
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The convergent total synthesis and antibacterial profile of the natural product streptothricin F†
Matthew G.
Dowgiallo
 a,
Brandon C.
Miller
a,
Mintesinot
Kassu
a,
Kenneth P.
Smith
ef,
Andrew D.
Fetigan
a,
Jason J.
Guo
acd,
James E.
Kirby
ef and
Roman
Manetsch
*abc
a,
Brandon C.
Miller
a,
Mintesinot
Kassu
a,
Kenneth P.
Smith
ef,
Andrew D.
Fetigan
a,
Jason J.
Guo
acd,
James E.
Kirby
ef and
Roman
Manetsch
*abc
aDepartment of Chemistry and Chemical Biology, Northeastern University, Boston, MA, USA. E-mail: r.manetsch@northeastern.edu
bDepartment of Pharmaceutical Sciences, Northeastern University, Boston, MA, USA
cCenter for Drug Discovery, Northeastern University, Boston, MA, USA
dBarnett Institute for Chemical and Biological Analysis, Northeastern University, Boston, MA, USA
eDepartment of Pathology, Beth Israel Deaconess Medical Center, Boston, MA, USA
fHarvard Medical School, Boston, MA, USA
First published on 25th February 2022
Abstract
A convergent, diversity-enabling total synthesis of the natural product streptothricin F has been achieved. Herein, we describe the potent antimicrobial activity of streptothricin F and highlight the importance of a total synthesis that allows for the installation of practical divergent steps for medicinal chemistry exploits. Key features of our synthesis include a Burgess reagent-mediated 1,2-anti-diamine installation, diastereoselective azidation of a lactam enolate, and a mercury(II) chloride-mediated desulfurization-guanidination. The development of this chemistry enables the synthesis and structure–activity studies of streptothricin F analogs.
Introduction
The streptothricins are a class of natural products exhibiting potent antimicrobial activity against multidrug-resistant, Gram-negative bacteria. Streptothricins were first isolated in 1942 by Waksman and Woodruff from Streptomyces lavendulae1 and have since been identified under a variety of pseudonyms from other Streptomyces species.2–9 Isolates of streptothricin generally exist as complex mixtures of homologs A–F, and X (Fig. 1). These mixtures are typically referred to as “nourseothricin” and contain varying ratios of the component streptothricins, with streptothricin F (1) being the principal component. Nourseothricin attracted initial interest because of the impressive Gram-positive and Gram-negative antimicrobial activity1,6,10–13 and high water solubility10,14–16 of the streptothricins. However, this natural product class has not been pursued as a therapeutic due to inherent toxicity.7,17–20 Additionally, isolation of the individual streptothricin components of nourseothricin has proven to be challenging, with limited reports of biological characterization on demonstrated pure material.6,19,21 The streptothricin backbone consists of a carbamoylated gulosamine sugar core (Fig. 1, black) affixed with a streptolidine lactam moiety (red) and β-lysine homopolymer (blue) attached to the C7 and C8 amines, respectively. Streptolidine is an unusual guanidine-containing amino acid that has been isolated as a streptothricin hydrolysis product and appears to be unique to this natural product class.22 Additionally, the rarity of β-amino acids adds another layer of structural peculiarity, synthetic challenge, and a unique opportunity for medicinal chemistry discovery.23 | ||
| Fig. 1 Structures of the streptothricins A–F, and X. The streptolidine lactam is shown in red, the gulosamine core is shown in black, and the β-lysine homopolymer is shown in blue. | ||
 | ||
| Scheme 1 Retrosynthetic analysis of streptothricin F (1). | ||
Streptothricins were discovered to be vulnerable to resistance through two mechanisms. The β-amine of the β-lysine moiety is susceptible to an acylation-based resistance mechanism in bacteria containing streptothricin acetyl transferases24–30 while enzymatic hydrolysis of the streptolidine moiety proceeds through a less-prominent resistance pathway.31 Streptothricin F was previously found to inhibit prokaryotic ribosomal translocation and also induce significant miscoding. That is, like aminoglycosides, they cause incorrect amino acids to be added to the growing peptide chains during protein synthesis and thereby poison the bacterial cell leading to cell death. Experimentally streptothricin F was previously found not to effect nucleic acid synthesis.32,33 Our main attraction to this natural product class derives from previous reports that have demonstrated streptothricin F (1) to be less toxic than other streptothricins. Studies of purified streptothricins in mice indicate that toxicity is influenced directly by the unit length (n) of the β-lysine homopolymer. Streptothricin F, (n = 1, LD50: 300 mg kg−1) shows remarkably less toxicity than streptothricin E (n = 2, LD50: 26 mg kg−1), streptothricin D (n = 3, LD50: ∼10 mg kg−1), and streptothricin C (n = 4, LD50: ∼10 mg kg−1).6,15,34 Conversely, antimicrobial activity favors longer β-lysine homopolymer chains, with nourseothricin (K. pneumoniae Nevada strain AR-0636 MIC: 0.15 μg mL−1) and streptothricin D (MIC: 0.19 μg mL−1) exhibiting approximately 4-fold more activity (per mol) than streptothricin F (MIC: 1 μM).34
