
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton decay mechanism in DNA single strands: back-electron transfer and ultrafast base motions†
Benjamin
Bauer
a,
Rahul
Sharma
 b,
Majed
Chergui
a and
Malte
Oppermann
*a
b,
Majed
Chergui
a and
Malte
Oppermann
*a
aLaboratory of Ultrafast Spectroscopy (LSU), Lausanne Centre for Ultrafast Science (LACUS), École Polytechnique Fédérale de Lausanne, ISIC-FSB, CH-1015 Lausanne, Switzerland. E-mail: malte.oppermann@epfl.ch
bLaboratory for Computation and Visualization in Mathematics and Mechanics, École Polytechnique Fédérale de Lausanne, MATH-FSB, CH-1015 Lausanne, Switzerland
First published on 19th April 2022
Abstract
The photochemistry of DNA systems is characterized by the ultraviolet (UV) absorption of π-stacked nucleobases, resulting in exciton states delocalized over several bases. As their relaxation sensitively depends on local stacking conformations, disentangling the ensuing electronic and structural dynamics has remained an experimental challenge, despite their fundamental role in protecting the genome from potentially harmful UV radiation. Here we use transient absorption and transient absorption anisotropy spectroscopy with broadband femtosecond deep-UV pulses (250–360 nm) to resolve the exciton dynamics of UV-excited adenosine single strands under physiological conditions. Due to the exceptional deep-UV bandwidth and polarization sensitivity of our experimental approach, we simultaneously resolve the population dynamics, charge-transfer (CT) character and conformational changes encoded in the UV transition dipoles of the π-stacked nucleotides. Whilst UV excitation forms fully charge-separated CT excitons in less than 0.3 ps, we find that most decay back to the ground state via a back-electron transfer. Based on the anisotropy measurements, we propose that this mechanism is accompanied by a structural relaxation of the photoexcited base-stack, involving an inter-base rotation of the nucleotides. Our results finally complete the exciton relaxation mechanism for adenosine single strands and offer a direct view into the coupling of electronic and structural dynamics in aggregated photochemical systems.
Introduction
The photochemical functions of natural and synthetic molecular aggregates are characterized by so-called exciton states that result from the electrostatic interactions of their constituent chromophores.1,2 An important example are DNA systems, where the exciton transitions of π-stacked nucleobases dominate the absorption cross-section in the near ultraviolet (UV) region and thus play a crucial role in the formation of harmful photolesions and radical species, which may lead to skin cancer for example.3 Indeed, whilst isolated nucleobase monomers efficiently dissipate their excitation energy on a sub-picosecond time-scale, single- (ssDNA) and double-stranded (dsDNA) DNA oligomers display excited state species with lifetimes up to the millisecond time scale.4,5 However, whilst the initial exciton formation and ensuing charge separation processes across π-stacked bases have been studied in various model systems, achieving a complete picture of the exciton relaxation dynamics has remained challenging, because of the complex ground and excited state interactions between multiple bases in solution-phase DNA.Due to the close proximity of π-stacked bases in B-form DNA (≈3.4 Å), the resulting electrostatic interactions generally comprise contributions from dipole–dipole, orbital overlap and electron exchange interactions.6 Consequently, the exciton states are characterized by two attributes:7 a delocalization length corresponding to the number of coupled bases and the degree of charge separation or charge-transfer (CT) character across them.‡ Importantly, the excitonic interactions sensitively depend on the geometrical arrangement of the coupled bases, such that the initially excited excitons are mostly determined by the ground state conformational ensemble. Their relaxation dynamics, however, crucially depend on the inter-base coupling interactions in the photoexcited state and may therefore also involve local conformational changes of the oligomer.8
To this end, deoxyadenosine monophosphate ssDNA (denoted as dAn with n nucleotides) has emerged as a convenient helical model system with high stacking ratios of around 80% at room-temperature.9 The monomer (dA1) in the gas phase displays three close-lying lowest-energy transitions consisting of two optically bright 1ππ* transitions, La and Lb, and an optically dark 1nπ* transition.10 In aqueous solution, La is strongly stabilized, becoming the lowest energy LUMO singlet state, which also carries most of the oscillator strength.11 From the Franck–Condon region, nearly barrierless pathways lead to conical intersections with the ground state surface, which rapidly deactivate the excited state on the sub-picosecond time scale forming a vibrationally hot ground state.12 In ssDNA, excitonic interactions between stacked bases cause a prominent decrease in oscillator strength (hypochromism) and a slight blue-shift (hypsochromism) of the 1ππ* band maximum compared to the non-aggregated bases. These features are consistent with those of self-assembled H-aggregates of π-conjugated organic molecules.13,14 However, even though these effects are well known since the 1960s,15,16 the exact delocalization length, CT character and energetic positions of the exciton states contributing to the static absorption of DNA oligomers remain somewhat controversial. In dAn, La and Lb transitions on different bases may couple and several theoretical treatments have predicted significant contributions from excitons delocalized over more than two stacked bases.17–20 Nevertheless, strand-length dependent circular dichroism (CD) measurements, which are directly sensitive to dipolar couplings in the ground state, show that nearest-neighbor interactions strongly dominate in the 1ππ* band.21 This implies that despite the pronounced stacking disorder in dAn, stacked domains with large dipolar coupling terms are limited to two bases.22 Indeed, there is growing consensus that the lowest-energy absorption band in dAn contains two exciton species: a neutral two-base exciton with zero CT character carrying most of the oscillator strength and a two-base CT exciton with nonzero CT character and a much weaker absorption coefficient.8,19,23 Strong solvent interactions are expected to inhomogeneously broaden and red-shift the absorption band of the CT exciton state and it is thus assumed to be the lowest-energy excited state, causing the enhanced low-energy absorption of dAn above 275 nm, compared to monomeric dA1.24,25
Naturally, ultrafast studies on ssDNA have been instrumental in mapping out the exciton dynamics, albeit with often conflicting interpretations. Whilst the first optical experiments suggested excitons to be formed dynamically from photoexcited monomers,26 this was later ruled out in favour of a direct excitation process. Here, early suggestions of average delocalization lengths of up to 4 bases27 were contested by mounting evidence for the exclusive excitation of two-base excitons, obtained from single-wavelength transient absorption (TA)22,28 and time-resolved fluorescence experiments.23 These studies broadly agreed on a mechanism where optically bright neutral two-base excitons are excited first, followed by an ultrafast internal conversion to the optically dark minimum of the exciton band. These states would then acquire a CT character on the picosecond scale, possibly enabled by a reduction of the inter-base distance. Recently, Borrego-Varillas and co-workers were able to measure the upper limits of the involved time scales, finding that internal conversion to the neutral exciton band minimum is completed in 100 fs, followed by a full charge-separation associated with an adenosine radical-ion pair in at most 3 ps.29
In a parallel development, the vibrational marker bands of nucleotide radical-ions were identified and tracked in a series of ultrafast spectroscopy studies on UV-excited ssDNA model systems.30,31 This corroborated the central role of CT excitons in the relaxation dynamics of ssDNA as the longest-living species dominating the dynamics beyond the first 10 ps. In addition, it has been established that a relevant fraction of the radical ions formed in adenosine stacks in both ss- and dsDNA survive well into the millisecond timescale, indicating that UV excitation may provide an important contribution to the oxidative damage of DNA.32,33 However, whilst the role of CT excitons as gateway states to DNA photodamage is now mostly settled, several aspects of their dynamic evolution have remained open. Importantly, a specific deactivation mechanism has not yet been established for dAn. To this end, several possibilities have been considered, most notably a charge recombination process either via back-electron transfer19 or intra-strand proton-transfer,34 a decay to a monomer-like state followed by internal conversion to the ground state,35 and the dynamic formation of a bonded two-base exciton species with a strongly reduced inter-base distance.36 Even though such excited state conformational changes have been theoretically predicted to play an important role in their relaxation,19,23,34,37 their direct observation has remained an experimental challenge, due to the current limitations of established spectroscopic techniques with combined time and structural resolution in the solution phase. Especially in the case of subtle motions of local chromophores or side chains, time-resolved X-ray solution scattering does currently not provide the required spatial resolution,38 whereas nuclear magnetic resonance spectroscopy does not provide the necessary time resolution.39
Here we pursue an alternative approach and employ a polarization-controlled pump-probe setup (see Methods) with femtosecond deep-UV probe pulses (250–360 nm) to conduct the first ultrafast spectroscopy experiments with broadband coverage of the 1ππ* band of dA20 < 300 nm. By combining transient absorption (TA) with transient absorption anisotropy (TAA) measurements, we simultaneously resolve the population dynamics, CT character and conformational changes encoded in the coupled UV transition dipoles of the photoexcited excitons. By modulating the strand-length, excitation wavelength and solvent environment we are then able to develop a complete reaction scheme. Quite remarkably, we find that CT excitons are already formed within the first 0.3 ps from initially excited neutral two-base excitons, excluding any conformational dynamics in the charge-separation process. However, the transient anisotropy data suggests that ultrafast inter-base rotations drive the CT exciton to its minimum energy configuration in about 40 ps. Finally, by comparing the exciton dynamics in H2O and D2O buffer solution, we are able to provide conclusive evidence for a back-electron transfer as the deactivation mechanism of the structurally relaxed CT excitons.
To establish these findings, we compare a large number of investigated sample systems, measurement conditions and experimental techniques. For achieving a clear presentation of the results without losing track of the discussion, we present and interpret each experiment in a separate section. We begin with the TA measurements of dA20 in H2O buffer solution and compare the kinetics observed for 266 nm and 285 nm excitation, thereby resolving different exciton formation mechanisms. Furthermore, a direct comparison with the TA kinetics of dA1 and dA2 highlights the close packing of bases in dA20 as a crucial factor in determining the exciton dynamics. In the following section we present molecular dynamics simulations to estimate the dipolar coupling strength between stacked bases in dA20, thereby establishing a link between its conformational ensemble and the photoexcited exciton states. Moving forward we present the TA measurements of the samples in D2O buffer solution, where the observed kinetic isotope effects suggest a back-electron transfer as the primary exciton relaxation mechanism. The TAA measurements in H2O and D2O buffer solutions are presented in the final section and corroborate this mechanism, whilst also providing insights into the structural dynamics that accompany the exciton decay. Finally, we combine the above findings to establish a complete reaction scheme in the Concluding section.
