 for DMAP and Et3N, respectively. In other words, the activating role of Et3N is significantly less effective compared to DMAP, notwithstanding Et3N is the stronger base. To further study the differences between the DMAP-mediated and Et3N-mediated triflylation of piperidine, NCI analyses were performed on their transition states (for theoretical background, see ESI Section 8.2†).46 Remarkably, the 3D NCI isosurface of the DMAP-mediated transition state and bond length analysis reveals an attractive non-classical CH⋯O hydrogen bond connecting DMAP with CF3SO2F (Fig. 1C, purple arrow and ESI Section 8.1†). The synergy between this CH⋯O hydrogen bond and Lewis adduct formation between DMAP and piperidine favourably align both reactants in the transition state. Furthermore, the isosurface of the Et3N-mediated transition state is characterized by larger repulsive (red) surfaces compared to the DMAP-mediated transition state, especially between Et3N and CF3SO2F. From the number of peaks present in the plot of s against ρsign(λ2), it can also be inferred that the Et3N-mediated transition state contains considerably more noncovalent interactions (Fig. 1C). Based on these results, we believe that the activating role of the base in the CF3SO2F-triflylation of piperidine transcends beyond deprotonation of the amine. Clearly, intricate non-covalent interactions such as hydrogen bonding or steric repulsion due to the bulkiness of all reactants involved play an important role in the stability of the termolecular transition state.
After establishing plausible reaction pathways for the activated triflylation of piperidine, we reconsidered the mechanism for the non-activated reaction (A, I). We reasoned that, besides acting as the nucleophile, a second equivalent of piperidine could activate the reaction, similar to an added base. Such a mechanistic picture would also coincide with the non-activating triflylation of 55 yielding 49% of product (Fig. 1B entry 1). Indeed, a maximum of 50% would be expected when the substrate acts as its own base. To our delight, we obtained an energetically more reasonable mechanism for the non-activated triflylation of piperidine when a second piperidine molecule was added to the simulation box, resulting in a ΔF‡ of 18 ± 4 kcal mol−1 (A, IV). In this mechanism, a second equivalent of piperidine forms a Lewis adduct with the piperidine nucleophile and a termolecular transition state is observed. A notable difference with the activated pathways (A, II and III), is that herein substitution preferably proceeds through frontside attack of the nucleophile. To further strengthen our hypothesis, the relative amount of phenylpiperazine with respect to CF3SO2F was increased (2:1 ratio). As expected, the experimental yield of the reaction increased to 79% (entry 4), suggesting that indeed a second equivalent of piperidine plays an active part in the reaction. Intriguingly, when the water content is gradually increased, as little as 1.5 equivalent shuts down the reaction completely (entries 5–7).
Based on these mechanistic insights, we propose an explanation for the observed chemoselectivity when comparing the triflylation of amines and phenols. When performing the reaction in MeCN:H2O (3:1), phenols are selectively triflylated, while amines remain unaffected (compounds 19 and 28). On the other hand, in dry MeCN (0.33 M), both phenols and amines are converted (compounds 60 and 70). We believe that the influence of H2O on chemoselectivity can be explained through the difference in mechanism. A trialkylamine (pKaH ∼11) will partially deprotonate the phenol (pKaH ∼10) towards the phenolate, which is likely to undergo triflylation via a bimolecular SN2 type mechanism, as shown by Zuilhof and co-workers.35 In contrast, our simulations showed that under the same conditions, amines would undergo an SN3 type mechanism, in which a hydrogen bond driven Lewis adduct between the nucleophile and the base is formed (Scheme 6). We assume H2O to disrupt these essential hydrogen bonds, explaining why the reaction in MeCN:H2O is selective towards phenols, while in dry MeCN both phenols and amines showcase a high reactivity towards triflylation.
