
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile post-functionalisation strategy for the formation of modular organic–inorganic polyoxometalate hybrids†
David E.
Salazar Marcano
 a,
Mhamad Aly
Moussawi
a,
Alexander V.
Anyushin
a,
Sarah
Lentink
a,
Luc
Van Meervelt
b,
Ivana
Ivanović-Burmazović
c and
Tatjana N.
Parac-Vogt
*a
a,
Mhamad Aly
Moussawi
a,
Alexander V.
Anyushin
a,
Sarah
Lentink
a,
Luc
Van Meervelt
b,
Ivana
Ivanović-Burmazović
c and
Tatjana N.
Parac-Vogt
*a
aLaboratory of Bioinorganic Chemistry, KU Leuven Department of Chemistry, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: tatjana.vogt@kuleuven.be
bBiomolecular Architecture, KU Leuven Department of Chemistry, Celestijnenlaan 200F, 3001 Leuven, Belgium
cDepartment of Chemistry, Ludwig-Maximilian-University, Butenandtstr. 5-13, Haus D, 81377 Munich, Germany
First published on 8th February 2022
Abstract
Hybrid structures incorporating different organic and inorganic constituents are emerging as a very promising class of materials since they synergistically combine the complementary and diverse properties of the individual components. Hybrid materials based on polyoxometalate clusters (POMs) are particularly interesting due to their versatile catalytic, redox, electronic, and magnetic properties, yet the controlled incorporation of different clusters into a hybrid structure is challenging and has been scarcely reported. Herein we propose a novel and general strategy for combining multiple types of metal-oxo clusters in a single hybrid molecule. Two novel hybrid POM structures (HPOMs) bis-functionalised with dipentaerythritol (R–POM1–R; R = (OCH2)3CCH2OCH2C(CH2OH)) were synthesised as building-blocks for the formation of heterometallic hybrid triads (POM2–R–POM1–R–POM2). Such a modular approach resulted in the formation of four novel heterometallic hybrids combing the Lindqvist {V6}, Anderson–Evans {XMo6} (X = Cr or Al) and trisubstituted Wells–Dawson {P2V3W15} POM structures. Their formation was confirmed by multinuclear Nuclear Magnetic Resonance (NMR), infrared (IR) and UV-Vis spectroscopy, as well as Mass Spectrometry, Diffusion Ordered Spectroscopy (DOSY) and elemental analysis. The thermal stability of the hybrids was also examined by Thermogravimetric Analysis (TGA), which showed that the HPOM triads exhibit higher thermal stability than comparable hybrid structures containing only one type of POM. The one-pot synthesis of these novel compounds was achieved in high yields in aqueous and organic media under simple reflux conditions, without the need of any additives, and could be translated to create other hybrid materials based on a variety of metal-oxo cluster building-blocks.
Introduction
The synthesis of intricate hybrid organic–inorganic structures for the formation of new functional materials remains a major challenge. This problem can be addressed by exploiting well-known building blocks with established catalytic, electronic, conductive, optical or biological properties, and combining them to form hybrid materials that will benefit from the synergy of their combined properties.1,2 Polyoxometalates (POMs), which are discrete anionic metal–oxygen nanoclusters of group 5 and 6 transition metals in their highest oxidation states, are promising inorganic building-blocks for the formation of such hybrid materials.3 This family of compounds includes a wide variety of structures such as (1) the Lindqvist structure with general formula [M6O19]n−, (2) the Anderson–Evans structure with general formula [(Xn+)M6O24](6−n)−, and (3) the Wells–Dawson structure with general formula [X2M18O62]n− (M is typically Mo6+, W6+ or V5+ depending on the structure; X is a heteroatom such as P, Cr, Al, etc.). POMs have many interesting physical and chemical properties due to their tuneable size, charge density, acidity, and redox potential. As a result, they have been extensively investigated for applications in many fields such as catalysis, materials science, energy storage, supramolecular chemistry and medicine.4–9 Moreover, some archetypal POM structures can be functionalised with organic ligands to form organic–inorganic hybrid polyoxometalates (HPOMs), further expanding their physical and chemical properties.10–12POMs covalently functionalised with triols ((HOCH2)3C-R), in which three of the bridging oxo ligands are replaced by the triol, are one of the most important classes of HPOMs.12,13 Whether a POM can be functionalised with triol ligands and how many triols can be attached, highly depends on the POM structure. The Lindqvist hexavanadate structure (V6) is typically bis-functionalised (Fig. 1(a)) unless it is reduced,14 while the Anderson–Evans structure (XM6) can be either mono- or bis-functionalised depending on the nature of the heteroatom present at its centre (Fig. 1(b and c)).15 On the other hand, the vanadium trisubstituted Wells–Dawson structure (P2V3W15) can only be mono-functionalised (Fig. 1(d)).16 All these HPOM structures are highly versatile since they can be easily post-functionalised with a wide variety of organic moieties.17–20 However, the use of polyols for the functionalisation of POMs with other species aside from organic molecules – such as functional surfaces,21–24 nanoparticles25,26 and even other POMs – is challenging and remains scarcely explored.
