
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric addition of Grignard reagents to ketones: culmination of the ligand-mediated methodology allows modular construction of chiral tertiary alcohols†
Claudio
Monasterolo
 *,
Ryan
O'Gara
,
Saranna E.
Kavanagh
,
Sadbh E.
Byrne
,
Bartosz
Bieszczad
,
Orla
Murray
,
Michael
Wiesinger
,
Rebecca A.
Lynch
,
Kirill
Nikitin
and
Declan G.
Gilheany
*
*,
Ryan
O'Gara
,
Saranna E.
Kavanagh
,
Sadbh E.
Byrne
,
Bartosz
Bieszczad
,
Orla
Murray
,
Michael
Wiesinger
,
Rebecca A.
Lynch
,
Kirill
Nikitin
and
Declan G.
Gilheany
*
Centre for Synthesis and Chemical Biology, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: claudio.monasterolo@ucd.ie; declan.gilheany@ucd.ie
First published on 20th April 2022
Abstract
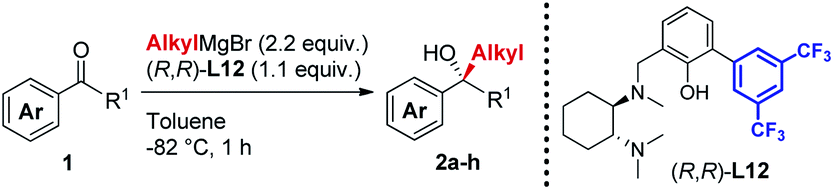
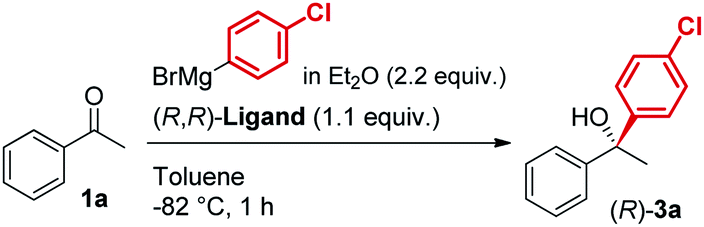
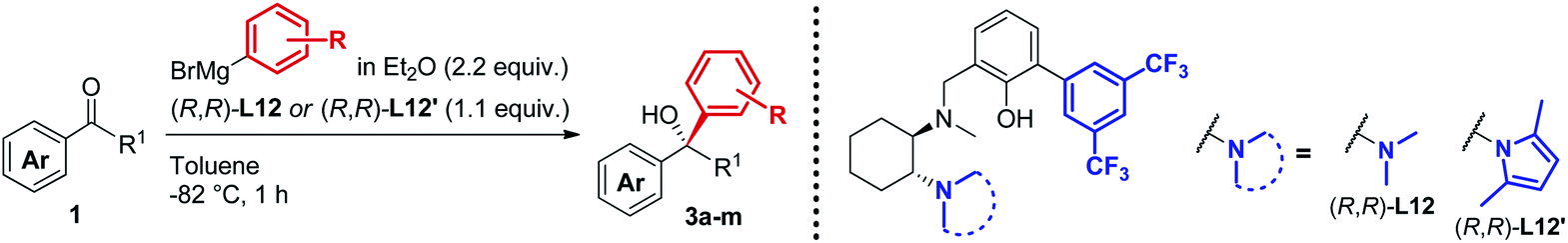
A new class of biaryl chiral ligands derived from 1,2-diaminocyclohexane (1,2-DACH) has been designed to enable the asymmetric addition of aliphatic and, for the first time, aromatic Grignard reagents to ketones for the preparation of highly enantioenriched tertiary alcohols (up to 95% ee). The newly developed ligands L12 and L12′ together with the previously reported L0 and L0′ define a set of complementary chiral promoters, which provides access to the modular construction of a broad range of structurally diverse non-racemic tertiary alcohols, bearing challenging quaternary stereocenters. The present advancements bring to completion our asymmetric Grignard methodology by expanding the scope to aromatic organomagnesium reagents, while facilitating its implementation in organic synthesis thanks to improved synthetic routes for the straightforward access to the chiral ligands. The synthetic utility of the method has been demonstrated by the development of a novel and highly enantioselective formal synthesis of the antihistamine API clemastine via intermediate (R)-3a. Exploiting the power of the 3-disconnection approach offered by the Grignard synthesis, (R)-3a is obtained in 94% ee with ligand (R,R)-L12. The work described herein marks the finalization of our ongoing effort towards the establishment of an effective and broadly applicable methodology for the asymmetric Grignard synthesis of chiral tertiary alcohols.