 | ||
| Scheme 2 Synthesis of streptolidine isothiocyanate 3. | ||
Results and discussion
To fully explore the therapeutic potential of this promising scaffold, it is important to develop an efficient and robust total synthesis to produce significant quantities of streptothricins and analogs. A single total synthesis of streptothricin F has been reported in the literature by Shiba and co-workers,35 and no other streptothricin has been attained through synthetic means exclusively. While a landmark for its time, Shiba's synthesis contains over 46 total steps with a longest linear sequence of 25 steps and an overall yield of less than 0.28%.36 While Shiba's synthesis contains elements of convergence, 12 synthetic steps take place after the first fragment coupling, including the installation of a stereocenter. Drawing inspiration from Shiba's efforts and the promising attributes of streptothricin F, we have designed a total synthesis of streptothricin F that readily enables SAR exploration. Through the incorporation of late-stage fragment coupling, we believe independent modification of the three structural components of streptothricin F is possible. The intended design of our synthesis is to facilitate rapid, combinatorial-like library generation of streptothricin F analogs that are targeted to evade known resistance pathways and maintain, or further reduce, low toxicity. Herein, we report our highly convergent, diversity-enabling streptothricin F total synthesis consisting of 35 total steps, with a longest linear sequence of 19 steps and an overall yield of 0.40%. | ||
| Scheme 3 Synthesis of the gulosamine 7. | ||
Retrosynthetically, our synthesis stems from two key disconnections at the C7 and C8 amines on the gulosamine core (Scheme 1). This approach hedged the production of streptothricin F (1) on a Lewis acid-catalyzed guanidine closure of thiourea 2, followed by global, stepwise deprotection. We envisioned thiourea 2 as the product of fragment couplings of isothiocyanate 3, partially protected β-lysine 6, and gulosamine 7. The first of these three fragments, isothiocyanate 3, can be generated through stereoselective azidation and tandem Staudinger–aza-Wittig thioisocyanation of lactam 4. Preparation of lactam 4 relied on nitromethylation of protected aspartic acid 5, followed by a formic acid-promoted deprotective-lactamization. Prior to coupling with isothiocyanate 3, partially protected β-lysine 6 and gulosamine 7 would be coupled at the C8 amine. We anticipated that access to gulosamine 7 could arise from nucleophilic ring-opening of β-sulfamidate 8. Burgess reagent-sulfamidation to obtain β-sulfamidate 8 was readily performed following stereoinversion and dihydroxylation of glucal sugar 9, which is obtained in decagram quantities from commercially available starting material.
Forward synthesis began with construction of the streptolidine moiety of streptothricin F (Scheme 2). Commercially available N-Cbz-L-aspartic acid 4-tert-butyl ester 5 was treated with carbonyl diimidazole producing an activated anhydride that reacted smoothly with a solution of excess nitromethane and stoichiometric t-BuOK37 to yield nitroketone 10.38,39 Diastereoselective reduction was carried out under Felkin–Ahn conditions, allowing hydride attack from the less hindered face of 10 resulting in >9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr of the desired erythro (3S,4R) nitroalcohol 11.39–41 Nitro reduction through generation of nickel boride followed by immediate addition of di-tert-butyl dicarbonate in one pot produced dicarbamate 12 as a mixture of diastereomers.42,43 Warming in formic acid deprotected the previously installed Boc group and promoted lactamization to afford 13, where the diastereomeric mixture could be separated. Silylation of 13 with TBS triflate, followed by hydrogenation to remove benzyl carbamate and subsequent Boc protection produced dicarbamate lactam 4 in good yield over three steps. Deprotonation of 4 to lactam enolate 14 and treatment with trisyl azide provided α-azidolactam 15 as a single diastereomer.44,45 The complete selectivity of this reaction is likely directed through the synergistic steric effects of a congested top face of enolate 14 and the bulky nature of trisyl azide. A moderate yield of 15 was observed as a consequence of enolate formation and substrate stability. To avoid stepwise isothiocyanate formation and risk lactam hydrolysis, we looked to the tandem Staudinger–aza-Wittig method for a one pot conversion of azides to isothiocyanates.46,47 Adhering to this protocol, α-azidolactam 15 was treated with triphenylphosphine and excess carbon disulfide to yield isothiocyanate 3, with conservation of our lactam ring.46,47