Results and discussion
Transient absorption in H2O buffer solution
This section compares three samples of dAn in aqueous phosphate buffer solution with different strand lengths (for sample preparation see Methods): the monomer dA1, the dimer dA2, and the multimer dA20. Fig. 1 in the ESI† displays their steady-state absorption spectra, which are normalized to the respective band maxima to highlight differences in the band shapes. Compared to dA1, dA20 shows a pronounced blue-shift of the band maximum which can be attributed to the dipole–dipole interaction between stacked bases resulting in neutral exciton states. Below 275 nm, dA20 displays an enhanced absorption, which has been attributed to a CT exciton band.8,25 Whilst photoexcitation at 266 nm thus mainly populates neutral exciton states, photoexcitation at 285 nm is expected to predominantly excite CT excitons. Also note that whilst dA2 displays qualitatively similar spectral differences with dA1, these features are much less pronounced compared to dA20.Fig. 1 displays normalized time- and wavelength-resolved TA maps for the three investigated samples with the UV excitation wavelength indicated in brackets: dA1 (266 nm), dA20 (266 nm), dA20 (285 nm), and dA2 (266 nm). The original data sets are displayed in Fig. 4 and 9 in the ESI.† All four TA maps display a negative TA amplitude <275 nm, associated with a ground state bleach (GSB), and a positive amplitude >280 nm, associated mostly with an excited state absorption (ESA). The direct comparison between the samples reveals striking differences in the GSB and ESA band shapes and decay kinetics. Whilst dA1 displays a narrow ESA maximum near 290 nm that decays on a picosecond timescale, the oligomer samples display a broad ESA band that decays two orders of magnitude more slowly. Because of the picosecond kinetics, we can identify these ESA bands with the long-lived CT exciton states that are exclusively formed in the oligomer systems. However, different ESA bandwidths are observed for the oligomer samples, which suggests that the precise nature and evolution of the formed CT excitons strongly depend on both the excitation wavelength and the number of nucleotides in the strand. In the following we will first focus on the TA data for pump-probe delays <1 ps to discuss the CT exciton formation mechanism. We will then focus on the TA data for pump-probe delays >1 ps to discuss their relaxation mechanism.
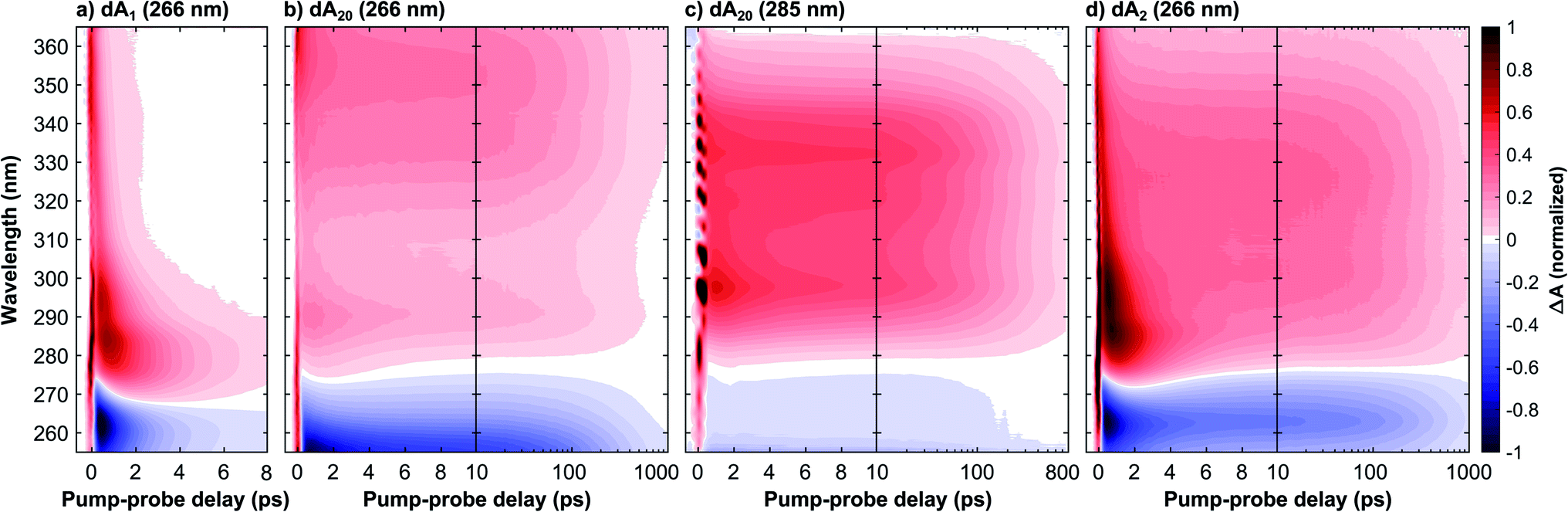 | ||
| Fig. 1 Transient absorption time–wavelength maps of a deoxyadenosine monophosphate ssDNA with n nucleotides in aqueous phosphate buffer solution and UV excitation wavelength as indicated in brackets: (a) monomer dA1 (266 nm), (b) 20-mer dA20 (266 nm), (c) 20-mer dA20 (285 nm), and (d) dimer dA2 (266 nm). The experimental time-resolution was <300 fs and maps (a, b and d) are normalized to the ground state bleach minimum near 265 nm, whereas as map (c) was scaled to 70% of its excited state absorption maximum near 300 nm for improved comparability. Whilst the monomer's excited state absorption (ESA, positive signal) decays in <10 ps, the multimer systems's ESA band decays on the 100 ps time scale and is associated with a CT exciton state. Their different band shapes >10 ps indicate that the formed CT exciton depends on both the excitation wavelength and strand length. A global analysis of the data assigns a 190 ps time constant to its decay in dA20, independent of the excitation wavelength, whereas a much longer time constant of 340 ps is observed in dA2. | ||
To this end, Fig. 2 compares the early-time TA spectra of the adenosine samples at a pump-probe delay of 0.5 ps. Whilst dA1 (266 nm) and dA2 (266 nm) display mostly identical GSB bands, dA20 (266 nm) shows a pronounced blue-shift of >10 nm. In comparison, the GSB band of dA20 (285 nm) is extremely weak and likely extends further towards the mid-UV region. In the ESA region, dA1 and dA2 show relatively similar spectral shapes, albeit with different amplitudes, dominated by a pronounced band centred near 290 nm and a tail decaying towards 370 nm. In contrast, dA20 (266 nm) shows only a very weak ESA band at 290 nm, whereas such a band is not observable in dA20 (285 nm). Both 20-mers display a broad ESA band, however dA20 (266 nm) has an additional ESA band near 370 nm.
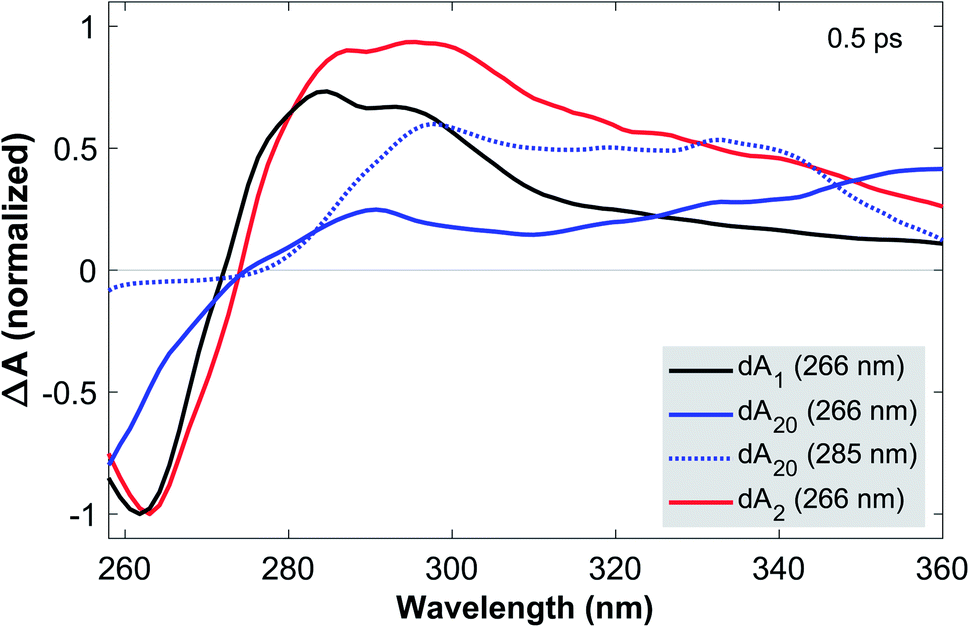 | ||
| Fig. 2 TA spectra of dA1, dA20, and dA2 in aqueous phosphate buffer solution at a pump-probe delay of 0.5 ps. All samples were photoexcited at 266 nm, whereas dA20 was also excited at 285 nm, as indicated. For 266 nm excitation, the TA spectra are normalized to the GSB minimum near 265 nm, whereas the normalization factor for dA20 was adapted in accordance with the curvature of this feature. The TA spectrum for dA20 (285 nm) was scaled to 70% of the ESA maximum. The comparison of the GSB bands suggests that whilst dA2 is dominated by monomeric excitations, these are mostly absent in dA20, where excitonic transitions dominate. Comparing dA20 (266 nm) and dA20 (285 nm) shows that two different exciton species are excited, resulting in strong spectral differences in the GSB and ESA bands. | ||
Starting with dA1, we can assign its GSB to the La transition and the main ESA band to the hot ground state, consistent with the literature.12 The tail extending into the near-UV has previously been assigned to the almost fully decayed La state located near 370 nm.40,41 The strongly blue-shifted GSB in dA20 (266 nm) is consistent with its blue-shifted steady state absorption spectrum and confirms that at 266 nm, a direct photoexcitation channel to high-energy bright neutral excitons dominates. The corresponding ESA region therefore only shows a residual contribution from a hot monomeric ground state near 290 nm, likely resulting from monomer-like excitations of unstacked nucleotides within the strand.22 Instead, the ESA band is dominated by an additional maximum near 370 nm, which decays rapidly on the sub-picosecond time scale (see Fig. 1c). We thus assign this band to the neutral exciton ESA from coupled La transition dipoles, which rapidly decays via internal conversion to the neutral exciton band minimum, consistent with recent theoretical and experimental studies.29,40 In direct contrast, dA20 (285 nm) does not display any of the spectral signatures of the neutral exciton or any monomeric excitation. However, its weak GSB band is consistent with the direct excitation of a (mostly) optically dark CT exciton with a strongly inhomogeneously broadened absorption band, as suggested in the literature. The observed ESA band can be attributed entirely to a CT exciton, which thus corresponds to the lowest-energy excited state in dA20. Finally, the GSB in dA2 suggests that 266 nm excitation mostly populates monomeric La states without any clear evidence for the direct excitation of neutral or CT excitons. However, whilst the ESA is dominated by a hot monomeric ground state, the differences in ESA amplitudes between dA1 and dA2 suggest the presence of an additional broad ESA band that is not observed in the monomer.
