
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of vicinal 4°/3°-carbons via reductive Cope rearrangement†
Kristin M.
Sobie
,
Matthew
Albritton
,
Yinuo
Yang
,
Mariana M.
Alves
,
Adrian
Roitberg
* and
Alexander J.
Grenning
 *
*
Department of Chemistry, University of Florida, PO Box 117200, Gainesville, FL 32611, USA. E-mail: grenning@ufl.edu; roitberg@ufl.edu
First published on 21st January 2022
Abstract
Herein reported is a strategy for constructing vicinal 4°/3° carbons via reductive Cope rearrangement. Substrates have been designed which exhibit Cope rearrangement kinetic barriers of ∼23 kcal mol−1 with isoenergetic favorability (ΔG ∼ 0). These fluxional/shape-shifting molecules can be driven forward by chemoselective reduction to useful polyfunctionalized building blocks.
Constructing sterically congested vicinal quaternary–tertiary carbons (4°/3° carbons) via Cope rearrangement is currently quite limited with only a handful of papers on the subject published over the past 40 years. This stands in stark contrast to the plethora of other methods for establishing sterically congested vicinal carbons.1–5 Central to the challenge are kinetic and thermodynamic issues associated with the transformation. In the simplest sense, Cope rearrangements proceed in the direction that results in highest alkene substitution (Fig. 1).6,7 To forge 4°/3° motifs by Cope rearrangement, additional driving forces must be introduced to reverse the [3,3] directionality and compensate for the energetic penalty associated with the steric and torsional strain of the targeted vicinal 4°/3° motif. With limited reports in all cases, oxy-Cope substrates (Scheme 1, eqn (1)),8–14 divinylcyclopropanes (Scheme 1, eqn (2)),15–20 and vinylidenecyclopropane-based 1,5-dienes21 (Scheme 1, eqn (3)) have demonstrated favourability for constructing vicinal 4°/3° carbons. Malachowski et al. put forth a series of studies on the construction of quaternary centers via Cope rearrangement driven forward by a conjugation event (Scheme 1, eqn (4)).22–25 In their work, a single example related to the construction of vicinal 4°/3° centers was disclosed, though kinetic (180 °C) and thermodynamic (equilibrium mixtures) challenges are also observed.23 And of particular relevance to this work, Wigfield et al. demonstrated that 3,3-dicyano-1,5-dienes with the potential to generate vicinal 4°/3° carbons instead react via an ionic mechanism yielding the less congested products (Scheme 1, eqn (5)).26
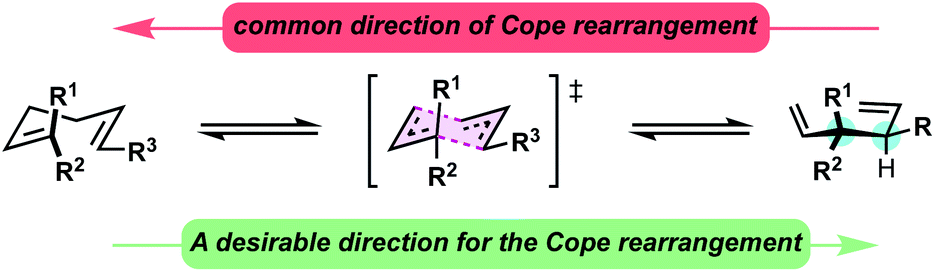 | ||
| Fig. 1 Cope equilibrium of 1,1,6-trisubstituted 1,5-dienes. | ||
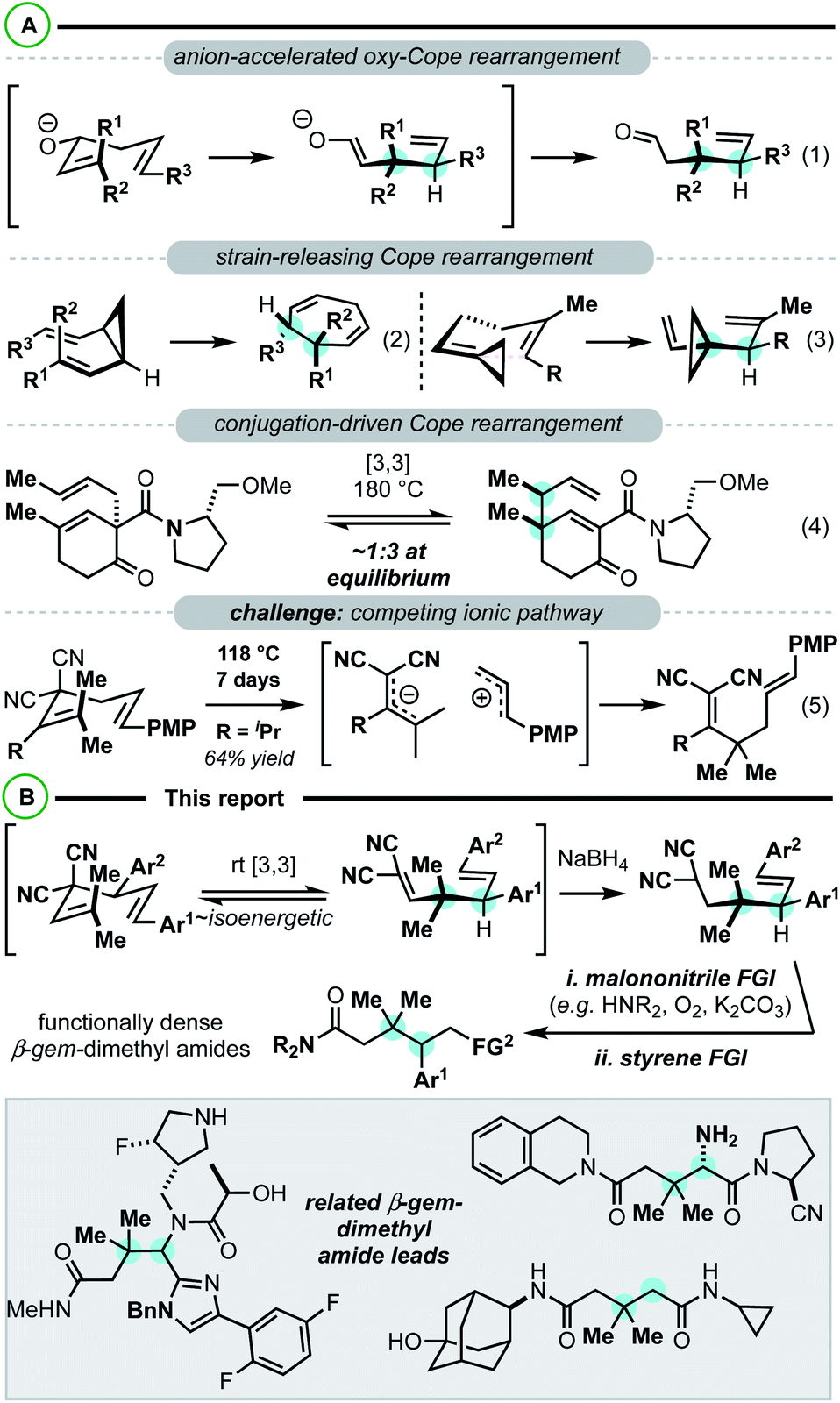 | ||
| Scheme 1 (A) Cope rearrangements for constructing vicinal 4°/3°-centers (B) this report. | ||
Our group has been examining strategies to decrease kinetic barriers and increase the thermodynamic favourability of 3,3-dicyano-1,5-diene-based Cope substrates.27–31 Beyond the simplest, unsubstituted variants, this class of 1,5-diene is not particularly reactive in both a kinetic and thermodynamic sense (e.g.Scheme 1, eqn (5)).26,32 Reactivity issues aside, these substrates are attractive building blocks for two main reasons: (1) they have straightforward accessibility from alkylidenemalononitriles and allylic electrophiles by deconjugative