:1 dr of the desired erythro (3S,4R) nitroalcohol 11.39–41 Nitro reduction through generation of nickel boride followed by immediate addition of di-tert-butyl dicarbonate in one pot produced dicarbamate 12 as a mixture of diastereomers.42,43 Warming in formic acid deprotected the previously installed Boc group and promoted lactamization to afford 13, where the diastereomeric mixture could be separated. Silylation of 13 with TBS triflate, followed by hydrogenation to remove benzyl carbamate and subsequent Boc protection produced dicarbamate lactam 4 in good yield over three steps. Deprotonation of 4 to lactam enolate 14 and treatment with trisyl azide provided α-azidolactam 15 as a single diastereomer.44,45 The complete selectivity of this reaction is likely directed through the synergistic steric effects of a congested top face of enolate 14 and the bulky nature of trisyl azide. A moderate yield of 15 was observed as a consequence of enolate formation and substrate stability. To avoid stepwise isothiocyanate formation and risk lactam hydrolysis, we looked to the tandem Staudinger–aza-Wittig method for a one pot conversion of azides to isothiocyanates.46,47 Adhering to this protocol, α-azidolactam 15 was treated with triphenylphosphine and excess carbon disulfide to yield isothiocyanate 3, with conservation of our lactam ring.46,47
 | ||
| Scheme 4 The convergent total synthesis of streptothricin F. | ||
To assemble the gulosamine core, the first task was to generate the rare gulal sugar core from readily obtained starting material while enabling discrete alcohol functionalization (Scheme 3). Construction of gulal sugars has not been widely explored; however, two reliable methods have been reported. The first, pioneered by the Danishefsky laboratory, used a thio-phenol Ferrier-type/Mislow–Evans [2,3] sigmatropic rearrangement starting from tri-O-acetyl-D-galactal.48–50 The second method, reported by Crotti and co-workers, started from tri-O-acetyl-D-glucal and relied on a regioselective epoxide ring-opening to invert both the 9- and 10-position alcohols.51,52 Our route hewed more closely to the Crotti approach due to scalability and availability of inexpensive starting material. Deacylation of 16 and regioselective benzylation of the primary alcohol using Taylor's aminoethyl diphenylborinate catalyst53 and silver oxide as an activator gave benzyl glucal 9. Silylation with good regioselectivity and subsequent mesylation produced fully functionalized glucal 17. Treatment with TBAF yielded our stereoinversion precursor 18, which was treated with t-BuOK to form epoxide 19. When exposed to freshly prepared tetrabutylammonium trimethylsilanolate,52 epoxide 19 is selectively opened at the allylic position to give TMS alcohol 20 which is hydrolyzed upon work-up to give 6-O-benzyl-D-gulal 21. Silylation of 21 was performed with high regioselectivity and subsequent treatment with freshly prepared 2,4-dimethoxybenzyl isocyanate49,54 yielded the carbamoylated gulal 22.
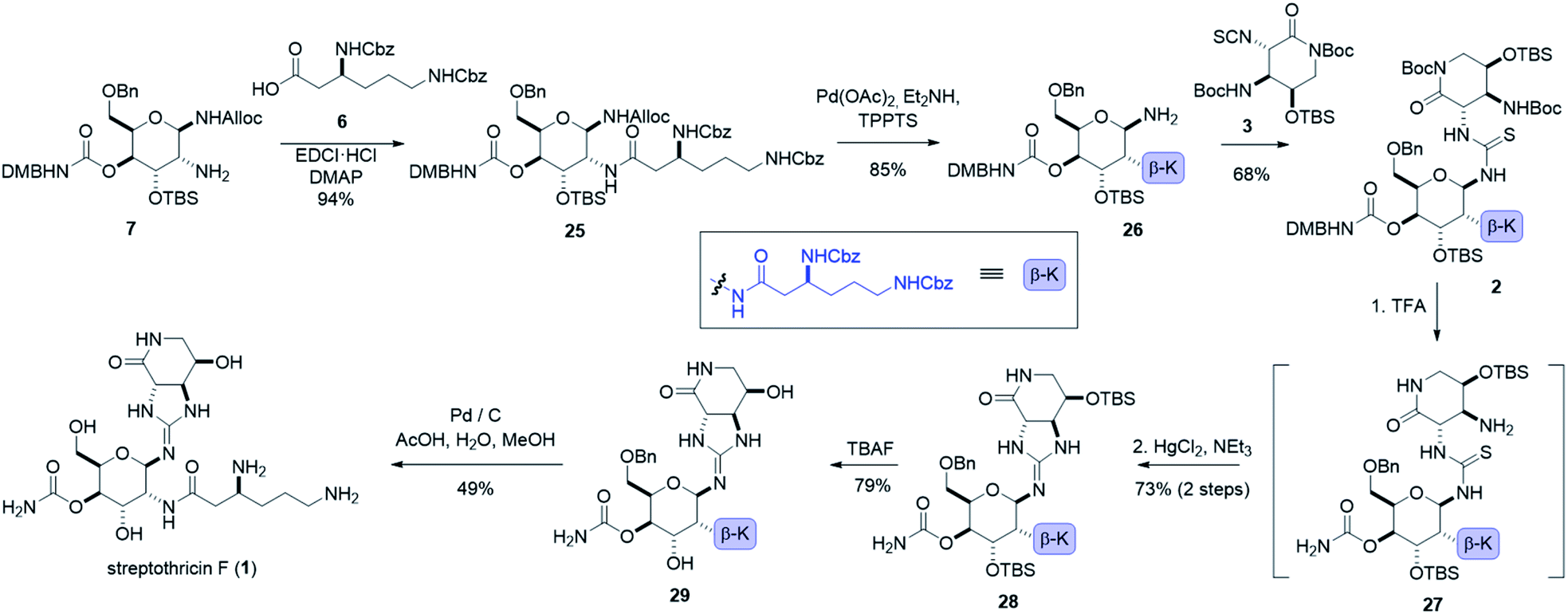
To install our 1,2-diamine functionality on the gulal sugar, we employed Burgess reagent chemistry pioneered by Nicolaou and co-workers.55 Among other uses for the Burgess reagent, a method for preparation of 1,2-diamino sugars was reported, which we adopted for our synthesis. Dihydroxylation of gulal 22 gave diol 23 as an 8:1 mixture of anomers favoring the α-anomer, as implicated by the coupling constant of the anomeric proton (J1,2 = 11.3 Hz). Addition of the alloc-modified Burgess reagent 24 produced a disulfamate intermediate that reacts via C2 delivery to give β-sulfamidate 8 (>10:1 dr), (see ESI†).55 Ring opening of β-sulfamidate 8 with sodium azide followed by Staudinger reduction yielded gulosamine 7.55,56 To generate our C8 linkage, gulosamine 7 was coupled to partially protected β-lysine 6 (attained through homologation of α-lysine, see the ESI†) to give protected β-lysyl-gulosamine 25 (Scheme 4). Allyl carbamate deprotection of 25 proceeded through a catalytic allyl transfer mechanism to avoid anomerization. This mild deprotection method uses the palladium–TPPTS complex in tandem with diethyl amine as an allyl acceptor to generate β-lysyl-gulosamine 26.55,57 Coupling of β-lysyl-gulosamine 26 with previously prepared isothiocyanate 3 yielded thiourea 2, and completed our C7 linkage. Coupling of β-lysyl-gulosamine 26 with previously prepared isothiocyanate 3 yielded thiourea 2, and completed our C7 linkage.