We now proceed to the discussion of the TA data for pump-probe delays >1 ps, as displayed in Fig. 1. In dA1, the hot ground state's ESA feature decays on the picosecond timescale due vibrational cooling, consistent with the time-dependent blue-shift of its spectral weight and the simultaneous recovery of the ground state. In contrast, dA20 (266 nm) shows a negligible contribution from this channel and instead displays an ESA decay in >10 ps. Following the work by Borrego-Varillas and co-workers,29 the ESA band on this scale is due to the absorption of a two-base CT exciton, associated with a fully separated adenosine radical ion pair. This implies that the long-lived ESA band in dA20 (285 nm) must describe a different CT exciton species, either with a different delocalization length or a reduced CT character. To this end, the comparison with dA2 offers further insights. Whilst the vibrational cooling of the hot monomeric ground state dominates the kinetics <10 ps, the ESA decay >10 ps is slower than in both experiments on dA20. Importantly, the long-lived ESA in dA2 takes a band shape that is remarkably similar to dA20 (285 nm). Indeed, in Fig. 9d in the ESI,† we show that the early-time TA spectrum of dA2 (266 nm) can be very well reproduced by a sum of the scaled TA spectra of dA1 and dA20 (285 nm) at the same pump-probe delay of 0.5 ps. We thus suggest that a two-base CT exciton with a partial CT character is formed in both in dA2 and dA20 (285 nm).
To quantify the TA kinetics, we performed global fits on each of the presented TA data sets. To this end, we employed a sequence of multi-exponential functions convoluted with a Gaussian instrument response function (for details see Methods and Section 3 in the ESI†). The resulting decay associated spectra (DAS) and residuals are displayed in Fig. 4 and 9 in the ESI† and the obtained decay constants are displayed in Table 1. Each decay constant is reported with an error of 10%. The kinetics of dA1 can be accurately fitted with a minimum of two exponential functions, with time constants τ1(1) = 0.7 ± 0.1 ps and τ2(1) = 1.9 ± 0.2 ps. Even though we obtain a high accuracy for the performed fits, it is well known that the spectral shifts caused by vibrational cooling dynamics cannot be modeled adequately by globally fitted multi-exponential functions.42 We therefore neglect a detailed analysis of the short-time dynamics. Nevertheless, this has no impact on the fitting accuracy of the kinetics that are orders of magnitude slower and thus unaffected by the cooling dynamics. The fits for the remaining oligomers display similar kinetic components <10 ps, but notably require two additional slower decay components to achieve an adequate fit of the data: a time constant τ3 on the 100 ps scale, associated with the deactivation of the CT exciton, and τ4, which is much longer than the maximum pump-probe delay and is associated with a long-lived photo-product.32 First of all we note that the value obtained for τ3(20,266) = 190 ± 20 ps agrees well with previously published data.22 Remarkably, we find that in dA20 this decay constant is mostly independent of the excitation wavelength, as we obtain τ3(20,285) = 180 ± 20 ps for dA20 (285 nm). This is in contrast with the much longer decay constant τ3(2) = 340 ± 30 ps obtained for dA2 (266 nm). In addition, Fig. 9e† compares the DAS for τ3 for the three oligomer samples and shows mostly identical spectral shapes for dA20 (285 nm) and dA2 (266 nm) in the ESA region, whereas the corresponding DAS for dA20 (266 nm) extends further beyond 360 nm.
| Sample | τ 1 (ps) | τ 2 (ps) | τ 3 (ps) | τ 4 (ps) | ρ 1 (ps) | ρ 2 (ps) | ρ 3 (ps) |
|---|---|---|---|---|---|---|---|
| dA1, H2O, 266 nm | 0.7 ± 0.1 | 2.0 ± 0.2 | — | — | — | — | — |
| dA2, H2O, 266 nm | 0.7 ± 0.1 | 2.0 ± 0.2 | 340 ± 30 | ≫1000 | 2.3 ± 0.5 | — | 240 ± 30 |
| dA20, H2O, 266 nm | 1.0 ± 0.1 | 2.1 ± 0.2 | 190 ± 20 | ≫1000 | 1.8 ± 0.5 | 44 ± 10 | 1200 ± 200 |
| dA20, H2O, 285 nm | — | 4.0 ± 0.4 | 180 ± 20 | ≫1000 | — | — | — |
| dA1, D2O, 266 nm | 1.0 ± 0.2 | 2.5 ± 0.5 | — | — | — | — | — |
| dA2, D2O, 266 nm | 1.0 ± 0.1 | 3.1 ± 0.3 | 730 ± 70 | ≫1000 | 3.1 ± 0.5 | — | 270 ± 30 |
| dA20, D2O, 266 nm | 1.0 ± 0.1 | 3.8 ± 0.4 | 330 ± 30 | ≫1000 | 1.9 ± 0.5 | 35 ± 10 | 1300 ± 200 |
On the basis of these observations, we thus propose two different excitation pathways in dA20: (1) the dynamic formation of a fully charge-separated CT exciton via an optically bright neutral two-base exciton, and (2) the direct excitation of a CT exciton, which displays a reduced CT character at 285 nm excitation. Since the observed CT exciton decay time is independent of the formation pathway, there is likely a single CT exciton band in dA20, which can be accessed via an internal conversion process from the neutral exciton band. Note that whilst the absorption of the CT exciton band is sufficiently broad to be excited at 266 nm as well, its contribution to the total yield is expected to be negligible, due to its low oscillator strength. The same reasoning thus applies to dA2, even though an ESA signature of neutral excitons is not resolved in the TA data. This is surprising, since the non-zero CD spectrum of dA2 clearly shows the presence of dipolar couplings between the nucleobases.35 However, we propose that the number of strongly coupled bases is much lower in dA2 than in dA20, because the greater steric freedom of dA2 is expected to lead to a much broader conformational ensemble. Since the dipolar coupling strength J between neighbouring bases has an R−3 dependence (with R denoting the inter-base distance) and is maximised for parallel transition dipoles (see eqn (2) in Methods), the average dipolar coupling strength is thus expected to be reduced compared to dA20.
Molecular dynamics simulations
In order to rationalize the discussion of the TA data, we conducted molecular dynamics simulations of the ground state configurational ensemble of dA20 and dA2 (for computational details see Methods). Fig. 3, displays the probability distribution of nearest-neighbor configurations as a function of their dipolar coupling strength, for three different dimer classes: (1) an isolated dA2, (2) the two-base stacks at the ends of a strand (dA20 ends), and (3) the average of all two-base stacks in the strand (dA20 mean). As expected, the probability distribution for a free dA2 in solution is strongly shifted towards lower values of J compared to the mean of dimer configurations within dA20, whilst also showing a much larger number of configurations with J close to zero. Note that the probability distribution of the stacks at the end of the strand describes an intermediate case, since only one of the bases has two nearest neighbors. Even though this simulation is based on a simplified definition of the dipolar coupling strength, it strongly suggests that the close packing of nucleotides in dA20 reduces the conformational heterogeneity of inter-base geometries and thus increases the number of neutral exciton states at the expense of monomeric excited states. Similarly, we propose that electron exchange and orbital overlap interactions are increased, such that the CT exciton in dA20 (266 nm) displays a larger CT character than in dA2 (266 nm). In this view, the CT character of the formed exciton is determined by the base stack conformation upon photo-excitation. We thus speculate that the lowest energy CT exciton excited in dA20 (285 nm) corresponds to a strongly solvent-stabilized conformation with an increased inter-base distance, for example.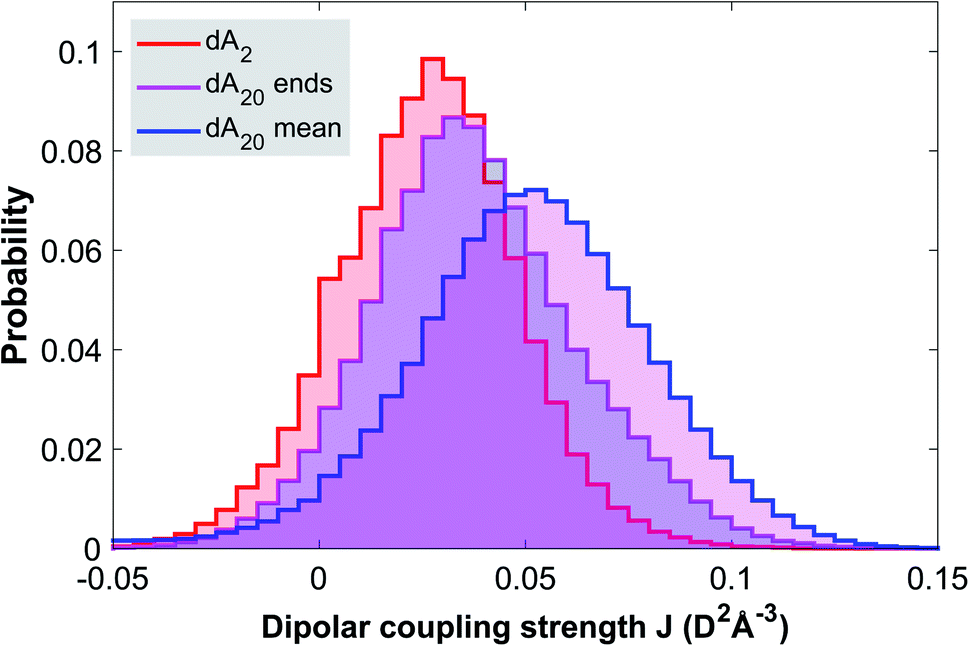 | ||
| Fig. 3 Molecular dynamics simulation of the ground state ensembles of solvated dA2 and dA20. The probability distributions are obtained by binning the nearest-neighbor base stack configurations according to their calculated dipolar coupling strength J (see Methods) for an isolated dimer (dA2), for the end-pairs in the strand (dA20 ends) and for all base-pairs in the strand (dA20 mean). The results show that the closer packing in dA20 leads to a significant increase of nearest-neighbor stacks with a strong dipolar coupling. | ||
Transient absorption in D2O buffer solution