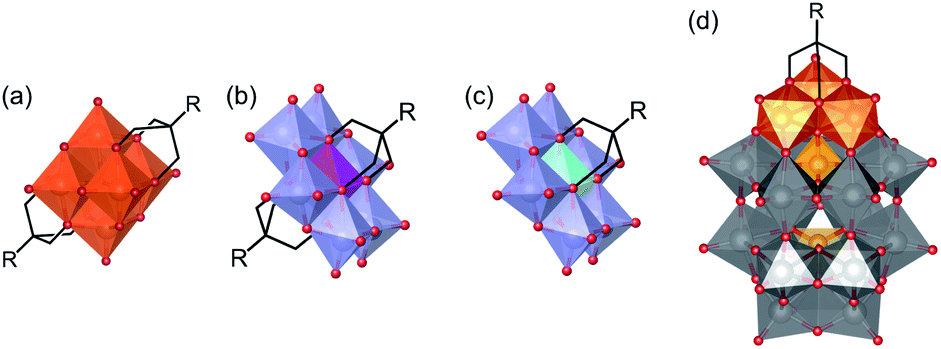 | ||
| Fig. 1 Representations of different hybrid polyoxometalate (HPOM) structures: (a) the bis-functionalised [V6O13{(OCH2)3C-R}2]2− Lindqvist hexavanadate, (b) the bis-functionalised δ-[{Xn+}M6O18{(OCH2)3C-R}2](6−n)− and (c) mono-functionalised δ-[{Xn+}(OH)3M6O18{(OCH2)3C-R}](6−n)− Anderson–Evans as well as (d) the mono-functionalised [P2V3W15O59{(OCH2)3C-R}]6− vanadium trisubstituted Wells–Dawson. Oxygen in red, vanadium in orange, metal addenda atom (Mo or W) in purple or grey, phosphorous in yellow, heteroatom X in magenta or cyan and organic carbon chain in skeletal form in black. | ||
To date, only a few examples of POM structures connected together to form POM–POM hybrids have been reported. In general, POM–POM hybrid structures have been obtained via self-assembly of POMs functionalised with pyridyl ligands promoted by metal ion coordination or via covalent attachment employing an organic linker to form more robust POM–POM hybrids.27,28 The latter can be achieved through two main strategies. The first approach is based on triol functionalisation using polyol ligands, while the second uses the replacement of some of the terminal oxo ligands of the POM by a nitrogen atom on the linker to form an organoimido POM derivative. The method employing polyols has been mostly reported for linking together P2V3W15 POMs to form POM–POM “dumbbell”, “triangular” and “dendritic” hybrids, which are catalytically active or can self-assemble to form vesicles, a process that can be controlled by coordination of metal ions depending on the nature of the organic linker.29–33 Linking XM6 POMs with polyol ligands has also been used to form both “dumbbell” and polymeric structures.34,35 Similarly, 1,3,5-benzenetricarboxamide ligands with three triol groups have been used to form extended networks of V6 POMs, which showed promising catalytic activity.36 On the other hand, the second method of covalent attachment of POM structures based on organoimido POM derivatives involves linking Lindqvist hexamolybdate (Mo6) POM structures to form “dumbbell” hybrids with the same Mo6 POMs at either end of an organic linker.37,38 Furthermore, nanorod triads, in which POMs bis-functionalised with tris(hydroxymethyl)aminomethane ((HOCH2)3CNH2) were used as the linker between two Mo6, have also been reported and displayed redox activity and photochromism.39–41 However, the formation of POM–POM hybrids using this approach is currently mostly limited to hexamolybdates and requires more complex reaction conditions involving the use of additional reagents, such as N,N′-dicyclohexylcarbodiimide (DCC).42 In contrast, the formation of triol-functionalised HPOMs can be achieved for a much wider range of POM structure types via a simpler one-pot synthesis that just involves mixing the starting materials under reflux. Nevertheless, the use of polyols to link POMs together has so far been mainly limited to the formation of hybrid structures involving only one kind of POM.
A very limited number of discrete structures that contain more than one type of POM have been reported even though combining different types of POM structures in one molecule could result in interesting catalytic, redox, electronic, and magnetic properties, as these are highly dependent on the structure and composition of the POMs. Such POM–POM hybrids would not only benefit from the synergistic contributions of the organic and inorganic components but also from the association of different inorganic species. This can be seen from a recently reported 2D hybrid containing [V10O28]6− and [XV12O38]12− (X = Ni4+, Mn4+) POMs non-covalently linked by 4,4′-bipyridine-N,N′-dioxide and lanthanide ions to form a novel extended structure which displayed better catalytic activity than the individual POMs.43 However, the reported synthetic approach has a limited general applicability and offers restricted versatility and control over the resulting mixed POM hybrids. Consequently, the formation of molecular hybrids containing more than one type of metal-oxo cluster presents a synthetic challenge, and to the best of our knowledge a general strategy for combing different metal-oxo structure types into a discrete hybrid structure has not been reported so far. Therefore, since an accessible post-functionalisation strategy for the combination of different types of metal-oxo clusters would be a valuable asset in the directed formation of novel functional materials, in this paper we propose a novel approach based on the controlled design and combination of POM building-blocks with polyols resulting in the formation of mixed heterometallic POM–POM hybrids.
Results and discussion
Herein, we report the synthesis and characterisation of the tetrabutylammonium (TBA) and sodium salts of two novel Lindqvist and Anderson–Evans HPOMs bis-functionalised with dipentaerythritol (HOCH2)3CCH2OCH2(CH2OH)3 (dP): [V6O13{(OCH2)3CCH2OCH2C(CH2OH)3}2]2− (L) and [CrMo6O18{(OCH2)3CCH2OCH2C(CH2OH)3}2]3− (C). These novel symmetrical HPOMs, functionalised on either side of the POM core (POM1) with a ligand containing free triol groups (R), can be used as building blocks for coupling different types of POM structures (POM2) to form POM2–R–POM1–R–POM2 hybrids. One key requirement for the successful formation of such a POM–POM hybrid structure is the presence of anchoring points on the second POM onto which the free triol “claws” can be grafted during a POM post-functionalisation reaction. Therefore, for this purpose, the vanadium trisubstituted Wells–Dawson ([P2V3W15O62]9−; D) and the aluminium Anderson–Evans ([Al(OH)6Mo6O18]3−; A) POM structures were selected since D can be mono-functionalised with triols through the V3 cap,16 while mono-functionalisation of A can be achieved by attachment of the triol on one side of the central {AlO6} octahedron.15,35 Upon binding of the triol ligand to one side of the AlMo6 core, the OH groups on the other side of the core become less labile, thus preventing further functionalisation that would result in the formation of infinite chains: (–R–POM1–R–POM2–)n.35 Hence, the ability of dP-functionalised POMs to form discrete novel structures by covalently connecting POMs with different compositions and properties was established by post-functionalising L and C with D or A as shown in Fig. 2.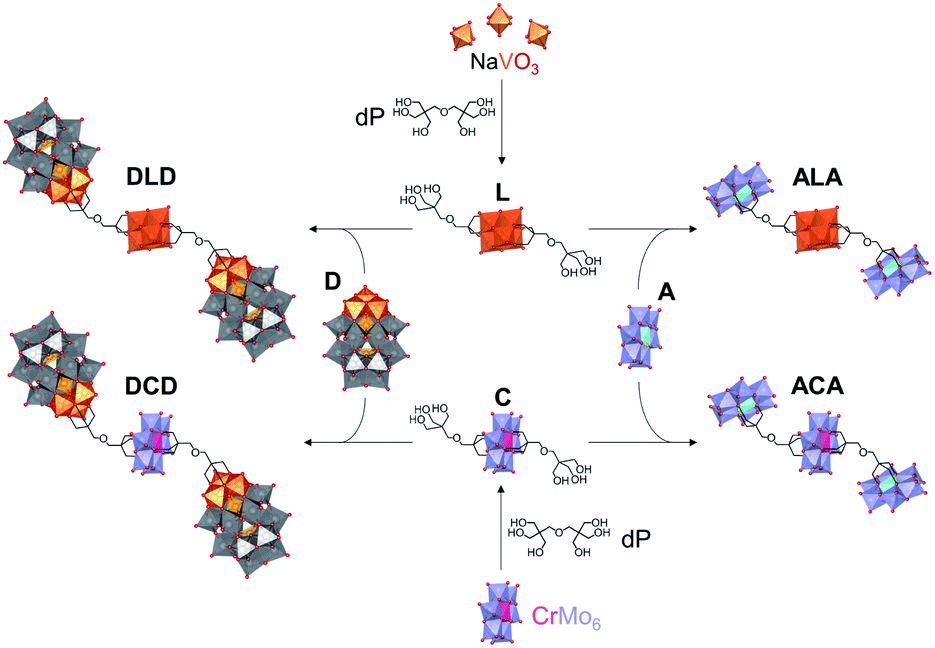 | ||