allylic alkylation.33 (2) The 1,5-diene termini are substantially different (malononitrile vs. simple alkene) thus allowing for orthogonal functional group interconversion facilitating target and analogue synthesis.34 Herein we report that a combination of 1,5-diene structural engineering28,31 and reductive conditions (the reductive Cope rearrangement29,30) can result in the synthesis of building blocks containing vicinal gem-dimethyl 4°/3° carbons along with orthogonal malononitrile and styrene functional groups for interconversion (Scheme 1B). On this line, malononitrile can be directly converted to amides34 yielding functionally dense β-gem-dimethylamides, important pharmaceutical scaffolds.35
This project began during the Covid-19 pandemic lockdown (ca. March–May 2020). As such, we were not permitted to use our laboratory out of an abundance of caution. We took this opportunity to first computationally investigate a Cope rearrangement that could result in vicinal 4°/3° carbons (Scheme 2). Then, when permitted to safely return to the lab, we would experimentally validate our findings (vide infra). From our previous work, it is known that by adding either a 4-aromatic group28 or a 4-methyl group31 to a 3,3-dicyano-1,5-diene, low barrier (rt – 80 °C) diastereoselective Cope rearrangements can occur. Notably, the 4-substituent was found to destabilize the starting material (weaken the C3–C4 bond, conformationally bias the substrate for [3,3]), and stabilize the product side of the equilibrium via resonance (phenyl group) or hyperconjugation (methyl group). In this study, we modelled substrates 1, 3, and 5 that have variable 4-substitution and would result in vicinal gem-dimethyl- and phenyl-containing 4°/3° carbons upon Cope rearrangement to 2, 4, or 6, respectively. We chose to target this motif due to likely synthetic accessibility from simple starting materials but also because of the important and profound impact that gem-dimethyl groups impart on pharmaceuticals.35 Substrate 1 lacking 4-substitution had an extremely unfavourable kinetic and thermodynamic profile (ΔG‡ = 31.6; ΔG = +5.3 kcal mol−1). When a 4-methyl group was added, the kinetic barrier (ΔG‡) dropped appreciably to 28.2 kcal mol; however, the thermodynamics were still quite endergonic (ΔG = +4.4 kcal mol−1). Most excitingly, it was uncovered that the 4-phenyl group dramatically impacted the kinetics and thermodynamics: the [3,3] has a barrier of 22.9 kcal mol−1 (ΔG‡) and is ∼isoenergetic (ΔG = +0.17 kcal mol−1). Thus, the reaction appears to be fluxional/shape-shifting at room temperature.36–40 For this substrate, we also modelled the dissociative pathway (Scheme 2D). It was found that bond breakage to two allylic radical intermediates is a higher energy process than the concerted transition state (Scheme 2Cvs.Scheme 2D). Specifically, the dissociative pathway was found to be kinetically less favourable (ΔG‡ ∼ 27.6 kcal mol; ΔG = 26.2 kcal mol−1) than the concerted process (ΔG‡ = 22.9 kcal mol−1). While the dissociative pathway is less favourable than the concerted transformation, we surmised that the two-step process becomes accessible at elevated temperature (vide infra). Finally, the ionic pathway was calculated to be significantly higher for this substrate (see the ESI†).
