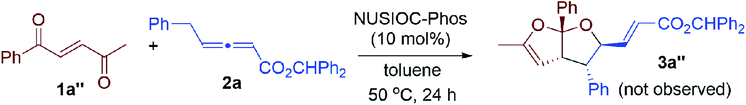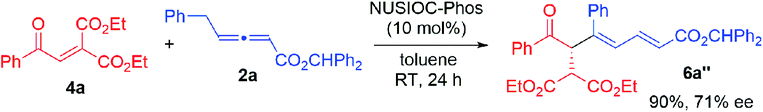DOI:
10.1039/D1SC06364B
(Edge Article)
Chem. Sci., 2022,
13, 3161-3168
Phosphine-catalyzed divergent domino processes between γ-substituted allenoates and carbonyl-activated alkenes†
Received
16th November 2021
, Accepted 10th February 2022
First published on 11th February 2022
Abstract
Highly enantioselective and chemodivergent domino reactions between γ-substituted allenoates and activated alkenes have been developed. In the presence of NUSIOC-Phos, triketone enone substrates smoothly reacted with γ-substituted allenoates to form bicyclic furofurans in good yields with high stereoselectivities. Alternatively, the reaction between diester-activated enone substrates and γ-substituted allenoates formed chiral conjugated 1,3-dienes in good yields with excellent enantioselectivities. Notably, by employing substrates with subtle structural difference, under virtually identical reaction conditions, we were able to access two types of chiral products, which are of biological relevance and synthetic importance.
Introduction
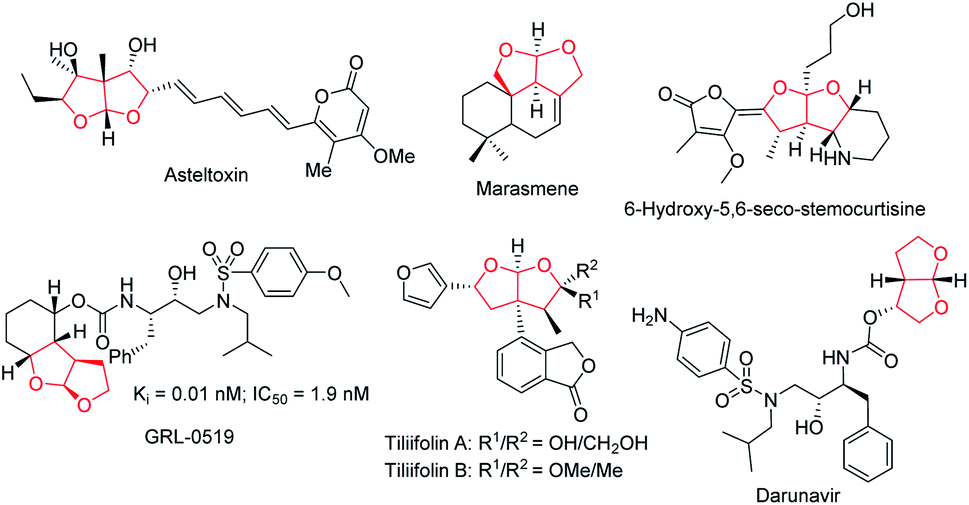
Ever since Lu's seminal report1 on phosphine-catalyzed [3 + 2] annulation and Trost's disclosure2 of phosphine-mediated “Umpolung” addition in the mid 1990s, the past few decades have seen tremendous progress of asymmetric phosphine catalysis.3 Phosphine-mediated reactions have attracted attention from synthetic chemists and have been widely used for the creation of a broad range of molecular architectures.4 Phosphine-catalyzed annulation reactions are the most common reaction types, and various [3 + 2],5 [4 + 1],6 and [4 + 2]7 annulation reactions are found to be useful for building up five- or six-membered ring systems. Moreover, phosphine-catalyzed γ-additions,8 Michael additions,9 Morita–Baylis–Hillman (MBH) reactions,10 and Rauhut–Currier (RC) reactions11 have also been shown to be synthetically useful. In our continuous pursuit of phosphine catalysis, we are particularly interested in the effective asymmetric creation of chiral scaffolds of biological significance. Fused bicyclic chiral acetals are widely present in nature. Some representative examples are illustrated in Fig. 1, including asteltoxin,12 marasmene,13 6-hydroxy-5,6-seco-stemocurtisine,14 GRL-0519,15 tiliifolin A/B,16 and darunavir.17 The biological activities of these compounds are often dependent on the substitution pattern of the chiral acetal moiety, thus the asymmetric synthesis of these molecules is highly desirable. While there are a number of reports on the synthesis of benzofused acetals,18 efficient catalytic asymmetric preparation of bicyclic furofurans was barely explored.19 We recently introduced a C2-symmetric chiral phosphine catalyst NUSIOC-Phos, which effectively promoted the formation of tricyclic γ-lactams via an enantioselective domino process.20 We therefore wondered whether a tandem reaction employing suitable substrates may be developed to access chiral bicyclic furofuran structural motifs.
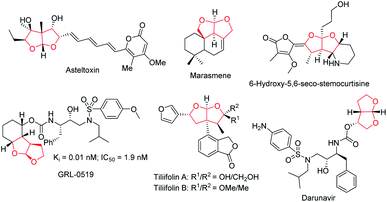 |
| | Fig. 1 Natural products and bioactive molecules containing a fused bicyclic acetal. | |
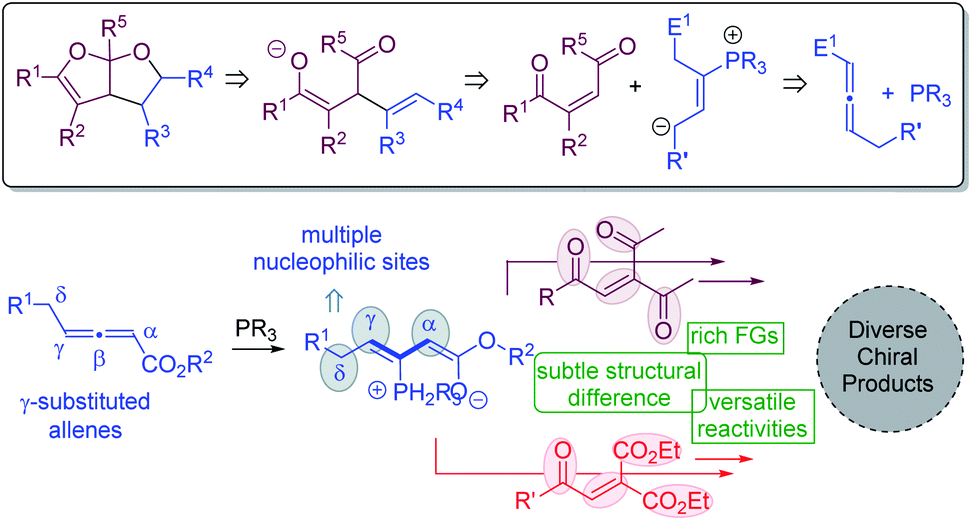
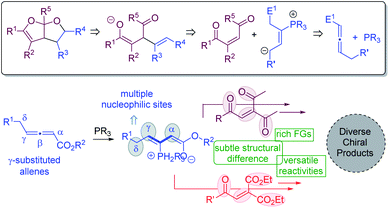
The Paal–Knorr synthesis is a classic method for the preparation of furans from 1,4-diketones upon treatment with strong mineral acids.21 We envisioned that a modified Paal–Knorr synthesis may be utilized to construct bicyclic furofuran cores (Fig. 2). If the oxygen anion resulting from the initial enolate-induced ring closure is trapped by an intramolecular electrophilic reaction partner, then bicyclic furofurans can be created. To generate the enolate species for the furofuran formation, substrates bearing a double bond flanked by two carbonyl groups appear to be ideal. The zwitterionic intermediate derived from a phosphine catalyst and an allene substrate is anticipated to play two distinct roles: (1) to undergo a nucleophilic attack on the enone to form the key enolated species; (2) to electrophilically trap the oxygen anion to form bicyclic furofurans. Moreover, if γ-substituted allenes22 are employed, upon phosphine attack, an intermediate with three potential nucleophilic sites at the α, γ, and δ-positions is generated, which will add in great flexibility in creating molecular complexity. For the alkene substrates bearing 1,4-dicarbonyl groups, different carbonyl functional groups may be utilized, and such subtle structural differences may lead to different reactivities, making it feasible to create diverse chiral products. Herein, we document phosphine-catalyzed divergent domino reactions between γ-substituted allenoates and alkenes that are activated by carbonyl groups. Hinging on the nature of electron-withdrawing groups, the reaction proceeded through different pathways, and either fused bicyclic furofurans or 1,3-conjugate dienes were obtained in high yields with good to excellent stereoselectivities.