| Fig. 2 Reaction scheme showing the formation of [V6O13{(OCH2)3C-R}2]2− (L) and [CrMo6O18{(OCH2)3C-R}2]3− (C) functionalised with dipentaerythritol (R = CH2OCH2C(CH2OH)3) as building blocks for the formation of 4 heterogeneous POM–POM hybrids via post-functionalisation with [P2V3W15O62]9− (D) or [Al(OH)6Mo6O18]3− (A): DLD, DCD, ALA, ACA. | ||
Synthesis & characterisation of bis-dipentaerythritol HPOMs
The formation of the dP-functionalised building-blocks, C and L, was achieved by heating the POM precursors (i.e. [Cr(OH)6Mo6O18]3− {CrMo6} or metavanadate) with dP in aqueous solution. Then, the desired TBA-salts were obtained by precipitation with tetrabutylammonium bromide. The successful synthesis of L through the attachment of only one of the triol groups of each dP ligand to either side of the V6 POM core, thereby breaking the symmetry of dP, was unambiguously confirmed by 1H and 13C NMR (Fig. S4–S5†). In the 1H NMR spectrum, the V–O–CH2– protons attached to the POM are highly deshielded, giving rise to a peak at 5.04 ppm, while a peak at 3.46 ppm was observed for the free –CH2-OH ethyl protons as expected for POM-bound and free –OCH2– groups.44,45 Similarly, the peak due to the bridging –CH2–O–CH2– ether group was split into two peaks at 3.31 and 3.29 ppm due to the formation of the functionalised POM. The 13C NMR spectrum showed a similar break in the symmetry of dP, with peaks at 85.94 and 63.10 ppm assigned to the V–O–CH2– and –CH2–OH carbons respectively, and two peaks observed at 73.21 and 71.79 ppm corresponding to the –CH2–O–CH2– carbons. The formation of the V6 POM core was also confirmed by the presence of a single sharp peak at −497 ppm in the 51V NMR spectrum (Fig. S6†), which is typical of trans-functionalised hexavanadates,45,46 as well as by the characteristic peaks in the IR spectrum (Fig. S10†) at 953 (ν V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, vs.), 810 (ν V–O–V, s), 791 (ν V–O–V, s), and 709 (ν V–O–V, vs.) cm−1.47
O, vs.), 810 (ν V–O–V, s), 791 (ν V–O–V, s), and 709 (ν V–O–V, vs.) cm−1.47
Due to the paramagnetic nature of Cr3+, C could not be fully characterised by 1H NMR as very broad peaks were obtained (Fig. S8†).34 Similarly, in the 13C NMR spectrum only three peaks corresponding to carbon environments furthest away from the Cr3+ could be observed, with the peaks becoming weaker and broader with increasing proximity to the POM (Fig. S9†). Nevertheless, the integrity of the POM core was confirmed from the characteristic peaks in the IR spectrum (Fig. S12†) at 937 (ν MoO, s), 912 (ν MoO, s), 897 (ν MoO, s) and 650 (ν Mo–O–Mo, vs.) cm−1.48,49 Furthermore, the peaks at 1130 and 1016 cm−1 due to C–O stretching vibrations are indicative of attachment of the tripodal anchor of dP onto the POM core, while the peaks in the region 400–600 cm−1 are characteristic of a bis-functionalised CrMo6 and can be distinguished from the all-inorganic POM as well as from the mono-functionalised form.50,51 In addition, the CrMo6 POM core gives rise to characteristic peaks in the UV-Vis absorbance spectrum (Fig. S13†) at 214 nm (ε = 5.77 ×105 M−1 cm−1) and 512 nm (ε = 8.96 M−1 cm−1), which are due to the ligand to metal charge transfer (LMCT) from the ligand-centred μ3-OR π orbitals to the metal-centred Mo6+ t2g orbitals (O → Mo6+) and the d–d transition from the HOMO t2g to the LUMO eg of Cr3+ respectively. Furthermore, functionalisation of the POM clearly results in a hypochromic shift in the absorbance bands with respect to those of the all-inorganic CrMo6 precursor, which gives peaks at 239 nm and 540 nm, as has been previously reported for mono-functionalised CrMo6 species.51,52
Moreover, the composition of the pure compound for both L and C was further confirmed by elemental analysis, negative mode ESI-MS (Fig. S3 & S7†) and single crystal X-ray diffraction. Single crystals were obtained by ether diffusion into acetonitrile/methanol or ethanol solutions of L and C respectively. Both compounds crystallised forming a hydrogen bonding network in the centrosymmetric P21/c space group and display the expected bis-functionalised Lindqvist and Anderson–Evans structures (Fig. 3). The crystal structures, parameters and refinement details are discussed in more detail in the ESI (Table S1†).
 | ||
| Fig. 3 Mixed ball-stick and polyhedral representation of the structures of L (left) and C (right) as determined by single crystal X-ray diffraction. TBA counterions and the positional disorder were omitted for clarity. | ||
It should be noted that C, with two dP ligands attached to either side of the POM core (R–POM–R), represents a new member of a family of related compounds based on the Anderson–Evans structure functionalised with dP that were recently reported by Wei et al. and, therefore, it further demonstrates the versatility of this POM scaffold.34 The formation of a dumbbell-like structure (POM–R–POM) was reported to be obtained when the ratio of CrMo6 and dP was set to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 while changing the ratio to 1:1.4 gave rise to the mono-functionalised CrMo6 (POM-R). However, isolation of the symmetrical bis-functionalised CrMo6 HPOM was not reported by Wei et al. Nevertheless, through the procedure reported herein, using a higher temperature and a 1:4 ratio of CrMo6 to dP, it was possible to synthesize C without any side products, showing that controlling the ratio of starting materials and the temperature allows for the formation of different functionalised structures.