Introduction
Since Victor Grignard's early work at the beginning of the 20th century,1 organomagnesium reagents have been established as an essential tool in organic chemistry, gaining a central role in a broad range of transformations and synthetic applications,2 and remain the privileged subject of ongoing research interest today.3 Among the variety of applications offered by organomagnesium species,4 the asymmetric 1,2-addition of Grignard reagents to prochiral ketones has emerged as a powerful strategy providing access to non-racemic tertiary alcohols,5 widespread structural motifs and valuable synthetic intermediates for the preparation of complex chiral scaffolds, bioactive targets and APIs.6 Owing to the synthetic flexibility of the 3-disconnection approach, the Grignard synthesis enables the straightforward and modular construction of molecular complexity from simple and cheap substrates (Scheme 1). However, the 1,2-addition of organomagnesium nucleophiles to ketones entails substantial challenges, due to the reduced reactivity of ketones and the presence of competing enolization pathways,7 which have posed a major limit to the development of effective synthetic methods to date. Recent reports successfully overcame these issues by modulating the reactivity of the Grignard reagent by the action of additional metal species,8 producing highly enantioenriched tertiary alcohols. Despite the excellent stereoselectivities achieved by the mixed-metals strategies, the need for additional metal catalysts and additives affects the atom economy of the processes and limits the potential efficiency and sustainability offered by the Grignard approach. Consequently, the development of effective ligand-mediated, single metal based methods (i.e. requiring only a chiral ligand, without the need for additional metals and/or additives) is highly desirable, as it would give access to greener and more cost-effective methodologies featuring improved atom-economy and reduced waste production, in accordance with the modern green-chemistry requirements.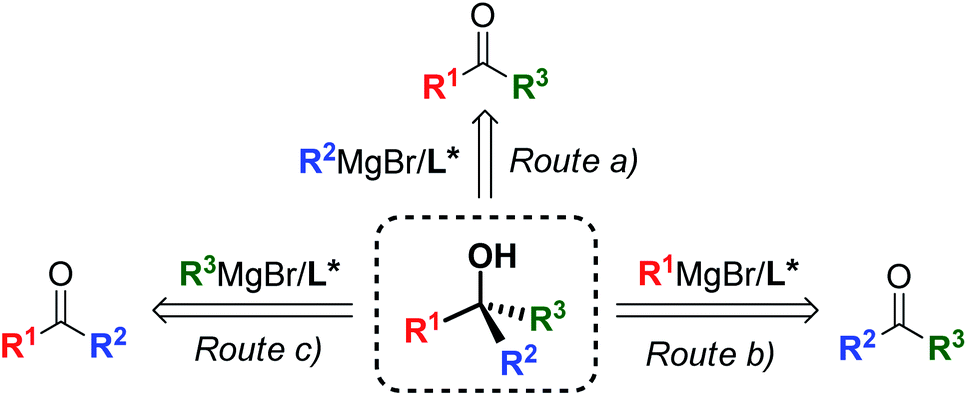 | ||
| Scheme 1 Modular construction of chiral tertiary alcohols via ligand-mediated, single metal based asymmetric Grignard synthesis. | ||
As a result of the challenges associated with asymmetric Grignard synthesis, only a few useable direct methods have been reported to date. The first example by Weber and Seebach in 1992,9 making use of TADDOL as chiral ligand, has been followed by the BINOL-mediated strategy developed by Osakama and Nakajima in 2016.10 Both methods suffered from drawbacks which limited their general implementation, such as the need for extremely low temperatures and limited scope. To overcome these limitations, we designed a conceptually new N,N,O-tridentate ligand L0 derived from trans-1,2-diaminocyclohexane (1,2-DACH), which enabled the development of an effective ligand-mediated, single metal based approach to the asymmetric Grignard synthesis of chiral tertiary alcohols, via an operationally simple and cost-effective process (Scheme 2).11 Fine tuning of L0 led to the design of the N-pyrrole analogue L0′12 enhancing the stereoselectivity and broadening the scope to new classes of tertiary alcohols (Scheme 2a). The application of the asymmetric Grignard method to the synthesis of valuable targets provided new effective and step-economical entries to a range of challenging products, significantly shortening the previously established preparations, including: (i) natural products, e.g. tocopherols (vitamin-E) and tocotrienols13 and sesquiterpenes (gossonorol, boivinianin A and boivinianin