 |
| | Fig. 2 Our hypothesis. | |
Results and discussion
Construction of bicyclic furofurans
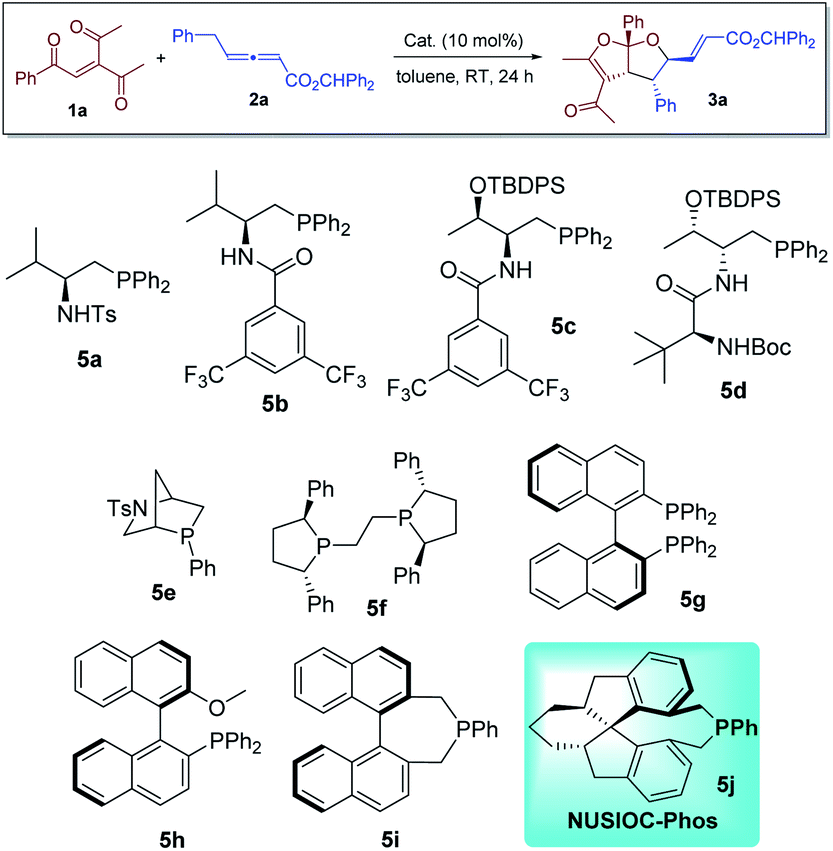
We first evaluated the reaction between 3-acetyl-1-aryl-2-pentene-1,4-diones 1a and γ-substituted allenoate 2a in the presence of different phosphine catalysts (Table 1). Amino acid-based bifunctional phosphines bearing different hydrogen bond donors (5a–5d) failed to yield the desired product (entries 1–4). While other mono-functional phosphines (5e–5h) at most led to the formation of the desired products in low yields (entries 5–8), we were delighted to discover that our earlier developed23 NUSIOC-Phos (5j) was a good catalyst; bicyclic furofuran 3a was obtained in moderate yield with good diastereoselectivity and excellent enantioselectivity (entry 10). When the reaction was performed at 50 °C, the chemical yield was substantially improved, with virtually maintained stereoselectivities (entry 11). Different solvents were also screened (entries 12–17), and toluene remained to the solvent of choice.
Table 1 Reaction screeninga
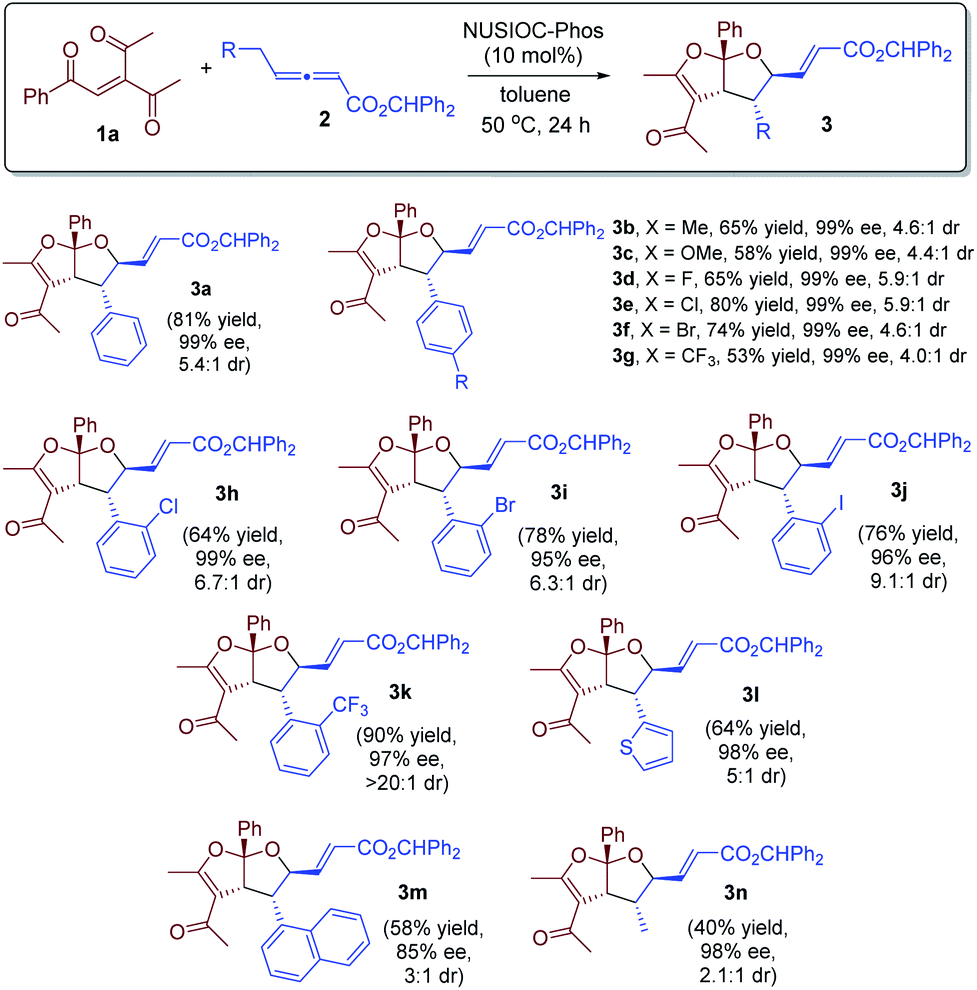
With the optimized reaction conditions in hand, we investigated the scope of this domino process by employing different γ-substituted allenoates 2, and the results are summarized in Scheme 1. γ-Benzyl-substituted allenoates bearing a para-substituent on the phenyl are well-tolerated, and the desired products were obtained in high yields with excellent enantioselectivities and good diastereoselectivities (65% to 81% yields, 99% ee, up to >5.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 3a–3g). The allenoates containing an ortho-substituted phenyl proved to be excellent substrates; excellent ee values and higher dr ratios were attainable (3h–3j). In particular, when an ortho-CF3-phenyl containing γ-benzylic allenoate was employed, only one diastereoisomer was observed (3k). Allenoates bearing a thiophene (3l) or a naphthyl substituent (3m) were also found to be suitable substrates, although the diastereoselectivity was slightly lower for the latter. Moreover, the substituent of γ-allenoates could also be aliphatic, and the desired acetal was obtained in 40% yield with 98% ee and 2:1 dr (3n). The absolute configurations of the fused bicyclic furofurans were assigned on the basis of X-ray crystallographic analysis of 3d.24