:1 while changing the ratio to 1:1.4 gave rise to the mono-functionalised CrMo6 (POM-R). However, isolation of the symmetrical bis-functionalised CrMo6 HPOM was not reported by Wei et al. Nevertheless, through the procedure reported herein, using a higher temperature and a 1:4 ratio of CrMo6 to dP, it was possible to synthesize C without any side products, showing that controlling the ratio of starting materials and the temperature allows for the formation of different functionalised structures.
Synthesis & characterisation of D-terminal POM–POM hybrids
The POM–POM hybrids formed via post-functionalisation of L or C with D were obtained by refluxing the POM linker with TBA5H4[P2V3W15O62] for one week in dry acetonitrile under an inert argon atmosphere while the reaction mixture was kept in the dark to prevent photo-induced side reactions. In this way, TBA11.4H2.6[V6O13{(OCH2)3CCH2OCH2C(CH2O)3(P2V3W15O59)}2] (DLD) and TBA12.4H2.6[CrMo6O18{(OCH2)3CCH2OCH2C(CH2O)3(P2V3W15O59)}2] (DCD) were obtained in high yields from L and C respectively. The reactions were left running for a week to ensure the POM linker fully reacted, but evidence of the formed POM–POM hybrid with only trace amounts of the starting materials was observed by multinuclear NMR after refluxing for just over an hour, which suggests that a shorter reaction time is possible.As shown in Fig. 4, evidence for the attachment of the free triol group of L to D could be clearly seen from changes in the chemical shift of the peaks in the 1H, 13C, 51V and 31P NMR spectra of DLD (Fig. S16–S18†) with respect to the starting materials. By 1H NMR a significant downfield shift from 3.46 ppm to 5.45 ppm was observed for the –CH2–O– protons of the triol group, corresponding to binding of the tripodal anchor to the V3 cap of D. To a lesser extent, a shift was also observed for the peaks of the adjoining ethyl protons in the bridging –CH2–O–CH2– ether group. Similar changes were also observed in the 13C NMR spectrum of DLD with respect to L. In addition, 51V NMR showed a major downfield shift from −585 ppm to −538 ppm for the peak belonging to the V3 cap of D, as a result of functionalisation, while the peak of the V6 core remained relatively unchanged at −497 ppm. The integration of both peaks in the 51V NMR spectrum was approximately the same, which is indicative of the formation of a symmetrical POM–POM hybrid with two V3 caps attached to either side of the V6 core. Moreover, 31P NMR showed only two peaks at −7.51 ppm and −13.55 ppm, which were observed to shift slightly from the typical chemical shifts of D, further confirming functionalisation and the structural integrity of the Wells–Dawson POM structure. The integrity of the POM cores was also confirmed by IR spectroscopy (Fig. S15†) from the peak at 1084 cm−1 due to the {PO4} template of the Wells–Dawson structure and the peaks in the range 400–1000 cm−1 due to ν MO and ν M–O–M vibrations (M = V or W).47,53,54 In addition, the UV-Vis absorbance spectrum of DLD (Fig. S19†) showed a significantly higher absorbance compared to the 1:2 mixture of L and D with the same total concentration. However, after normalisation with respect to the maximum absorbance, the spectra matched very well, further confirming that DLD is indeed a 1:2 combination of L and D. Hence, distinct spectroscopic changes evidence the formation of a novel POM–POM hybrid via the covalent attachment of the Lindqvist and Wells–Dawson POM structures, thereby combining their characteristic properties in a single discrete hybrid structure.16,30,36,55
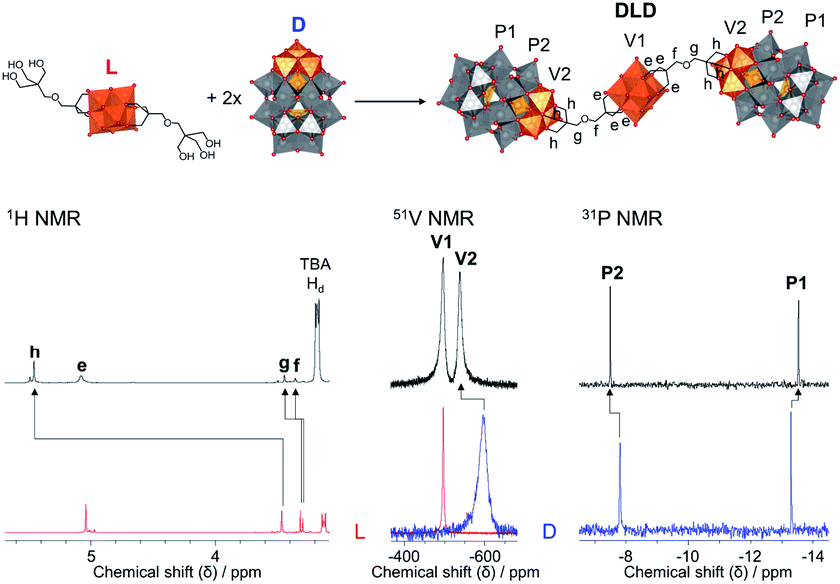 | ||
| Fig. 4 Synthesis of DLD from L and D (top) and the corresponding 1H, 51V and 31P NMR spectra (bottom; ordered from left to right) of the POM–POM hybrid (black) and the starting materials (red for L and blue for D) illustrating the changes in the chemical shifts due to POM post-functionalisation of L. | ||
The formation of DCD was also observed by multinuclear NMR and the integrity of the POM cores was confirmed by IR spectroscopy. IR showed peaks in the range 400–1000 cm−1 due to ν MO and ν M–O–M vibrations (M = Mo, V or W) which are clearly due to both the Anderson–Evans and Wells–Dawson POM cores (Fig. S21†). Furthermore, as observed with DLD, the UV-Vis absorbance spectrum of DCD (Fig. S24†) was dominated by the O → W6+ and O → V5+ LMCT bands of D.32 Similarly, DCD also displayed a higher absorbance than a 1:2 mixture of C and D with the same total concentration, but the normalised spectra gave an excellent match, indicating that DCD is indeed a result of a 1:2 combination of C and D. Additionally, a major shift in the peak corresponding to the V3 cap from −585 ppm to −539 ppm was also observed by 51V NMR as a result of functionalisation (Fig. 5; S23†). Likewise, 31P NMR gave only two peaks at −7.58 ppm and −13.51 ppm corresponding to the functionalised Wells–Dawson structure (Fig. 5; S22†). Moreover, the peak at −7.58 ppm, which is due to the 31P environment closest to the V3 cap, had a half-width (Δν1/2) of 11.1 Hz, which was significantly broader than the second 31P peak at −13.51 ppm, and was also much broader than the peaks in the 31P NMR spectra of DLD and D, which had a Δν1/2 of around 4–7 Hz. This broadening of the most deshielded peak is caused by the paramagnetic Cr3+ centre of the Anderson–Evans structure and clearly indicates the proximity of the POMs through their covalent attachment. Moreover, this suggests that the combination of C and D could potentially be exploited to probe biomolecules, such as proteins, due to 1H paramagnetic relaxation enhancement resulting from C and the favourable interactions which can take place between proteins and the Wells–Dawson structure.56,57