B);14 (ii) chiral O-heterocycles (2,2-disubstituted THFs and THPs) and (iii) biologically active compounds, such as the anticonvulsant and hypnotic γ-ethyl-γ-phenyl butyrolactone and the antimalarial yingzhaosu C.10,12 Nevertheless, the efficient implementation of aromatic organomagnesium nucleophiles has remained elusive to date. This prompted us to undertake a comprehensive investigation of the ligand structure, with the aim to deepen our understanding of its impact over the stereoselectivity, and to address the compatibility issues hampering the use of aromatic Grignard reagents. Herein we report the development of a new class of biaryl ligands L12 and L12′ which, in addition to the previously established L0 and L0′, complete our asymmetric Grignard methodology allowing the modular synthesis of a broad range of chiral tertiary alcohols. The new ligands enabled, for the first time, the effective implementation of aromatic organomagnesium nucleophiles, expanding the scope and synthetic utility of the strategy (Scheme 2b). The study is complemented by the development of new short, protection-free and purification-free synthetic routes for the preparation of the chiral ligands, to promote the straightforward implementation of the method in organic synthesis.
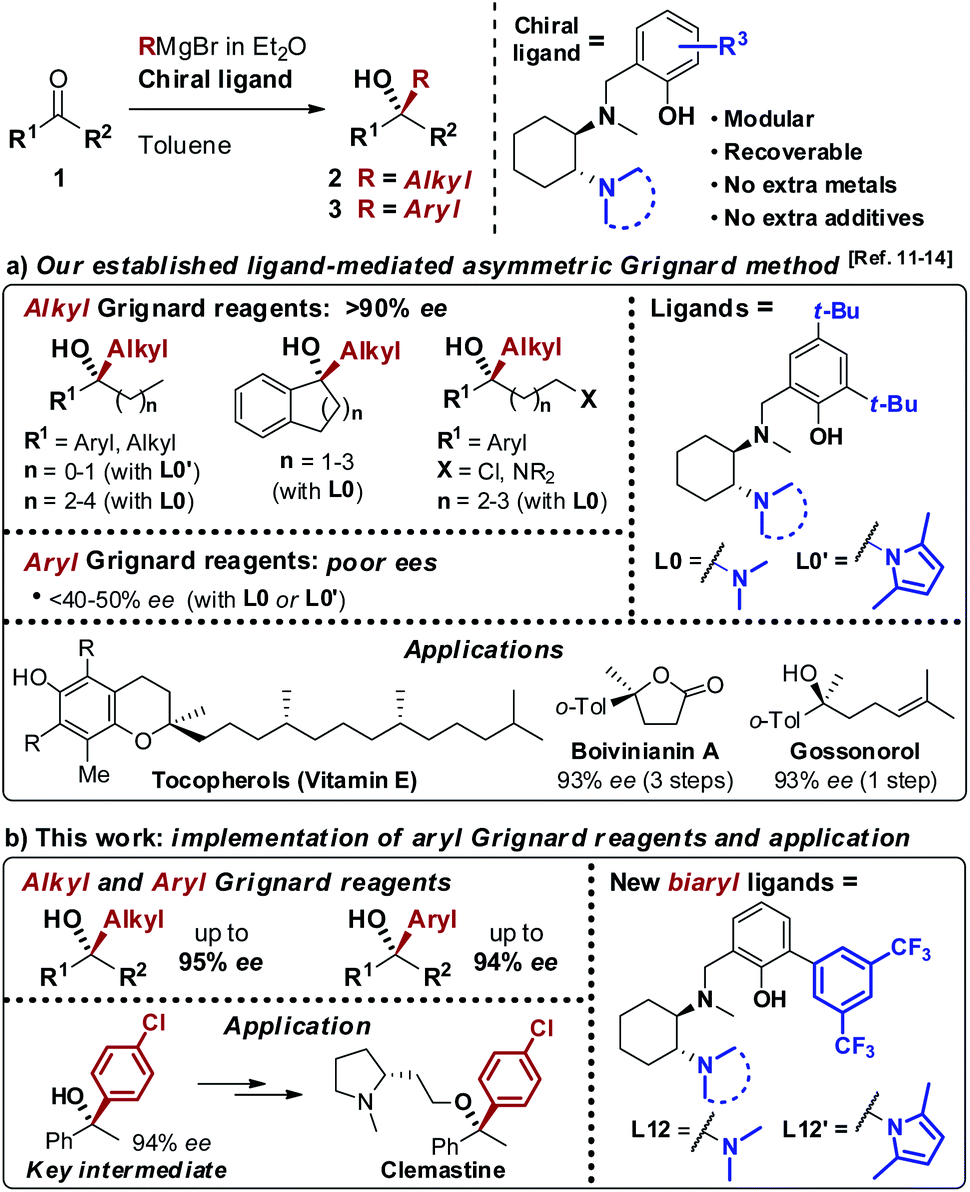 | ||
| Scheme 2 (a) Our asymmetric Grignard synthesis of chiral tertiary alcohols mediated by DACH-derived ligands L0 and L0′ and application to the preparation of valuable natural products and APIs. (b) Newly designed biaryl ligands L12 and L12′ for the enantioselective addition of both alkyl and aryl Grignard reagents to ketones. | ||
Results and discussion
At the onset of the study, we focused on the preparation of the new DACH-derived ligands to be screened L1–L14. First, we developed an improved one-pot synthetic route for the preparation of L0–L3 from commercially available 1,2-DACH hydrochloride 4·HCl.15 The new route was designed to provide an operationally simple and scaleable access to DACH-derived ligands, by means of a shorter and more cost-effective process, avoiding the need for N-protecting groups and the purification of intermediates. Reductive amination of 4·HCl with substituted salicylaldehydes 5a–d, followed by exhaustive Eschweiler–Clarke methylation of intermediates 6a–d, efficiently delivered ligands L0–L3 in two steps (Scheme 3, Route (a)), significantly shortening the previous four-step preparations.16 Next, we prepared the new biaryl ligands L7–L14. To get quick access to a range of ortho-aryl substituted analogues, a divergent route was designed involving the late-stage diversification of the common aryl bromide intermediate L6 (Scheme 3, Route (b)).17 A convenient one-pot sequence was optimized to deliver halogenated intermediates L4–L6via reductive amination of N-Boc-1,2-DACH 7 with halogenated salicylaldehydes 5e–g (X = F, Cl, Br), followed by deprotection and N-methylation, without the need for purification. Subsequent palladium-catalyzed cross-coupling of aryl bromide L6 with different aryl boronic acids delivered the desired ligands L7–L14, obtained in high overall yields over 2 steps (Scheme 3, Route (b)). With ligands L1–L14 in hand, we proceeded with the screening of their efficacy in the asymmetric Grignard reaction.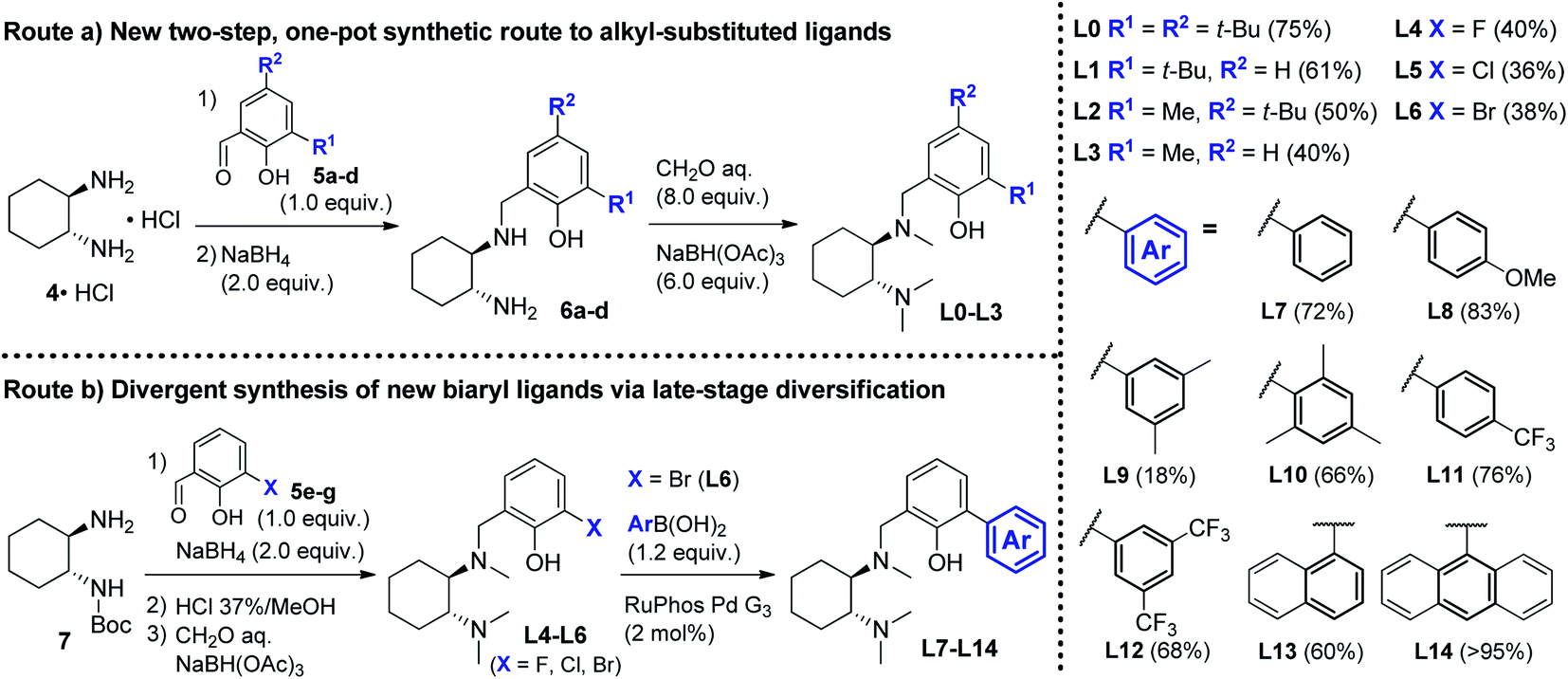 | ||
| Scheme 3 Synthetic routes for the preparation of DACH-derived chiral ligands. Route (a) new two-step, one-pot synthesis of alkyl-substituted ligands L0–L3. Route (b) preparation of halogenated ligands L4–L6 (X = F, Cl, Br) and divergent synthesis of novel biaryl ligands L7–L14via late-stage diversification of the common intermediate L6via cross-coupling arylation with aryl boronic acids. | ||