:1 dr, 3a–3g). The allenoates containing an ortho-substituted phenyl proved to be excellent substrates; excellent ee values and higher dr ratios were attainable (3h–3j). In particular, when an ortho-CF3-phenyl containing γ-benzylic allenoate was employed, only one diastereoisomer was observed (3k). Allenoates bearing a thiophene (3l) or a naphthyl substituent (3m) were also found to be suitable substrates, although the diastereoselectivity was slightly lower for the latter. Moreover, the substituent of γ-allenoates could also be aliphatic, and the desired acetal was obtained in 40% yield with 98% ee and 2:1 dr (3n). The absolute configurations of the fused bicyclic furofurans were assigned on the basis of X-ray crystallographic analysis of 3d.24
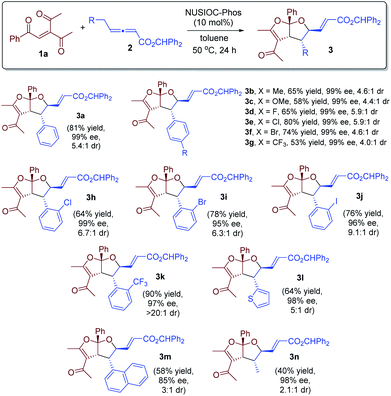 |
| | Scheme 1 Reaction scope. Conditions: 1a (0.1 mmol), 2 (0.15 mmol), and NUSIOC-Phos (0.01 mmol) in toluene (1.0 mL) at 50 °C for 24 hours. The dr values were determined by crude 1H NMR analysis. Yields given are the isolated yields of the main isomer. The ee values were determined by HPLC analysis on a chiral stationary phase. For 3g–3k, 20% catalyst and 40% 4-methoxyphenol were used. For 3n, 20% catalyst was employed. | |
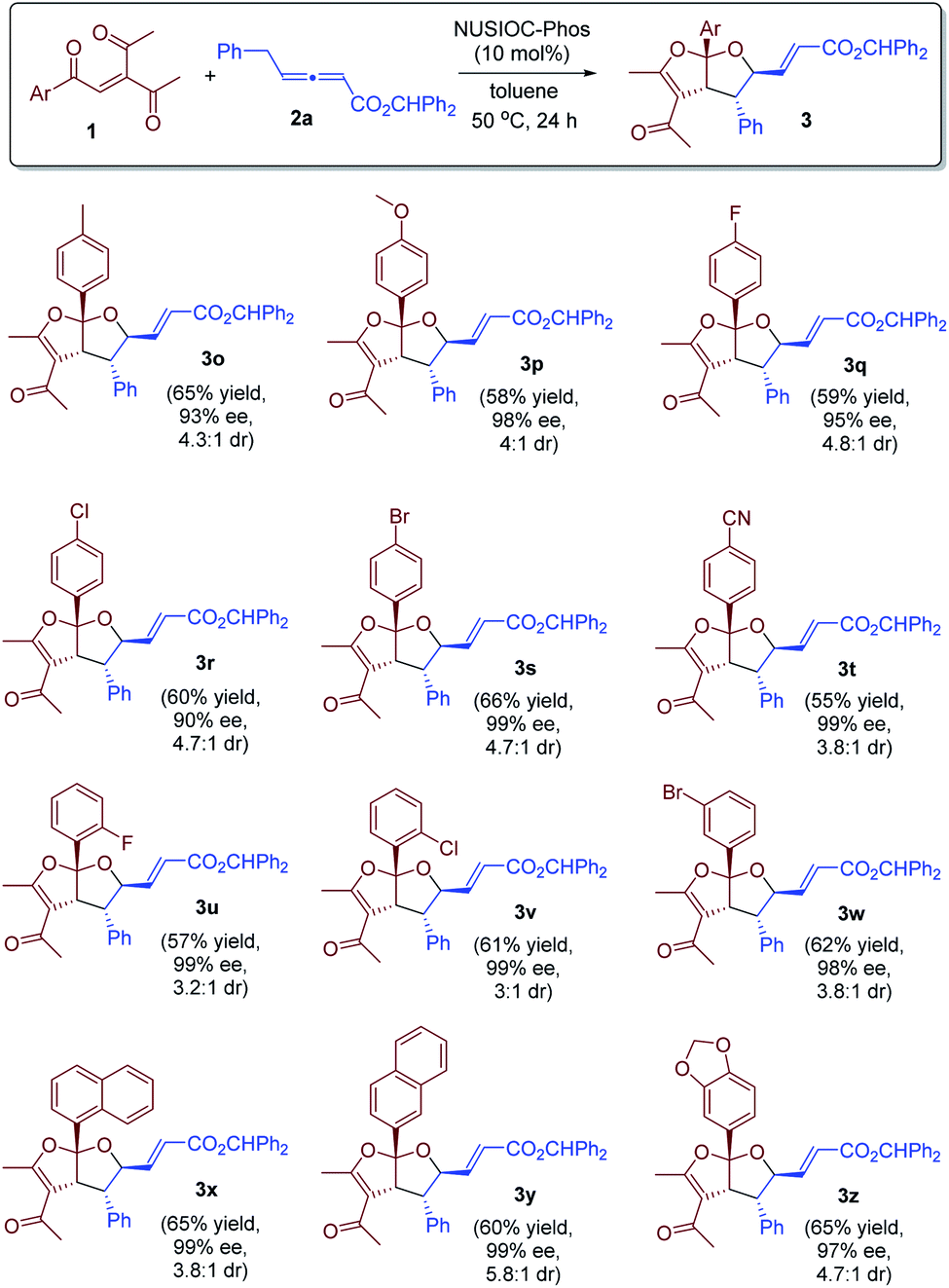
We further examined the reaction scope by utilizing a range of enone substrates 1 bearing different substituents (Scheme 2). In general, the electronic nature and substitution pattern of different phenyl groups had little influence on the reaction, and the desired bicyclic furofurans were obtained in reasonable yields, with excellent enantioselectivities and good diastereoselectivities (3o–3w). Moreover, enones containing 1-naphthyl, 2-naphthyl, or 5-piperonyl groups were all suitable substrates, and the desired products were derived in satisfactory yields and with high stereoselectivities (3x–3z).
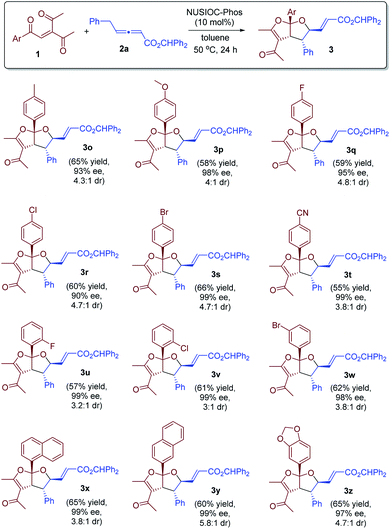 |
| | Scheme 2 Further scope exploration. Conditions: 1 (0.1 mmol), 2a (0.15 mmol), 4-methoxyphenol (0.02 mmol), and NUSIOC-Phos (0.02 mmol) in toluene (1.0 mL) at 50 °C for 24 hours. The dr values were determined by crude 1H NMR analysis. Yields given are the isolated yields of the main isomer. The ee values were determined by HPLC analysis on a chiral stationary phase. | |
Synthesis of conjugated 1,3-dienes
From the synthetic viewpoint, divergent pathways from the same/similar substrates, under the catalysis of similar/same catalysts, would be quite ideal. In this context, we have keen interest in developing divergent asymmetric synthetic approaches.25 At the outset, we wanted to examine the domino reaction of γ-substituted allenoates with other carbonyl-activated alkenes. Such structural variation is mechanistically interesting; for instance, the ester-derived enolate will unlikely undergo intramolecular cyclization with another ester moiety and thus offer opportunities for deriving different products.