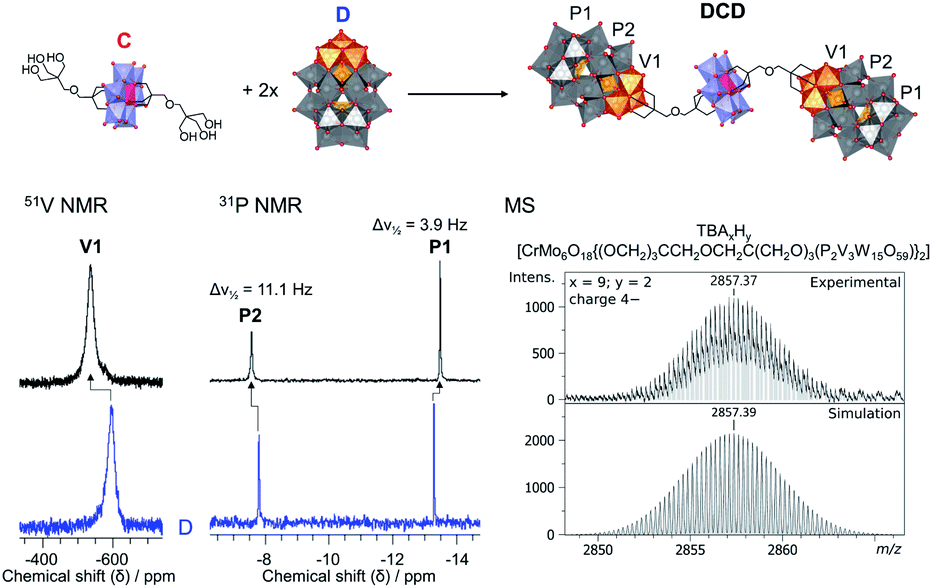 | ||
| Fig. 5 Synthesis of DCD from C and D (top) together with the corresponding 51V and 31P NMR spectra (bottom left) of the POM–POM hybrid (black) and of D (blue) as well as the experimental and simulated cryo-MS spectra for DCD (bottom right). | ||
The successful formation of both DLD and DCD was also confirmed by Ultra High Resolution cryo-mass spectrometry (UHR cryo-MS), which gave several peaks corresponding to the intact POM–POM hybrid structures with different numbers of protons and TBA as counter-cations (Fig. 5; Tables S2–S3; Fig. S14 & S20†). Their composition was also further confirmed by elemental analysis. Crystallisation of these novel POM–POM hybrids was also attempted in various solvents and solvent mixtures, but unfortunately crystals of suitable quality for X-ray diffraction could not be obtained due to the flexibility of the dP linker, as has been previously reported for other POM–POM hybrids.30 To the extent of our knowledge, these novel heterometallic POM–POM hybrids represent the first examples of the covalent attachment of other POM structures onto Wells–Dawson type POMs, and DCD in particular demonstrates the high number of different metal centres that can be combined in this way.
Synthesis & characterisation of A-terminal POM–POM hybrids
Similar to the formation of DLD and DCD, the synthesis of POM–POM hybrids with Anderson–Evans terminal groups was performed by refluxing the dP-functionalised POM linker and A. In this way, an additional two new POM–POM hybrids were obtained: TBA8[V6O13{(OCH2)3CCH2OCH2C(CH2O)3(Al(OH)3Mo6O18)}2] (ALA) and TBA9[CrMo6O18{(OCH2)3CCH2OCH2C(CH2O)3(Al(OH)3Mo6O18)}2] (ACA). However, the syntheses were performed in water due to the higher reported reactivity of the labile OH groups of A towards substitution in aqueous media with respect to that in organic solvents.35 Hence, taking into account the insolubility of the TBA salts of C and L in water, an ion metathesis procedure – involving precipitation of the Na-salt by addition of NaClO4 to the TBA-salts in acetonitrile – was employed to obtain the water-soluble sodium salts of these compounds, which were characterised by multinuclear NMR, UV-Vis and/or IR spectroscopy (Fig. S10–S13†).58The synthesis of ALA was confirmed by multinuclear NMR, MS, UV-Vis, IR, and elemental analysis. Functionalisation of L was observed by 1H NMR, which showed a downfield shift of the –OCH2– protons from around 3.46 ppm for L to 4.50 ppm for ALA (Fig. 6; S28†). Similar changes were also observed from the 13C NMR spectra, which showed a downfield shift in the corresponding carbon environment from 63.70 ppm for L to 76.89 ppm for ALA (Fig. S29†). In addition, the peaks in negative mode ESI-MS of ALA in acetonitrile could be assigned to ALA with different numbers of protons and TBA as counter-cations giving rise to species with charges of 2- or 3- (Fig. S25; Table S4†). However, the spectra obtained by cryo-MS suggest that ALA does not ionise well or is not particularly stable under the measurement conditions used since only one profile centred at m/z 3383.87 (Fig. S26†) could be tentatively assigned to the mono-anionic species with 2 TBA and 5 protons as counter ions ([TBA2H5ALA]−; calcd. m/z = 3384.05). The difference between the calculated and observed m/z is also significantly larger than expected for ultra-high resolution measurements preventing a confident assignment of the cryo-MS spectra. Nevertheless, similar to DLD and DCD, the UV-Vis absorbance spectrum of ALA (Fig. S32†) displayed a higher absorbance than a 1:2 mixture of L and A with the same total concentration while the normalised spectra matched fairly well, with the exception of an additional peak at 218 nm. This extra peak at 218 nm is due to the functionalisation of A, which causes a hypochromic shift in the absorbance peak originating from the O → Mo6+ LMCT that was also observed for the functionalisation of CrMo6 to form C. This further confirms that ALA is composed of a 1:2 combination of L and Avia covalent attachment. The peaks at 1132 and 1035 cm−1 in the IR spectrum (Fig. S27†) due to C–O stretching vibrations are also indicative of functionalisation, and the peaks in the region 400–1000 cm−1 originating from ν MO and ν M–O–M vibrations (M = V or Mo) confirm the presence of both POM cores. The integrity of the V6 and AlMo6 POM cores after post-functionalisation were also evidenced from single sharp peaks at −497 ppm and 15.69 ppm in the 51V NMR and 27Al NMR spectra, respectively (Fig. S30–S31†). Moreover, the attachment of A to L resulted in a decrease in the translational diffusion coefficient (Ddiff) from 921 μm2 s−1 (L) to 472 μm2 s−1 (ALA) as determined from all peaks corresponding to the POM–POM hybrid by 1H 2D Diffusion Ordered NMR spectroscopy (DOSY), which shows that all peaks correspond to the same species and that ALA is significantly larger than L (Fig. 6). It is worth noting that a similar decrease in Ddiff compared to L was also observed for DLD, but DLD gave an even lower Ddiff due to the larger size of D with respect to A. In fact, the hydrodynamic radius (r) determined from the observed Ddiff using the Stokes–Einstein equation showed the expected increase in size from 0.69 ± 0.01 nm for L to 1.34 ± 0.03 nm for ALA and 1.60 ± 0.06 nm for DLD. These values also match fairly well with the crystal structure of L and the simulated structures of ALA and DLD, which were based on a combination of the structure of L with the crystal structures of A (ICSD 415139) or D (CCDC 675452), from which r was estimated to be 0.96, 1.19, and 1.99 nm respectively.55,59