In line with our previous studies the addition of ethylmagnesium bromide to acetophenone 1a to produce chiral tertiary alcohol 2a was used as a model reaction (Table 1). Unlike ortho-alkyl substituted L1–L3 and ortho-halogenated L4–L6 ligands, the introduction of a biaryl functionality in L7–L14 enhanced the efficiency of the transformation, with the 3,5-bis(trifluoromethyl)phenyl substituted ligand (R,R)-L12 resulting the most effective, delivering the desired tertiary alcohol (S)-2a in 87% ee (Table 1, entry 14). The high selectivity achieved by L12 in the model reaction prompted us to further investigate the scope of the transformation. To our delight, the new ligand provided high to excellent enantioselectivities over a range of functionalized short-chain phenones (Table 2). With the aim to overcome the selectivity issues we previously observed in the addition of aryl Grignard reagents to ketones using ligand L0,18 the new ligands L7–L14 were screened in the addition of para-chlorophenylmagnesium bromide to acetophenone 1a (Table 3).19 While most of the new ligands failed to improve the enantioselectivity of the process (Table 3, entries 1–5 and 8), the use of (R,R)-L12 showed unprecedented enantiocontrol, delivering the product (R)-3a in 89% ee (Table 3, entry 6). Taking into consideration the previously demonstrated positive effect exerted by replacing the N-dimethyl group of ligand L0 with N-pyrrole L0′,12 we extended the investigation of (R,R)-L12 to the N-pyrrole analogue (R,R)-L12′ (Table 3, entry 7).20 Thus, a range of substituted aryl Grignard reagents were reacted with functionalized phenones in the presence of (R,R)-L12 and (R,R)-L12′ (Table 4). Both ligands demonstrated a remarkable improvement in asymmetric induction compared to the modest level of stereocontrol of L0 and L0′, providing access to diaryl-alkyl chiral tertiary alcohols 3a–m in good to excellent enantioselectivities (Table 4). Complementary behaviors were observed in relation to the electronic demand of the aryl Grignard reagents and the nature of the functional groups present on the aromatic ring, as L12 gave better results in the case of electron withdrawing groups, while L12′ best performed in combination with electron donating substituents (Table 4).21
|
|
|||
|---|---|---|---|
| Entry | (R,R)-ligand | (S)-2a conversionb (%) | (S)-2a eeb (%) |
a Acetophenone 1a 0.5 M in dry toluene (0.1 mmol), ligand (R,R)-L0–L14 (0.11 mmol), EtMgBr 3.0 M in Et2O (0.22 mmol), in dry toluene (1.2 mL). The Grignard reagent in Et2O was diluted with dry toluene (0.4 mL, ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6), see ESI for detailed procedures.
b Conversion and ee determined by HPLC analysis on chiral stationary phase of the crude reaction mixture after work-up. The crude mixture contained only product and starting material. :6), see ESI for detailed procedures.
b Conversion and ee determined by HPLC analysis on chiral stationary phase of the crude reaction mixture after work-up. The crude mixture contained only product and starting material.
|
|||
| 1 | L0 | 55 | 78 |
| 2 | L0′ | 80 | 93 |
| 3 | L1 | 77 | 23 |
| 4 | L2 | 47 | 67 |
| 5 | L3 | 43 | 67 |
| 6 | L4 | 40 | 51 |
| 7 | L5 | 53 | 63 |
| 8 | L6 | 50 | 65 |
| 9 | L7 | 50 | 81 |
| 10 | L8 | 70 | 79 |
| 11 | L9 | 50 | 85 |
| 12 | L10 | 63 | 82 |
| 13 | L11 | 40 | 78 |
| 14 | L12 | 63 | 87 |
| 15 | L13 | 70 | 81 |
| 16 | L14 | 20 | 50 |
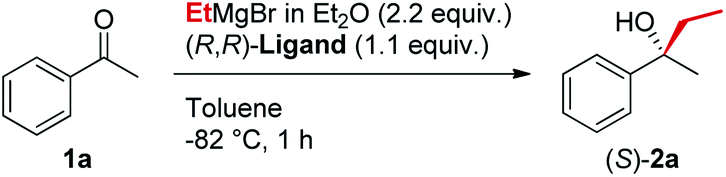
|
|
|---|