At this stage (four steps from the completion of the synthesis), all streptothricin F stereocenters are set, and our three synthetic routes have converged. Treatment of thiourea 2 with TFA removed both Boc groups as well as the 2,4-dimethoxybenzyl moiety to yield thiourea 27 which was unstable to purification methods. The crude reaction mixture containing 27 was therefore directly cyclized to guanidine 28 through mercury(II) chloride mediated desulfurization with triethylamine (for detailed optimization attempts and alternative approaches to prepare guanidine 28 see the ESI†). Two consecutive deprotections from guanidine 28 would complete our total synthesis. Silyl removal with TBAF provided partially deprotected streptothricin F 29 and under conditions similar to the Shiba total synthesis, benzyl and carboxybenzyl groups were removed in a single step through hydrogenolysis in acidic solvent.35 This hydrogenolysis provided streptothricin F (1) as an acetate salt; however, we desired to convert streptothricin F acetate to the sulfate to compare the activity and spectral data of synthetic streptothricin F more accurately to streptothricin F sulfate isolated from commercially available nourseothricin sulfate. Purified streptothricin F acetate was acidified to a pH of 2 in H2SO4, precipitated from methanol-diethyl ether, and collected via centrifugation.58
The comparison of antimicrobial activity of synthetic streptothricin F sulfate to isolated streptothricin F sulfate purified in our laboratory is shown in Table 1. Our purification method was adopted from Taniyama et al. with modifications to column length and flow rate.6 Upon loading a ∼300 mg sample of commercially available nourseothricin sulfate onto a glass column (150 × 2.4 cm) packed with Sephadex LH-20 afforded ∼75 mg of pure streptothricin F and ∼15 mg of pure streptothricin D, as well as mixed fractions. The microbes highlighted in Table 1 represent a diverse panel of Gram-positive and Gram-negative pathogens, many of which have been designated as either urgent or serious threats by the CDC and the WHO for their resistance capabilities,59–61 or are surrogates for CDC category A or B biothreat pathogens. Many of these species are also members of the so-called ESKAPE pathogens62 for which emerging antibiotic resistance threatens to eliminate effectiveness of all currently available antibiotics. Of note, both natural and synthetic streptothricin F were found to be active against the following: vancomycin-resistant Staphylococcus aureus; Bacillus anthracis (the cause of anthrax); multi-drug-resistant species of Gram-negative pathogens including the pan-resistant K. pneumoniae,63Escherichia coli expressing the colistin resistance gene mcr-1, and Acinetobacter baumannii; Yersinia pestis (the cause of bubonic plague); and Francisella tularensis (the cause of tularemia). However, there was low activity against Burkholderia and Pseudomonas strains tested. As a useful metric of comparison, the minimum inhibitory concentration (MIC) values for ampicillin, gentamicin, and tetraycline were 2, 0.5, and 2 μg mL−1 for Escherichia coli; 16, 0.5, and 8 μg mL−1 for Acinetobacter baumannii; and 128 (defined for benzylpenicillin), 0.125, and 0.5 μg mL−1 for methicillin-resistant Staphylococci aureus, respectively.64 MIC values for Gram-negative multidrug-resistant pathogens are typically much higher and are dependent upon the antibiotic and speicies of interest.65–67 Importantly, the MIC values of synthetic streptothricin F and streptothricin F purified from nourseothricin natural product were essentially identical within the expected experimental error, providing biological confirmation of the anticipated activity for our synthetic natural product.