 | ||
| Fig. 6 Overlayed 1H 2D DOSY NMR spectra of L, ALA and DLD labelled with the translational diffusion coefficient (Ddiff) used to determine the hydrodynamic radius (r) for each structure, as shown on the right, and the corresponding 1H 1D NMR spectra shown at the top. | ||
Based on the successful isolation of DCD and following a similar procedure used for the synthesis of ALA, the synthesis of a triple Anderson–Evans POM–POM hybrid structure based on C with terminal A was also attempted. Negative mode ESI-MS (Fig. S33†) of the isolated product gave a peak centred around m/z 1931.61 which can be attributed to the di-anionic POM with 2 TBA and 5 protons as counter ions ([TBA2H5ACA + (ACN) + 2(H2O)]2−; calcd m/z = 1931.40) confirming the successful synthesis of ACA. However, ACA exhibited a similar behaviour to that observed for ALA under the cryo-MS measurement conditions since only one profile centred at m/z 4027.85 (Fig. S34†) could correspond to the mono-anionic species with 3 TBA and 5 protons as counter ions ([TBA3H5ACA]−; calcd m/z = 4028.03) but, like with ALA, the difference between the calculated and observed m/z is significantly larger than expected for ultra-high resolution measurements and, therefore, this profile could not be unambiguously assigned. Furthermore, due to the paramagnetic nature of the Cr3+ centre, NMR analysis was limited to 13C and 27Al NMR (Fig. S36–S37†). 13C NMR of ACA showed that grafting of the free triol group of C onto A resulted in the disappearance of the peak at 64.21 ppm, which is due to the –CH2OH groups of C, and the appearance of a peak at 77.31 ppm corresponding to the formation of –CH2O–Al, as seen for ALA. However, only one additional peak due to ACA at 42.06 ppm, corresponding to the quaternary carbon furthest from the CrMo6 core, could be observed since the other carbon environments are too close to the Cr3+ centre and, therefore, did not give rise to noticeable peaks. Nevertheless, 27Al NMR gave a single peak at 15.64 ppm due to the functionalised AlMo6 POM core, which also closely matches with the 27Al NMR spectrum of ALA. Additionally, the characteristic peaks of the Anderson–Evans structure in the IR spectrum at 937 (ν MoO, s), 916 (ν MoO, s), 899 (ν MoO, s) and 650 (ν Mo–O–Mo, vs.) cm−1 confirm the integrity of the POM structures (Fig. S35†). Moreover, the UV-Vis absorbance spectrum (Fig. S38†) shows that ACA consists of a 1:2 combination of C and Avia covalent attachment since it displayed a higher absorbance than a 1:2 mixture of C and A with the same total concentration while the normalised spectra matched fairly well, with only slight differences due to the functionalisation of A. Furthermore, similar to DLD and DCD, single crystals suitable for X-ray diffraction could not be obtained for ALA or ACA, in accordance with previous reports,35 yet the elemental composition of the isolated compounds displayed the expected stoichiometry as shown in the ESI.†
Thermal stability
The thermal stability of all new HPOMs reported in this work was evaluated by Thermogravimetric Analysis (TGA) under an N2 atmosphere between 30 °C and 630 °C. All HPOMs were observed to remain stable below 180 °C, which is comparable to the previously reported stability of similar structures.34,35,60 Between 180 and 500 °C a weight loss was observed in the TGA profiles of the HPOMs, indicating their decomposition due to loss of the organic components (Fig. 7). L was the least stable as it began to significantly decompose just above 180 °C while, in contrast, C was much more stable and only started decomposing above 250 °C. Furthermore, bis-functionalisation to form C results in a higher thermal stability compared to the corresponding mono-functionalised form and having Cr3+ as the heteroatom improves the stability with respect to the equivalent bis-functionalised structure with Mn3+.34 The higher thermal stability of the Anderson–Evans building block was also reflected in the higher stability of ACA compared to ALA, each of which had similar stabilities compared to their respective building-blocks. However, the highest stability was observed for the DLD and DCD, which only decomposed above 250 °C. As previously observed for the other HPOMs based on CrMo6, DCD was also slightly more stable than DLD with no major decomposition until 265 °C. In addition, DCD demonstrated a higher thermal stability than the homogenous “dumbbell” and “triangular” POM–POM hybrids based on P2V3W15 POMs covalently linked with purely organic linkers, which showed significant decomposition just above 200 °C.30,33 Moreover, for all POM–POM hybrids the residual weight at 630 °C after complete loss of the organic part was in good agreement with the calculated values for the loss of all TBA counter ions and the dP linker as shown in Table 1, further confirming their structures. Hence, the number of counter-cations for all compounds was determined based on a multi-technique approach combining NMR, elemental analysis and TGA.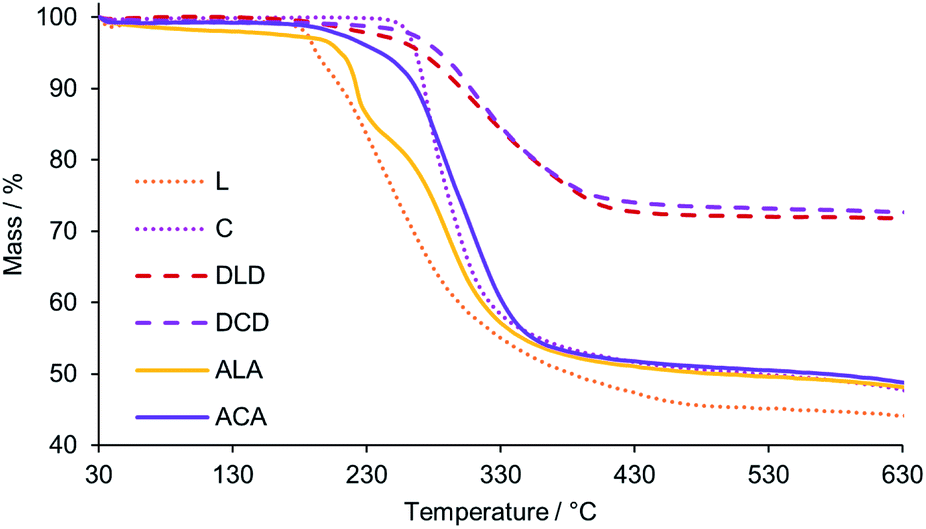 | ||