Since both dA20 (266 nm) and dA20 (285 nm) display a significantly faster CT exciton decay time than dA2 (266 nm), it is likely that the close packing of dA20 also plays a role in its deactivation mechanism. This suggests a process that is sensitive to the spatial configuration of the base-stack, such as a back-electron transfer and an intra-strand proton transfer, for example. We thus conducted TA experiments in heavy water (D2O) phosphate buffer solution to obtain further insights into the CT decay mechanism. Fig. 6 in the ESI† displays the obtained TA data sets for dA1 (266 nm), dA20 (266 nm), and dA2 (266 nm) and the results of their global analysis. The obtained decay constants are reported in Table 1. Whilst the observed vibrational cooling dynamics are slightly slower as expected, a significant kinetic isotope effect (KIE) is observed for the CT exciton decay constant τ3. Defining K = τD3/τH3 = kH3/kD3, we obtain moderate KIEs of K(20) ≈ 1.7 for dA20 (266 nm) and K(2) ≈ 2.1 for dA2 (266 nm). This is somewhat surprising, as previous studies on dsDNA have only detected KIEs for alternating base-sequences and no deuteration effects in non-alternating sequences (such as (dTdA)n).43,44 Nevertheless, the observed KIEs were assigned to a sequential process with an initial intra-strand exciton formation followed by quenching through inter-strand proton transfer.45,46 Taking into account the lack of base-pairing interactions, we thus note that the values for K(20) and K(2) are consistent with KIEs commonly observed for a back-electron transfer in radical-ion pairs.47 Whilst an intra-strand proton transfer might not be excluded on this basis, the average inter-base distance of ≈3.4 Å in B-form DNA is expected to lead to a high reaction barrier34 and a significantly higher KIE.48In this view, it is important to consider two ways in which the solvent deuteration may alter the electronic properties of dAn. First, hydrogen-bonding is a key factor in mediating base-stacking,49 such that its weakening through deuteration may affect the ground state conformational ensemble and thus the excitonic coupling interactions. However, the CD spectra for dA20 and dA2 in H2O and D2O buffer solution (see Fig. 2) display nearly identical shapes, suggesting a negligible impact on the inter-base dipolar couplings. Second, it is well known that solvation in D2O buffer solution leads to deuteration of the amino group of dA1.50 Here, solvent-dependent studies on structurally similar naphthylalkylamines have shown that amino-deuteration and the increase of its ionization potential through the weakened hydrogen-bonding network lead to a reduced CT character of the intra-molecular CT exciton state.51 Similar arguments have also been employed by Mataga and coworkers to explain the solvent-dependent CT exciton decays of hydrogen-bonded molecular ion-pairs involving amino groups as proton donors.52
Transient absorption anisotropy in H2O and D2O buffer solution
In transient absorption anisotropy (TAA), a linearly polarized excitation pulse creates a partially aligned ensemble of photoexcited molecules. Note that for a given angle θ between the excited transition dipole and the pump polarization, the photoexcitation probability has a cos2(θ) dependence. Analogously to fluorescence anisotropy, TAA measures the TA difference of a parallel and perpendicular pump and probe pulse polarization configuration, according to eqn (1) in Methods. As a consequence, the TAA of the ensemble average takes a maximum value of r = 0.4 if the probed transition dipole is parallel to the one initially excited and it takes a minimum value of r = −0.2 if they are perpendicular. Note that this assignment only applies to single dipole transitions, whereas typical TAA spectra usually contain overlapping contributions from different GSB, ESA and stimulated emission bands. Whilst this may complicate the interpretation of the measured TAA values, broadband TAA and TA experiments can be combined to mitigate this.53 In the GSB region of the reported experiments, TAA probes the time-dependent orientation of the bleached transition dipoles with respect to the originally excited ground state transition dipole. In the ESA region, TAA then probes the relative orientation between the excited state transition dipole and the original photoexcitation axis within the molecule's frame of reference.To this end, Fig. 10† displays time- and wavelength-resolved TAA maps of dA2, dA20 (266 nm), and dA20 (285 nm) in H2O buffer solution, whereas Fig. 12† displays the TAA maps of dA2 and dA20 (266 nm) in D2O buffer solution. All maps display positive TAA values in both the GSB and ESA bands, which decay to zero in a global fashion due to rotational diffusion. Note that due to its mathematical definition, the calculated TAA diverges to infinity when the TA signal approaches zero, which takes place both for zero-crossings between the GSB and ESA bands and for low excited state populations at long pump-probe delays. However, except near the zero-crossings, the TAA bands do not display any significant spectral dependence. The uniform TAA bands of dA2 and dA20 in the ESA region therefore provide further evidence that the CT exciton is the only excited species probed in this region, as the presence of an additional ESA transition dipole would likely lead to a variation of the TAA value.53 As for the TA data, we first compare the spectral shapes of the TAA spectra at early pump-probe delays and then proceed to discuss the time-evolution of the TAA signal.
Fig. 4a displays the TAA spectra in the ESA region for a pump-probe delay of 3 ps, where CT excitons are expected to be fully formed and any ESA from a hot monomeric ground state mostly decayed. In dA20 (266 nm) and dA2 (266 nm), most CT excitons are formed through neutral excitons and thus the excitation of coupled La transition dipoles within a base-stack. This implies that the initially excited transition dipole is directed parallel to the nucleobase plane. As a charge separation across π-stacked bases creates a transition dipole moment perpendicular to the nucleobase planes, any increase in CT character would lead to a decrease of the measured TAA. On this basis, the comparison of the TAA spectra confirms that the CT character of the CT exciton in dA20 (266 nm) is indeed higher than in dA2 (266 nm). Quite remarkably, however, changing the solvent from H2O to D2O has no appreciable effect on the measured TAA in any of the samples. Therefore, the amino group does not play a role in the CT exciton formation and we may rule out any contribution as a proton donor in an intra-strand proton transfer. We thus conclude that in dAn, CT excitons are quenched via an electron transfer, such that the observed KIEs result from a reduced vibrational coupling upon solvent deuteration. The longer CT exciton decay times in dA2 are then likely caused by a combination of less favorable structural parameters. First, the broader ground state conformational ensemble likely includes a higher proportion of inter-base geometries with lower transfer rates. Second, the increased solvent accessibility compared to the strand may require larger changes in the solvent coordinate for efficient back-electron transfer.
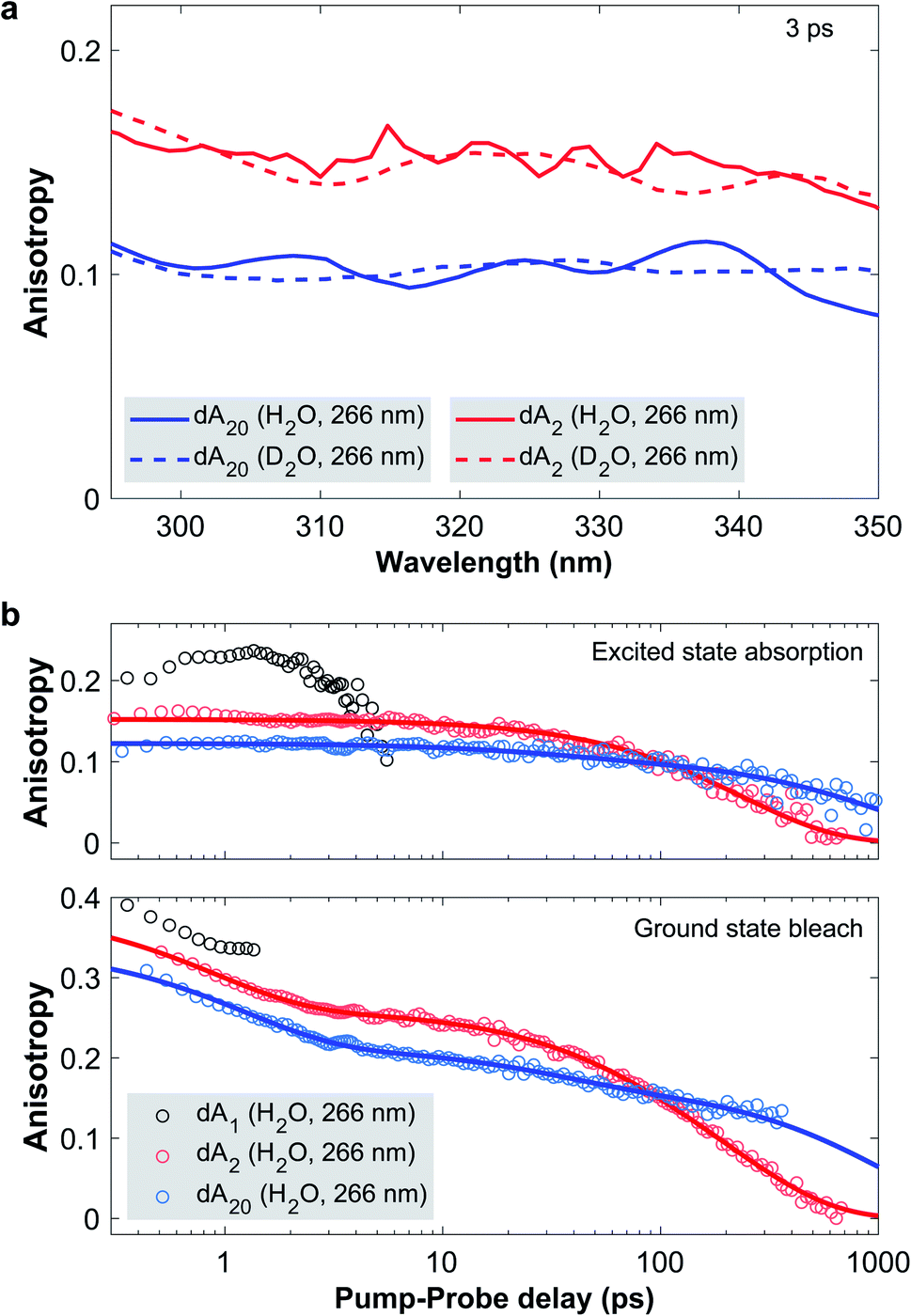 | ||
| Fig. 4 (a) Transient absorption anisotropy (TAA) spectra in the ESA region of dA2 (266 nm) and dA20 (266 nm) in H2O and D2O phosphate buffer solution at a pump-probe delay of 3 ps. In the ESA region, TAA probes the orientation of the transition dipole of the CT exciton and provides an indirect marker of its CT character. This implies that solvent deuteration has no impact on the CT character of the excitons, thus ruling out the involvement of the amino group as a proton donor. (b) TAA decay traces of dA1, dA2 and dA20 in H2O phosphate buffer solution, photoexcited at 266 nm. The data points (open circles) are obtained by spectrally averaging the TAA over the GSB and ESA bands with the exact boundaries listed in the ESI.† Global fits of the spectrally resolved TAA data are presented in detail in the ESI† with representative fit curves included in the figure as solid lines. In the GSB region, TAA is sensitive to the distribution of bleached ground state transition dipoles and reveals a conformational change of the CT exciton in dA20 with a 40 ps time constant. | ||
We now proceed to discuss the time-evolution of the TAA data. To this end, Fig. 4b displays the TAA of dA1, dA2, and dA20 (266 nm) as a function of pump-probe delay. The kinetic traces were obtained by averaging the TAA in the GSB and ESA regions with the exact spectral boundaries reported in Table 1 in the ESI.† Starting with the ESA region, the TAA probes the relative orientation between the photoexcited ground and the excited state transition dipoles. In dA1, this is the hot ground state, whereas in the two oligomers it is associated with the CT exciton. In dA1 the TAA reaches a maximum value of approximately 0.23 and then decays to zero, which we attribute to rotational diffusion. In dA2 the anisotropy takes a value of 0.15 at the earliest pump-probe delay of 0.3 ps and decays mono-exponentially to zero on the 100 ps timescale. In direct comparison, dA20 displays a lower initial TAA value of about 0.12 and decays bi-exponentially to zero with a minor component on the 100 ps timescale and a major component on the 1000 ps timescale. In each oligomer, the slowest decay component has to be attributed to rotational diffusion. In addition, two processes may change the orientation of the transition dipole of the CT exciton: a conformational change of the nucleobases in the stack and a change in CT character. Importantly, neither dA2 nor dA20 display any change in anisotropy during the first 3 ps. This implies, that neither a change in base stack conformation, nor in CT character are taking place on this time scale. Instead, we conclude that the CT character of the exciton is fully formed in <0.3 ps, which corresponds to the time-resolution of our setup (see Fig. 3 in the ESI†). This implies that CT exciton formation in ssDNA is unlikely to involve a conformational change of the base stack, such as a continuous approach of the bases, as has been suggested by several authors.23,29 Instead, we propose that whilst some CT excitons may be formed directly upon photoexcitation, the majority are formed via an indirect channel, where the CT exciton is formed from a neutral exciton through an inter-base electron transfer on the sub-picosecond timescale, preceding any conformational change.