| Organism | Phenotype | MIC values (μg mL−1) | ||
|---|---|---|---|---|
| Isolated S-F | Synthetic S-F | |||
| Gram-positive | S. aureus ATCC 29213 | Pan-susceptible control | 4 | 4 |
| S. aureus (VRSA) NR46422 | Vancomycin-resistant | 4 | 8 | |
| S. aureus (VRSA) NR49120 | Vancomycin-resistant | 4 | 16 | |
| S. aureus (VRSA) NR46420 | Vancomycin-resistant | 4 | 16 | |
| Gram-negative | A. baumannii ATCC 17978 | Pan-susceptible control | 2 | 4 |
| A. baumannii MSRN 1450 | Extensively-drug resistant | 16 | 8 | |
| B. cenocepacia clinical isolate K56-2 (ET-12 clone) | Cystic fibrosis pathogen | >64 | >64 | |
| B. thailandensis NR9908 | Surrogate for biothreat B. mallei/pseudomallei | >64 | >64 | |
| B. thailandensis NR9909 | Surrogate for biothreat B. mallei/pseudomallei | >64 | >64 | |
| E. coli ATCC 25922 | Pan-susceptible control | 1 | 2 | |
| E. coli FDA-CDC 346 (MCR-1) | Plasmid-borne colistin resistance | 1 | 4 | |
| B. anthracis Sterne 9131 | Surrogate for biothreat anthrax | 8 | 8 | |
| F. tularensis LVS | Surrogate for biothreat tularemia | 0.125 | 0.125 | |
| K. pneumoniae Nevada strain AR-0636 | Pan-drug resistant | 1 | 1 | |
| P. aeruginosa ATCC 27853 | Pan-susceptible control | 64 | >64 | |
| Y. pestis Yokahama NR4693 | Surrogate for biothreat bubonic plague | 1 | 4 | |
Conclusions
The streptothricin scaffold has been overlooked by the synthetic chemistry community until now. Our disclosed work represents the second total synthesis of streptothricin F (1), and the first through a diversity-enabling convergent route. We have prepared streptothricin F in a longest linear sequence of 19 steps, with 35 total steps, and 0.40% overall yield. Additionally, our convergent total synthesis includes the ability to install practical, divergent synthetic steps and facilitate manipulations of each of the three independent structural moieties. We have shown that streptothricin F has great promise as a broadly active antibiotic scaffold ripe for optimization. Accordingly, through the fully developed, diversity-enabling total synthesis of streptothricin F, we are pursuing medicinal chemistry-guided analog generation of streptothricin F to explore synthetic analogs for potential therapeutic development.Author contributions
R. M. and J. E. K. conceptualized and supervised the work as well as edited the manuscript. M. G. D., B. C. M., M. K., K. P. S., and A. D. F designed and performed experiments. J. J. G. performed some NMR spectral acquisition. The first draft of the manuscript was written by M. G. D. and B. C. M. All authors contributed to manuscript review.Conflicts of interest
The authors declare no competing financial interest. The HP D300 digital dispenser and TECAN M1000 used for MIC analysis in Table 1 were provided for our use by TECAN (Morrisville, NC). TECAN had no role in study design, data collection/interpretation, manuscript preparation, or decision to publish.Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under awards R21AI140212 and R01AI157208. K. P. S. was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number F32 AI124590. We gratefully acknowledge and thank Clarissa Santori and G. Marco Rossi for contributions to the synthetic studies early in development. We thank Donna Baldisseri from Bruker Biospin Corporation for recording NMR data of select late-stage intermediates. We thank Charles Sheahan from Harvard Medical School for assistance in recording NMR data of select late-stage intermediates.Notes and references
- S. A. Waksman and H. B. Woodruff, Streptothricin, a new selective bacteriostatic and bactericidal agent, particularly active against gram-negative bacteria, Proc. Soc. Exp. Biol. Med., 1942, 49(2), 207–210 CrossRef CAS.
- M. Lumb, N. Chamberlain, T. Cross, P. E. Macey, J. Spyvee, J. M. Uprichard and R. D. Wright, An antibiotic complex (A4788) containing streptothricin and having activity against powdery mildews, J. Sci. Food Agric., 1962, 13(6), 343–352 CrossRef CAS.
- A. W. Johnson and J. W. Westley, The streptothricin group of antibiotics. Part I. the general structural pattern, J. Chem. Soc., 1962, 1042, 1642 RSC.
- 1R. K. Sinha and P. N. Nandi, A new antibacterial agent produced by streptomyces Sp. AC6-569, Experientia, 1968, 24(8), 795–796 CrossRef PubMed.
- R. K. Sinha, Boseimycin-a new streptothricin-like antibiotic, J. Antibiot., 1970, 23(7), 360–364 CrossRef CAS.
- H. Taniyama, Y. Sawada and T. Kitagawa, Characterization of racemomycins, Chem. Pharm. Bull., 1971, 19(8), 1627–1634 CrossRef CAS.
- J. Fried and O. Wintersteiner, Crystalline reineckates of streptothricin and streptomycin, Science, 1945, 101(2633), 613–615 CrossRef CAS PubMed.
- S. Hosoya, M. Soeda, N. Komatsu, S. Imamura, M. Iwasaki, Y. Sonoda and K. Okada, The antibiotic substances produced by streptomyces, Jpn. J. Exp. Med., 1949, 20, 121–133 CAS.