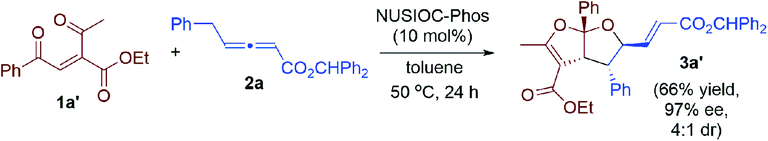
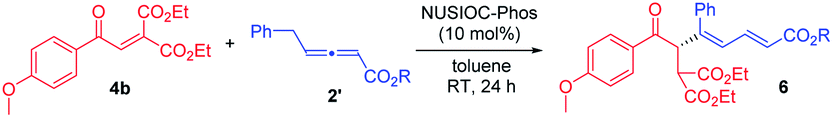
We examined this domino process by employing other types of activated alkenes, under the optimal conditions that we have established for the synthesis of bicyclic furofurans. When one ketone moiety was replaced by an ester group, both 1a′-Z and 1a′-E were found to be suitable for the reaction, forming acetal product 3a′ in slightly decreased yield with similar stereoselectivities (eqn (1)). However, if one ketone moiety was deleted (1a′′), the corresponding bicyclic furofuran was not formed (eqn (2)). Interestingly, when enone 4a bearing two ester groups was treated with allene 2a in the presence of NUSIOC-Phos, conjugated 1,3-diene 6a was obtained in 90% yield and with 71% ee (eqn (3)). Apparently, a different mechanistic pathway is in operation, due to the structural and electronic differences of the activating groups in the enones. The conjugated 1,3-dienes are useful structural motifs that are often found in natural products and bioactive molecules,26 and they are also of special importance in polymer chemistry and materials science.27 Synthetically, 1,3-dienes are extremely valuable, and thus intensive efforts have been devoted to their efficient synthesis.28 We were then wondering whether we could establish an efficient asymmetric synthetic approach to access chiral building blocks containing conjugated 1,3-dienes.
| |  | (1) |
| |  | (2) |
| | 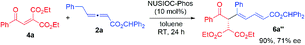 | (3) |
We examined the reaction between diester-activated alkene 4b and allenoates 2′, under the catalysis of NUSIOC-Phos, and the results are summarized in Table 2. As we anticipated that certain proton transfer processes are likely to be involved during the formation of 1,3-diene products (vide infra for the proposed reaction mechanism), we thus evaluated the influence of adding a number of proton donors to the reaction system. The addition of phenol led to a substantial increase in enantioselectivity (entry 2). While p-methoxylphenol and benzoic acid were both effective, o-methoxylphenol appeared to be slightly better (entries 3–5). A quick solvent screening confirmed that toluene was most ideal (entries 6–9). We next explored allenoates with different ester moieties and discovered that the employment of phenyl allenoate led to best enantioselectivity (entries 10–13). When the reaction was performed at lower temperature under the above optimal conditions, the desired 1,3-diene 5 was obtained in 80% yield with 93% ee (entry 14).
Table 2 Optimization for the δ-carbon Michael addition/isomerization reactiona
|
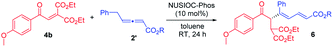
|
| Entry |
Solvent |
Additive |
R |
Yieldb [%] |
eec [%] |
|
Reactions were performed with 4b (0.1 mmol), 2′ (0.15 mmol), the additive (0.05 mmol) and NUSIOC-Phos (0.01 mmol) in the solvent specified (1.0 mL) at 25 °C for 24 h.
Isolated yield.
Determined by HPLC analysis on a chiral stationary phase.
The reaction mixture was first stirred at 0 °C for 6 h and then at 25 °C for 24 h.
|
| 1 |
Toluene |
No |
CHPh2 |
90 |
71 |
| 2 |
Toluene |
Phenol |
CHPh2 |
86 |
80 |
| 3 |
Toluene |
p-OMePhOH |
CHPh2 |
87 |
81 |
| 4 |
Toluene |
o-OMePhOH |
CHPh2 |
87 |
83 |
| 5 |
Toluene |
PhCO2H |
CHPh2 |
73 |
79 |
| 6 |
Mesitylene |
o-OMePhOH |
CHPh2 |
88 |
82 |
| 7 |
MTBE |
o-OMePhOH |
CHPh2 |
71 |
81 |
| 8 |
PhCl |
o-OMePhOH |
CHPh2 |
86 |
80 |
| 9 |
Dioxane |
o-OMePhOH |
CHPh2 |
82 |
82 |
| 10 |
Toluene |
o-OMePhOH |
Me |
86 |
87 |
| 11 |
Toluene |
o-OMePhOH |
Ph |
82 |
88 |
| 12 |
Toluene |
o-OMePhOH |
Bn |
83 |
85 |
| 13 |
Toluene |
o-OMePhOH |
t
Bu |
76 |
77 |
| 14d |
Toluene
|
o-OMePhOH
|
Ph
|
80
|
93
|
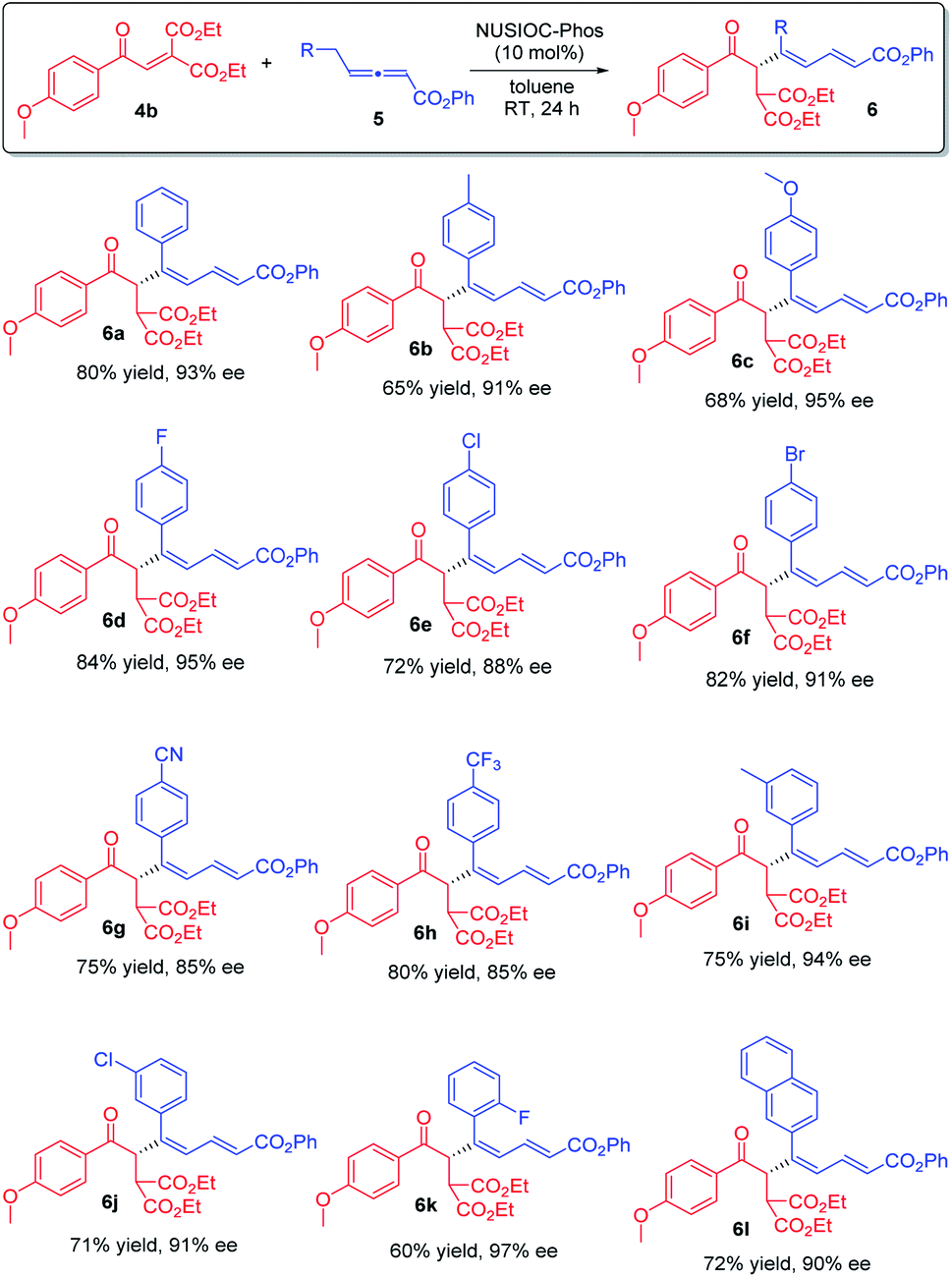
We subsequently applied the optimum conditions to establish the reaction scope (Scheme 3). In general, different benzylic types of γ-substituted allenoates 5 could be utilized, and regardless of the electronic nature and substitution patterns of the aryl moieties, conjugated 1,3-dienes 6 were obtained in decent yields and with excellent enantioselectivities (6a–6l).