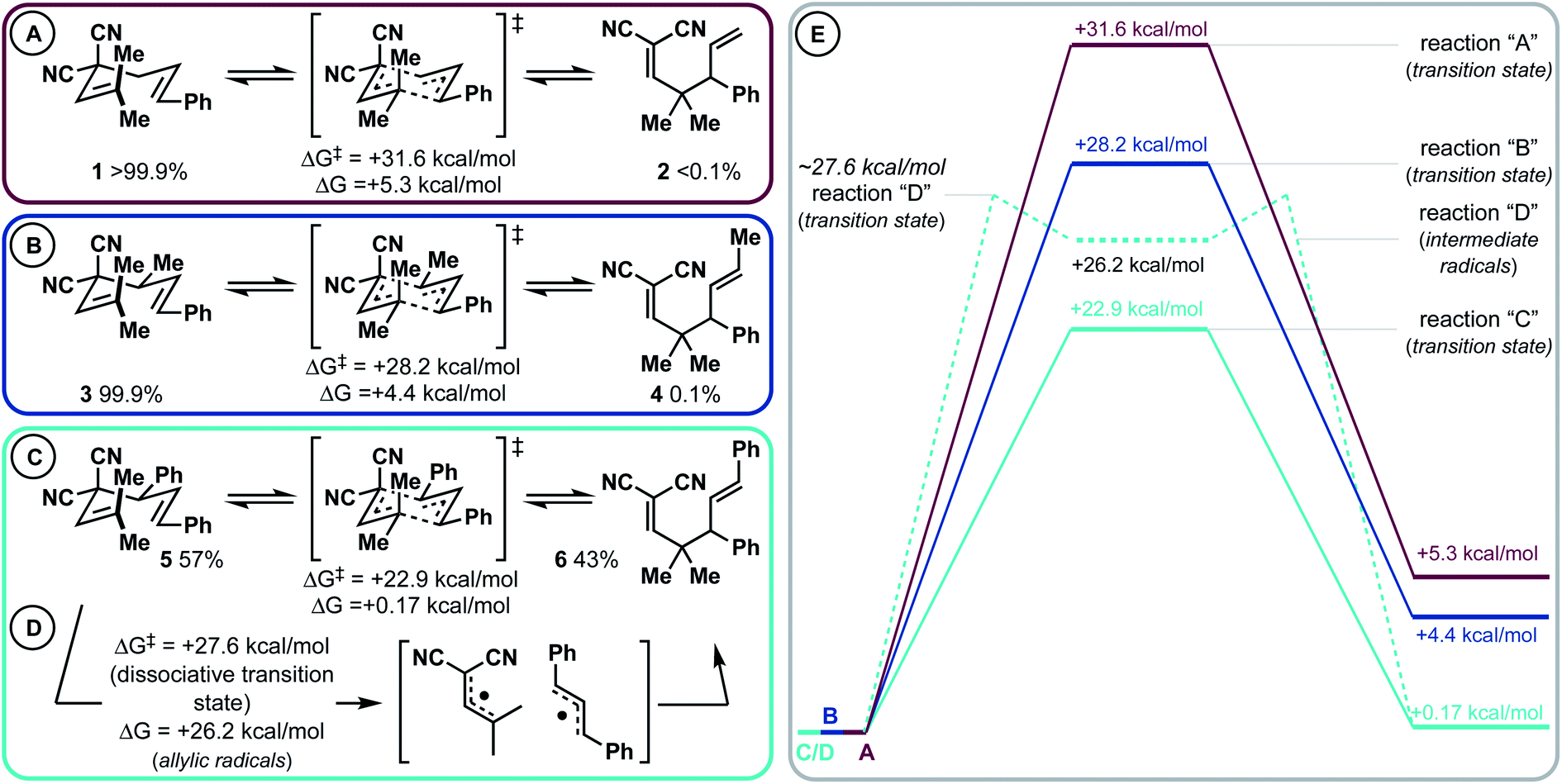 | ||
| Scheme 2 Computational analysis of 3,3-dicyano-1,5-diene that in theory could result in vicinal 4°/3° carbons. (A) 4-Unsubstituted 3,3-dicyano-1,5-diene. (B) 4-Methyl 3,3-dicyano-1,5-diene. (C) 4-Phenyl 3,3-dicyano-1,5-diene. (D) The dissociative mechanism for substrate 5 is higher than the closed transition state. (E) visualization of the kinetic- and thermodynamic differences of transformations (A–D). | ||
The class of substrate uncovered from our computational investigation could be accessed from γ,γ-dimethyl-alkylidenemalononitrile (7a) and 1,3-diarylallyl electrophiles (such as 8a) by Pd-catalyzed deconjugative allylic alkylation (Scheme 3A).33 As such, model 1,5-diene 5a was prepared to verify the computational results. It was found that upon synthesis of 5a, an inseparable 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :79 mixture of 1,5-diene 5a and the 1,5-diene 6a was observed. The predicted ratio of 5a to 6a was 57:43 (Scheme 2C). These two results are within the error of the calculations (predicted; slightly endergonic, observed; slightly exergonic). To determine whether the transformation was progressing through the predicted concerted pathway (Scheme 2C) over the dissociative pathway (Scheme 2D), substrate 5b was prepared by an analogous deconjugative allylic alkylation reaction. Similarly, two Cope equilibrium isomers 5b and 6b are observed at room temperature in a 12:88 ratio. Upon heating at 100 °C for 3 h, the 1,5-dienes “scramble” (e.g. iso-6b is observed; 0.2:1.0:1.5 ratio of 5b:6b:iso-6b) indicating that the dissociative pathway is only accessible at elevated temperature. This is all in good agreement with the calculated kinetics and thermodynamics of this system (Scheme 2).
:79 mixture of 1,5-diene 5a and the 1,5-diene 6a was observed. The predicted ratio of 5a to 6a was 57:43 (Scheme 2C). These two results are within the error of the calculations (predicted; slightly endergonic, observed; slightly exergonic). To determine whether the transformation was progressing through the predicted concerted pathway (Scheme 2C) over the dissociative pathway (Scheme 2D), substrate 5b was prepared by an analogous deconjugative allylic alkylation reaction. Similarly, two Cope equilibrium isomers 5b and 6b are observed at room temperature in a 12:88 ratio. Upon heating at 100 °C for 3 h, the 1,5-dienes “scramble” (e.g. iso-6b is observed; 0.2:1.0:1.5 ratio of 5b:6b:iso-6b) indicating that the dissociative pathway is only accessible at elevated temperature. This is all in good agreement with the calculated kinetics and thermodynamics of this system (Scheme 2).