- S. Hosoya, M. Soeda, N. Komatsu and S. Imamura, Antibiotic substances produced by a streptomyces H-277 strain, Jpn. J. Exp. Med., 1950, 20, 481–488 CAS.
- H. J. Robinson, O. E. Graessle and D. G. Smith, Studies on the toxicity and activity of streptothricin, Science, 1944, 99(2583), 540–542 CrossRef CAS PubMed.
- D. M. Reynolds, A. Schatz and S. A. Waksman, Grisein, a new antibiotic produced by a strain of streptomyces griseus, Proc. Soc. Exp. Biol. Med., 1947, 64(1), 50–54 CrossRef CAS PubMed.
- K. Akasaki, H. Abe, A. Seino and S. Shirato, Yazumycin, a new antibiotic produced by streptomyces lavendulae, J. Antibiot., 1968, 21(2), 98–105 CrossRef CAS PubMed.
- A. S. Khokhlov, Streptothricins and related antibiotics, J. Chromatogr. Libr., 1978, 15, 617–713 CAS.
- Y. Inamori, K. Morimoto, K. Morisaka, G. Saito, Y. Sawada, H. Taniyama and H. Matsuda, Toxicological approaches to streptothricin antibiotics. II. The developmental mechanism of delayed toxicity in mice and rats, Chem. Pharm. Bull., 1979, 27(1), 230–234 CrossRef CAS PubMed.
- Y. Inamori, Y. Kato, K. Morimoto, K. Morisaka, G. Saito, Y. Sawada and H. Taniyama, Toxicological approaches to streptothricin antibiotics. I. Implications of delayed toxicity in mice, Chem. Pharm. Bull., 1978, 26(4), 1147–1152 CrossRef CAS PubMed.
- H. Taniyama, Y. Sawada and S. Tanaka, Chemical modification of streptothricin group antibiotics. II. Some amino derivatives in racemomycin-A and their biological activity, Chem. Pharm. Bull., 1974, 22(2), 337–341 CrossRef CAS PubMed.
- Y. Kono, S. Makino, S. Takeuchi and H. Yonehara, Sclerothricin, a new basic antibiotic, J. Antibiot., 1969, 22(12), 583–589 CrossRef CAS PubMed.
- H. E. Carter, R. K. Clark, P. Kohn, J. W. Rothrock, W. R. Taylor, C. A. West, G. B. Whitfield and W. G. Jackson, Streptothricin. I. preparation, properties and hydrolysis products, J. Am. Chem. Soc., 1954, 76(2), 566–569 CrossRef CAS.
- M. I. Horowitz and C. P. Schaffner, Paper chromatography of streptothricin antibiotics. Differentiation and fractionation studies, Anal. Chem., 1958, 30(10), 1616–1620 CrossRef CAS.
- M. Gan, X. Zheng, Y. Liu, Y. Guan and C. Xiao, Three new 12-carbamoylated streptothricins from streptomyces Sp. I08A 1776, Bioorg. Med. Chem. Lett., 2012, 22(19), 6151–6154 CrossRef CAS PubMed.
- Z. Ji, M. Wang, J. Zhang, S. Wei and W. Wu, Two new members of streptothricin class antibiotics from streptomyces qinlingensis Sp. Nov, J. Antibiot., 2007, 60(12), 739–744 CrossRef CAS PubMed.
- B. W. Bycroft and T. J. King, Crystal structure of streptolidine, a guanidine-containing amino-acid, J. Chem. Soc., Chem. Commun., 1972, 81(11), 652 RSC.
- F. Kudo, A. Miyanaga and T. Eguchi, Biosynthesis of natural products containing β-amino acids, Nat. Prod. Rep., 2014, 31(8), 1056–1073 RSC.
- R. M. Burckhardt and J. C. Escalante-Semerena, Insights into the function of the N-acetyltransferase SATa that detoxifies streptothricin in Bacillus subtilis and Bacillus anthraci, Appl. Environ. Microbiol., 2019, 85(6), 1–20 CrossRef PubMed.
- K. R. Farley and W. W. Metcalf, The streptothricin acetyltransferase (SAT) gene as a positive selectable marker for methanogenic archaea, FEMS Microbiol. Lett., 2019, 366(17), 1689–1699 CrossRef PubMed.
- J. Jacob, S. Evers, K. Bischoff, C. Carlier and P. Courvalin, Characterization of the SAT4 gene encoding a streptothricin acetyltransferase in Campylobacter coli be/g4, FEMS Microbiol. Lett., 1994, 120(1–2), 13–17 CAS.
- S. Keeratipibul, M. Sugiyama and R. Nomi, Mechanism of resistance to streptothricin of a producing microorganism, Biotechnol. Lett., 1983, 5(7), 441–446 CrossRef CAS.
- K. Smalla, R. Prager, M. Isemann, R. Pukall, E. Tietze, J. D. Elsas and H. Tschäpe, Distribution of streptothricin acetyltransferase encodi determinants among environmental bacteria, Mol. Ecol., 1993, 2(1), 27–33 CrossRef CAS.
- H. E. Webb, F. J. Angulo, S. A. Granier, H. M. Scott and G. H. Loneragan, Illustrative examples of probable transfer of resistance determinants from food animals to humans: streptothricins, glycopeptides, and colistin, F1000Research, 2017, 6, 1805 Search PubMed.