In the GSB region, TAA probes the ensemble of the bleached ground state 1ππ* transition dipoles that are parallel to the molecular plane of the adenosine nucleotide. As a benchmark case, we note that the TAA of dA1 takes an initial value close to 0.4, as expected for a single bleached transition dipole. During the first 3 ps, all three samples experience a rapid TAA decay with a time constant denoted as ρ1. For dA1, the TAA diverges beyond 2 ps, due to a low TA signal (see Fig. 10 in the ESI†). Just as in the ESA region the oligomers display additional decay components: whilst the TAA of dA20 decays biexponentially with decay constants ρ2 and ρ3, dA2 decays monoexponentially. To extract the associated time constants, we performed global fits of the complete time–wavelength TAA maps instead of individual single-wavelength traces. As the probed spectral window generally contains contributions from different transition dipoles, the observed TAA kinetics may vary strongly as a function of probe wavelength. In addition, the divergence observed at zero-crossings between GSB and ESA bands and low TA signals may further distort the observed TAA kinetics, especially when analyzing only single-wavelength kinetic traces. To mitigate the described effects, we performed global fits of a sum of exponential decays, convoluted with the IRF. As described in detail in Section 4,† probe wavelengths near zero-crossings and band edges were excluded, along with pump-probe delays associated with a low TA amplitude. For dA20 (266 nm) and dA2 in H2O and D2O, Fig. 10 and 12† display the time–wavelength maps of the obtained residuals, whereas Fig. 10 and 12† display the resulting DAS and representative kinetic traces from the GSB and ESA region. The obtained time constants are displayed in Table 1. The individual errors were estimated by systematically varying the spectral boundaries and cut-offs in the pump-probe delays included in the fit and noting the impact on the fit results.
Starting with the TAA data obtained in H2O buffer solution, we note that the slowest decay constants ρ3(2) and ρ3(20) for dA2 and dA20, respectively, must denote their rotational diffusion times. To assign the fastest decay constant ρ1, we observe that the associated DAS for dA2 and dA20 only displays non-zero values <310 nm, with a band shape that is consistent with the ESA of the hot ground state in dA1. Taking into account that an identical sub-picosecond decay is observed in dA1, we conclude that ρ1 likely describes the depolarization of the hot monomeric ground state's transition dipole due to vibrational cooling. The contribution of ρ1 to the TAA kinetics in the GSB region is thus likely caused by the overlapping monomeric ESA band.
The most interesting aspect of the TAA decay is the intermediate time constant ρ2(20) = 44 ± 10 ps obtained for dA20 (266 nm) in H2O buffer solution. First of all we note that the associated DAS spectrum shows a uniform, but much lower contribution of the decay component in the ESA region, compared to the GSB region. This suggests that ρ2(20) contributes to both TAA decays and is not simply caused by an overlap with the ESA band of the CT exciton. With global rotational diffusion of the strand accounted for by ρ3(20), the orientation of a bleached transition dipole can only change if it moves from the position where it was initially photoexcited. We thus identify four possible causes for the observed intermediate TAA decay component in dA20: (1) local conformational fluctuations of the base stack, (2) non-exponential TAA kinetics due to the stacking disorder within the strand, (3) exciton migration along the strand, and (4) a conformational change of the photoexcited base stack. In the following we evaluate each of the potential explanations. (1) Fluorescence anisotropy experiments have shown that the local conformational fluctuations of the central base within a tri-nucleotide in aqueous solution displays an anisotropy decay time constant of approximately 80 ps.54 The time constant measured here is notably shorter and is associated with two stacked bases, which thus suggests that a local fluctuation process is not a suitable explanation. (2) Due to the pronounced stacking disorder within dA20, exciton states with different delocalization lengths may lead to an ensemble of diffusion times. The resulting non-exponential TAA kinetics may then appear to contain additional exponential decay components. However, as exciton states in dA20 are known to be limited to two stacked bases, we can exclude the presence of an ensemble of exciton states with different delocalization lengths. Nevertheless, it is plausible that the rotational diffusion of a two-base exciton depends on its position within the strand, which would also lead to an ensemble of diffusion times. However, the associated time constants are all expected to be slower than ρ3(2) measured for the freely diffusing dA2. This means that the positional disorder of the CT excitons within the strand cannot cause the TAA decay on the 10 to 200 ps time scale. (3) In dA20, energy transfer of neutral excitons along the oligomer chain could in principle contribute to a decay of the GSB anisotropy. However, our previous discussion showed that internal conversion to the neutral exciton band minimum and charge separation of neutral excitons are completed in <0.3 ps. These processes efficiently localize and trap the initially formed neutral excitons, which therefore excludes any intra-chain hopping on the picosecond timescale. (4) The most probable origin of ρ2(20) is thus an inter-base conformational change associated with the relaxation of the CT exciton. Note that the observed time scale is consistent with results from a similar study, where the structural relaxation of exciton states in π-stacked perylene-bis(dicarboximide) (PBI) H-aggregates was measured via TAA and fluorescence anisotropy.55
In order to determine the nature of this conformational change it is important to note how the motion of a photoexcited nucleotide impacts the anisotropy in the GSB and the ESA region. In the GSB region, the TAA decay tracks the motion of the bleached 1ππ* transition dipoles, which are initially parallel to the nucleobase planes. In comparison, the transition dipole of the CT exciton in the ESA region has an additional component perpendicular to the nucleobase planes. The DAS of ρ2 shows that its kinetic contribution is significantly more pronounced in the TAA decay in the GSB rather than the ESA region. This suggests that the dominant conformational motion takes place in a plane parallel to the nucleobase planes, thus leading to a stronger TAA change in the GSB relative to the ESA region. Whilst we cannot exclude an additional reduction in inter-base distance as a possible structural relaxation pathway, we note that this would likely lead to a change of CT character and thus a pronounced change of the TAA in the ESA region. We therefore propose that ρ2 is dominated by a reduction of the twisting angle ϕ between the coupled nucleotides around the helix axis from the typical B-form value of approximately ϕ = 36° (corresponding to 10 bases per helix turn) towards a parallel arrangement (ϕ = 0°). This motion thus drives the CT exciton to its minimum energy conformation. A similar structural relaxation pathway was proposed by Son and co-workers for the exciton evolution in PBI aggregates.55 Furthermore, an inter-base rotation towards a parallel nucleotide alignment is qualitatively consistent with recent QM/MM simulations, which calculated minimum energy conformations of excited adenosine dimers incorporated into a solvated dsDNA geometry.36 We thus note that whilst the presented experiments cannot provide unambiguous evidence for the proposed base twisting motion, it currently represents the most plausible explanation for the observed TAA kinetics in dA20.
In D2O buffer solution, the same qualitative behavior of the TAA decay is observed and comparable time constants are obtained from the global fits. First of all, this shows that the fitting procedure is robust across several data sets. Second, it shows that solvent deuteration has only a small impact on the rotational diffusion, as expected. Third, ρ2(20) is unaffected by the solvent change within its error range, which is consistent with a change in inter-base conformation. In this view, however, it is somewhat surprising that ρ2 is not observed in dA2. First of all, we found that it is possible to perform a global fit with three exponential decays on the TAA data in H2O and obtain a small amplitude contribution from an intermediate decay with ρ2(2) ≈ 50 ps (fits not displayed). However, this could neither be achieved for repeated experiments, nor for the data set in D2O, where the associated global fits did not converge to meaningful results. We thus speculate that whilst a structural relaxation of the CT exciton may also be present in dA2, it is likely to be less pronounced due to the weaker average electronic coupling of its nucleotides, compared to dA20.