| a Procedure as for Table 1, EtMgBr 3.0 M in Et2O, MeMgBr 3.0 M in Et2O. b Isolated yields, unless otherwise stated. c Enantiomeric excess determined by HPLC analysis on chiral stationary phase of the crude reaction mixture after work-up. d Absolute configuration determined by comparison with literature. e Conversion determined by 1H-NMR analysis of the crude reaction mixture. f Absolute configuration determined by X-ray crystallographic analysis of the carbamate derivative, see ESI for details. |
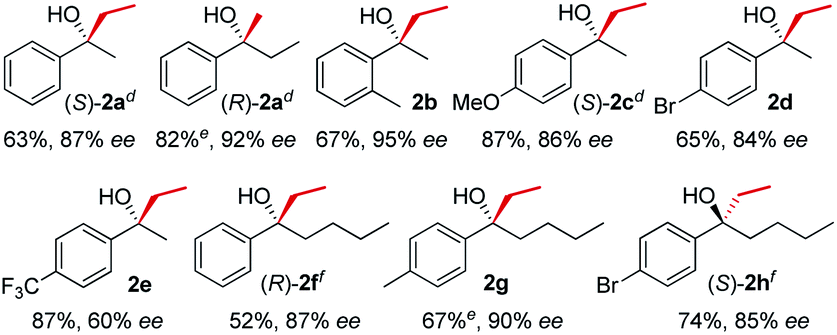
|
|
|
|||
|---|---|---|---|
| Entry | (R,R)-ligand | (R)-3a conversionb (%) | (R)-3a eeb (%) |
| a Procedure as for Table 1, using para-chlorophenylmagnesium bromide 1.0 M in Et2O. b Conversion and ee determined by HPLC analysis on chiral stationary phase of the crude reaction mixture after work-up. The crude mixture contained only product and returned starting material. c Isolated yield. n.d. = not determined. | |||
| 1 | L0 | 51 | 21 |
| 2 | L7 | 46 | 39 |
| 3 | L8 | 38 | 43 |
| 4 | L9 | n.d. | 31 |
| 5 | L11 | n.d. | 36 |
| 6 | L12 | 78c | 89 |
| 7 | L12′ | 44 | 82 |
| 8 | L14 | n.d. | 14 |
|
|
|---|
| a Procedure as for Table 1. b Isolated yields, unless otherwise stated. c Enantiomeric excess determined by HPLC analysis on chiral stationary phase of the crude reaction mixture after work-up. d Absolute configuration determined by comparison with previously reported results. e Using alternative order of addition optimized for the synthesis of clemastine, see ESI. f Conversion determined by 1H-NMR analysis of the crude reaction mixture after work-up. |
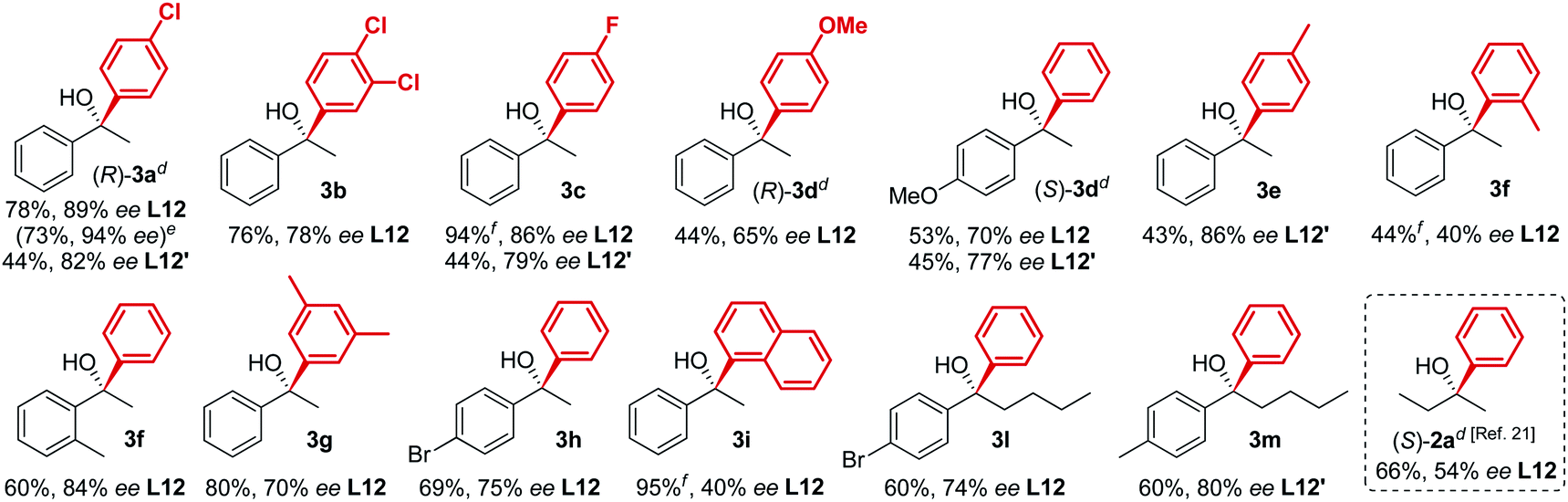
|
The new biaryl ligands L12 and L12′ for the first time enabled excellent asymmetric control over both alkyl and aryl Grignard nucleophiles, and thus allowed the implementation of the methodology in the preparation of novel classes of bioactive targets, as shown by the synthesis of the previously inaccessible highly enantioenriched alcohol (R)-3a, key intermediate in the synthesis of the antihistamine drug clemastine (Scheme 4).22 Following a comprehensive screening of conditions, addition of para-chlorophenylmagnesium bromide to acetophenone 1a, in the presence of (R,R)-L12, delivered the product (R)-3a in 94% ee and 73% isolated yield (Scheme 4, Route (c)).23 The newly developed highly enantioselective formal synthesis of clemastine further demonstrated the broad versatility and applicability of the methodology to the preparation of valuable APIs, and its compatibility with a wide range of structurally diverse ketone substrates and Grignard reagents.24,25
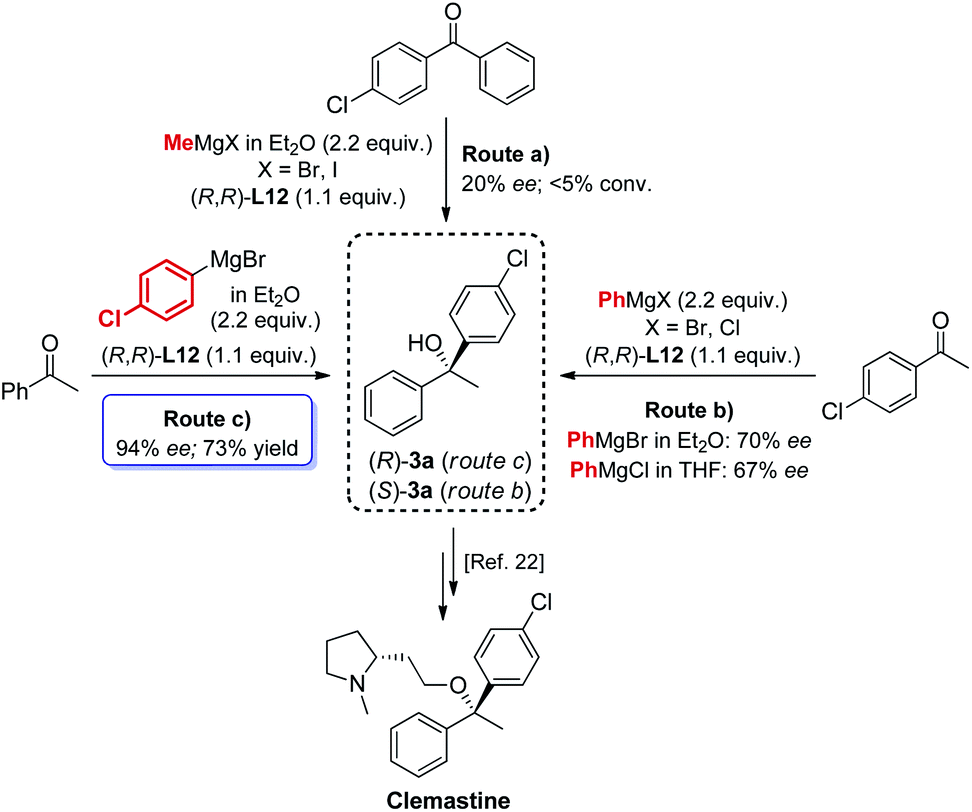 | ||
| Scheme 4 Modular access to highly enantioenriched (R)-3a, key intermediate in the synthesis of the antihistamine API clemastine. | ||
Excited about the results delivered by the novel class of N,N,O-tridentate ligands derived from 1,2-DACH, from the early design of L0 and the following structural variations to L0′, to the most recent development of the biaryl analogues L12 and L12′ presented herein, we were curious to identify the source of the enantioselectivity. At this point, we decided to step back and consider possible mononuclear and dinuclear hexacoordinated complexes involving the historical L0.11 While the original rationale behind the design of the DACH tridentate ligands invoked the dinuclear model, the formation of highly active mononuclear ligand-magnesium species with the participation of electron-donating Lewis basic and anionic functionalities, i.e. N,N,O-tridentate ligand, halide, ethereal solvent and the carbonyl of the ketone substrate, was an alternative worth considering (Scheme 5).