| Fig. 7 TGA profiles (top) showing the weight loss with increasing temperature for the building blocks, L and C, and their corresponding POM–POM hybrids: DLD, DCD, ALA, and ACA. | ||
| HPOM | Calcd res. mass/% | Observed res. mass/% |
|---|---|---|
| DLD | 71.89 | 71.85 |
| DCD | 71.39 | 72.63 |
| ALA | 49.59 | 48.01 |
| ACA | 51.1 | 48.63 |
Conclusions
In summary, a set of novel metal-oxo cluster building-blocks were synthesised for the formation of a wide variety of hybrid structures combining different inorganic cluster types. The first examples of POM–POM hybrid structures combining the Wells–Dawson POM structure with Lindqvist and Anderson–Evans POMs were successfully obtained as confirmed by multinuclear NMR, IR, UV-Vis, MS and elemental analysis. Furthermore, it was also shown that this method can be used to link together Anderson–Evans and Lindqvist POMs, as well as Anderson–Evans structures with different heteroatoms. All POM triads were thermally stable until 180 °C, and showed in general higher thermal stability compared to the HPOM structures linked by only organic ligands. Moreover, the proposed post-functionalisation strategy for the formation of these heterometallic hybrid structures composed of multiple inorganic components is relatively simple and results in high yields without the need of additional reagents. The same strategy could be applied to an even wider range of metal oxo-clusters and, in principle, it could also be employed to produce more complex structures such as polymers containing different POMs linked together, as well as asymmetric POM–POM hybrids containing two or more different types of POM structures (POM1–POM2–POM3---POMn) allowing for the controlled design of compounds with the desired composition. For example, the Anderson–Evans structure can accommodate a large variety of different central heteroatoms, including most first-row transition metals and even some p-block elements (e.g., X = Mn, Fe, Co, Ni, Cu, Zn, Ga, Al, Te, etc.), resulting in the formation of either bis- or mono-functionalised structures that can be combined to tune the properties of the material based on the choice of the metal centre.15 The Lindqvist structure also offers additional structural variability since it can be mono-, bis-, tri- or tetra- functionalised, depending on the oxidation state and composition of the metal centres ([V3Mo3O16{(OCH2)3C-R}]; [V6O19-3x−y(OH)y{(OCH2)3C-R}x] where x = 2, 3, and 4).14,17,61 Furthermore, the vanadium and nickel substituted Keggin structures ([XM12O40]n−) can also be functionalised with polyols in the same way as for the Wells–Dawson structure.62,63 In addition, tailoring of the structures formed via this strategy is not limited to the inorganic components, since employing different polyols with other functional groups as organic linkers could also be used for added functionality. Therefore, the novel strategy proposed here based on metal-oxo clusters functionalised with polyols could be used as a platform for the combination of a wide variety of other metal-oxo structures via covalent attachment. Ideally, this strategy could also be extended to functional surfaces or nanoparticles and even to the non-covalent self-assembly of species via the free triol groups.21–26,64,65Data availability
Crystallographic data for compounds C and L has been deposited at the CCDC and allocated the deposition numbers CCDC-2114104 and 2114105, respectively. All other data supporting the results discussed in this work is available within the paper and its ESI.†Author contributions
D. E. S. M., M. A. M., A. V. A., and S. L. performed the synthesis and characterisation of the novel compounds. The cryo-MS measurements were performed and interpreted with the assistance of I. I. B. The crystal structures were solved by L. V. M. T. N. P. V. helped with the design and interpretation of the experiments. The manuscript was written through contributions of D. E. S. M., M. A. M., A. V. A., S. L. and T. N. P. V. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank KU Leuven (grant C14/19/076) and the Research Foundation Flanders (FWO) for funding. D. E. S. M. (83523/1183021N) and M. A. M. (203282/1279721N) thank the FWO for fellowships. L. V. M. thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035.Notes and references
- L. Rozes and C. Sanchez, Chem. Soc. Rev., 2011, 40, 1006–1030 RSC
.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC
-
M. Hutin, M. H. Rosnes, D.-L. Long and L. Cronin, in Comprehensive Inorganic Chemistry II, Elsevier, 2013, vol. 2, pp. 241–269 Search PubMed
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed
-
Y.-F. Song, Polyoxometalate-Based Assemblies and Functional Materials, Springer International Publishing, Cham, 2018, vol. 176 Search PubMed
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC
- M. Stuckart and K. Y. Monakhov, Chem. Sci., 2019, 10, 4364–4376 RSC
- Y. Ji, L. Huang, J. Hu, C. Streb and Y.-F. Song, Energy Environ. Sci., 2015, 8, 776–789 RSC
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed
-
O. Linnenberg, A. Kondinski and K. Y. Monakhov, in Supramolecular Systems: Chemistry, Types and Applications, 2017, pp. 39–66 Search PubMed