Conclusion
In summary, we are now able to present a complete reaction mechanism for the exciton dynamics in adenosine ssDNA, which is displayed in Fig. 5. Upon photoexcitation, predominantly neutral excitons delocalized over two stacked bases are formed within the oligomer, with a stack conformation selected from the ground state conformational ensemble. Within <0.3 ps, internal conversion and charge separation takes place without any significant conformational change, resulting in a CT exciton. As a consequence, the CT character is mostly determined by the inter-base configuration upon photoexcitation. Afterwards, an ultrafast inter-base rotation drives the CT exciton to its minimum energy configuration in about 40 ps, from where a back-electron transfer leads to a charge recombination back to the ground state. Importantly, this mechanism takes both ground and excited state structural properties of the oligomer into account and finally explains why short oligomers such as dimers display excitons with a lower CT character and slower recombination times: their broader conformational ensemble leads to a weaker electronic coupling and less efficient back-electron transfer on average. In addition, our results suggest a structural mechanism in the ultrafast photochemistry of ssDNA, opening a novel perspective on its photodamage control processes. In a broader context, our results highlight the key role of ground and excited state structural features in the ultrafast photochemistry of aggregated organic molecular systems. This is expected to contribute to a more detailed understanding of the related energy and charge transfer processes in synthetic molecular aggregates.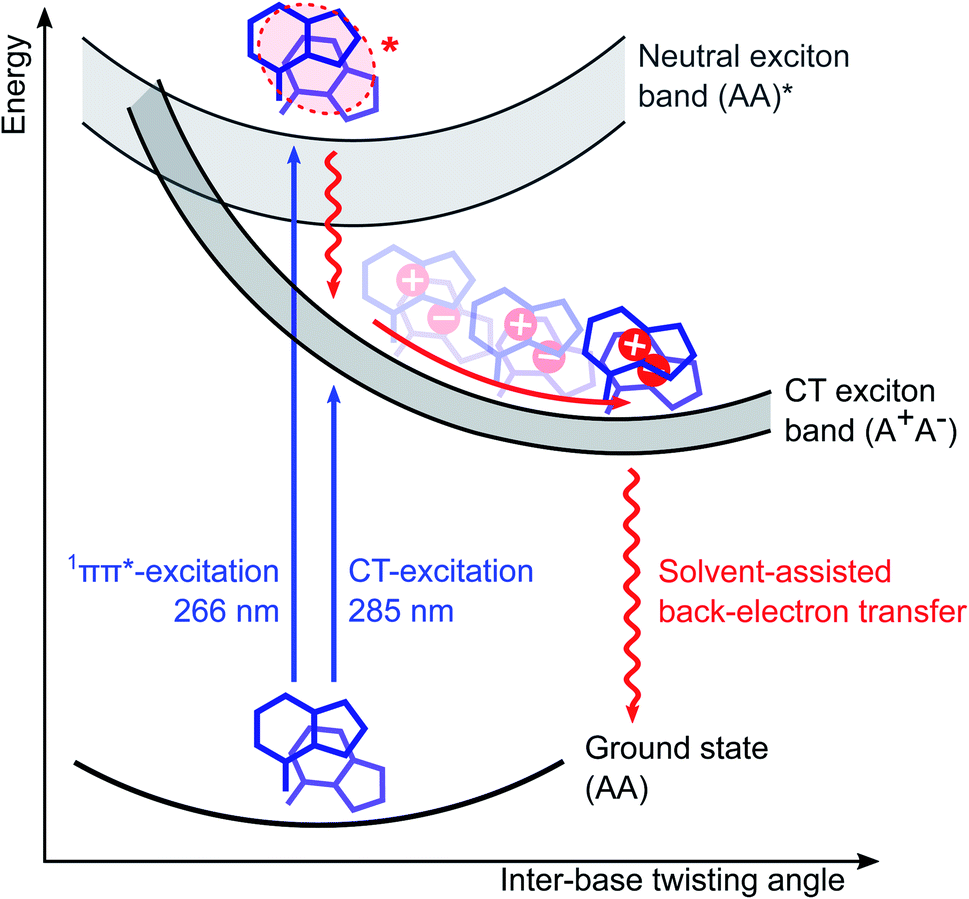 | ||
| Fig. 5 Schematic illustration of the photochemical relaxation mechanism in adenosine ssDNA (dAn). As inter-base electronic couplings are mostly restricted to nearest-neighbour stacked bases, sufficiently long dAn can be approximated as a chain of two-base stacks. Photoexcitation of dAn thus selects a two-base stack conformation from the fluctuating ground state conformational ensemble and predominantly forms neutral exciton states delocalized over two bases at the top of the exciton band. Most of these states undergo rapid internal conversion and charge separation to form charge-transfer (CT) excitons in <0.3 ps, without any significant change in conformation. Ultrafast base rotations then drive the CT excitons to the minimum energy conformation of the CT exciton band in about 40 ps. We propose that this motion reduces the twisting angle ϕ between the coupled nucleotides around the helix axis from the typical B-form value of approximately ϕ = 36° towards a parallel arrangement (ϕ = 0°). From there a back-electron transfer deactivates the exciton back to the ground state. Note that a small but significant fraction of the charge-separated states do not recombine and display a lifetime on the millisecond scale. | ||
Methods
Broadband transient absorption and anisotropy in the deep-UV
The laser setup for generating the deep-UV probe pulses has been described in detail elsewhere.56,57 Briefly, a customized cryogenic Ti:Sapphire amplifier, with an exceptional shot-to-shot stability of 0.1% root-mean-square, pumps a commercial non-collinear optical parametric amplifier (NOPA) at 20 kHz. The broadband visible femtosecond pulses are achromatically doubled in a thin BBO crystal58 covering 250–360 nm. The probe pulse is set to S-polarization with an achromatic half-wave plate before splitting it into a probe and a reference beam with a 1 mm thick reflective neutral density filter (0.3 OD). The probe is focused into the sample with a 10 cm focal-length off-axis parabolic mirror, resulting in a focal spot diameter of approximately 30 μm at full-width half maximum (FWHM). The probe polarization is cleaned with a high-quality Glan-Laser polarizer prior to detection with a fiber-coupled imaging spectrograph, which is equipped with two CMOS chips for recording the probe and reference simultaneously shot-to-shot. The sample is delivered via a wire-guided liquid jet with an approximate pathlength of 300 μm (design adapted from ref. 59), in which a total volume of about 10 ml is circulated in a closed loop. Solvent evaporation is accounted for by continuously replenishing it with a syringe pump. Two pump pulse configurations were employed: (1) a pump centered at 266 nm (FWHM ≈ 2 nm), derived from the Ti:Sapphire amplifier via third harmonic generation, and (2) a pump centered at 285 nm (FWHM ≈ 2 nm), obtained via narrowband second harmonic generation of the NOPA. The pump beam is focused to a FWHM diameter of 80 μm resulting in an estimated fluence 125 μJ cm−2 on target. This was determined to be in the linear excitation regime via fluence-dependent transient absorption measurements. Note that the absorption of photoinduced solvated electrons is negligible in the deep-UV,60 such that their impact on the experiments can be excluded. Due to a relatively large angle of about 10° between the pump and probe beams and the use of a free sample jet, any pump scatter could be prevented from reaching the probe detector. The repetition rate of the pump is reduced to 10 kHz via an optical chopper and its linear polarization is switched between S- and P-polarization via a motorized zero-order half-wave plate. At a given pump-probe delay t, 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 consecutive probe shots were acquired for each pump-polarization state, resulting in a TA spectrum for parallel pump and probe polarizations (TA∥) and a second for perpendicular polarizations (TA⊥). The isotropic magic-angle TA and the TAA spectrum were then calculated as:
000 consecutive probe shots were acquired for each pump-polarization state, resulting in a TA spectrum for parallel pump and probe polarizations (TA∥) and a second for perpendicular polarizations (TA⊥). The isotropic magic-angle TA and the TAA spectrum were then calculated as: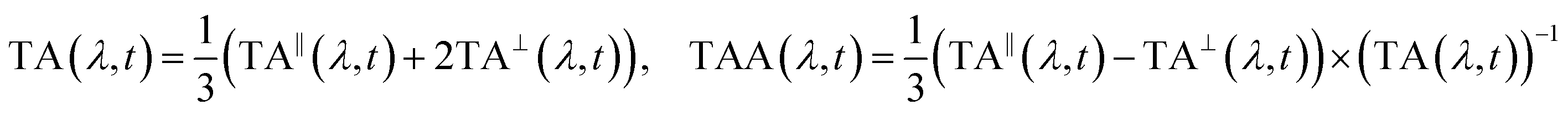 | (1) |
The instrument response function (IRF) of the setup was determined by recording the two-photon absorption signal in the neat buffer solution. The data is displayed in Fig. 3 in the ESI† and displays the extracted FWHM of the Gaussian fits as a function of probe wavelength. For both pump configurations we find that the IRF is limited to about 0.3 ps in the GSB region (<280 nm) and to 0.2 ps in the ESA region (>280 nm). To quantify the TA and TAA kinetics, we performed a global analysis for each data set with the OPTIMUS software package,61 simultaneously fitting multi-exponential functions convoluted with the Gaussian IRF over the probed spectral window.