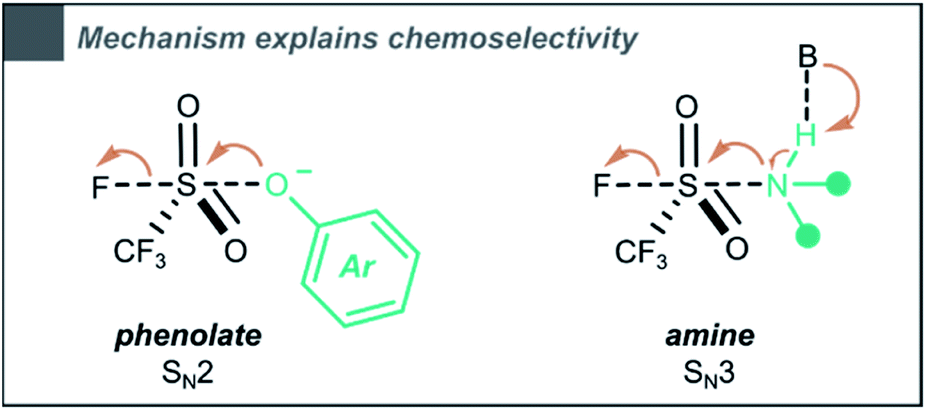 | ||
| Scheme 6 The CF3SO2F triflylation of phenols (phenolate as reactive species) and amines occurs through different pathways. | ||
Conclusions
To summarize, we designed a two-chamber procedure for the safe and efficient ex situ handling of triflyl fluoride gas (CF3SO2F) as a new type of SuFEx connector. Herewith, a diverse library of triflates and triflamides was built straightforwardly, often without the need for further purification. Comparing with literature triflation methods, CF3SO2F consistently furnished higher yields and selectivities. A particularly interesting finding was the lack of reactivity of carboxylic acids and amines in the presence of water, allowing a completely chemoselective triflation of phenolic nucleophiles. In a more in-depth study of this phenomenon, ab initio metadynamics (AIMtD) simulations offered insight into the reactivity of the CF3SO2F triflylation with secondary amine nucleophiles. In contrast to phenolates reacting in a bimolecular fashion, the simulations for amines suggested a formal SN3 mechanism with a termolecular transition state that relies on hydrogen bond formation between base and nucleophile. Due to the absence of such H-bonds in aqueous media, we believe this mechanism explains the observed difference in reaction outcome. The formation of aryl triflates proved amenable to peptide functionalization and reaction telescoping into one-pot Suzuki–Miyaura cross-coupling. In addition, the sulfonylation chemistry developed for triflyl fluoride CF3SO2F was found to be fully translatable to triflimidoyl fluorides CF3SO(NR)F. These aza-analogous SuFEx hubs provided an efficient route to aryl triflimidate esters, a barely reported class of compounds with three-dimensional, potentially chiral character and unknown biological properties. Overall, we believe that the ex situ gas generation method will lead to increased use of CF3SO2F in chemoselective, lab-scale synthesis of valuable aryl triflates and triflamides. Also, process chemistry may benefit from the clean reaction profiles demonstrated here, when using gaseous CF3SO2F directly as a low-MW progenitor to current standard Tf2O. Ultimately, we believe the insights derived from high-quality ab initio calculations form the next step in understanding the fundamental interactions during SVI–F chemistry, and provide a better-informed basis for future applications.Data availability
All experimental data, procedures for data analysis and pertinent data sets are provided in the ESI.†Author contributions
J. D., W. M. D. B., S. H. L. V., and B.-Y. L conceived the formulation and evolution of overarching research goals and aims. B.-Y. L, L. V, F. H and J. D. performed the experiments. R. V. L and M. A performed computational calculations. J. D., B.-Y. L and R. V. L wrote the original draft. All the authors discussed the results and contributed to edit the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Bart Van Huffel and Luc Baudemprez for the assistance with NMR measurements and to Bart Van Huffel for the elemental analysis. We kindly acknowledge Marcus Frings for HPLC analyses. B.