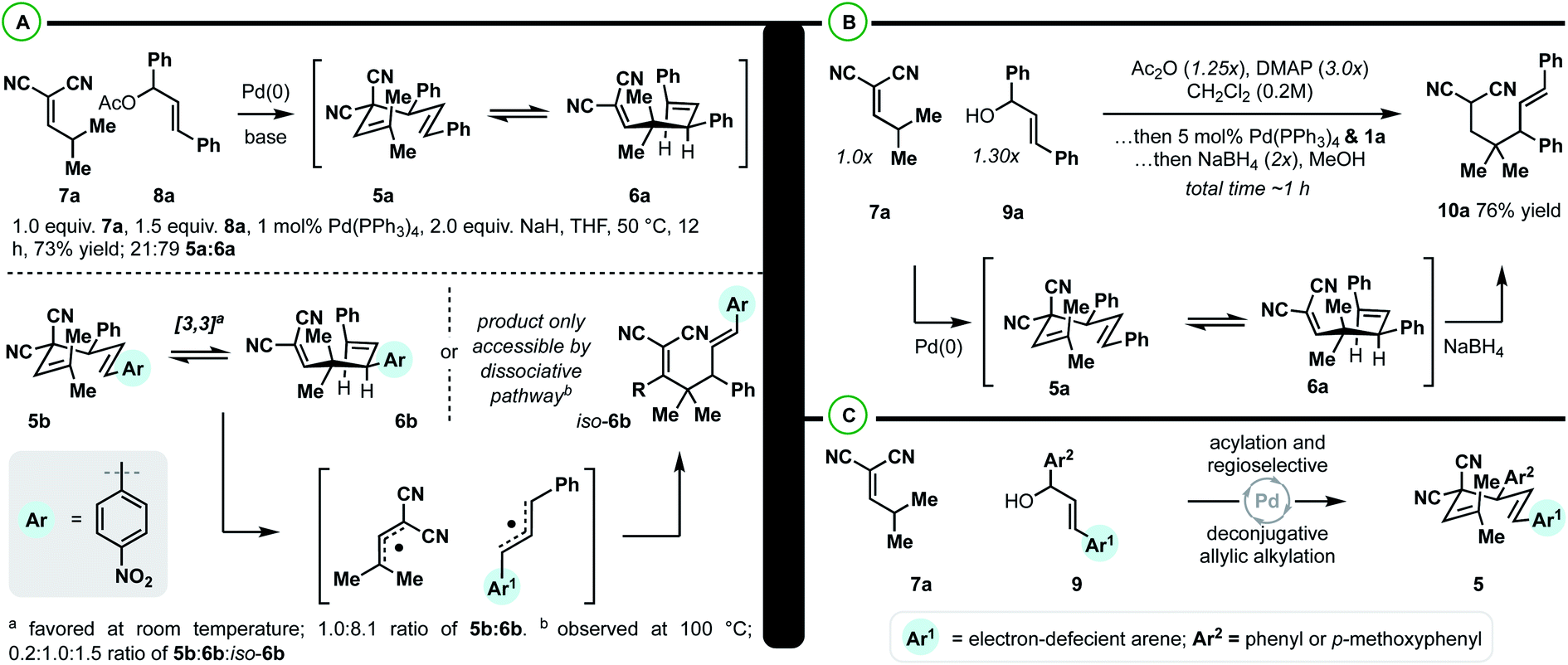 | ||
| Scheme 3 (A) Observation of fluxional [3,3] and confirmation of calculated predictions. (B) Optimization of a reductive Cope rearrangement protocol for constructing vicinal 4°/3° centers. (C) The Pd-catalyzed deconjugative allylic alkylation must be regioselective. | ||
With respect to the synthetic methodology, we aimed to increase the overall efficiency and applicability of the sequence (Scheme 3B). Specifically, we wanted to avoid [3,3] equilibrium mixtures and sensitive/unstable substates and intermediates. It was found that the direct coupling of 7a with diphenylallyl alcohol 9a could take place in the presence of DMAP, Ac2O, and Pd(PPh3)4. When the coupling was complete, methanol and NaBH4 were added to drive the Cope equilibrium forward, yielding the reduced Cope rearrangement product 10a in 76% isolated yield. In terms of practicality and efficiency, this method utilizes diphenylallyl alcohols, which are more stable and synthetically accessible than their respective acetates, and the [3,3] equilibrium mixture can be directly converted dynamically to a single reduced product.
With an efficient protocol in hand for constructing malononitrile–styrene-tethered building blocks featuring central vicinal 4°/3° carbons, we next examined the scope of the transformation (Scheme 4). We chose diarylallyl alcohols with the propensity to react regioselectively via an electronic bias (Scheme 3C).41,42 The combination of p-nitrophenyl and phenyl (10b) or p-methoxyphenyl (10c) yielded regioselective outcomes with the electron-deficient arene at the allylic position. This is consistent with the expected regiochemical outcome where the nucleophile reacts preferentially at the α-position and the electrophile reacts at the allylic position bearing the donor-arene (Scheme 3C).41,42 Then, reductive Cope rearrangement occurs to position the electron-deficient arene adjacent to the gem-dimethyl quaternary center. This is an exciting outcome as many pharmaceutically relevant (hetero)arenes are electron deficient. Thus, fluorinated arenes were installed at the allylic position of products 10d–10k. While the phenyl group resulted in poor regioselectivity (1:1–3:1), the p-methoxyphenyl group enhanced the regiomeric ratios in all cases (3:1–15:1). The degree of selectivity is correlated with the number and position of fluorine atoms. N-Heterocycles could be incorporated with excellent regioselectivity, generally speaking (10l–10q). For example, 3-chloro-4-pyridyl (10l/10m) groups were installed at the allylic position with >20:1 rr. 4-Chloro-3-pyridyl was poorly regioselective (10n), but the combination of 4-trifluomethyl-3-pyridyl/p-methoxyphenyl (10o) gave good regioselectivity of 11:1. 2-Pyridyl/p-methoxyphenyl (10q) was also a regioselective combination. We also examined a few other heterocycles including quinoline (10s) and thiazole (10t and 10u) with excellent and modest regioselectivity observed, respectively. As a general trend, when the arenes on the allylic electrophile become less polarized, poor regioselectivity is observed in the Pd-catalyzed allylic alkylation. For example, the combination of p-chlorophenyl and p-methoxyphenyl (10v) or phenyl (10w) yields regioisomeric mixtures of products. This can be circumvented by utilizing symmetric electrophiles (to 10x).