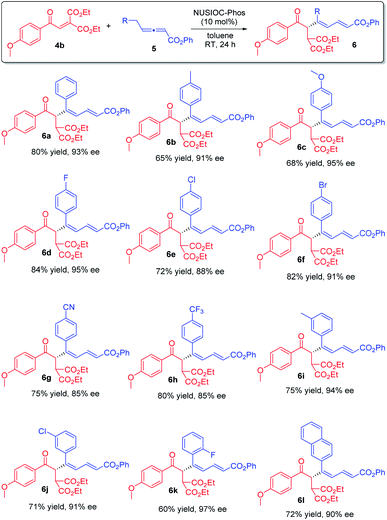 |
| | Scheme 3 Synthesis of chiral 1,3-dienes: reaction scope. Conditions: 4b (0.1 mmol), 5 (0.15 mmol), 2-methoxyphenol (0.05 mmol) and NUSIOC-Phos (0.01 mmol) in toluene (1.0 mL) at 0 °C for 6 h, and subsequently at 25 °C for 24 h; yields given were isolated yields; the ee values were determined by HPLC analysis on a chiral stationary phase. | |
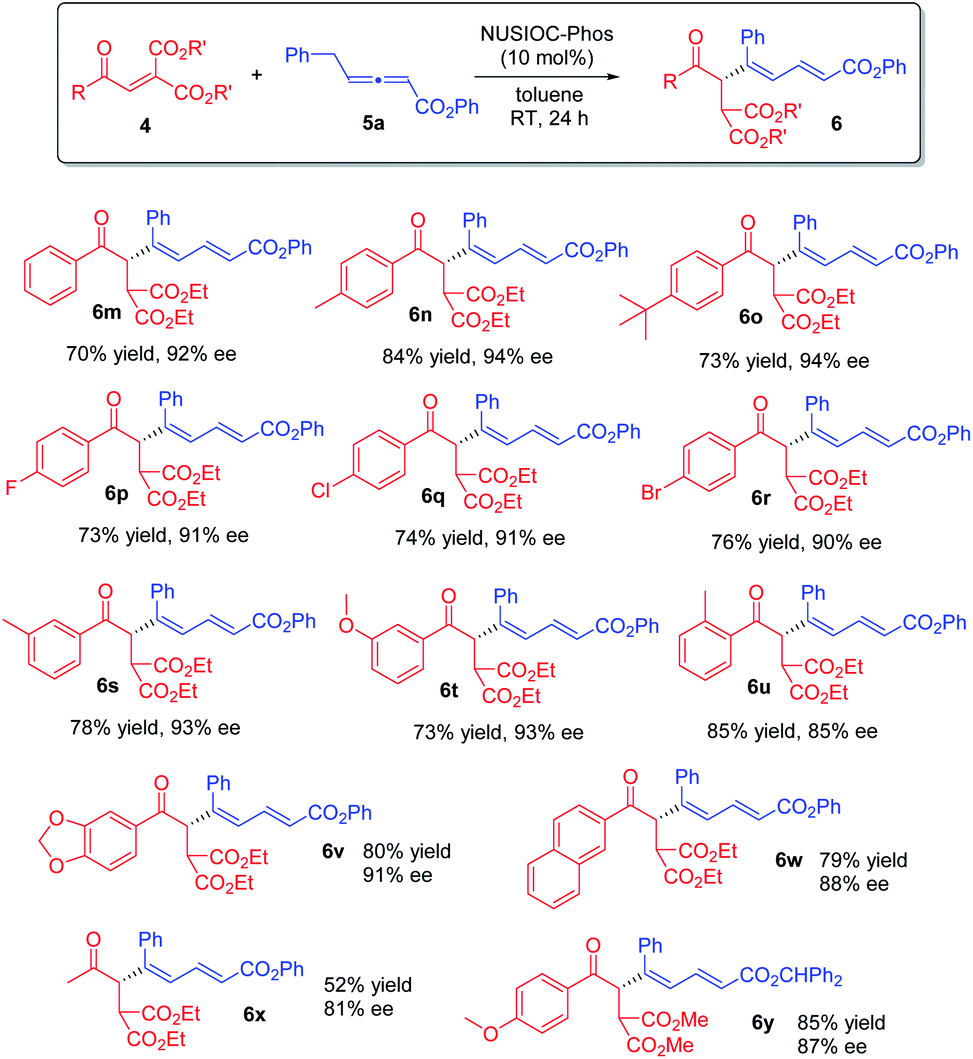
Furthermore, we examined the reaction scope by employing different diester enones 4 (Scheme 4). Enones containing an electron donating group on the phenyl ring with different substitution patterns were well-tolerated (6n, 6o, and 6s–6u), and high enantioselectivities were obtained. Similarly, aryl enones bearing different halogen atoms were also found to be suitable (6p–6r). Moreover, the reaction was applicable to 5-piperonyl and 2-naphthyl enone substrates, and the desired 1,3-dienes were obtained in good yields with high enantioselectivities (6v & 6w). Finally, when an aliphatic enone was employed, the reaction proceeded smoothly, affording product 6x in moderate yield with good enantioselectivity. The absolute configuration of 1,3-dienes 6 was assigned on the basis of X-ray crystallographic analysis of 6y (see the ESI† for more details).