-Y. L thanks the CSC (Chinese Scholarship Council) for her fellowship received. J. D., L. V, R. V. L, M. A and W. M. D. B. thank FWO Vlaanderen (Research Foundation – Flanders) for fellowships and grants received (12ZL820N, 1SA1121N, 1185221N, 12F4416N, G0D6221N). M. A. thanks Vrije Universiteit Brussel (VUB) for financial support. W. M. D. B. thanks KU Leuven for financial support via Project DOA/2020/013. S. H. L. V. acknowledges financial support from the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin-inkl. Wissenschaft und Forschung, and the Bundesministerium für Bildung und Forschung. Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225–7). Tier2 computational resources and services were provided by the Shared ICT Services Centre funded by the VUB, the Flemish Supercomputer Center (VSC) and FWO. Tier1 computational resources and services that were used in this work were provided by the VSC, funded by the FWO and the Flemish Government-department EWI.Notes and references
- (a) J. C. Brendel, L. Martin, J. Zhang and S. Perrier, Polym. Chem., 2017, 8, 7475–7485 RSC; (b) H. Wang, F. Zhou, G. Ren, Q. Zheng, H. Chen, B. Gao, L. Klivansky, Y. Liu, B. Wu, Q. Xu, J. Lu, K. B. Sharpless and P. Wu, Angew. Chem., Int. Ed., 2017, 56, 11203–11208 CrossRef CAS PubMed; (c) T. Hmissa, X. Zhang, N. R. Dhumal, G. J. McManus, X. Zhou, H. B. Nulwala and A. Mirjafari, Angew. Chem., Int. Ed., 2018, 57, 16005–16009 CrossRef CAS PubMed; (d) S. Park, H. Song, N. Ko, C. Kim, K. Kim and E. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 33785–33789 CrossRef CAS PubMed; (e) B. Yang, H. Wu, P. D. Schnier, Y. Liu, J. Liu, N. Wang, W. F. DeGrado and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11162–11167 CrossRef CAS PubMed; (f) G. Laudadio, A. d. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed; (g) W. Liu, S. Zhang, S. Liu, Z. Wu and H. Chen, Macromol. Rapid Commun., 2019, 40, 1900310 CrossRef CAS PubMed.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed; (b) C. Veryser, J. Demaerel, V. Bieliūnas, P. Gilles and W. M. De Borggraeve, Org. Lett., 2017, 19, 5244–5247 CrossRef CAS PubMed; (c) J. D. Randall, D. J. Eyckens, F. Stojcevski, P. S. Francis, E. H. Doeven, A. J. Barlow, A. S. Barrow, C. L. Arnold, J. E. Moses and L. C. Henderson, ChemPhysChem, 2018, 19, 3176–3181 CrossRef CAS PubMed; (d) J. Kwon and B. M. Kim, Org. Lett., 2018, 21, 428–433 CrossRef PubMed; (e) C. Lee, N. D. Ball and G. Sammis, Chem. Commun., 2019, 55, 14753–14756 RSC; (f) Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 201909972 Search PubMed.
- (a) S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 2903–2908 CrossRef CAS PubMed; (b) B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS PubMed.
- (a) Q. Chen, P. Mayer and H. Mayr, Angew. Chem., Int. Ed., 2016, 55, 12664–12667 CrossRef CAS PubMed; (b) Q. Zheng, J. Dong and K. B. Sharpless, J. Org. Chem., 2016, 81, 11360–11362 CrossRef CAS PubMed; (c) H. L. Qin, Q. Zheng, G. A. Bare, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2016, 55, 14155–14158 CrossRef CAS PubMed; (d) J. Leng and H.-L. Qin, Chem. Commun., 2018, 54, 4477–4480 RSC; (e) J. Chen, B. Q. Huang, Z. Q. Wang, X. J. Zhang and M. Yan, Org. Lett., 2019, 21, 9742–9746 CrossRef CAS PubMed; (f) Y.-P. Meng, S.-M. Wang, W.-Y. Fang, Z.-Z. Xie, J. Leng, H. Alsulami and H.-L. Qin, Synthesis, 2020, 52, 673–687 CrossRef CAS.