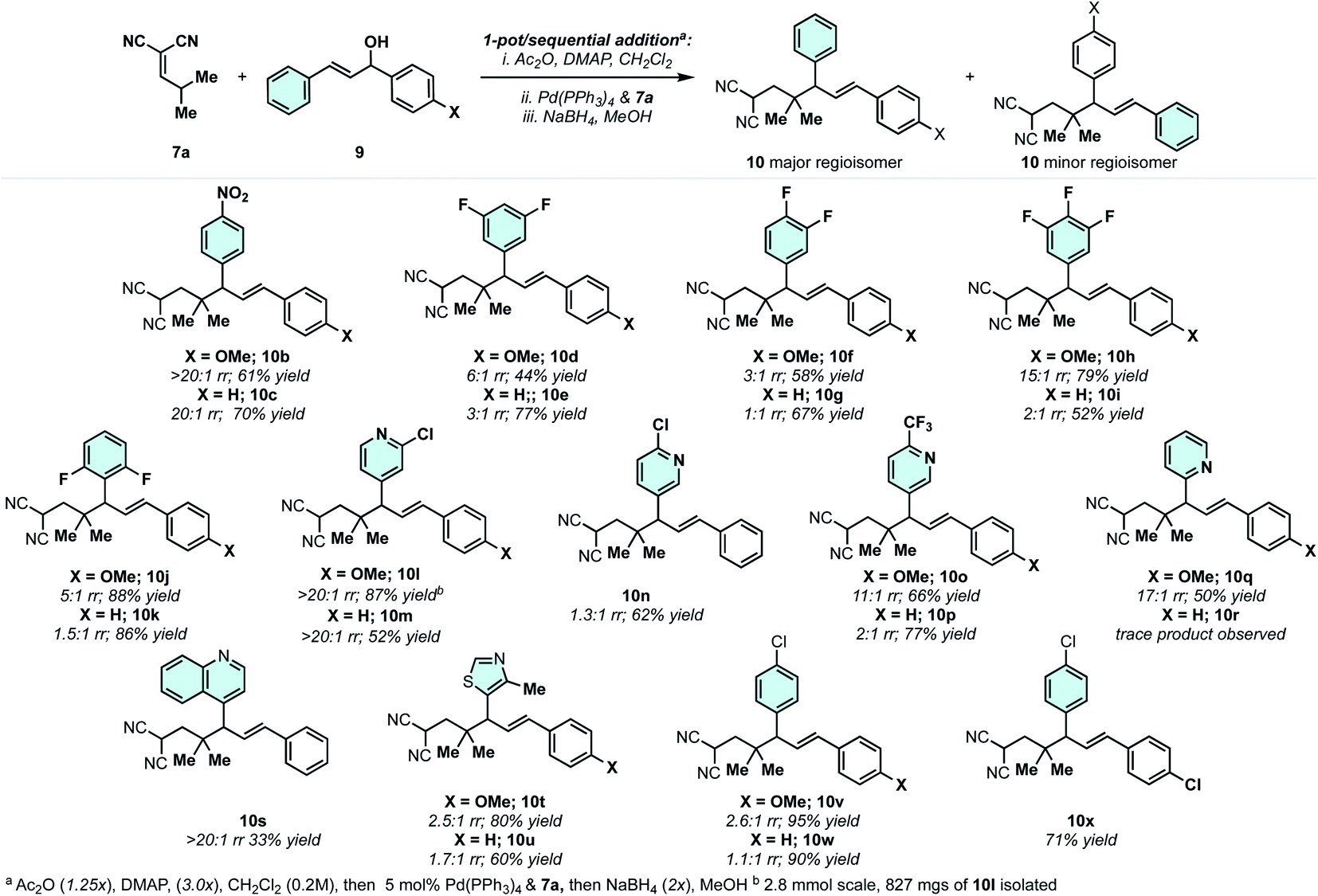 | ||
| Scheme 4 Scope of the 4°/3°-center-generating reductive Cope rearrangement. | ||
The phenyl or the p-methoxyphenyl group is necessary to achieve the 4°/3° carbon-generating Cope rearrangement: it functions as an “activator” by lowering the kinetic barrier and increasing thermodynamic favourability. These activating groups can be removed through alkene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C cleavage reactions (e.g. metathesis (Scheme 5) and ozonolysis (Scheme 6B)). In this regard, highly substituted cycloheptenes 11 were prepared by allylation and metathesis (Scheme 4).28,43 The yields were modest to excellent over this two-step sequence. In many cases, where 10 exists as a mixture of regioisomers, the major allylation/RCM products 11 could be chromatographically separated from their minor constituents. As shown in Scheme 6A, the malononitrile can be transformed via oxidative amidation34 to products 12 containing a dense array of pharmaceutically relevant functionalities (amides, gem-dimethyl, fluoroaromatics, and heteroaromatics). Following this transformation, ozonolysis terminated with a NaBH4 quench installs an alcohol moiety on small molecule 13a.
C cleavage reactions (e.g. metathesis (Scheme 5) and ozonolysis (Scheme 6B)). In this regard, highly substituted cycloheptenes 11 were prepared by allylation and metathesis (Scheme 4).28,43 The yields were modest to excellent over this two-step sequence. In many cases, where 