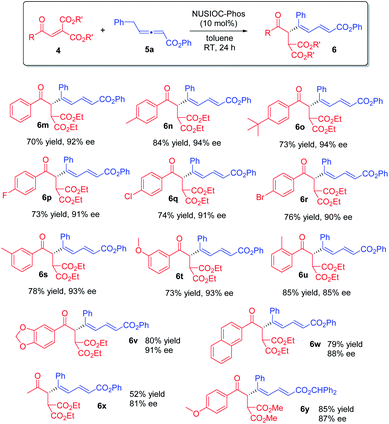 |
| | Scheme 4 Further scope. Conditions: 4 (0.1 mmol), 5a (0.15 mmol), 2-methoxyphenol (0.04 mmol), and NUSIOC-Phos (0.01 mmol) in toluene (1.0 mL) at 0 °C for 6 h, and subsequently at 25 °C for 24 h; yields given were isolated yields; the ee values were determined by HPLC analysis on a chiral stationary phase. | |
Proposed reaction mechanisms
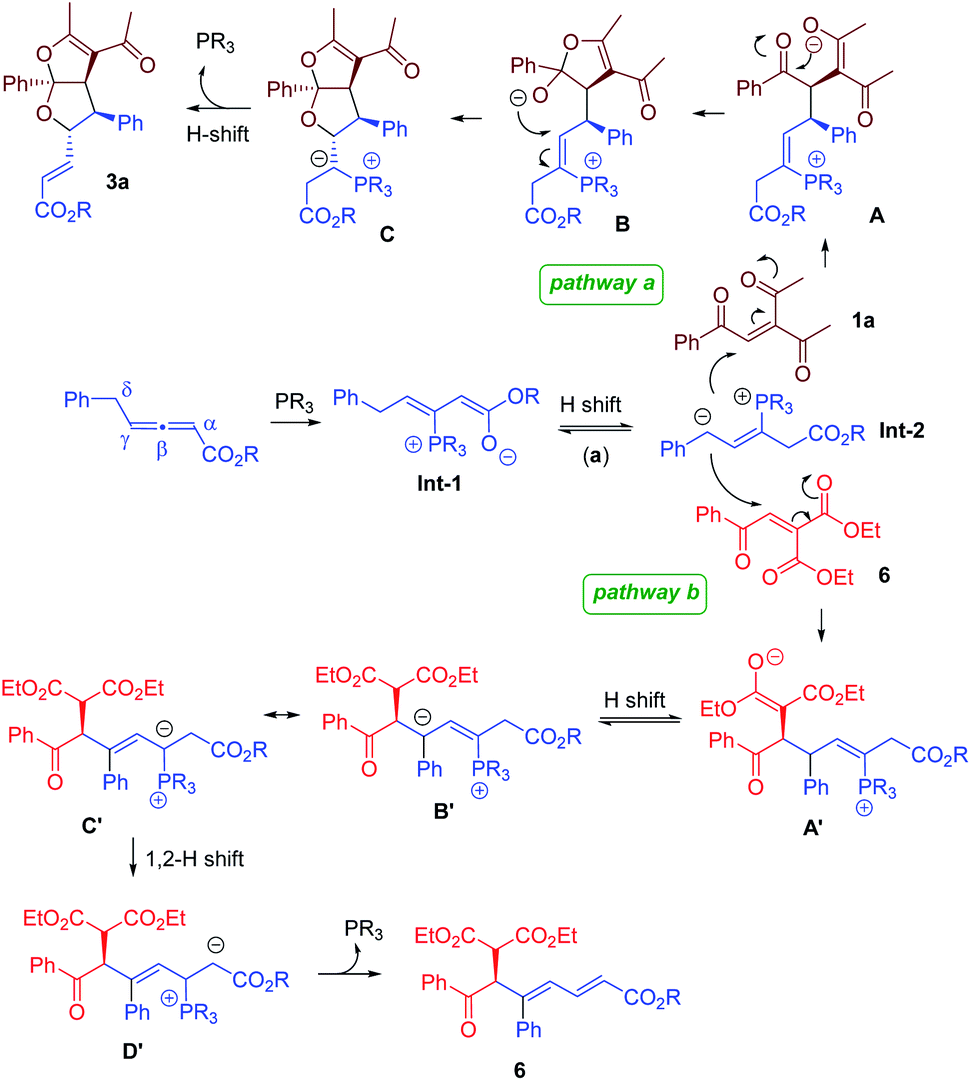
Plausible mechanistic pathways are proposed in Fig. 3. Phosphine attack on the γ-substituted allenoate generates zwitterionic intermediate Int-1, which is nucleophilic at either the α- or the γ- position. Subsequently, a proton shift leads to the formation of δ-anionic Int-2. When the triketone enone 1a is used, the conjugate addition of Int-2 to 1a forms enolate A, which attacks the ketone function intramolecularly to yield B. A second intramolecular addition then takes place to afford the bicyclic acetal core structure C. Another proton shift, followed by the elimination and re-generation of phosphine, forms the bicyclic furofuran product 3a (pathway a). With the employment of diester-containing enone 6, the reaction proceeds through different mechanistic steps. The conjugate addition of Int-2 to 6 creates intermediate A′, which undergoes a proton transfer to form B′. Finally, a 1,2-proton shift, followed by re-generation of the phosphine catalyst, furnishes chiral 1,3-diene 6 (pathway b).| | 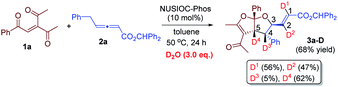 | (4) |
| |  | (5) |
| |  | (6) |
| | 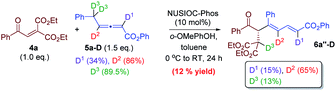 | (7) |
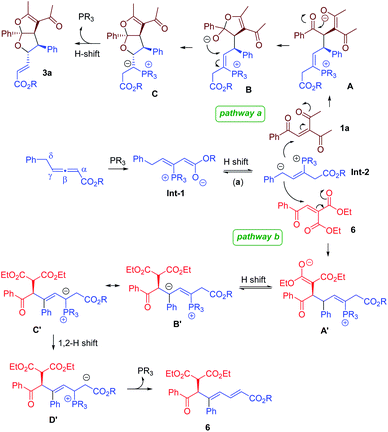 |
| | Fig. 3 Proposed mechanism. | |
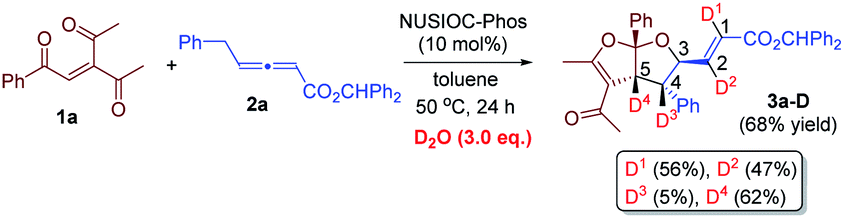
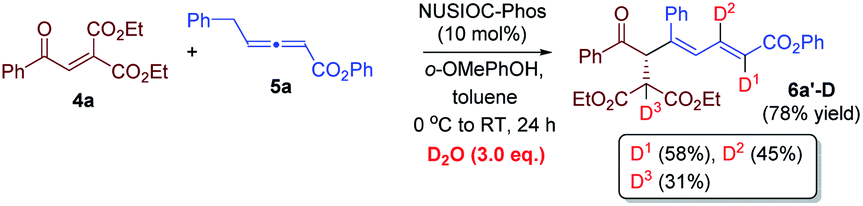
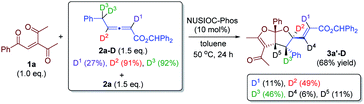
We have performed a few deuterium-labelling experiments to further probe our proposed reaction mechanisms. When bicyclic acetal 3a was synthesized in the presence of D2O and under otherwise identical reaction conditions, deuterated product 3a-D with multiple deuterium incorporations was obtained (eqn (4)). Notably, 62% deuterium incorporation at the C5 position of 3a-D was observed, which is likely due to the facile tautomerization of ketone intermediate A to its enol form. When the reaction leading to the formation of conjugated 1,3-dienes was performed in the presence of D2O, similar deuterium incorporations into 6a′-D were observed (eqn (5)). Notably, the sites where the deuterium atoms were incorporated into the products are at the proposed positions whereby the proton transfers take place, consistent with our proposed mechanistic pathways. When enone 1a was reacted with equal amounts of allenoate 2a and deuterated 2a-D, around half deuterium incorporation in bicyclic acetal 3a′-D was observed (eqn (6)), suggesting that both deuterated and non-deuterated allenes have similar reaction rates. For the 1,3-diene forming reaction, if a deuterated allenoate (5a-D) was employed under otherwise the same reaction conditions, product 6a′′-D with much less deuterium incorporation was obtained in only 12% yield (eqn (7)), and this is in stark contrast to a similar reaction of employing non-deuterated 5a, whereby 78% of 6a′-D was attainable (eqn (5)).29 The kinetic isotope effects observed in the above experiment clearly suggest that the rate-determining step for the conjugated 1,3-diene formation involves the proton transfer process.
Conclusions
In summary, we have developed phosphine-catalyzed divergent domino processes between γ-substituted allenoates and carbonyl-activated alkenes. When triketone enone substrates were reacted with allenoates in the presence of NUSIOC-Phos, a domino reaction process led to the formation of diastereoselective bicyclic furofurans in decent yields with excellent enantioselectivities. If enone substrates bearing two ester groups are utilized under similar reaction conditions, a different cascade sequence was in operation, forming chiral conjugated 1,3-dienes in high yields with excellent enantioselectivities. Notably, through careful design and precise control of catalytic processes, by employing substrates with subtle structural differences, we were able to develop highly efficient asymmetric synthetic methodologies that allow for quick access to important chiral molecular architectures. We are currently applying the synthetic methodologies disclosed herein to the synthesis of molecules of biological significance and shall report our findings in due course.