- (a) C. J. Smedley, M.-C. Giel, A. Molino, A. S. Barrow, D. J. D. Wilson and J. E. Moses, Chem. Commun., 2018, 54, 6020–6023 RSC; (b) B. Moku, W.-Y. Fang, J. Leng, L. Li, G.-F. Zha, K. P. Rakesh and H.-L. Qin, iScience, 2019, 21, 695–705 CrossRef CAS PubMed; (c) H. Zhou, P. Mukherjee, R. Liu, E. Evrard, D. Wang, J. M. Humphrey, T. W. Butler, L. R. Hoth, J. B. Sperry, S. K. Sakata, C. J. Helal and C. W. am Ende, Org. Lett., 2018, 20, 812–815 CrossRef CAS PubMed; (d) M. F. Khumalo, E. D. Akpan, P. K. Chinthakindi, E. M. Brasil, K. K. Rajbongshi, M. M. Makatini, T. Govender, H. G. Kruger, T. Naicker and P. I. Arvidsson, RSC Adv., 2018, 8, 37503–37507 RSC.
- (a) T. A. Fattah, A. Saeed and F. Albericio, J. Fluorine Chem., 2018, 213, 87–112 CrossRef; (b) A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- (a) H. Vorbrüggen, Synthesis, 2008, 1165–1174, DOI:10.1055/s-2008-1067006; (b) M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS PubMed; (c) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed.
- (a) M. Epifanov, J. Y. Mo, R. Dubois, H. Yu and G. M. Sammis, J. Org. Chem., 2021, 86, 3768–3777 CrossRef CAS PubMed; (b) J. Y. Mo, M. Epifanov, J. W. Hodgson, R. Dubois and G. M. Sammis, Chem. –Eur. J., 2020, 26, 4958–4962 CrossRef CAS PubMed; (c) P. J. Foth, F. Gu, T. G. Bolduc, S. S. Kanani and G. Sammis, Chem. Sci., 2019, 10, 10331–10335 RSC; (d) M. Epifanov, P. J. Foth, F. Gu, C. Barrillon, S. S. Kanani, C. S. Higman, J. E. Hein and G. M. Sammis, J. Am. Chem. Soc., 2018, 140, 16464–16468 CrossRef CAS PubMed; (e) C. Zhao, G.-F. Zha, W.-Y. Fang, N. S. Alharbi and H.-L. Qin, Tetrahedron, 2019, 75, 4648–4656 CrossRef CAS.
- (a) P. J. Foth, T. C. Malig, H. Yu, T. G. Bolduc, J. E. Hein and G. M. Sammis, Org. Lett., 2020, 22, 6682–6686 CrossRef CAS PubMed; (b) S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087–4101 RSC; (c) C. J. Smedley, A. S. Barrow, C. Spiteri, M.-C. Giel, P. Sharma and J. E. Moses, Chem. –Eur. J., 2017, 23, 9990–9995 CrossRef CAS PubMed.
- V. Gembus, F. Marsais and V. Levacher, Synlett, 2008, 1463–1466, DOI:10.1055/s-2008-1078407.
- A. Ishii, T. Ishimaru, T. Yamazaki and M. Yasumoto, Process for Producing Fluorosulfuric Acid Aromatic-ring Esters, US Pat., 9040745B2, 2015 Search PubMed.
- A concise literature discussion can be found in ESI Section 15.†.
- Examples using triflic anhydride as a [Tf] source: A. G. Martínez, L. R. Subramanian, M. Hanack, S. J. Williams and S. Régnier, Journal, 2016, 1–17, DOI:10.1002/047084289X.rt247.pub3.