10 exists as a mixture of regioisomers, the major allylation/RCM products 11 could be chromatographically separated from their minor constituents. As shown in Scheme 6A, the malononitrile can be transformed via oxidative amidation34 to products 12 containing a dense array of pharmaceutically relevant functionalities (amides, gem-dimethyl, fluoroaromatics, and heteroaromatics). Following this transformation, ozonolysis terminated with a NaBH4 quench installs an alcohol moiety on small molecule 13a.
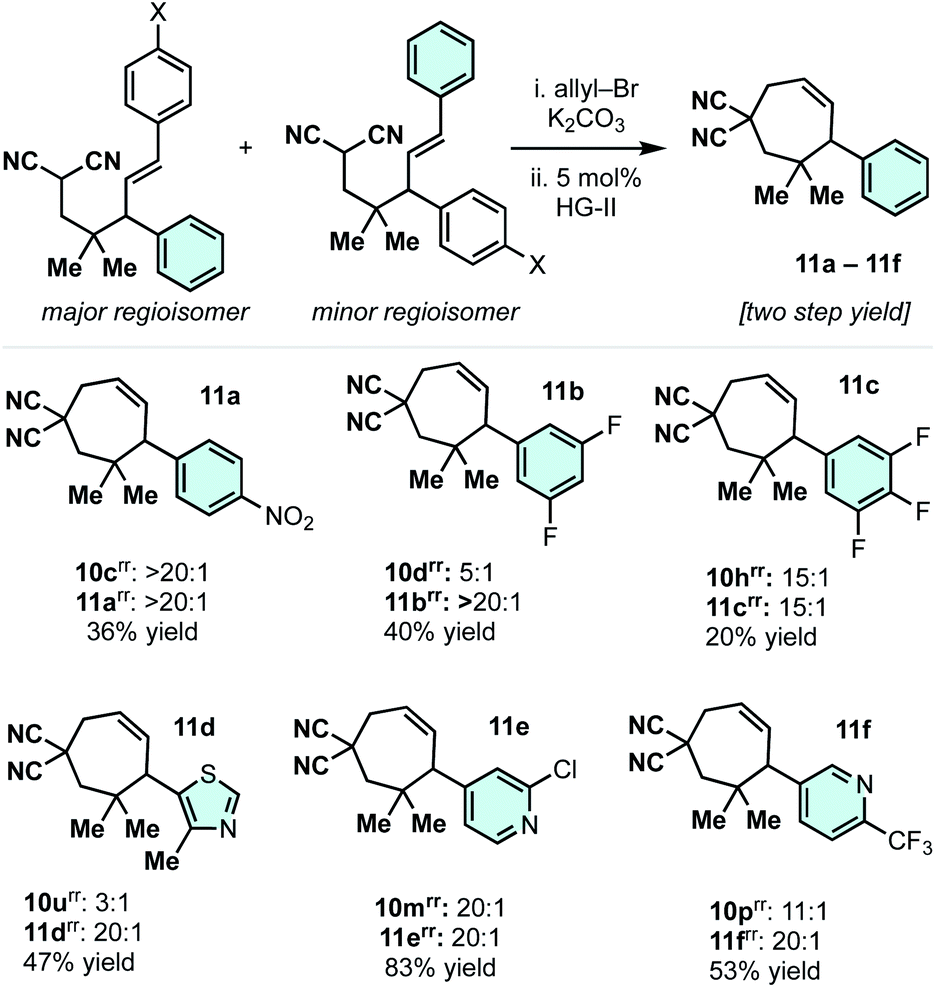 | ||
| Scheme 5 Removal of the “activating group” by ring-closing metathesis. | ||
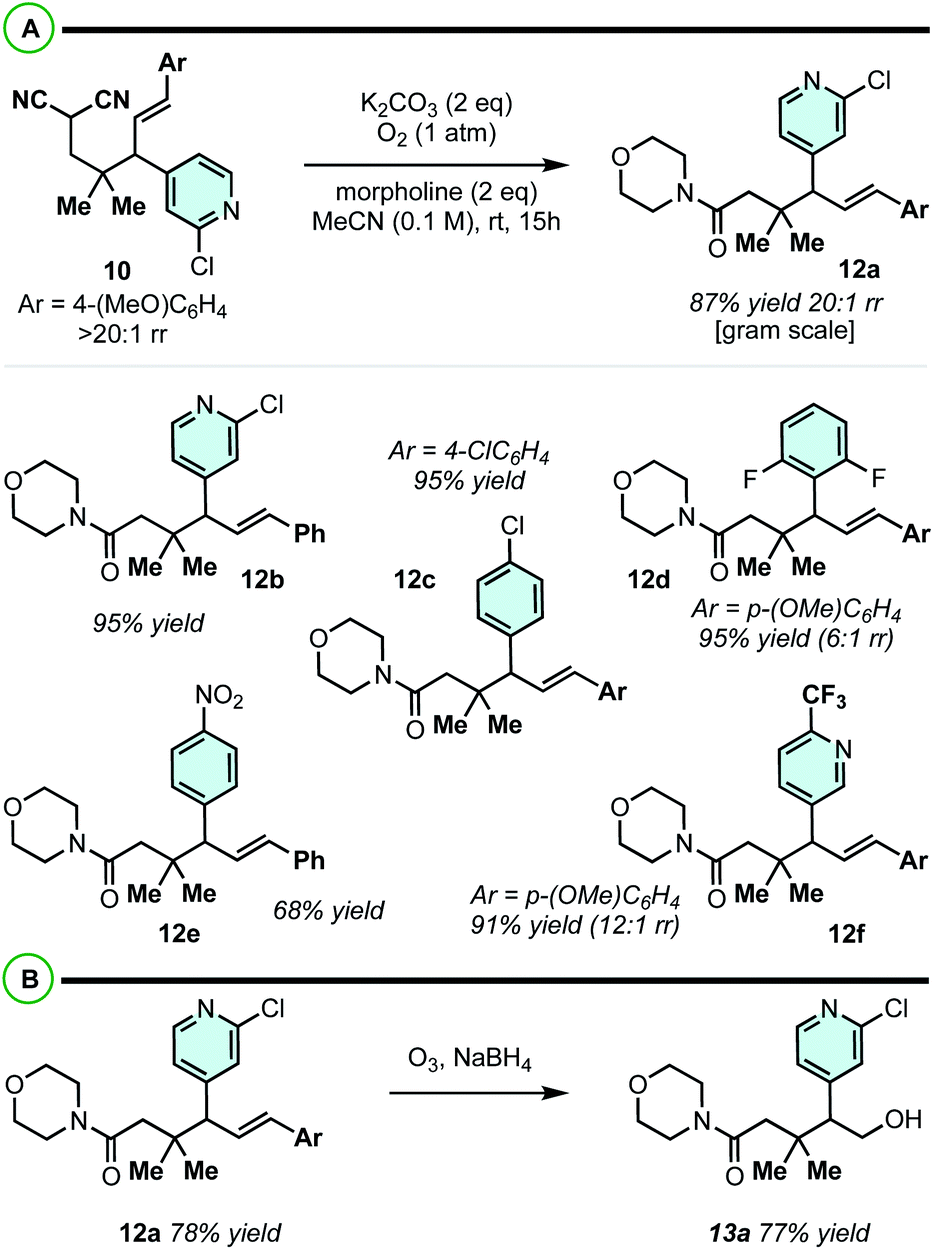 | ||
| Scheme 6 (A) oxidative amidation of malononitrile. (B) Removal of “activating group” by ozonolysis. | ||
These first computational and experimental studies utilizing 3,3-dicyano-1,5-dienes as substrates for constructing vicinal 4°/3° centers sets the stage for much further examination and application. For example, while we focused our efforts on gem-dimethyl-based quaternary carbons, it is likely that other functionality can be installed at this position. For example, while unoptimized, it appears the protocol is reasonably effective at incorporating a piperidine moiety in addition to heteroarenes from the allylic electrophile (7b + 9f → 14a; Scheme 7A). Similar functional group interconversion chemistry as described in Schemes 5 and 6 can thus yield functionally dense building blocks 15 and 16 in good yields.