Sample preparation
2′-Deoxyadenosine 5′-monophosphate (dA1) was purchased from Sigma-Aldrich and dA2 and dA20 from (https://biomers.net). The dimer and multimer consist of dA1 as building blocks with the phosphate group covalently linked to the deoxyribose at the 3′ carbon of the neighbouring dA1. From there a single strand is formed with a sugar phosphate backbone. The samples were used without any further purification and were dissolved in a phosphate-buffered saline purchased from Sigma-Aldrich, which was diluted with highly pure Milli-Q water or spectroscopy-grade deuterated water purchased from Sigma-Aldrich. This resulted in a buffer solution with a buffer concentration of 0.01 M, a sodium chloride concentration of 0.154 M and a pH of 7.4. The sample concentration was chosen to have an absorbance of 0.75 OD at 260 nm in a 300 μm path length. The sample integrity was characterized and confirmed before and after any experiment with a static UV-vis spectrometer (UV-3600, Shimadzu) and a static circular dichroism spectrometer (J-810, Jacso).Molecular dynamics simulations
For the molecular dynamics (MD) simulations, initial single-stranded structures for both dA2 and dA20 were designed using the leap program of AMBERTOOLS 18.62 Both molecules were solvated in a truncated octahedral box of explicit TIP3P63 water molecules, maintaining the water layer thickness greater than 11 Å surrounding the DNA molecule. First, Na+ ions were added to neutralize the system, following the Joung and Cheatham ion model.64 Second, Na+ and Cl− ion-pairs were added to reach an approximate overall salt concentration of 150 mM. The ions in the simulation box were placed randomly with the constraint that the ions should be at least 5 Å away from the DNA molecule and at least 3.5 Å from each other. The solvated systems thus obtained were first equilibrated to minimize the energy of the solvent and then subjected to a slow thermalization to reach room temperature followed by a further equilibration of 500 ps. Finally, 500 ns of production simulations were carried out in the NPT ensemble using a time-step of 2 fs and storing the trajectory at a picosecond interval. All simulations were performed using the PARMBSC1 force field65 in AMBER18. The SHAKE algorithm66 was used to constrain the bond involving hydrogen. For the long-range electrostatic interactions, the particle mesh Ewald method67 was used with a 9 Å real space cutoff. The short-range Lennard–Jones interactions were truncated at 9 Å. Finally, AMBERTOOLS 18 was used to analyze the simulation results and to compute the electronic coupling element, J = |![[J with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004a_20d1.gif) |.68 For two identical bases with subscripts 1 and 2, it is defined as:
|.68 For two identical bases with subscripts 1 and 2, it is defined as: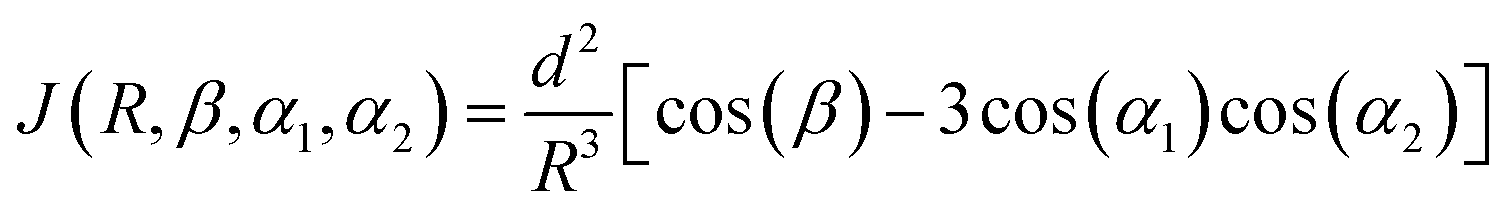 | (2) |
![[d with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0064_20d1.gif) | is the transition dipole moment of the base, R = |
| is the transition dipole moment of the base, R = |![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) | is the separation between two neighboring bases (defined as the vector between the centroids of the two bases), β is the angle between the transition dipole moments of the two bases, and αi is the angle between
| is the separation between two neighboring bases (defined as the vector between the centroids of the two bases), β is the angle between the transition dipole moments of the two bases, and αi is the angle between 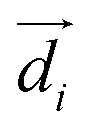 and . The direction and the absolute value of for adenine (2.56 D) are taken from ref. 69.
and . The direction and the absolute value of for adenine (2.56 D) are taken from ref. 69.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
B. B. performed the experiments. B. B. and M. O. analyzed the data, discussing the results regularly with M. C. R. S. performed the MD simulations. M. O. designed and supervised the research and wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. John H. Maddocks from EPFL for his invaluable advice and support for the project. This work was supported by the Swiss NSF through the NCCR MUST and M. O. in particular by a fellowship within the Postdoc-Program of the German Academic Exchange Service (DAAD). R. S. would like to acknowledge SCITAS for computational facilities and the Swiss National Science Foundation grant 200020 182184.References
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Exciton Transport in Molecular Aggregates – From Natural Antennas to Synthetic Chromophore Systems, Adv. Energy Mater., 2017, 7, 1700236, DOI:10.1002/aenm.201700236.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, The exciton model in molecular spectroscopy, Pure Appl. Chem., 1965, 11, 371–392, DOI:10.1351/pac196511030371.
- W. J. Schreier, P. Gilch and W. Zinth, Early Events of DNA Photodamage, Annu. Rev. Phys. Chem., 2015, 66, 497–519, DOI:10.1146/annurev-physchem-040214-121821.
- J. Chen, Y. Zhang and B. Kohler, Excited states in DNA strands investigated by ultrafast laser spectroscopy, in Photoinduced Phenomena in Nucleic Acids II: DNA Fragments and Phenomenological Aspects, ed. M. Barbatti, A. C. Borin and S. Ullrich, Springer International Publishing, Cham, 2015, pp. 39–87, DOI:10.1007/128_2014_570.
- D. Markovitsi, UV-induced DNA Damage: The Role of Electronic Excited States, Photochem. Photobiol., 2016, 92, 45–51, DOI:10.1111/php.12533.
- F. Plasser and H. Lischka, Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789, DOI:10.1021/ct300307c.
- L. Blancafort and A. A. Voityuk, Exciton delocalization, charge transfer, and electronic coupling for singlet excitation energy transfer between stacked nucleobases in DNA: An MS-CASPT2 study, J. Chem. Phys., 2014, 140, 095102, DOI:10.1063/1.4867118.
- R. Improta, F. Santoro and L. Blancafort, Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases, Chem. Rev., 2016, 116, 3540–3593, DOI:10.1021/acs.chemrev.5b00444.
- C. S. M. Olsthoorn, L. J. Bostelaar, J. F. M. D. Rooij, J. H. V. Boom and C. Altona, Circular Dichroism Study of Stacking Properties of Oligodeoxyadenylates and Polydeoxyadenylate, Eur. J. Biochem., 1981, 115, 309–321, DOI:10.1111/j.1432-1033.1981.tb05240.x.
- M. Barbatti and S. Ullrich, Ionization potentials of adenine along the internal conversion pathways, Phys. Chem. Chem. Phys., 2011, 13, 15492–15500, 10.1039/c1cp21350d.
- T. Gustavsson, N. Sarkar, I. Vayá, M. C. Jiménez, D. Markovitsi and R. Improta, A joint experimental/theoretical study of the ultrafast excited state deactivation of deoxyadenosine and 9-methyladenine in water and acetonitrile, Photochem. Photobiol. Sci., 2013, 12, 1375–1386, 10.1039/c3pp50060h.
- J.-M. L. Pecourt, J. Peon and B. Kohler, Ultrafast Internal Conversion of Electronically Excited RNA and DNA Nucleosides in Water, J. Am. Chem. Soc., 2000, 122, 9348–9349, DOI:10.1021/ja0021520.
- F. C. Spano, Excitons in conjugated oligomer aggregates, films, and crystals, Annu. Rev. Phys. Chem., 2006, 57, 217–243, DOI:10.1146/annurev.physchem.57.032905.104557.
- L. Hu, Y. Zhao, F. Wang, G. Chen, C. Ma, W.-M. Kwok and D. L. Phillips, Are Adenine Strands Helical H-Aggregates?, J. Phys. Chem. B, 2007, 111, 11812–11816, DOI:10.1021/jp070403m.
- I. Tinoco, Hypochromism in Polynucleotides 1, J. Am. Chem. Soc., 1960, 82, 4785–4790, DOI:10.1021/ja01503a007.
- J. Eisinger, M. Guéron, R. G. Shulman and T. Yamane, Excimer fluorescence of dinucleotides, polynucleotides, and DNA, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 1015–1020 CrossRef CAS PubMed.
- S. Tonzani and G. C. Schatz, Electronic Excitations and Spectra in Single-Stranded DNA, J. Am. Chem. Soc., 2008, 130, 7607–7612, DOI:10.1021/ja7103894.
- Y.-J. Ai, G.-L. Cui, Q. Fang, W.-H. Fang and Y. Luo, Exploring concerted effects of base pairing and stacking on the excited-state nature of DNA oligonucleotides by DFT and TD-DFT studies, Int. J. Quantum Chem., 2011, 111, 2366–2377, DOI:10.1002/qua.22524.
- R. Improta and V. Barone, Interplay between “Neutral” and “Charge-Transfer” Excimers Rules the Excited State Decay in Adenine-Rich Polynucleotides, Angew. Chem., Int. Ed., 2011, 50, 12016–12019, DOI:10.1002/anie.201104382.
- J. J. Nogueira, F. Plasser and L. González, Electronic delocalization, charge transfer and hypochromism in the UV absorption spectrum of polyadenine unravelled by multiscale computations and quantitative wavefunction analysis, Chem. Sci., 2017, 8, 5682–5691, 10.1039/c7sc01600j.
- U. Kadhane, A. I. S. Holm, S. V. Hoffmann and S. B. Nielsen, Strong coupling between adenine nucleobases in DNA single strands revealed by circular dichroism using synchrotron radiation, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 021901, DOI:10.1103/physreve.77.021901.
- C. Su, C. T. Middleton and B. Kohler, Base-Stacking Disorder and Excited-State Dynamics in Single-Stranded Adenine Homo-oligonucleotides, J. Phys. Chem. B, 2012, 116, 10266–10274, DOI:10.1021/jp305350t.
- A. Banyasz, T. Gustavsson, D. Onidas, P. Changenet-Barret, D. Markovitsi and R. Improta, Multi-Pathway Excited State Relaxation of Adenine Oligomers in Aqueous Solution: A Joint Theoretical and Experimental Study, Chem.–Eur. J., 2013, 19, 3762–3774, DOI:10.1002/chem.201202741.
- F. J. Avila Ferrer, R. Improta, F. Santoro and V. Barone, Computing the inhomogeneous broadening of electronic transitions in solution: a first-principle quantum mechanical approach, Phys. Chem. Chem. Phys., 2011, 13, 17007–17012, 10.1039/c1cp22115a.
- H. Yin, Y. Ma, J. Mu, C. Liu and M. Rohlfing, Charge-Transfer Excited States in Aqueous DNA: Insights from Many-Body Green's Function Theory, Phys. Rev. Lett., 2014, 112, 228301, DOI:10.1103/PhysRevLett.112.228301.
- W.-M. Kwok, C. Ma and D. L. Phillips, Femtosecond Time- and Wavelength-Resolved Fluorescence and Absorption Spectroscopic Study of the Excited States of Adenosine and an Adenine Oligomer, J. Am. Chem. Soc., 2006, 128, 11894–11905, DOI:10.1021/ja0622002.
- I. Buchvarov, Q. Wang, M. Raytchev, A. Trifonov and T. Fiebig, Electronic energy delocalization and dissipation in single-and double-stranded DNA, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4794–4797 CrossRef CAS PubMed.