- Examples using triflyl imidazole as a [Tf] source: F. Effenberger and K. E. Mack, Tetrahedron Lett., 1970, 11, 3947–3948 CrossRef.
- Examples using PhNTf2 as a [Tf] source: (a) J. B. Hendrickson and R. Bergeron, Tetrahedron Lett., 1973, 14, 4607–4610 CrossRef; (b) J. E. Mc Murry and W. J. Scott, Tetrahedron Lett., 1983, 24, 979–982 CrossRef CAS; (c) W. E. Zeller and R. Schwörer, N-Phenyltrifluoromethanesulfonimide, in Encyclopedia of Reagents for Organic Synthesis, 2009, DOI:10.1002/047084289X.rp142.pub2.
- Examples using Comins' reagent as a [Tf] source: D. L. Comins and A. Dehghani, Tetrahedron Lett., 1992, 33, 6299–6302 CrossRef CAS.
- Apart from phenolic starting materials, aryl triflates have also been made using triflic acid (TfOH) in diazonium or benzyne chemistry, or using metal triflate salts in cross coupling chemistry: (a) O. Planas, V. Peciukenas and J. Cornella, J. Am. Chem. Soc., 2020, 142, 11382–11387 CrossRef CAS PubMed; (b) C. Picherit, F. Wagner and D. Uguen, Tetrahedron Lett., 2004, 45, 2579–2583 CrossRef CAS; (c) T. Pages and B. R. Langlois, J. Fluorine Chem., 2001, 107, 321–327 CrossRef CAS; (d) C. Ge, G. Wang, P. Wu and C. Chen, Org. Lett., 2019, 21, 5010–5014 CrossRef CAS PubMed; (e) A. Pialat, B. Liégault and M. Taillefer, Org. Lett., 2013, 15, 1764–1767 CrossRef CAS PubMed.
- T. Gramstad and R. N. Haszeldine, J. Chem. Soc., 1956, 173–180, 10.1039/JR9560000173.
- Y. Wang, Z. Gao, B. Wang, W. Zhou, P. Yu and Y. Luo, Ind. Eng. Chem. Res., 2019, 58, 21913–21920 CrossRef CAS.
- (a) L. Conte, G. Gambaretto, G. Caporiccio, F. Alessandrini and S. Passerini, J. Fluorine Chem., 2004, 125, 243–252 CrossRef CAS; (b) M. Kobayashi, T. Inoguchi, T. Iida, T. Tanioka, H. Kumase and Y. Fukai, J. Fluorine Chem., 2003, 120, 105–110 CrossRef CAS; (c) T. J. Brice and P. W. Trott, Fluorocarbon Sulfonic Acids and Derivatives, US. Pat., US2732398A,. 1956 Search PubMed.
- T. Kume, T. Morinaka, T. Nanmyo and S. Sakai, Process for Preparation of Trifluoromethanesulfonyl Fluoride, European Patent, EP2216325A1, 2010 Search PubMed.
- Y. Morino and Y. Tateishi, Method for Producing Trifluoromethanesulfonyl Fluoride, Japan Patent, 2008285419A, 2008 Search PubMed.
- For applications of CF3SO2F in synthetic chemistry, see: (a) D. M. Barnes, V. S. Chan, T. B. Dunn, T. S. Franczyk, A. R. Haight and S. Shekhar, Process for Preparing Antiviral Compounds, US Pat., 20130224149A1, 2013 Search PubMed; (b) I. Akihisa, U. Koji and Y. Manabu, Method for Producing Acid Fluorides, Japan Patent, 2009155248A, 2009 Search PubMed; (c) N. Masashi and N. Satoshi, Method for Producing Perfluoroalkanesulfonic Acid Ester, JP2007119355A, 2007; (d) I. Akihiro, U. Mikio, K. Yokusu and T. Mitsuru, Process for Producing Binaphthol Bistriflate, US Pat., 6399806B1, 2002 Search PubMed.