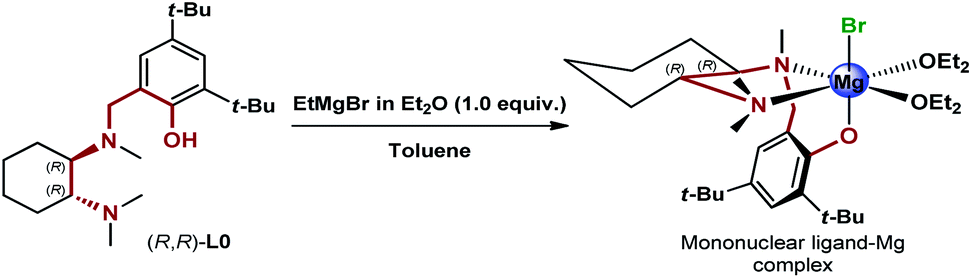 | ||
| Scheme 5 Proposed mononuclear ligand–Mg complex generated via deprotonation of (R,R)-L0 with EtMgBr. | ||
In order to shed light on the nature of the chiral active species involved in the asymmetric Grignard reaction, the structure of the ligand–magnesium complex derived from L0 and EtMgBr solution in Et2O, was investigated. The X-ray crystallographic analysis of the material obtained by deprotonation of (S,S)-L0 with EtMgBr, in toluene and in the absence of ketone, indicated the presence of an unexpected pseudo-mirror symmetrical trinuclear complex C1 (molecular structure shown in Fig. 1, see ESI† for further details)26 whereby the two hexacoordinated magnesium centres were bridged by bromide anions and featured the coordination with two separate deprotonated L0 moieties. Careful examination of C1 reveals that the stereochemical configurations of the benzylated chiral nitrogens N1 of each L0 (“key nitrogens”), are opposite in such a fashion that the hydroxylated aromatic groups on the two L0 moieties have the same orientation, pointing “up” from the nominal plane of the DACH ring. Although the structure of complex C1 crystallized from toluene may not be representative of the executive active species operating in solution, it clearly shows fac-coordination of the N,N,O-tridentate ligand L0 to the hexacoordinated magnesium centre and the bridging halide, in good agreement with our original hypothesis.11 The variability of the key nitrogen N1 stereochemical configuration, however, points towards potential equilibration of multiple ligand–magnesium species in solution, owing to the molecular flexibility of the DACH-derived moieties. Indeed, 1H-NMR analysis of the mixture resulting from the deprotonation of (R,R)-L0 in toluene-d8, via treatment with 1.0 equivalent of EtMBr in Et2O, clearly demonstrated the presence of multiple species deriving from L0 in solution. The 1H–1H COSY correlations observed for the non-equivalent benzylic methylene protons (N1–CH2Ar), indicated the generation of at least five L0-derived species following deprotonation (see ESI† for details). Unfortunately, it was not possible to get further structural information due to extensive signal overlapping, further complicated by the presence of residual Et2O deriving from the Grignard reagent solution.
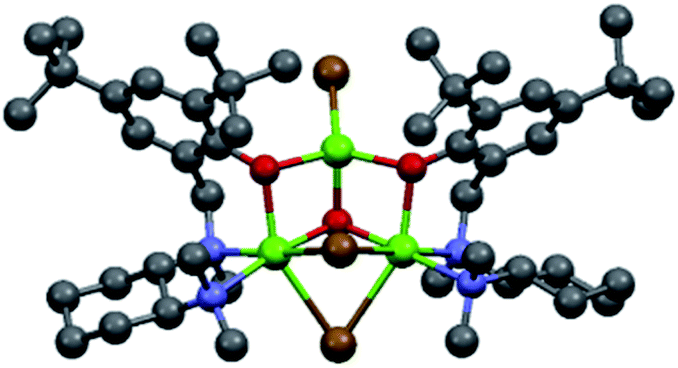 | ||
| Fig. 1 Molecular structure of the ligand–Mg complex C1, precipitated from toluene following deprotonation of (S,S)-L0 with EtMgBr in Et2O. | ||
These results suggested a substantially complex system being at play in the ligand-mediated asymmetric Grignard process, characterized by the concomitant presence of multiple potentially active ligand–magnesium species. On the basis of the findings provided by the X-ray and NMR analyses, we propose the establishment of an equilibrium in solution involving several stereoisomeric ligand-magnesium mononuclear complexes, along with possible dinuclear species. To this end, a further DFT computational study of mononuclear complexes C2, featuring magnesiated ligand (R,R)-L0 and Et2O solvent molecules, showed that the arrangement of the bromide anion opposite to the phenolic oxygen represents the energetically more stable orientation (Scheme 6). It also transpired that, among the two trans-species in equilibrium, R(N1)-C2 and S(N1)-C2, the S(N1) configuration of the key chiral nitrogen is more stable than the R(N1) (ca. 2 kcal mol−1 at the 6-31G* level, see ESI†), thus making complex S(N1)-C2 a potentially likely intermediate in the stereoselective process (Fig. 2).