- T. Takaya, C. Su, K. de La Harpe, C. E. Crespo-Hernández and B. Kohler, UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10285–10290 CrossRef CAS PubMed.
- R. Borrego-Varillas, G. Cerullo and D. Markovitsi, Exciton Trapping Dynamics in DNA Multimers, J. Phys. Chem. Lett., 2019, 10, 1639–1643, DOI:10.1021/acs.jpclett.9b00450.
- D. B. Bucher, B. M. Pilles, T. Carell and W. Zinth, Charge separation and charge delocalization identified in long-living states of photoexcited DNA, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4369–4374, DOI:10.1073/pnas.1323700111.
- Y. Zhang, J. Dood, A. A. Beckstead, X.-B. Li, K. V. Nguyen, C. J. Burrows, R. Improta and B. Kohler, Efficient UV-induced charge separation and recombination in an 8-oxoguanine-containing dinucleotide, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11612–11617, DOI:10.1073/pnas.1404411111.
- A. Banyasz, T.-M. Ketola, A. Muñoz-Losa, S. Rishi, A. Adhikary, M. D. Sevilla, L. Martinez-Fernandez, R. Improta and D. Markovitsi, UV-Induced Adenine Radicals Induced in DNA A-Tracts: Spectral and Dynamical Characterization, J. Phys. Chem. Lett., 2016, 7, 3949–3953, DOI:10.1021/acs.jpclett.6b01831.
- A. Banyasz, T. Ketola, L. Martínez-Fernández, R. Improta and D. Markovitsi, Adenine radicals generated in alternating AT duplexes by direct absorption of low-energy UV radiation, Faraday Discuss., 2018, 207, 181–197, 10.1039/c7fd00179g.
- I. Conti, A. Nenov, S. Höfinger, S. F. Altavilla, I. Rivalta, E. Dumont, G. Orlandi and M. Garavelli, Excited state evolution of DNA stacked adenines resolved at the CASPT2//CASSCF/Amber level: from the bright to the excimer state and back, Phys. Chem. Chem. Phys., 2015, 17, 7291–7302, 10.1039/c4cp05546b.
- J. Chen and B. Kohler, Base Stacking in Adenosine Dimers Revealed by Femtosecond Transient Absorption Spectroscopy, J. Am. Chem. Soc., 2014, 136, 6362–6372, DOI:10.1021/ja501342b.
- V. A. Spata and S. Matsika, Photophysical deactivation pathways in adenine oligonucleotides, Phys. Chem. Chem. Phys., 2015, 17, 31073–31083, 10.1039/c5cp04254b.
- F. Plasser and H. Lischka, Electronic excitation and structural relaxation of the adenine dinucleotide in gas phase and solution, Photochem. Photobiol. Sci., 2013, 12, 1440–1452, 10.1039/c3pp50032b.
- J. S. Hub, Interpreting solution X-ray scattering data using molecular simulations, Curr. Opin. Struct. Biol., 2018, 49, 18–26, DOI:10.1016/j.sbi.2017.11.002.
- T. Sugiki, N. Kobayashi and T. Fujiwara, Modern Technologies of Solution Nuclear Magnetic Resonance Spectroscopy for Three-dimensional Structure Determination of Proteins Open Avenues for Life Scientists, Comput. Struct. Biotechnol. J., 2017, 15, 328–339, DOI:10.1016/j.csbj.2017.04.001.
- J. Segarra-Martí, V. K. Jaiswal, A. J. Pepino, A. Giussani, A. Nenov, S. Mukamel, M. Garavelli and I. Rivalta, Two-dimensional electronic spectroscopy as a tool for tracking molecular conformations in DNA/RNA aggregates, Faraday Discuss., 2018, 207, 233–250, 10.1039/c7fd00201g.
- U. C. Stange and F. Temps, Ultrafast electronic deactivation of UV-excited adenine and its ribo- and deoxyribonucleosides and -nucleotides: A comparative study, Chem. Phys., 2018, 515, 441–451, DOI:10.1016/j.chemphys.2018.08.031.
- C. Ruckebusch, M. Sliwa, P. Pernot, A. de Juan and R. Tauler, Comprehensive data analysis of femtosecond transient absorption spectra: A review, J. Photochem. Photobiol., C, 2012, 13, 1–27, DOI:10.1016/j.jphotochemrev.2011.10.002.
- C. E. Crespo-Hernández, B. Cohen and B. Kohler, Base stacking controls excited-state dynamics in A·T DNA, Nature, 2005, 436, 1141–1144, DOI:10.1038/nature03933.
- K. de La Harpe, C. E. Crespo-Hernández and B. Kohler, Deuterium Isotope Effect on Excited-State Dynamics in an Alternating GC Oligonucleotide, J. Am. Chem. Soc., 2009, 131, 17557–17559, DOI:10.1021/ja9076364.
- D. B. Bucher, A. Schlueter, T. Carell and W. Zinth, Watson–Crick Base Pairing Controls Excited-State Decay in Natural DNA, Angew. Chem., Int. Ed., 2014, 53, 11366–11369, DOI:10.1002/anie.201406286.
- Y. Zhang, K. de La Harpe, A. A. Beckstead, R. Improta and B. Kohler, UV-Induced Proton Transfer between DNA Strands, J. Am. Chem. Soc., 2015, 137, 7059–7062, DOI:10.1021/jacs.5b03914.
- I. R. Gould and S. Farid, Specific deuterium isotope effects on the rates of electron transfer within geminate radical-ion pairs, J. Am. Chem. Soc., 1988, 110, 7883–7885, DOI:10.1021/ja00231a053.
- L. I. Krishtalik, The mechanism of the proton transfer: an outline, Biochim. Biophys. Acta, Bioenerg., 2000, 1458, 6–27, DOI:10.1016/S0005-2728(00)00057-8.
- V. R. Smith, E. Samoylova, H.-H. Ritze, W. Radloff and T. Schultz, Excimer states in microhydrated adenine clusters, Phys. Chem. Chem. Phys., 2010, 12, 9632–9636, 10.1039/C003967E.
- A. Kettani, M. Guéron and J.-L. Leroy, Amino Proton Exchange Processes in Mononucleosides, J. Am. Chem. Soc., 1997, 119, 1108–1115, DOI:10.1021/ja962022k.
- R. A. Beecroft, R. Stephen Davidson, D. Goodwin, J. E. Pratt, L. A. Chewter and D. A. Phillips, A solvent isotope effect upon exciplex photochemistry, Chem. Phys. Lett., 1982, 93, 468–473, DOI:10.1016/0009-2614(82)83222-3.
- N. Mataga, H. Chosrowjan and S. Taniguchi, Ultrafast charge transfer in excited electronic states and investigations into fundamental problems of exciplex chemistry: Our early studies and recent developments, J. Photochem. Photobiol., C, 2005, 6, 37–79, DOI:10.1016/j.jphotochemrev.2005.02.003.
- T. Takaya, H.-o. Hamaguchi and K. Iwata, Femtosecond time-resolved absorption anisotropy spectroscopy on 9,9’-bianthryl: Detection of partial intramolecular charge transfer in polar and nonpolar solvents, J. Chem. Phys., 2009, 130, 014501, DOI:10.1063/1.3043368.
- J. M. Jean and B. P. Krueger, Structural Fluctuations and Excitation Transfer between Adenine and 2-Aminopurine in Single-Stranded Deoxytrinucleotides, J. Phys. Chem. B, 2006, 110, 2899–2909, DOI:10.1021/jp054755+.
- M. Son, K. H. Park, C. Shao, F. Würthner and D. Kim, Spectroscopic Demonstration of Exciton Dynamics and Excimer Formation in a Sterically Controlled Perylene Bisimide Dimer Aggregate, J. Phys. Chem. Lett., 2014, 5, 3601–3607, DOI:10.1021/jz501953a.
- G. Auböck, C. Consani, F. v. Mourik and M. Chergui, Ultrabroadband femtosecond two-dimensional ultraviolet transient absorption, Opt. Lett., 2012, 37, 2337–2339, DOI:10.1364/ol.37.002337.
- G. Auböck, C. Consani, R. Monni, A. Cannizzo, F. van Mourik and M. Chergui, Femtosecond pump/supercontinuum-probe setup with 20 kHz repetition rate, Rev. Sci. Instrum., 2012, 83, 093105, DOI:10.1063/1.4750978.
- P. Baum, S. Lochbrunner and E. Riedle, Tunable sub-10-fs ultraviolet pulses generated by achromatic frequency doubling, Opt. Lett., 2004, 29, 1686–1688, DOI:10.1364/ol.29.001686.
- A. Picchiotti, V. I. Prokhorenko and R. J. D. Miller, A closed-loop pump-driven wire-guided flow jet for ultrafast spectroscopy of liquid samples, Rev. Sci. Instrum., 2015, 86, 093105, DOI:10.1063/1.4929860.
- F.-Y. Jou and G. R. Freeman, Temperature and isotope effects on the shape of the optical absorption spectrum of solvated electrons in water, J. Phys. Chem., 1979, 83, 2383–2387, DOI:10.1021/j100481a016.
- C. Slavov, H. Hartmann and J. Wachtveitl, Implementation and Evaluation of Data Analysis Strategies for Time-Resolved Optical Spectroscopy, Anal. Chem., 2015, 87, 2328–2336, DOI:10.1021/ac504348h.
- D. Case, I. Ben-Shalom, S. Brozell, D. Cerutti, T. Cheatham III, V. Cruzeiro, T. Darden, R. Duke, D. Ghoreishi and M. Gilsonet al., Amber 2018, University of California, San Francisco, 2018 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- I. S. Joung and T. E. Cheatham III, Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS PubMed.
- I. Ivani, P. D. Dans, A. Noy, A. Pérez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goñi and A. Balaceanu, et al. Parmbsc1: a refined force field for DNA simulations, Nat. Methods, 2016, 13, 55 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, On the geometry dependence of molecular dimer spectra with an application to aggregates of perylene bisimide, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- D. Svozil, P. Jungwirth and Z. Havlas, Electron binding to nucleic acid bases. experimental and theoretical studies. a review, Collect. Czech. Chem. Commun., 2004, 69, 1395–1428 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1sc06450a |
| ‡ Owing to Kasha's original definition, some authors have used the term ‘exciton’ to exclusively denote exciton states without any CT character. We explicitly include the possibility of a non-zero CT character in our use of the term, due to the presence of significant orbital overlap and electron exchange interactions in DNA systems. |
| This journal is © The Royal Society of Chemistry 2022 |