Data availability
All experimental procedures, characterization, copies of NMR spectra for all new compounds related to this article can be found in the ESI.†
Author contributions
M. W. discovered and performed most of the experiments and wrote the first draft of the manuscript. Z. H. and H. N contributed to some experiments (synthesis of some NUSIOC-Phos intermediates). N. W. analyzed the 2-D NMR spectrum and discussed the results. K. D. and Y. L. supervised the work, finalized the manuscript, and coordinated the overall project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Y. L. thanks the Singapore National Research Foundation, Prime Minister's Office for the NRF Investigatorship Award (A-0004067-00-00). Financial support from the Ministry of Education of Singapore (A-0008481-00-00) is also gratefully acknowledged.
Notes and references
- C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906–2908 CrossRef CAS.
- B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 3167–3168 CrossRef CAS.
- H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed.
- H. Guo, Y. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- For selected phosphine-catalyzed [3 + 2] annulations, see
(a) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726–1729 CrossRef CAS PubMed;
(b) M. G. Sankar, M. Garcia-Castro, C. Golz, C. Strohmann and K. Kumar, Angew. Chem., Int. Ed., 2016, 55, 9709–9713 CrossRef CAS PubMed;
(c) X. Han, W.-L. Chan, W. Yao, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2016, 55, 6492–6496 CrossRef CAS PubMed;
(d) W. Zhou, H. Wang, M. Tao, C.-Z. Zhu, T.-Y. Lin and J. Zhang, Chem. Sci., 2017, 8, 4660–4665 RSC;
(e) W.-L. Chan, X. Tang, F. Zhang, G. Quek, G.-J. Mei and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 6260–6330 CrossRef CAS PubMed;
(f) K. Li, T. P. Gonçalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5481–5485 CrossRef;
(g) B. Tan, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 4672–4675 CrossRef CAS PubMed;
(h) S. Y. Lee, Y. Fujiwara, A. Nishiguchi, M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 4587–4591 CrossRef CAS PubMed;
(i) H. Ni, Z. Yu, Y. Lan, N. Ullah and Y. Lu, Chem. Sci., 2017, 8, 5699–5704 RSC;
(j) W. Yao, Z. Yu, S. Wen, H. Ni, N. Ullah, Y. Lan and Y. Lu, Chem. Sci., 2017, 8, 5196–5200 RSC;
(k) K. Li, T. P. Gonçalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS PubMed;
(l) H. Wang, J. Zhang, Y. Tu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 5422–5426 CrossRef CAS PubMed.
- For selected phosphine-catalyzed [4 + 1] annulations, see
(a) Q. Zhang, L. Yang and X. Tong, J. Am. Chem. Soc., 2010, 132, 2550–2551 CrossRef CAS PubMed;
(b) X. Han, W. Yao, T. Wang, Y. R. Tan, Z. Yan, J. Kwiatkowski and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 5643–5647 CrossRef CAS PubMed;
(c) D. T. Ziegler, L. Riesgo, T. Ikeda, Y. Fujiwara and G. C. Fu, Angew. Chem., Int. Ed., 2014, 53, 13183–13187 CrossRef CAS PubMed;
(d) S. Kramer and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 3803–3806 CrossRef CAS PubMed;
(e) X. Tang, H. Ni and Y. Lu, Org. Chem. Front., 2021, 8, 4485–4489 RSC.
- For selected phosphine-catalyzed [4 + 2] annulations, see
(a) X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS PubMed;
(b) R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234–12235 CrossRef CAS PubMed;
(c) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632–12633 CrossRef CAS PubMed;
(d) W. Yao, X. Dou and Y. Lu, J. Am. Chem. Soc., 2015, 137, 54–57 CrossRef CAS PubMed;
(e) X. Tang, C. Tan, W.-L. Chan, F. Zhang, W. Zheng and Y. Lu, ACS Catal., 2021, 11, 1361–1367 CrossRef CAS.
- For selected phosphine-catalyzed γ-addition reactions see:
(a) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 10819–10820 CrossRef CAS;
(b) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2011, 131, 14231–14233 CrossRef PubMed;
(c) D. T. Ziegler and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 12069–12072 CrossRef CAS PubMed;
(d) J. Sun and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 4568–4569 CrossRef CAS PubMed;
(e) R. Sinisi, J. Sun and G. C. Fu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20652–20654 CrossRef CAS PubMed;
(f) T. Wang, W. Yao, F. Zhong, F. G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964–2968 CrossRef CAS PubMed;
(g) T. Wang, D. L. Hoon and Y. Lu, Chem. Commun., 2015, 51, 10186–10189 RSC;
(h) T. Wang, Z. Yu, D. L. Hoon, K.-W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912–4922 RSC;
(i) H. Qiu, X. Chen and J. Zhang, Chem. Sci., 2019, 10, 10510–10515 RSC.
- For selected phosphine-catalyzed Michael addition reactions see:
(a) F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Angew. Chem., Int. Ed., 2013, 52, 943–947 CrossRef CAS PubMed;
(b) B. Huang, C. Li, H. Wang, C. Wang, L. Liu and J. Zhang, Org. Lett., 2017, 19, 5102–5105 CrossRef CAS PubMed.
- For selected Morita–Baylis–Hillman (MBH) reactions see:
(a) F. Zhong, Y. Wang, X. Han, K.-W. Huang and Y. Lu, Org. Lett., 2011, 13, 1310–1313 CrossRef CAS PubMed;
(b) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837–7841 CrossRef CAS PubMed;
(c) H. P. Deng, D. Wang, Y. Wei and M. Shi, Beilstein J. Org. Chem., 2012, 8, 1098–1104 CrossRef CAS PubMed;
(d) L. Zhang, H. Liu, G. Qiao, Z. Hou, Y. Liu, Y. Xiao and H. Guo, J. Am. Chem. Soc., 2015, 137, 4316–4319 CrossRef CAS PubMed;
(e) F. Zhong, J. Lou, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222–10227 CrossRef CAS PubMed;
(f) M. Shi and L.-H. Chen, Chem. Commun., 2003, 39, 1310–1311 RSC;
(g) M. Shi, L.-H. Chen and C.-Q. Li, J. Am. Chem. Soc., 2005, 127, 3790–3800 CrossRef CAS PubMed.
-
(a) K. Li, Z. Jin, W.-L. Chan and Y. Lu, ACS Catal., 2018, 8, 8810–8815 CrossRef CAS;
(b) W. Yao, X. Dou, S. Wen, J. Wu, J. J. Vittal and Y. Lu, Nat. Commun., 2016, 7, 13024–13032 CrossRef PubMed;
(c) W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853–14857 CrossRef CAS PubMed;
(d) X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874–6877 CrossRef CAS PubMed.
- S. L. Schreiber and K. Satake, J. Am. Chem. Soc., 1984, 106, 4186–4188 CrossRef CAS.
- H. Shi, L. Fang, C. Tan, L. Shi, W. Zhang, C.-C. Li, T. Luo and Z. Yang, J. Am. Chem. Soc., 2011, 133, 14944–14947 CrossRef CAS PubMed.
- P. Mungkornasawakul, S. G. Pyne, A. C. Willis, A. Jatisatienr, D. Phuthsuk and W. Lie, Phytochem. Lett., 2013, 6, 602–605 CrossRef CAS.
-
(a) H. Zhang, Y. F. Wang, C.-H. Shen, J. Agniswamy, K. V. Rao, X. Xu, A. K. Ghosh, R. W. Harrison and I. T. Weber, J. Med. Chem., 2013, 56, 1074–1083 CrossRef CAS PubMed;
(b) A. K. Ghosh, G. L. Parham, C. D. Martyr, P. R. Nyalapatla, H. L. Osswald, J. Agniswamy, Y. F. Wang, M. Amano, I. T. Weber and H. Mitsuya, J. Med. Chem., 2013, 56, 6792–6802 CrossRef CAS PubMed.