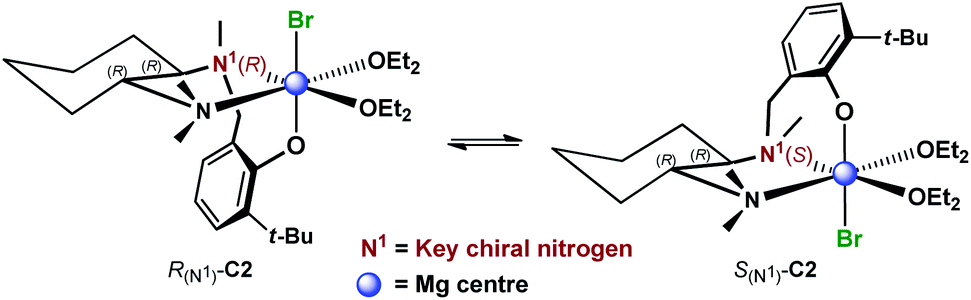 | ||
| Scheme 6 Proposed equilibrium involving diastereomeric mononuclear ligand–Mg complexes C2, resulting from deprotonation of (R,R)-L0 with EtMgBr in Et2O. | ||
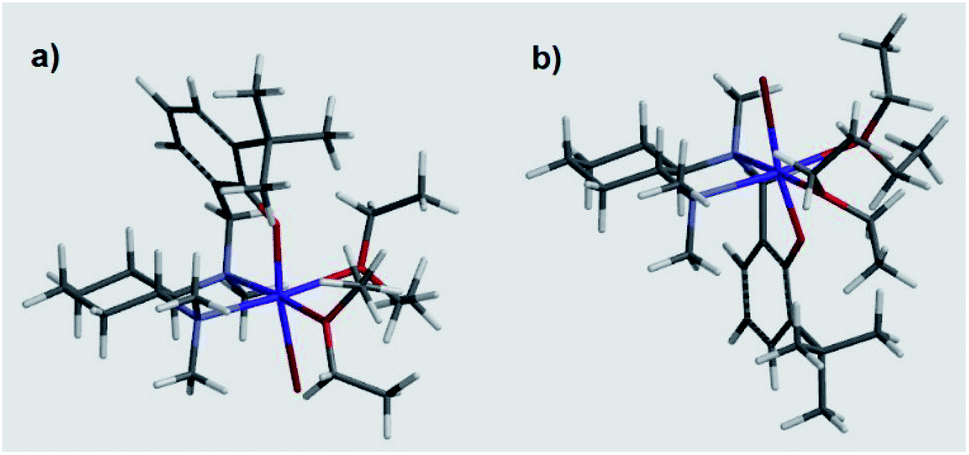 | ||
| Fig. 2 DFT calculated molecular structures of R(N1)-C2 (a) and S(N1)-C2 (b). | ||
The following step of the mechanism may involve coordination of the ketone substrate to the magnesium centre of C2 with replacement of one Et2O molecule, leading to diastereomeric ligand–Mg–ketone intermediates whose stability is directly dictated by the sterics of the large ortho-tert-butyl functionality present in L0. Subsequent attack by external Grignard nucleophile, via asymmetric 1,2-addition to the Mg-coordinated ketone, would then produce the intermediate tert-alkoxide products in unequal stereochemical ratio. A more dedicated computational study of the mechanistic aspects underpinning the asymmetric Grignard synthesis of chiral tertiary alcohols, aimed at elucidating the possible intermediate species participating in the reaction mechanism, is currently underway and will be reported in due course.
Conclusions
In conclusion, we have developed a new class of N,N,O-tridentate chiral ligands for single-metal controlled asymmetric addition of Grignard reagents to ketones. The design methodology involves the introduction of a biaryl fragment in the ligand's structure, crucial for the control of stereoselectivity. The new biaryl ligands L7–L14 have been derived from 1,2-DACH through a divergent synthetic route involving efficient late-stage diversification of the common aryl bromide precursor L6via palladium-catalyzed cross-coupling with a range of aryl boronic acids. The biaryl analogues L12 and L12′, featuring an ortho-3,5-(bis-trifluoromethyl)phenyl substitution, emerged as new efficient and versatile chiral ligands, providing high to excellent enantioselectivities in the asymmetric 1,2-addition of alkyl Grignard reagents to ketones. Most crucial, L12 and L12′ resulted equally effective in promoting the addition of challenging aromatic Grignard nucleophiles, as opposed to the limited success characterizing the previous attempts with ligands L0 and L0′. These findings enabled, for the first time, the implementation of aromatic organomagnesium reagents in our asymmetric Grignard methodology and, in turn, to expand its scope to previously inaccessible targets. The optimization of the ligands preparation completed the method's development, by means of new one-pot synthetic sequences featuring improved yields and step-economy, together with easier procedures and faster operations. The new routes make use of cheap and widely available materials, ensuring the straightforward and cost-effective preparation and application of the DACH-ligands. The present study marks the finalization of the stoichiometric version of our asymmetric Grignard method. The performances achieved herein establish it as a general and broadly applicable strategy for the synthesis of chiral tertiary alcohols, enabling the modular and highly enantioselective construction of challenging tetrasubstituted stereocenters without the need for additional metal catalysts or additives. It is our belief that the asymmetric Grignard approach will facilitate the preparation of valuable bioactive targets and foster the underdeveloped use of chiral tertiary alcohols as versatile substrates and synthetic intermediates. Further studies are currently underway for the development of an effective catalytic version of the method.Data availability
Synthetic procedures, NMR spectra, X-ray crystallographic data and supplementary computational details are provided as ESI.†Author contributions
Investigation: ROG, SEK, CM, SEB, BB, OM, MW, RAL. Methodology: ROG, SEK, KN, CM. Data curation: CM, SEK, KN. Supervision: CM, DGG. Visualization: CM. Writing – original draft: CM. Writing – review&editing: CM, SEK, KN, DGG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Irish Research Council is acknowledged for PhD funding for SEK (grant GOIPG/2017/1179) and SEB (grant GOIPG/2018/2256). Science Foundation Ireland is acknowledged for PhD funding for ROG through the Synthesis & Solid State Pharmaceutical Centre (grant 12/RC/2275) and for post-doctoral funding for CM (grant SFI-IFA/19/7427). Arran Chemicals, Athlone, Ireland is gratefully acknowledged for gifts of resolved trans-1,2-DACH.Notes and references
- V. Grignard, Compt. Rend. Hebd. Séances Acad. Sci., 1900, 130, 1322 CAS.
- P. Knochel, Organomagnesium and Organozinc Chemistry, in Organometallics in Synthesis, Third Manual, ed. Schlosser, M., Wiley & Sons, Hoboken, NJ, 2013; pp. 223–372 Search PubMed.
- For a recent comprehensive study of the mechanism of the Grignard reaction, see: R. M. Peltzer, J. Gauss, O. Eisenstein and M. Cascella, J. Am. Chem. Soc., 2020, 142, 2984 CrossRef CAS PubMed.
- (a) P. Knochel, A. Gavryushin and K. Brade, Functionalized organomagnesium compounds: Synthesis and reactivity, in The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Jonh Wiley & Sons, 2008, pp. 511–594 Search PubMed; (b) H. G. Richey, Grignard Reagents: New Developments, ed. H. G. Richey, Wiley & Sons, Chichester, U.K., 2000 Search PubMed; (c) B. J. Wakefield, Organomagnesium Methods in Organic Synthesis, in Best Synthetic Methods, ed. B. J. Wakefield, Academic Press, 1995 Search PubMed.