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS
- Y. Hou and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 11823–11830 CrossRef CAS
- A. V Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC
- S. Vanhaecht, T. Quanten and T. N. Parac-Vogt, Inorg. Chem., 2017, 56, 3095–3101 CrossRef CAS PubMed
- S. Vanhaecht, T. Quanten and T. N. Parac-Vogt, Dalton Trans., 2017, 46, 10215–10219 RSC
- S. Vanhaecht, J. Jacobs, L. Van Meervelt and T. N. Parac-Vogt, Dalton Trans., 2015, 44, 19059–19062 RSC
- R. J. Errington, S. S. Petkar, B. R. Horrocks, A. Houlton, L. H. Lie and S. N. Patole, Angew. Chem., Int. Ed., 2005, 44, 1254–1257 CrossRef CAS PubMed
- B. J. S. Johnson and A. Stein, Inorg. Chem., 2001, 40, 801–808 CrossRef CAS PubMed
- A. W. H. Lee and B. D. Gates, Langmuir, 2016, 32, 7284–7293 CrossRef CAS PubMed
- A. W. H. Lee and B. D. Gates, Langmuir, 2017, 33, 8707–8715 CrossRef CAS PubMed
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC
- H. J. H. Kang, R. F. Ali, M. T. Y. Paul, M. J. Radford, I. Andreu, A. W. H. Lee and B. D. Gates, Chem. Commun., 2019, 55, 10452–10455 RSC
- M.-P. Santoni, A. K. Pal, G. S. Hanan, M.-C. Tang, K. Venne, A. Furtos, P. Ménard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48, 200–202 RSC
- M. S. Centellas, M. Piot, R. Salles, A. Proust, L. Tortech, D. Brouri, S. Hupin, B. Abécassis, D. Landy, C. Bo and G. Izzet, Chem. Sci., 2020, 11, 11072–11080 RSC
- H. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., 2000, 112, 1841–1844 CrossRef
- C. P. Pradeep, M. F. Misdrahi, F.-Y. Li, J. Zhang, L. Xu, D.-L. Long, T. Liu and L. Cronin, Angew. Chem., 2009, 121, 8459–8463 CrossRef
- P. Yin, T. Li, R. S. Forgan, C. Lydon, X. Zuo, Z. N. Zheng, B. Lee, D. Long, L. Cronin and T. Liu, J. Am. Chem. Soc., 2013, 135, 13425–13432 CrossRef CAS PubMed
- T. Auvray, M.-P. Santoni, B. Hasenknopf and G. S. Hanan, Dalton Trans., 2017, 46, 10029–10036 RSC
- C. P. Pradeep, F.-Y. Li, C. Lydon, H. N. Miras, D.-L. Long, L. Xu and L. Cronin, Chem.–Eur. J., 2011, 17, 7472–7479 CrossRef CAS PubMed
- Q. Xu, S. Yuan, L. Zhu, J. Hao and Y. Wei, Chem. Commun., 2017, 53, 5283–5286 RSC
- H. Ai, Y. Wang, B. Li and L. Wu, Eur. J. Inorg. Chem., 2014, 2014, 2766–2772 CrossRef CAS
- K. P. Sullivan, W. A. Neiwert, H. Zeng, A. K. Mehta, Q. Yin, D. A. Hillesheim, S. Vivek, P. Yin, D. L. Collins-Wildman, E. R. Weeks, T. Liu and C. L. Hill, Chem. Commun., 2017, 53, 11480–11483 RSC
- M. Lu, Y. Wei, B. Xu, C. F.-C. Cheung, Z. Peng and D. R. Powell, Angew. Chem., Int. Ed., 2002, 41, 1566–1568 CrossRef CAS
- J. L. Stark, A. L. Rheingold and E. A. Maatta, J. Chem. Soc., Chem. Commun., 1995, 1165–1166 RSC
- J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS PubMed
- J. Zhang, P. Yin, J. Hao, F. Xiao, L. Chen and Y. Wei, Chem.–Eur. J., 2012, 18, 13596–13599 CrossRef CAS PubMed
- A. Bayaguud, J. Zhang, R. N. N. Khan, J. Hao and Y. Wei, Chem. Commun., 2014, 50, 13150–13152 RSC
- J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599–3615 RSC
- H. An, J. Zhang, S. Chang, Y. Hou and Q. Zhu, Inorg. Chem., 2020, 59, 10578–10590 CrossRef CAS PubMed
- A. Bayaguud, K. Chen and Y. Wei, CrystEngComm, 2016, 18, 4042–4045 RSC
- I. Fa Bamba, C. Falaise, G. K. Gbassi, P. Atheba, M. Haouas and E. Cadot, J. Coord. Chem., 2020, 73, 2567–2578 CrossRef CAS
- C. Qin and J. Zubieta, Inorg. Chim. Acta, 1992, 198–200, 95–110 CrossRef
- P. Wu, J. Chen, P. Yin, Z. Xiao, J. Zhang, A. Bayaguud and Y. Wei, Polyhedron, 2013, 52, 1344–1348 CrossRef CAS
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2003, 2406–2412 CrossRef
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 2002, 1081–1087 CrossRef
- C. G. Lin, W. Chen, D. L. Long, L. Cronin and Y. F. Song, Dalton Trans., 2014, 43, 8587–8590 RSC
- J. Zhang, Y. Huang, J. Zhang, S. She, J. Hao and Y. Wei, Dalton Trans., 2014, 43, 2722–2725 RSC
- J. Zhang, Z. Zhao, J. Zhang, S. She, Y. Huang and Y. Wei, Dalton Trans., 2014, 43, 17296–17302 RSC
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS
- L. E. Briand, H. J. Thomas and G. T. Baronetti, Appl. Catal., A, 2000, 201, 191–202 CrossRef CAS
- C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS PubMed
- J. Iwahara, C. D. Schwieters and G. M. Clore, J. Am. Chem. Soc., 2004, 126, 12800–12808 CrossRef CAS PubMed
- L. Vandebroek, Y. Mampaey, S. Antonyuk, L. Van Meervelt and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2019, 2019, 506–511 CrossRef CAS
- D. E. Salazar Marcano, S. Lentink, M. A. Moussawi and T. N. Parac-Vogt, Inorg. Chem., 2021, 60, 10215–10226 CrossRef CAS PubMed
- V. Shivaiah and S. K. Das, J. Chem. Sci., 2005, 117, 227–233 CrossRef CAS
- H. Jia, Q. Li, A. Bayaguud, S. She, Y. Huang, K. Chen and Y. Wei, Sci. Rep., 2017, 7, 12523 CrossRef PubMed
- S. Spillane, R. Sharma, A. Zavras, R. Mulder, C. A. Ohlin, L. Goerigk, R. A. J. O'Hair and C. Ritchie, Angew. Chem., Int. Ed., 2017, 56, 8568–8572 CrossRef CAS PubMed
- D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS
- X.-X. Li, S.-T. Zheng, W.-H. Fang and G.-Y. Yang, Inorg. Chem. Commun., 2011, 14, 1541–1545 CrossRef CAS
- C. Sperandio, J. Rodriguez and A. Quintard, Chem. Sci., 2020, 11, 1629–1635 RSC
- J. Teichert and M. Ruck, Eur. J. Inorg. Chem., 2019, 2019, 2267–2276 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2114104 and 2114105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06326j |
| This journal is © The Royal Society of Chemistry 2022 |