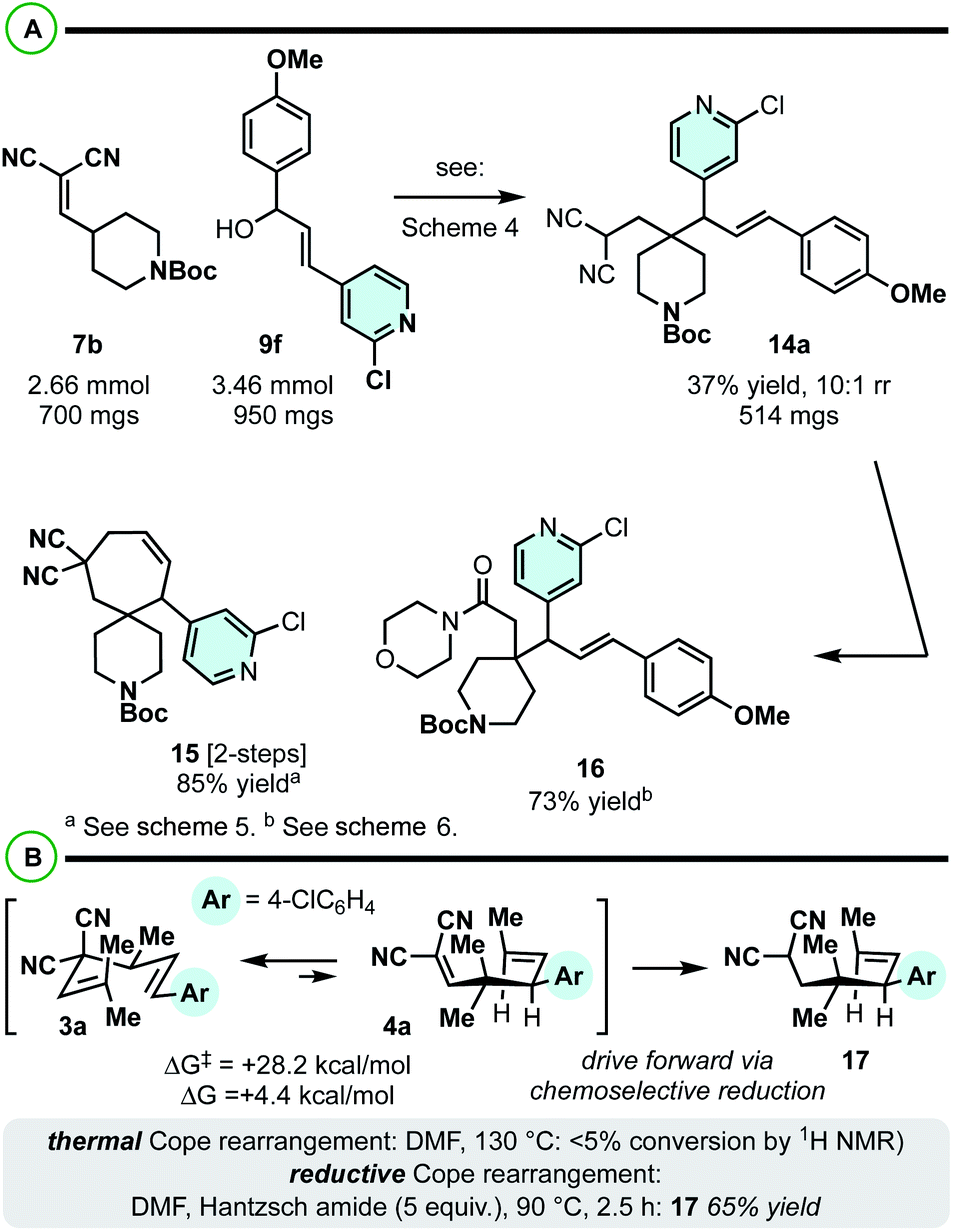 | ||
| Scheme 7 (A) The construction of 4/3° centres on piperidines. (B) Promoting endergonic [3,3] rearrangements is possible, assuming the [3,3] kinetic barrier is sufficiently low. | ||
While the 4,6-diaryl-3,3-dicyano-1,5-dienes offered the most attractive energetic profile (low kinetic barrier, isoenergetic [3,3] equillibrium; Scheme 2C), the 4-methyl analogue is also intriguing to consider as a viable substrate class for reductive Cope rearrangement (Scheme 2B). The challenge here is that the kinetics and thermodynamics are quite unfavourable (not observable by NMR), but potentially not prohibitively so. It is extremely exciting to find that Cope equilibria that are significantly endergonic in the desired, forward direction (e.g.3a to 4a) can be promoted by a related reductive protocol (Scheme 7B). While unoptimized, we were able to isolate product 17 in xx% yield by heating at 90 °C in the presence of Hantzsch ester in DMF.
Conclusions
We have developed a method to construct vicinal gem-dimethyl 4°/3° carbons via reductive Cope rearrangement. 1,5-Diene substrates bearing a key 4-phenyl or p-methoxyphenyl group have low kinetic barriers for Cope rearrangement. The 1,5-dienes are fluxional/shape-shifting (ΔG‡ ∼ 23 kcal mol−1, ΔG ∼ 0 kcal mol−1) and reductive conditions are utilized to drive the rearrangement forward, toward products containing a central gem-dimethyl/arene motif flanked on either side by a malononitrile and styrene moiety, respectively. These functional groups can be manipulated to construct unique molecular architectures such as cycloheptenes (Scheme 5) and amides (Scheme 6). Future directions involve expanding the scope of this transformation, developing enantioselective variants, and identifying specific opportunities for molecular synthesis and lead generation in medicinal chemistry campaigns.Data availability
The data is already shared in the ESI.†Author contributions
Kristin Sobie led the experimental studies outlined in this work. Matthew Albritton initiated the computational and experimental studies outlined in this work. Yinuo Yang performed computational analyses associated with this work. Mariana Alves performed experimental studies associated with this work. Adrian E. Roitberg led and mentored the computational team and co-wrote the manuscript. Alexander J. Grenning led and mentored the experimental team and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH NIGMS for financial support (R35 GM137893-01). This material is based upon work supported by the National Science Foundation (NSF CAREER 1844443). We thank the ACS Division of Organic Chemistry for a Summer Undergraduate Research Fellowship (SURF) (MA and AJG). We thank the Mass Spectrometry Research and Education Center and their funding source: NIH S10 OD021758-01A1. We acknowledge the University of Florida – Research Computing for providing computational resources and support that have contributed to the results reported in this publication.Notes and references
- G. Wu, J.-R. Wu, Y. Huang and Y.-W. Yang, Chem.–Asian J., 2021, 16, 1864–1877 CrossRef CAS PubMed.
- F. Zhou, L. Zhu, B.-W. Pan, Y. Shi, Y.-L. Liu and J. Zhou, Chem. Sci., 2020, 11, 9341–9365 RSC.