- M. Fan, Y. Bao, Z.-J. Zhang, H.-B. Zhang and Q.-S. Zhao, Fitoterapia, 2017, 123, 44–50 CrossRef CAS PubMed.
- K. McKeage, C. M. Perry and S. J. Keam, Drugs, 2009, 69, 477–503 CrossRef CAS PubMed.
-
(a) G. M. Borrajo-Calleja, V. Bizet and C. Mazet, J. Am. Chem. Soc., 2016, 138, 4014–4017 CrossRef CAS PubMed;
(b) B. M. Paz, L. Klier, L. Næsborg, L. V. H. Lauridsen, F. Jensen and K. A. Jørgensen, Chem.–Eur. J., 2016, 22, 16810–16818 CrossRef CAS PubMed;
(c) W.-L. Huang, A. Raja, B.-C. Hong and G.-H. Lee, Org. Lett., 2017, 19, 3494–3497 CrossRef CAS PubMed;
(d) J. Yang, G. Qiu, J. Jiang, Y. Hu, S. Chen, S. Zhang and Y. Zhang, Adv. Synth. Catal., 2017, 359, 2184–2190 CrossRef CAS.
-
(a) A. K. Ghosh and Y. Chen, Tetrahedron Lett., 1995, 36, 505–508 CrossRef CAS PubMed;
(b) A. K. Ghosh, C. X. Xu, K. V. Rao, A. Baldridge, J. Agniswamy, Y. F. Wang, I. T. Weber, M. Aoki, S. G. Miguel, M. Amano and H. Mitsuya, ChemMedChem, 2010, 5, 1850–1854 CrossRef CAS PubMed;
(c) S. G. Akakios, M. L. Bode and R. A. Sheldon, Green Chem., 2021, 23, 3334–3347 RSC;
(d) Y. Hayashi, T. Aikawa, Y. Shimasaki, H. Okamoto, Y. Tomioka, T. Miki, M. Takeda and T. Ikemoto, Org. Process Res. Dev., 2016, 20, 1615–1620 CrossRef CAS.
- M. Wu, Z. Han, K. Li, J. Wu, K. Ding and Y. Lu, J. Am. Chem. Soc., 2019, 141, 16362–16373 CrossRef CAS PubMed.
-
(a) C. Paal, Ber. Dtsch. Chem. Ges., 1884, 17, 2756–2767 CrossRef;
(b) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 2863–2870 CrossRef.
- For selected phosphine-catalyzed reactions using γ-substituted allenes, see:
(a) X. Meng, Y. Huang, H. Zhao, P. Xie, J. Ma and R. Chen, Org. Lett., 2009, 11, 991–994 CrossRef CAS PubMed;
(b) E. Li, Y. Huang, L. Liang and P. Xie, Org. Lett., 2013, 15, 3138–3141 CrossRef CAS PubMed;
(c) J. Zheng, Y. Huang and Z. Li, Org. Lett., 2013, 15, 5064–5067 CrossRef CAS PubMed;
(d) H. Zhao, X. Meng and Y. Huang, Chem. Commun., 2013, 49, 10513–10515 RSC;
(e) E. Li and Y. Huang, Chem. Commun., 2014, 50, 948–950 RSC;
(f) E. Li, P. Jia, L. Liang and Y. Huang, ACS Catal., 2014, 4, 600–603 CrossRef CAS;
(g) E. Li and Y. Huang, Chem.–Eur. J., 2014, 20, 3520–3527 CrossRef CAS PubMed;
(h) E. Li, H. Jin, P. Jia, X. Dong and Y. Huang, Angew. Chem., Int. Ed., 2016, 55, 11591–11594 CrossRef CAS PubMed;
(i) D. Wang, Z. Song, W. Wang and T. Xu, Org. Lett., 2019, 21, 3963–3967 CrossRef CAS PubMed;
(j) M. Gicquel, C. Gomez, P. Retailleau, A. Voituriez and A. Marinetti, Org. Lett., 2013, 15, 4002–4005 CrossRef CAS PubMed;
(k) D. Wang, Y. Lei, Y. Wei and M. Shi, Chem.–Eur. J., 2014, 20, 15325–15329 CrossRef CAS PubMed;
(l) S. Xu, L. Zhou, R. Ma, H. Song and Z. He, Chem.–Eur. J., 2009, 15, 8698–8702 CrossRef CAS PubMed;
(m) R. Ma, S. Xu, X. Tang, G. Wu and Z. He, Tetrahedron, 2011, 67, 1053–1061 CrossRef CAS.
- H. Ni, Y. L. Wong, M. Wu, Z. Han, K. Ding and Y. Lu, Org. Lett., 2020, 22, 2460–2463 CAS . Also see ref. 20..
- CCDC 1986649 contains the supplementary crystallographic data for this compound. See the ESI† for more details..
-
(a) T. Wang, Z. Yu, D. L. Hoon, C. Y. Phee, Y. Lan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 265–271 CAS;
(b) Y. Chen, K. Zhu, Q. Huang and Y. Lu, Chem. Sci., 2021, 12, 13564–13571 RSC . Also see ref. 5j..
-
(a) C. Thirsk and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2002, 999–1023 RSC;
(b) C. Souris, F. Frebault, A. Patel, D. Audisio, K. N. Houk and N. Maulide, Org. Lett., 2013, 15, 3242–3245 CrossRef CAS PubMed.
-
(a) H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007–1019 CrossRef CAS;
(b) Z. Zhang, D. Cui, B. Wang, B. Liu and Y. Yang, Struct. Bonding, 2010, 137, 49–108 CrossRef CAS.
-
(a) M. De Paolis, I. Chataigner and J. Maddaluno, Top. Curr. Chem., 2012, 327, 87–146 CrossRef CAS PubMed;
(b) Z. Huang and E. Negishi, J. Am. Chem. Soc., 2007, 129, 14788–14792 CrossRef CAS PubMed;
(c) V. T. Nguyen, H. T. Dang, H. H. Pham, V. D. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2018, 140, 8434–8438 CrossRef CAS PubMed;
(d) M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805–5813 CrossRef CAS PubMed;
(e) D. Fiorito, S. Folliet, Y. Liu and C. Mazet, ACS Catal., 2018, 8, 1392–1398 CrossRef CAS;
(f) J. E. Bäckvall and A. Ericsson, J. Org. Chem., 1994, 59, 5850–5851 CrossRef;
(g) K. M. Miller, T. Luanphaisarnnont, C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 4130–4131 CrossRef CAS PubMed;
(h) J. R. Kong, C. W. Cho and M. Krische, J. Am. Chem. Soc., 2005, 127, 11269–11276 CrossRef CAS PubMed;
(i) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317–1382 CrossRef CAS PubMed;
(j) R. Shintani, W.-L. Duan, S. Park and T. Hayashi, Chem. Commun., 2006, 42, 3646–3647 Search PubMed;
(k) S. Mannathan and C.-H. Cheng, Chem. Commun., 2010, 46, 1923–1925 CAS;
(l) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CAS;
(m) J.-R. Kong, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 718–719 CrossRef CAS PubMed;
(n) W. R. Brown, F. Zamani, M. G. Gardiner, H. Yu, S. G. Pyne and C. J. T. Hyland, Chem. Sci., 2019, 10, 9051–9056 RSC;
(o) B. M. Trost and U. Kazmaier, J. Am. Chem. Soc., 1992, 114, 7933–7935 CAS;
(p) C. Zhang and X. Lu, Synlett, 1995, 645–646 CrossRef CAS;
(q) S. Xu, L. Zhou, S. Zeng, R. Ma, Z. Wang and Z. He, Org. Lett., 2009, 11, 3498–3501 CrossRef CAS PubMed.
- The reaction described in eqn (7) was run in the presence of H2O (3 eq.), and similar deuterium incorporation in 6a′-D was observed..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1986649 and 2024027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06364b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a