- R. M. Burckhardt and J. C. Escalante-Semerena, In bacillus subtilis, the SATa (formerly YYAR) acetyltransferase detoxifies streptothricin via lysine acetylation, Appl. Environ. Microbiol., 2017, 83(21), 1–11 CrossRef PubMed.
- Y. Hamano, N. Matsuura, M. Kitamura and H. Takagi, A novel enzyme conferring streptothricin resistance alters the toxicity of streptothricin d from broad-spectrum to bacteria-specific, J. Biol. Chem., 2006, 281(25), 16842–16848 CrossRef CAS PubMed.
- I. Haupt, J. Jonák, I. Rychlik and H. Thrum, Action of streptothricin F on ribosomal functions, J. Antibiot., 1980, 33(6), 636–641 CrossRef CAS PubMed.
- I. Haupt, R. Hübener, H. Thrum and F. Streptothricin, an inhibitor of protein synthesis with miscoding activity, J. Antibiot., 1978, 31(11), 1137–1142 CrossRef CAS PubMed.
- K. P. Smith, Y.-S. Kang, A. B. Green, M. G. Dowgiallo, B. C. Miller, L. Chiaraviglio, K. A. Truelson, K. E. Zulauf, S. Rodriguez, R. Manetsch and J. E. Kirby, Profiling the in vitro and in vivo activity of streptothricin-F against carbapenem-resistant enterobacterales: A historic scaffold with a novel mechanism of action, bioRxiv, 2021 DOI:10.1101/2021.06.14.448463.
- S. Kusumoto, S. Imaoka, Y. Kambayashi and T. Shiba, Total synthesis of antibiotic streptothricin F, Tetrahedron Lett., 1982, 23(29), 2961–2964 CrossRef CAS.
- The step counts and overall yield of Shiba's synthesis are approximated, not all synthetic methods and purified yields were fully described..
- It was key that the nitroketone formation occur under dilute, ambient conditions to avoid racemization of the Cbz-protected amino stereocenter..
- J. Rudolph, F. Hannig, H. Theis and R. Wischnat, Highly efficient chiral-pool synthesis of (2S,4R)-4-hydroxyornithine, Org. Lett., 2001, 3(20), 3153–3155 CrossRef CAS PubMed.
- Y. Yuasa, Y. Yuasa and H. Tsuruta, A facile synthesis of (2R,3S)-1-amino-3-tert-butoxycarbonylamino-2-hydroxy-4-phenylbutane; a useful component block of HIV protease inhibitor, Synth. Commun., 1998, 28(3), 395–401 CrossRef CAS.
- C. H. Heathcock, S. D. Young, J. P. Hagen, M. C. Pirrung, C. T. White and D. VanDerveer, Acyclic stereoselection. 9. Stereochemistry of the addition of lithium enolates to α-alkoxy aldehydes, J. Org. Chem., 1980, 45(19), 3846–3856 CrossRef CAS.
- A. Guaragna, D. D'Alonzo, C. Paolella, C. Napolitano and G. Palumbo, Highly stereoselective de novo synthesis of L-hexoses, J. Org. Chem., 2010, 75(11), 3558–3568 CrossRef CAS PubMed.
- N. J. Adderley, D. J. Buchanan, D. J. Dixon and D. I. Lainé, Highly diastereoselective oxy-michael additions of enantiopure δ-lactol anions to nitroalkenes: asymmetric synthesis of 1,2-amino alcohols, Angew. Chem., Int. Ed., 2003, 42(35), 4241–4244 CrossRef CAS PubMed.
- Boc protection was only employed in this instance to increase the recovery of product from the aqueous phase as the unprotected amino alcohol of 12 had high water solubility..
- T. Stark, M. Suhartono, M. W. Göbel and M. Lautens, A palladium-catalyzed domino reaction as key step for the synthesis of functionalized aromatic amino acids, Synlett, 2013, 24(20), 2730–2734 CrossRef CAS.
- D. A. Evans, T. C. Britton, J. A. Ellman and R. L. Dorow, The asymmetric synthesis of α-amino acids. electrophilic azidation of chiral imide enolates, a practical approach to the synthesis of (R)- and (S)-α-azido carboxylic acids, J. Am. Chem. Soc., 1990, 112(10), 4011–4030 CrossRef CAS.
- M. I. García-Moreno, P. Díaz-Pérez, J. M. Benito, C. Ortiz Mellet, J. Defaye and J. M. García Fernández, One-step synthesis of non-anomeric sugar isothiocyanates from sugar azides, Carbohydr. Res., 2002, 337(21–23), 2329–2334 CrossRef.
- P. Singh, V. Mehra, A. Anand, V. Kumar and M. P. Mahajan, β-lactam-synthon-interceded diastereoselective synthesis of novel thioxo-imidazolines: a convenient access to functionally decorated 4,5-dihydro-imidazoles, Tetrahedron Lett., 2011, 52(39), 5060–5063 CrossRef CAS.
- M. D. Wittman, R. L. Halcomb, S. J. Danishefsky, J. Golik and D. Vyas, A route to glycals in the allal and gulal series: synthesis of the thiosugar of esperamicin A1, J. Org. Chem., 1990, 55(7), 1979–1981 CrossRef CAS.