- A. Pees, C. Sewing, M. J. W. D. Vosjan, V. Tadino, J. D. M. Herscheid, A. D. Windhorst and D. J. Vugts, Chem. Commun., 2018, 54, 10179–10182 RSC.
- J. Demaerel, C. Veryser and W. M. De Borggraeve, React. Chem. Eng., 2020, 5, 615–631 RSC.
- Y. Zou, L. Qin, X. Ren, Y. Lu, Y. Li and J. Zhou, Chem. –Eur. J., 2013, 19, 3504–3511 CrossRef CAS PubMed.
- D. E. Frantz, D. G. Weaver, J. P. Carey, M. H. Kress and U. H. Dolling, Org. Lett., 2002, 4, 4717–4718 CrossRef CAS PubMed.
- T. Avullala, P. Asgari, Y. Hua, A. Bokka, S. G. Ridlen, K. Yum, H. V. R. Dias and J. Jeon, ACS Catal., 2019, 9, 402–408 CrossRef CAS PubMed.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- A. S. Voisin-Chiret, A. Bouillon, G. Burzicki, M. Célant, R. Legay, H. El-Kashef and S. Rault, Tetrahedron, 2009, 65, 607–612 CrossRef CAS.
- J. Liu, B. Sun, X. Zhao, J. Xing, Y. Gao, W. Chang, J. Ji, H. Zheng, C. Cui, A. Ji and H. Lou, J. Med. Chem., 2017, 60, 7166–7185 CrossRef CAS PubMed.
- H. Wenschuh, M. Beyermann, E. Krause, M. Brudel, R. Winter, M. Schuemann, L. A. Carpino and M. Bienert, J. Org. Chem., 2002, 59, 3275–3280 CrossRef.
- R. Y. Garlyauskayte, A. V. Bezdudny, C. Michot, M. Armand, Y. L. Yagupolskii and L. M. Yagupolskii, J. Chem. Soc. Perkin Trans. 1, 2002, 1887–1889, 10.1039/B205169A.
- M. Wright, C. Martínez-Lamenca, J. E. Leenaerts, P. E. Brennan, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2018, 83, 9510–9516 CrossRef CAS PubMed.
- D. D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman and H. Zuilhof, Angew. Chem., Int. Ed., 2020, 59, 7494–7500 CrossRef CAS PubMed.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- J.-N. Luy and R. Tonner, ACS Omega, 2020, 5, 31432–31439 CrossRef CAS PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- (a) R. Van Lommel, J. Bock, C. G. Daniliuc, U. Hennecke and F. De Proft, Chem. Sci., 2021, 12, 7746–7757 RSC; (b) G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 2, 200–212 CrossRef; (c) K. M. Bal, S. Fukuhara, Y. Shibuta and E. C. Neyts, J. Chem. Phys., 2020, 153 Search PubMed; (d) R. VanLommel, S. L. C. Moors and F. De Proft, Chem. –Eur .J., 2018, 24, 7044–7050 CrossRef CAS PubMed.
- N-Phenylpiperazine was chosen a higher-MW substrate for the mechanistic experiments because N-triflyl piperidine (the product in the simulations) is volatile and less suited for isolated yield determination.
- (a) J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wires Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS; (b) J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comp. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315–1330 CrossRef CAS PubMed.
- (a) M. Iazykov, M. Canle, J. A. Santaballa and L. Rublova, Int. J. Chem. Kinetics, 2015, 47, 744–750 CrossRef CAS; (b) S. Yamabe, G. Zeng, W. Guan and S. Sakaki, J. Comput. Chem., 2014, 35, 1140–1148 CrossRef CAS PubMed; (c) T. W. Bentley, R. O. Jones, D. H. Kang and I. S. Koo, J. Phys. Org. Chem., 2009, 22, 799–806 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06267k |
| This journal is © The Royal Society of Chemistry 2022 |