- (a) J. F. Collados, R. Solà, S. R. Harutyunyan and B. Maciá, ACS Catal., 2016, 6, 1952 CrossRef CAS; (b) M. R. Luderera, W. F. Bailey, M. R. Luderer, J. D. Fair, R. J. Dancer and M. Bech Sommer, Tetrahedron: Asymmetry, 2009, 20, 981 CrossRef; (c) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873 RSC.
- (a) K. C. Nicolaou and T. Montagnon, Molecules That Changed the World, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) J. G. de Vries, Important Pharmaceuticals and Intermediates, in Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005, pp. 25–50 Search PubMed.
- (a) A. G. Pinkus and W. C. Servoss, J. Chem. Soc., Perkin Trans. 2, 1979, 1600 RSC; (b) E. C. Ashby, J. Laemmle and H. M. Neumann, Acc. Chem. Res., 1974, 7, 272 CrossRef CAS.
- (a) A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard, Angew. Chem., Int. Ed., 2012, 51, 3164 CrossRef CAS PubMed; (b) A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Chem. Commun., 2012, 48, 1478 RSC; (c) E. Fernández-Mateos, B. Maciá and M. Yus, Eur. J. Org. Chem., 2014, 2014, 6519 CrossRef.
- B. Weber and D. Seebach, Angew. Chem., Int. Ed., 1992, 31, 84 CrossRef.
- K. Osakama and M. Nakajima, Org. Lett., 2016, 18, 236 CrossRef CAS PubMed.
- B. Bieszczad and D. G. Gilheany, Angew. Chem., Int. Ed., 2017, 56, 4272 CrossRef CAS PubMed.
- S. Kavanagh and D. G. Gilheany, Org. Lett., 2020, 22, 8198 CrossRef CAS PubMed.
- B. Bieszczad and D. G. Gilheany, Org. Biomol. Chem., 2017, 15, 6483 RSC.
- C. Monasterolo, H. Müller-Bunz and D. G. Gilheany, Chem. Sci., 2019, 10, 6531 RSC.
- The use of 1,2-DACH monohydrochloride 4·HCl as substrate, in place of the N-Boc protected analogue 7, reduced the overall costs of the process and minimized the generation of waste over the course of the preparation of the ligands.
- It is worth noting that the new synthetic route for ligands L0–L3 could be performed one-pot and without the need for purification of the intermediate species.
- The synthesis of L4–L6 was facilitated by the use of starting material 7, due to the emergence of solubility issues affecting the halogenated intermediates.
- The previously attempted use of alkyl-functionalized ligands L0 and L0′ in the addition of aryl Grignard reagents to ketones delivered the corresponding tertiary alcohols 3 in <40–50% ee, see ESI† for further details.
- The addition of para-chlorophenylmagnesium bromide to acetophenone 1a was selected as the model transformation for the study, in light of the potential application of the product 3a as key intermediate in the synthesis of the API clemastine.
- The N-pyrrole ligand analogue (R,R)-L12′ was prepared in three steps from 4·HCl, see ESI†.
- Replacing the aryl alkyl ketone substrates 1 with methyl ethyl ketone 8, gave a reasonable control of the enantioselectivity, producing the tertiary alcohol (S)-2a in 54% ee, thus demonstrating the ability of L12 to exert effective chiral recognition in challenging substrates as 8, with almost symmetrical structures, i.e. methyl vs. ethyl.
- (a) For the first preparation of the four stereoisomers of clemastine, see: A. Ebnöther and H. Weber, Helv. Chim. Acta, 1976, 59, 2462 CrossRef PubMed; (b) For clemastine synthesis via invertive aryl migration, see: A. M. Fournier, R. A. Brown, W. Farnaby, H. Miyatake-Ondozabal and J. Clayden, Org. Lett., 2010, 12, 2222 CrossRef CAS PubMed; (c) via lithiation/borylation, see: J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (d) via rhodium-catalysis, see: L. Huang, J. Zhu, G. Jiao, Z. Wang, X. Yu, W. Deng and W. Tang, Angew. Chem., Int. Ed., 2016, 55, 4527 CrossRef CAS PubMed; (e) via phosphoramide-Zn(II) complex, see: M. Hatano, T. Miyamoto and K. Ishihara, Org. Lett., 2007, 9, 4535 CrossRef CAS PubMed.
- The development of the new formal synthesis of clemastine involved an extensive screening of conditions. The nature of the Grignard reagent RMgX was investigated, with regard to the halide functionality (X = Cl, Br and I) and the ethereal solvent. The Grignard reagents MeMgBr, MeMgI, para-chlorophenylMgBr and PhMgBr were used as Et2O solutions, while PhMgCl was tested as THF solution, delivering comparable results. Finally, the study identified an alternative order of addition for the preparation of the intermediate (R)-3a, enhancing the enantioselectivity from 89% ee to 94% ee, see ESI† for further details.
- Of particular interest is the possibility to access both enantiomers (R)-3a and (S)-3a with the use of a single ligand (R,R)-L12, by simply changing the synthetic disconnection, while keeping unchanged the stereochemical configuration of the source of chirality which, conversely, commonly accounts as a critical requirement in asymmetric synthesis.
- It is worth to note that the determination of the absolute configuration of several tertiary alcohols products (via X-ray crystallographic analysis or by comparison with HPLC and optical rotation data reported in the literature, Tables 2 and 4), showed the asymmetric induction of the new biaryl ligands L12/L12′ to be consistent with the outcomes provided by the parent DACH-ligands L0 and L0′, thus substantiating the previously proposed selectivity model (see ref. 12–14). In addition, the study validated the use of the method we previously developed for the determination of the absolute configuration of challenging chiral tertiary alcohols, via X-ray analysis of 13, solid para-bromophenyl carbamate O-derivative of alcohol 2h, see ESI† for further details. For the early development of the chiral tertiary alcohol O-derivatization strategy, see ref. 14. Crystallographic data for (R)-13 have been deposited with the Cambridge Crystallographic Data Centre [CCDC 2158994].
- Crystallographic data for C1 have been deposited with the Cambridge Crystallographic Data Centre [CCDC 2158472].
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, detailed procedures for the synthesis of ligands and the asymmetric Grignard synthesis, additional previous results, spectroscopic and structural data, NMR spectra. CCDC 2158472 and 2158994. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc06350b |
| This journal is © The Royal Society of Chemistry 2022 |