- S. Buttar, J. Caine, E. Goné, R. Harris, J. Gillman, R. Atienza, R. Gupta, K. M. Sogi, L. Jain, N. C. Abascal, Y. Levine, L. M. Repka and C. M. Rojas, Glycal metallanitrenes for 2-amino sugar synthesis: amidoglycosylation of gulal-, allal-, glucal-, and galactal 3-carbamates, J. Org. Chem., 2018, 83(15), 8054–8080 CrossRef CAS PubMed.
- O. Boutureira, M. A. Rodríguez, M. I. Matheu, Y. Díaz and S. Castillón, General method for synthesizing pyranoid glycals. A new route to allal and gulal derivatives, Org. Lett., 2006, 8(4), 673–675 CrossRef CAS PubMed.
- V. Di Bussolo, M. Caselli, M. Pineschi and P. Crotti, New stereoselective β-C-glycosidation by uncatalyzed 1,4-addition of organolithium reagents to a glycal-derived vinyl oxirane, Org. Lett., 2003, 5(12), 2173–2176 CrossRef CAS PubMed.
- V. Di Bussolo, M. Caselli, M. R. Romano, M. Pineschi and P. Crotti, Stereospecific uncatalyzed α-O-glycosylation and α-C-glycosidation by means of a new D-gulal-derived α vinyl oxirane, J. Org. Chem., 2004, 69(21), 7383–7386 CrossRef CAS PubMed.
- T. M. Beale, P. J. Moon and M. S. Taylor, Organoboron-catalyzed regio- and stereoselective formation of β-2-deoxyglycosidic linkages, Org. Lett., 2014, 16(13), 3604–3607 CrossRef CAS PubMed.
- The introduction of a primary carbamoyl as opposed to the DMB-protected carbamoyl reduced the yields of the dihydroxylation and the burgess reagent sulfamidation from 88% to 51% and 28% to 15%, respectively..
- K. C. Nicolaou, S. A. Snyder, D. A. Longbottom, A. Z. Nalbandian and X. Huang, New uses for the Burgess reagent in chemical synthesis: methods for the facile and stereoselective formation of sulfamidates, glycosylamines, and sulfamides, Chem.–Eur. J., 2004, 10(22), 5581–5606 CrossRef CAS PubMed.
- E. H. Ryu, A. Ellern and Y. Zhao, High guest inclusion in 3β-amino-7α,12α-dihydroxycholan-24-oic acid enabled by charge-assisted hydrogen bonds, Tetrahedron, 2006, 62(29), 6808–6813 CrossRef CAS.
- J. P. Genêt, E. Blart, M. Savignac, S. Lemeune and J. M. Paris, Palladium-catalyzed reactions in aqueous media. an efficient removal of allyloxycarbonyl protecting group from oxygen and nitrogen, Tetrahedron Lett., 1993, 34(26), 4189–4192 CrossRef.
- J. B. Aggen, P. Dozzo, A. A. Goldblum, D. J. Hildebrandt, T. R. Kane, M. J. Gliedt and M. S. Linsell, Antibacterial aminoglycoside analogs, WO 2011/044501 A2, 2011.
- Centers for Disease Control and Prevention, Antibiotic Resistance Threats in the United States, Atlanta, 2019 Search PubMed.
- World Health Organization, Global Action Plan on Antimicrobial Resistance, Geneva, 2015 Search PubMed.
- World Health Organization, Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics, Geneva, 2017 Search PubMed.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Bad bugs, no drugs: no ESKAPE! an update from the infectious diseases society of America, Clin. Infect. Dis., 2009, 48(1), 1–12 CrossRef PubMed.
- L. Chen, R. Todd, J. Kiehlbauch, M. Walters and A. Kallen, Notes from the field: pan-resistant New Delhi metallo-beta-lactamase-producing klebsiella pneumoniae –Washoe County, Nevada, 2016, Morb. Mortal. Wkly. Rep., 2017, 66(1), 33 CrossRef PubMed.
- Modal MICs for MIC distributions provided by eucast (european committee on antimicrobial susceptibility testing), https://mic.eucast.org/search/, accessed 1/29/2022 Search PubMed.
- A. D. Kang, K. P. Smith, G. M. Eliopoulos, A. H. Berg, C. McCoy and J. E. Kirby, In vitro apramycin activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa, Diagn. Microbiol. Infect. Dis., 2017, 88(2), 188–191 CrossRef CAS PubMed.
- T. Brennan-Krohn, K. A. Truelson, K. P. Smith and J. E. Kirby, Screening for synergistic activity of antimicrobial combinations against carbapenem-resistant enterobacteriaceae using inkjet printer-based technology, J. Antimicrob. Chemother., 2017, 72(10), 2775–2781 CrossRef CAS PubMed.
- K. P. Smith and J. E. Kirby, Verification of an automated, digital dispensing platform for at-will broth microdilution-based antimicrobial susceptibility testing, J. Clin. Microbiol., 2016, 54(9), 2288–2293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for all new compounds. See DOI: 10.1039/d1sc06445b |
| This journal is © The Royal Society of Chemistry 2022 |