- Z. Wang, Org. Chem. Front., 2020, 7, 3815–3841 RSC.
- Y. Li and S. Xu, Chem.–Eur. J., 2018, 24, 16218–16245 CrossRef CAS PubMed.
- J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed.
- C. Schneider and C. F. Weise, in Compr. Org. Synth, Elsevier B.V., 2nd edn, 2014, pp. 867–911 Search PubMed.
- M. Hiersemann and T. Jaschinski, in Compr. Chirality, Elsevier B.V., 2012, pp. 625–647 Search PubMed.
- L. A. Paquette, Tetrahedron, 1997, 53, 13971–14020 CrossRef CAS.
- M. Simek, K. Bartova, R. Pohl, I. Cisarova and U. Jahn, Angew. Chem., Int. Ed., 2020, 59, 6160–6165 CrossRef CAS PubMed.
- D.-S. Hsu and C.-C. Liao, Org. Lett., 2003, 5, 4741–4743 CrossRef CAS PubMed.
- E. J. Corey and R. S. Kania, Tetrahedron Lett., 1998, 39, 741–744 CrossRef CAS.
- W.-C. Liu and C.-C. Liao, Synlett, 1998, 912–914 CrossRef CAS.
- T.-H. Lee and C.-C. Liao, Tetrahedron Lett., 1996, 37, 6869–6872 CrossRef CAS.
- R. C. Gadwood and R. M. Lett, J. Org. Chem., 1982, 47, 2268–2275 CrossRef CAS.
- S. Kruger and T. Gaich, Beilstein J. Org. Chem., 2014, 10, 163–193 CrossRef PubMed.
- D. Garayalde, K. Krueger and C. Nevado, Angew. Chem., Int. Ed., 2011, 50, 911–915 CrossRef CAS PubMed.
- K. Miki, K. Ohe and S. Uemura, J. Org. Chem., 2003, 68, 8505–8513 CrossRef CAS PubMed.
- S. Yokoshima, H. Tokuyama and T. Fukuyama, Angew. Chem., Int. Ed., 2000, 39, 4073–4075 CrossRef CAS PubMed.
- T. Fukuyama and G. Liu, J. Am. Chem. Soc., 1996, 118, 7426–7427 CrossRef CAS.
- E. Piers, M. Jean and P. S. Marrs, Tetrahedron Lett., 1987, 28, 5075–5078 CrossRef CAS.
- R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagné, Nat. Chem., 2012, 4, 405–409 CrossRef CAS PubMed.
- Y. Qiao, S. Kumar and W. P. Malachowski, Tetrahedron Lett., 2010, 51, 2636–2638 CrossRef CAS.
- T. Paul, W. P. Malachowski and J. Lee, J. Org. Chem., 2007, 72, 930–937 CrossRef CAS PubMed.
- W. P. Malachowski, T. Paul and S. Phounsavath, J. Org. Chem., 2007, 72, 6792–6796 CrossRef CAS PubMed.
- T. Paul, W. P. Malachowski and J. Lee, Org. Lett., 2006, 8, 4007–4010 CrossRef CAS PubMed.
- D. C. Wigfield, S. Feiner, G. Malbacho and K. Taymaz, Tetrahedron, 1974, 30, 2949–2959 CrossRef CAS.
- E. Fereyduni and A. J. Grenning, Org. Lett., 2017, 19, 4130–4133 CrossRef CAS PubMed.
- E. Fereyduni, J. N. Sanders, G. Gonzalez, K. N. Houk and A. J. Grenning, Chem. Sci., 2018, 9, 8760–8764 RSC.
- S. K. Scott, J. N. Sanders, K. E. White, R. A. Yu, K. N. Houk and A. J. Grenning, J. Am. Chem. Soc., 2018, 140, 16134–16139 CrossRef CAS PubMed.
- P. Vertesaljai, R. Serrano, M. D. Mannchen, M. Williams, E. Semenova and A. J. Grenning, Org. Lett., 2019, 21, 5704–5707 CrossRef CAS PubMed.
- E. Fereyduni, O. Lahtigui, J. N. Sanders, B. M. Tomiczek, M. D. Mannchen, R. A. Yu, K. N. Houk and A. J. Grenning, J. Org. Chem., 2021, 86, 2632–2643 CrossRef CAS PubMed.
- E. G. Foster, A. C. Cope and F. Daniels, J. Am. Chem. Soc., 1947, 69, 1893–1896 CrossRef.
- H. Nakamura, H. Iwama, M. Ito and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 10850–10851 CrossRef CAS.
- J. Li, M. J. Lear and Y. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 9060–9064 CrossRef CAS PubMed.
- T. T. Talele, J. Med. Chem., 2018, 61, 2166–2210 CrossRef CAS PubMed.
- O. Yahiaoui, L. F. Pasteka, B. Judeel and T. Fallon, Angew. Chem., Int. Ed., 2018, 57, 2570–2574 CrossRef CAS PubMed.
- J. F. Teichert, D. Mazunin and J. W. Bode, J. Am. Chem. Soc., 2013, 135, 11314–11321 CrossRef CAS PubMed.
- O. Yahiaoui, L. F. Pasteka, C. J. Blake, C. G. Newton and T. Fallon, Org. Lett., 2019, 21, 9574–9578 CrossRef CAS PubMed.
- H. D. Patel, T.-H. Tran, C. J. Sumby, L. F. Pasteka and T. Fallon, J. Am. Chem. Soc., 2020, 142, 3680–3685 CrossRef CAS PubMed.
- Y.-Y. Ma, M. Yan, H.-R. Li, Y.-B. Wu, X.-X. Tian, H.-G. Lu and S.-D. Li, Sci. Rep., 2019, 9, 1–8 Search PubMed.
- M. Moreno-Manas and J. Ribas, Tetrahedron Lett., 1989, 30, 3109–3112 CrossRef CAS.
- M. Prat, J. Ribas and M. Moreno-Manas, Tetrahedron, 1992, 48, 1695–1706 CrossRef CAS.
- O. Lahtigui, F. Emmetiere, W. Zhang, L. Jirmo, S. Toledo-Roy, J. C. Hershberger, J. M. Macho and A. J. Grenning, Angew. Chem., Int. Ed., 2016, 55, 15792–15796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR, 13C NMR, and HRMS data and reprints. See DOI: 10.1039/d1sc06307c |
| This journal is © The Royal Society of Chemistry 2022 |